

Abstract
Dynamic epigenetic reprogramming occurs during mammalian germ cell development, although the targets of this process, including DNA demethylation and de novo methylation, remain poorly understood. We performed genome-wide DNA methylation analysis in male and female mouse primordial germ cells at embryonic days 10.5, 13.5, and 16.5 by whole-genome shotgun bisulfite sequencing. Our high-resolution DNA methylome maps demonstrated gender-specific differences in CpG methylation at genome-wide and gene-specific levels during fetal germline progression. There was extensive intra- and intergenic hypomethylation with erasure of methylation marks at imprinted, X-linked, or germline-specific genes during gonadal sex determination and partial methylation at particular retrotransposons. Following global demethylation and sex determination, CpG sites switched to de novo methylation in males, but the X-linked genes appeared resistant to the wave of de novo methylation. Significant differential methylation at a subset of imprinted loci was identified in both genders, and non-CpG methylation occurred only in male gonocytes. Our data establish the basis for future studies on the role of epigenetic modifications in germline development and other biological processes.
In post-implantation mammalian embryos, a population of pluripotent epiblast cells gives rise to primordial germ cells (PGCs), the precursors of spermatozoa and oocytes; the fate of these PGCs is specified during gastrulation (Mochizuki and Matsui 2010). In mice, PGCs initially form a small cluster of 30–50 alkaline phosphatase-positive cells in the extra-embryonic mesoderm on embryonic day 7.25 (E7.25) (Ginsburg et al. 1990). Once the fate of these PGCs has been determined, they start proliferating and migrating into the developing gonadal region (the genital ridge). Early PGCs migrate from the dorsal aspect of the hindgut between E9.0 (∼150 PGCs) and E9.5 (∼250 PGCs), separate into left and right groups of individual cells, and migrate laterally across the dorsal body wall. At E10.5, ∼1000 PGCs reach the genital ridges and continue to migrate, and by E12.5 (∼8000 PGCs), migration into the genital ridges is complete. Within the genital ridges, PGCs continue to proliferate, reaching about 26,000 cells by E13.5, at which point cell division stops, and they undergo male or female gametogenesis. Notably, PGCs are sexually bipotent at the migrating stage, and sex-specific differentiation begins after colonization of the genital ridges around E10.5 (Saga 2008). At E13.5, male germ cells undergo cell cycle arrest at G1/G0 and do not enter meiosis during the embryonic stages of development, whereas female germ cells enter meiotic arrest.
During migration and proliferation, PGCs undergo global epigenetic reprogramming, including exchange of histone variants, remodeling of histone modifications, and erasure of DNA methylation, which is thought to be complete around E13.5 in male and female embryos (Seki et al. 2005; Hajkova et al. 2008; Popp et al. 2010; Guibert et al. 2012). Prior to colonizing the genital ridges, PGCs exhibit parent-of-origin-specific imprinting methylation marks (called genomic imprinting), which enforce the mono-allelic expression of many imprinted genes. Most parental methylation imprints on paternal and maternal alleles are erased in nonmigrating PGCs (between E11.5 and E12.5) (Hajkova et al. 2002; Yamazaki et al. 2005). In addition, some germline-specific genes such as Ddx4, Dazl, and Sycp3 are initially expressed in PGCs between E10.5 and E11.5, and DNA demethylation concomitantly occurs in the regions flanking these genes (Maatouk et al. 2006). Following gonadal sex determination, germ cells acquire the ability for sex-specific, de novo methylation. In the male germline, G1-arrested male PGCs (usually called gonocytes) are highly methylated, with increased expression of DNA de novo methyltransferase genes during mitotic cell division at fetal stages (Sakai et al. 2004), and the DNA methyltransferase (Dnmt) families Dnmt3a, Dnmt3b, and Dnmt3l play essential roles in the establishment of retroviral methylation and paternal methylation imprints during spermatogenesis (Kaneda et al. 2004; Kato et al. 2007). In the female germline, increasing DNA methylation and establishment of maternal methylation imprints occur predominantly in meiotically arrested growing oocytes at postnatal stages (Lucifero et al. 2004; Hiura et al. 2006). Dnmt3a and Dnmt3l are also necessary for this process, while Dnmt3b seems dispensable (Kaneda et al. 2004, 2010; Smallwood et al. 2011).
These methylation patterns have been investigated by indirect immunostaining methods using antibodies against 5-methylcytosine (Seki et al. 2005; Hajkova et al. 2008; Abe et al. 2011) and/or locus-specific analyses using bisulfite conversion-based methods, in which unmethylated cytosine is converted to uracil (Hajkova et al. 2002; Lucifero et al. 2004; Yamazaki et al. 2005; Hiura et al. 2006; Maatouk et al. 2006; Kato et al. 2007; Kaneda et al. 2010). Recent studies combining analyses of DNA methylation and whole-genome microarrays or high-throughput sequencing technologies have revealed the characteristic DNA methylation profiles of various types of cells (called “DNA methylomes”) (Laird 2010). In particular, the combination of bisulfite treatment and high-throughput sequencing has allowed researchers to map every methylated and unmethylated cytosine in the genome (Cokus et al. 2008; Lister et al. 2009). However, since these methods require large amounts of DNA (typically, micrograms, i.e., at least over 106 mammalian cells), samples that are only available in small amounts, such as germline cells, are difficult to evaluate by these methods. A pilot study of high-throughput bisulfite sequencing in mouse PGCs (at E13.5) showed global reduction of CpG methylation in genomes or genomic compartments, but demethylation-targeted DNA sequences have not been fully characterized due to low coverage (Popp et al. 2010). In other sequencing-based assays, reduced representation bisulfite sequencing (RRBS) allows global DNA methylation analysis in oocyte genomes, but the targets of analysis are limited to CpG-rich sequences (Smallwood et al. 2011). Thus, in germline development, identifying functional DNA methylation loci is a fascinating issue that remains relatively unexplored.
In this study, we examined genome-wide methylation profiles in developing germ cells of mice using high-throughput shotgun sequencing of bisulfite-treated DNA (whole-genome shotgun bisulfite sequencing; WGSBS), which accurately quantifies whole-genome methylation levels at single-base resolution. Using Illumina sequencing libraries, we scaled down the construction and analysis to nanogram quantities of DNA by generating a new WGSBS library, termed the post-bisulfite adapter tagging (PBAT) method (Miura et al. 2012). Here, we provide complete maps of cytosine methylation in developing male and female PGCs during gonadal sex determination.
Results
At E10.5, the male and female PGCs of mouse embryos migrate directionally from the dorsal body wall, the mesentery of the hindgut, into the genital ridges and have a very similar morphology; in contrast, primary testes and ovaries can be distinguished morphologically at E13.5, the onset of sex differentiation (Fig. 1A; Sabour et al. 2011). To determine the sex of E10.5 mouse embryos, we carried out PCR-based sex genotyping using DNA from individual embryonic heads. We collected thousands of PGCs by fluorescence-activated cell sorting (FACS) purification from each dorsal mesentery or fetal gonad of Pou5f1-ΔPE-GFP male and female mouse embryos at E10.5, E13.5, and E16.5 (male PGCs: E10.5mPGC, E13.5mPGC, and E16.5mPGC; and female PGCs: E10.5fPGC, E13.5fPGC, and E16.5fPGC) (Supplemental Fig. 1; Yoshimizu et al. 1999). Green fluorescence protein (GFP) intensity was dramatically reduced but detectable in E16.5fPGC, consistent with previous results (Supplemental Fig. 1; Sabour et al. 2011). To obtain base-pair-resolution DNA methylomes, we performed WGSBS analysis using Illumina HiSeq 2000. WGSBS libraries were generated from 2000–5000 PGCs using a nonamplification technique termed PBAT (Supplemental Fig. 2; Miura et al. 2012). We generated 394 Gb of single-read (SR) and paired-end (PE) sequence data. Of these, 149 Gb (37.9%) were successfully aligned to either strand of the mouse genome, and each average read depth (i.e., the number of hits of reads that were mapped to a given position) was 9.05×–12.23× for the PGCs examined (Supplemental Table 1). In total, >91% of the genomic sequence was covered by at least one sequence read (Supplemental Fig. 3).
Figure 1.

Scheme of mouse developmental-germline DNA methylome analyses. (A) Morphology of Pou5f1-ΔPE-GFP PGCs in male and female fetal gonads at E10.5, E13.5, and E16.5. The sexes of embryos at E10.5 were determined by the presence or absence of the Zfy gene. At E13.5 and E16.5, male gonads could be distinguished by the presence of testicular cords. (B) Whole-genome methylation levels (mC/C ratios) in each PGC at CpG, CpHpG, and CpHpH sites (H = C, T, or A). (Aqua bars) E10.5mPGC; (orange bars) E10.5fPGC; (green bars) E13.5mPGC; (purple bars) E13.5fPGC; (blue bars) E16.5mPGC; (red bars) E16.5fPGC. (C) Percentage of methylcytosines in each sequence context in E16.5mPGC.
First, we calculated whole-genome methylation levels of each germ cell at CpG and non-CpG sites. Our methylation data showed that methylated cytosines (mCs) in almost all PGCs occurred mostly in the context of CpG dinucleotides, while non-CpG methylation was not present or found only at very low levels (mC/C ratio < 0.8%). Interestingly, only E16.5mPGC exhibited a relatively high level of non-CpG methylation (∼1.4%) (Fig. 1B; Supplemental Table 1), and ∼49% of mCs occurred in non-CpG contexts (Fig. 1C). Global CpG demethylation occurred in male and female PGCs during gonadal sex determination from E10.5 to E13.5, and average methylation levels of male PGCs were slightly higher than those of female PGCs (mC/C ratio = 16.9% and 16.1% in E10.5mPGC and E10.5fPGC, respectively, and mC/C ratio = 3.8% and 3.3% in E13.5mPGC and E13.5fPGC, respectively). Furthermore, in mitotic or meiotic arrested stages at E16.5, male PGCs showed dynamically increased methylation levels, whereas female PGCs remained undermethylated (mC/C ratio = 31.8% and 2.9% in E16.5mPGC and E16.5fPGC, respectively). These methylation ratios were much smaller than in mature sperm cells (89.4%), fully grown oocytes (40.0%), and embryonic stem (ES) cells (70.6%) (Kobayashi et al. 2012).
To elucidate the distribution of CpG methylation on regional and genome-wide scales, we created dot plots of average CpG methylation levels in sliding 200-kb windows throughout each chromosome. Interestingly, differences in the average methylation levels of these 200-kb windows in male and female PGCs were observed in autosomal chromosomes, rather than the X chromosome (Fig. 2; Supplemental Table 2). Global demethylation of each chromosome appeared between E10.5 and E13.5; however, certain chromosomal regions were partially methylated (≥10%) at E13.5 in male and female PGCs, and these regions were more highly methylated (20%–60%) than other regions at E10.5 (Fig. 2; Supplemental Fig. 4). In fact, while differences in the methylation of autosomal chromosomes were observed in male and female PGCs, the methylation levels of individual 200-kb windows were significantly correlated among the examined PGCs, except for those in E16.5mPGC (Supplemental Fig. 4). In addition, particular genomic compartments, e.g., L1/ERVK/ERV1 retrotransposons (see after the next paragraph) and satellite DNA were better represented in the demethylation-resistant regions (Supplemental Table 3). These observations indicated that particular regions in germ cell genomes avoided DNA methylation reprogramming (demethylation) during gonadal sex determination. The demethylation-resistant genomic compartments are described below. Furthermore, while female PGCs did not exhibit increased global methylation between E13.5 and E16.5, the methylation of male PGCs showed an obvious increase during this developmental period (Fig. 2; Supplemental Table 2). These results demonstrated that global CpG methylation levels throughout individual chromosomes of male PGCs were higher than in female PGCs during PGC development. Moreover, genome-wide de novo CpG and non-CpG methylation was acquired during fetal male germ cell development. Therefore, because non-CpG methylation appeared as a rare event during PGC development (except for E16.5mPGC), we focused our methylome analysis on CpG methylation.
Figure 2.
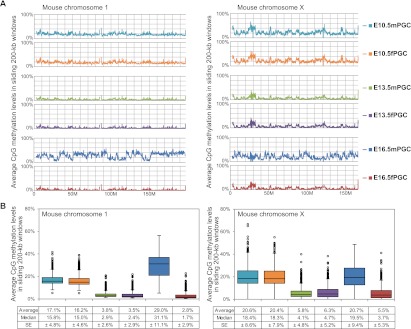
DNA methylome maps of chromosome 1 and chromosome X in mouse PGCs. (A) Chromosome-wide distribution of CpG methylation in sliding 200-kb windows. (Colored lines) The methylation levels in each PGC. (B) Box plots of each methylation level in 200-kb windows. Average and median methylation levels and standard errors (SEs) are shown below the graphs.
Since previous studies revealed a significant correlation between CpG frequency and methylation within intra- and intergenic regions in mammalian somatic and germ cells (Edwards et al. 2010; Kobayashi et al. 2012), we compared CpG densities and methylation levels to identify genome-wide differential methylation patterns in PGCs. Methylation levels of individual CpGs covered by at least three sequence reads (>85% of genomic CpGs) (Supplemental Fig. 5) were calculated, and CpG density was defined as the number of CpG dinucleotides per 200-nucleotide (nt) window (e.g., 1 CpG dinucleotide per 200 nt corresponded to a density of 0.005) (Fig. 3). At lower CpG densities (under 0.025, 64%–66% of genomic CpGs), the tendency toward average methylation levels was similar to that in previous reports (Fig. 1). However, at moderate CpG densities ranging from 0.030 to 0.080 (∼15% of genomic CpGs), except for exons, methylation levels were higher in both male and female PGCs at E10.5/E13.5. These results indicated that some CpGs, found in introns and intergenic regions at moderate CpG densities, remained partially methylated in PGCs after global demethylation. Furthermore, methylation levels fell off sharply at higher CpG densities (>0.085), consisting mostly of CpG-rich promoters and/or CpG islands. In fact, promoter regions around transcription start sites (TSSs) were hypomethylated in all investigated PGCs (Supplemental Fig. 6).
Figure 3.
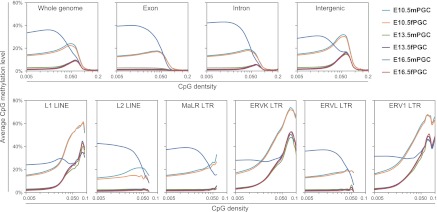
DNA methylation profiling of intra- and intergenic regions in mouse PGCs. CpG methylation levels are plotted as a function of CpG densities for whole-genome, exons, introns, intergenic regions (upper panels), and six major families of LINE/LTR retrotransposons (L1 LINEs, L2 LINEs, MaLR LTRs, ERVK LTRs, ERVL LTRs, and ERV1 LTRs [lower panels]).
Transposable elements (TEs) are mobile genetic sequences that comprise a large percentage of mammalian genomes; 37% of the mouse genome is made up of these elements (Waterston et al. 2002). Accordingly, we investigated the methylation of four major classes of transposable elements (long interspersed nuclear elements [LINEs], short interspersed nuclear elements [SINEs], long terminal repeats [LTRs], and DNA transposons) (Supplemental Fig. 7). At E10.5, higher levels of DNA methylation were observed at LINE and LTR regions with a relatively high CpG density (>0.030); thus, these repeated sequences were largely composed of methylated CpGs at moderate CpG densities in PGCs. Interestingly, at E13.5, partial CpG methylation (range, 20%–50%) in LINEs and LTRs at moderate CpG densities was observed, and almost all the other CpGs were hypomethylated in these elements. In addition, CpG methylation levels at lower CpG densities significantly increased, while those at relatively higher CpG densities were similar between E13.5 and E16.5 in male PGCs. We also investigated the methylation of major families of LINE/LTR retrotransposons: L1 LINEs, L2 LINEs, MaLR LTRs, ERVK LTRs, ERVL LTRs, and ERV1 LTRs. Interestingly, the demethylation-resistant CpGs with a higher CpG richness were observed only in L1 LINEs, ERVK LTRs, and ERV1 LTRs (Fig. 3; Supplemental Fig. 8). These results suggested that each PGC had unique sequence- and CpG-density-dependent methylation patterns, and particular subsets of LINE/LTR retrotransposons were resistant to global demethylation during PGC migration and sex determination.
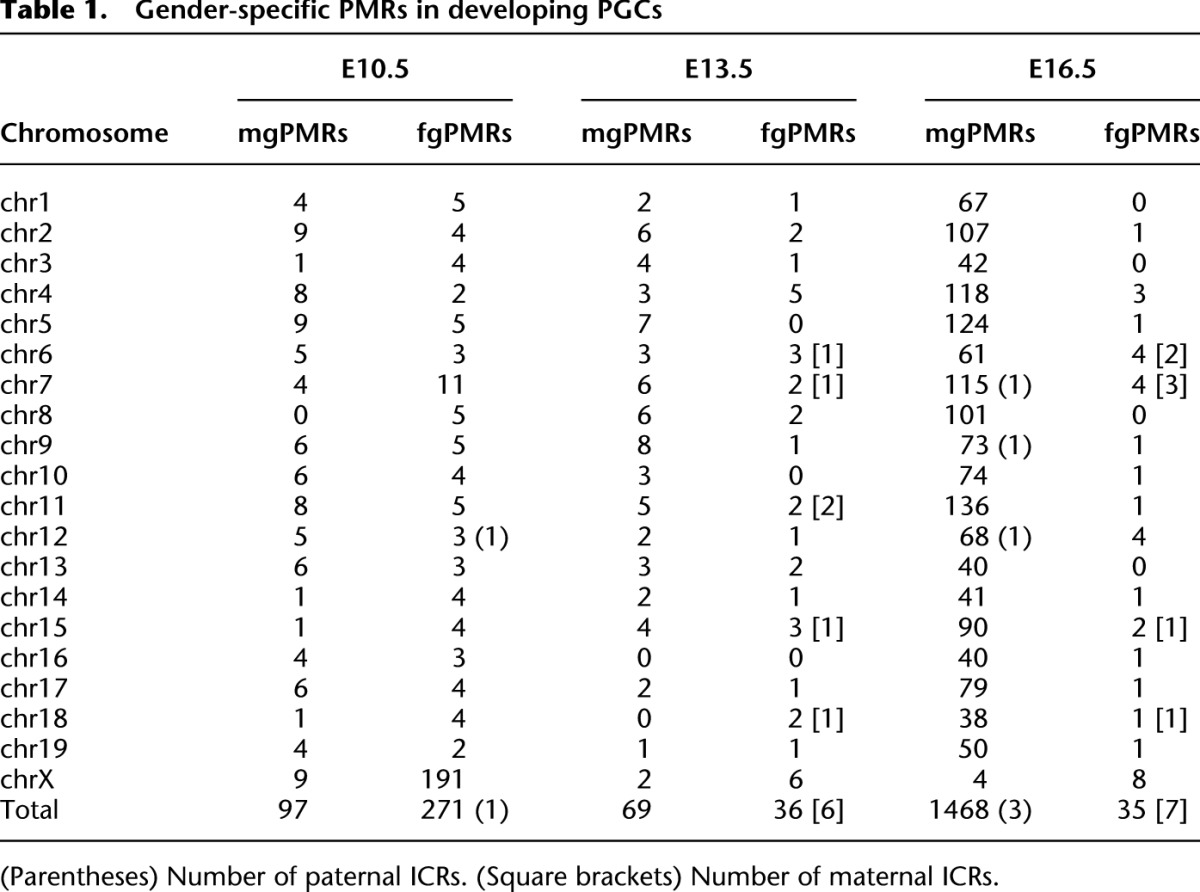
CpG islands (CGIs) are prominent in the mammalian genome due to their GC-rich base composition and high density of CpG dinucleotides and have been found within or near promoters of mammalian genes. Recently, Illingworth et al. (2010) identified 23,021 mouse CGIs by deep sequencing of isolated, zinc finger CXXC domain-binding unmethylated DNA clusters. Using these identified CGIs, we calculated the average methylation levels of each CGI to identify gender-specific differentially methylated regions (DMRs) in PGCs (Supplemental Table 4). Although ∼15% of CGIs were partially or highly methylated (≥10% methylation levels), most CGIs were hypomethylated (<10% methylation levels) at E10.5 in male and female PGCs (Supplemental Fig. 9). Moreover, while almost all CGIs were hypomethylated in PGCs after gonadal sex determination, a small percentage of these CGIs (∼4%) were only partially methylated in E16.5mPGC. In addition, ∼160–170 CGIs were incompletely demethylated (≥10% methylation levels) in male and female PGCs at E13.5, and many were located at introns or intergenic regions (Supplemental Fig. 10; Supplemental Table 5). Among the CGIs, demethylation-resistant sequences in PGCs, e.g., regions at Gm7120, Mid1, and Sfi1 loci (Guibert et al. 2012), were also re-identified. Typical germline DMRs were fully methylated (nearly 100%) in one mature gamete and unmethylated (nearly 0%) in the other; however, we could not identify such regions in PGCs at all investigated embryonic stages because of their undermethylated status (Smallwood et al. 2011; Kobayashi et al. 2012). Next, we identified male- and female-germ-cell preferentially methylated regions (mgPMRs and fgPMRs, respectively) from CGIs at each embryonic stage and determined the significance of differences using Mann-Whitney's U-test (Supplemental Table 6). Although average methylation levels in male germ cells were higher than in female germ cells, the number of fgPMRs (n = 271) was larger than the number of mgPMRs (n = 97) at E10.5, and more than half of the fgPMRs (n = 191) were located on chromosome X (X-DMRs) (Fig. 4A; Table 1). After gonadal sex determination, the number of fgPMRs decreased (n = 36 and 35 at E13.5 and E16.5, respectively), with hypomethylation observed on almost all X-DMRs (Supplemental Fig. 11). In contrast, the number of mgPMRs greatly increased in autosomal chromosomes but rarely in chromosome X (n = 1464, chromosomes 1–19; n = 4, chromosome X) at E16.5 (Table 1). Based on our previous DNA methylome data, more than half (48.4%, n = 709) of mgPMRs at E16.5 were hypermethylated with ≥80% methylation levels in mature sperm cells; of these, 292 mgPMRs were identified as germline DMRs (≥80% methylation in sperm and ≤20% methylation in fully grown oocytes) (Kobayashi et al. 2012). Further investigations of the methylation levels of CGIs on chromosome X are presented below. Notably, CGIs at imprinted germline DMRs, known to control the imprinting of a given domain as imprinting control regions (ICRs), were moderately methylated (near 40%) at E10.5 (Fig. 4B); however, a paternally imprinted Dlk1-Meg3 intergenic DMR (IG-DMR) was unexpectedly identified as an fgPMR. At E13.5, while these known ICRs were almost completely demethylated (<5%), some maternally imprinted ICRs exhibited partial methylation (5%–10%) in fPGCs; these were identified as fgPMRs. At E16.5, all three known paternally imprinted ICRs, i.e., H19, Rasgrf1, and Dlk1-Meg3, showed increased methylation levels (22.8%, 14.4%, and 8.4%, respectively) and were re-identified as mgPMRs. Methylation levels of a CGI on the Dlk1-Meg3 IG-DMR in male PGCs were lower than expected, based on a previous report of traditional bisulfite sequencing (conventional cloning and Sanger sequencing) by Henckel et al. (2012) (methylation levels; 30% at E15.5, 89% at E17.5 in male gonocytes) (Supplemental Fig. 12), but the reason for this discrepancy is unknown. Surprisingly, seven known maternally imprinted ICRs (among 20 maternally imprinted ICRs that were previously identified in mice), found within Peg10, Mest, Peg3, Snrpn, Kcnq1, Slc38a4, and Impact genes, were also identified as fgPMRs at E16.5, and some of these ICRs (i.e., Peg10, Peg3, and Impact) exhibited sex-differential methylation, even at E13.5. Female-germ-cell-specific partial methylation at Mest and Snrpn ICRs were confirmed in E16.5 by conventional bisulfite sequencing (Supplemental Fig. 13). Taken together, our results indicated that some maternally methylated imprinted regions contained partial methylation in primary oocytes during fetal stages. Next, we investigated the methylation profiles of germline-specific genes and pluripotency-associated genes containing CGIs. While some germline-specific genes, i.e., Ddx4, Dazl, Sycp3, and Figla, and PGC-specific genes (recently identified by microarray-based analysis) (Sabour et al. 2011), i.e., Fkbp6, Mov10l1, and Spo11, were partially or moderately methylated (∼20%–60%), most other genes were hypomethylated in male and female PGCs at E10.5 (Fig. 4B). Unexpectedly, at E13.5, Fkbp6 and Spo11 were identified as mgPMRs and fgPMRs, respectively, and all other investigated genes were hypomethylated at E13.5 and E16.5, and throughout PGC progression. Moreover, coefficient of variation analysis showed that male and female CGI methylation patterns on autosomal chromosomes in PGCs were significantly correlated during migration (R2 = 0.8616 at E10.5), but this correlation became much weaker after sex determination (R2 = 0.5563 at E13.5 and R2 = 0.0547 at E16.5) (Fig. 4C). Additionally, CGI methylation patterns on chromosome X were significantly correlated throughout the investigated fetal stages, especially at E16.5 (R2 = 0.946). These results indicated that sex differences in the CGI methylation of autosomes began to appear after gonadal sex determination through increased CpG methylation in male gonocytes, while X-linked genes were resistant to de novo methylation.
Figure 4.

DNA methylation profiles of mouse CGIs in developing PGCs. (A) Chromosome distribution of male and female germ cell–specific preferentially methylated regions (mgPMRs and fgPMRs, respectively) in E10.5–E16.5 PGCs. Mann-Whitney's U-test was performed to determine gender-specific PMRs between male and female CpG methylation profiles of CGIs (FDR-adjusted P-values < 0.05). The locations of mgPMRs and fgPMRs are indicated as light blue and light red bars, respectively. (B) CpG methylation profiles of paternal ICRs, maternal ICRs, germline-specific genes, PGC-specific genes, and pluripotency-associated genes during PGC progression. (†) CGIs identified as mgPMRs; (*) CGIs identified as fgPMRs. (C) Scattergraphs show the relationships between the methylation levels of individual CGIs on autosomes (gray) and chromosome X (red) in male and female PGCs. Linear approximations with squared correlation coefficient (R2) values are shown at the top of each graph.
Table 1.
Gender-specific PMRs in developing PGCs
Finally, we examined cytosine methylation in male gonocytes, in which the presence of non-CpG methylation has not been reported. We mapped CpG and non-CpG methylation data for each gene to a “gene model,” which contained annotated genomic features in the neighborhood of transcribed genes, including promoters/transcription start sites (TSSs, ±500 bp), upstream intergenic regions (−2 to −5 kb from TSSs), and downstream regions (+2 to +5 kb from TSSs). Methylation levels of individual cytosines, covered by at least five sequence reads, were calculated, covering 80% of the genome (Supplemental Fig. 3). Upstream and downstream regions showed increased methylation levels in both CpG and non-CpG sites, while promoter regions were deeply hypomethylated (Fig. 5A,B). Our results indicated that non-CpG methylation may be accompanied by intra-/intergenic CpG methylation in male gonocytes. Furthermore, sequence context analysis showed that, at non-CpG sites (Fig. 5C), the nucleotide immediately 3′ to highly methylated regions was most likely to be adenine (CpA), which is also a feature of non-CpG methylation in oocytes and embryonic stem (ES) cells (Tomizawa et al. 2011; Ziller et al. 2011).
Figure 5.
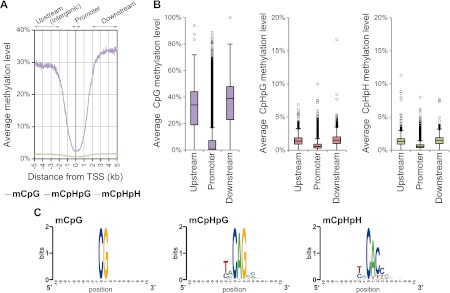
(A) The distribution of cytosine methylation in male gonocytes (E16.5mPGCs) is shown ±5 kb from the transcription start site (TSS). Methylation levels of CpG (light purple), CpHpG (red), and CpHpH (green) contexts. (B) Box plots of each methylation level for 20,854 RefSeq annotated genes at areas −2 to −5 kb from the TSS (upstream/intergenic), ±500 bp from the TSS (promoter), and +2 to +5 kb from the TSS (downstream). (C) WebLogo plots for sequences proximal to highly methylated cytosines (mC/C ≥ 50%) in all three sequence contexts in E16.5mPGCs.
Discussion
In this study, we performed WGSBS mapping with thousands of mammalian cells (equal to ∼20–50 ng of genomic DNA) using the PBAT method. This DNA methylome study demonstrated genome-wide DNA demethylation, with erasure of genomic imprinting during gonadal sex determination and gender-specific differences in genome-wide and gene-specific (a part of CGIs) DNA methylation levels in developing PGCs. Some of these global/local changes in DNA methylation during PGC progression were consistent with previous as well as more recent studies (Hajkova et al. 2002; Maatouk et al. 2006; Laird 2010; Popp et al. 2010; Seisenberger et al. 2012; Hackett et al. 2013). However, our complete DNA methylome maps revealed important and novel details of DNA methylation and demethylation processes during PGC development. Some of the new findings from this study include the following: (1) PGC DNA methylomes exhibited sex- and chromosome-specific differences in genome-wide CpG and CGI methylation during early to late PGC development; (2) LINE/LTR retrotransposons were resistant to DNA demethylation at high CpG densities during PGC migration; (3) some maternally imprinted regions (genes) remained partially methylated in primary oocytes during fetal stages; and (4) non-CpG methylation occurred in male gonocytes during mitotic arrest (Fig. 6; Supplemental Fig. 14). Our data and techniques can therefore serve as a platform for future studies to elucidate the role of epigenetic modifications in germline development and other biological processes.
Figure 6.

DNA methylome changes during gametogenesis and embryogenesis. Mouse PGCs emerge from precursor cells in the proximal epiblast at E7.25. They proliferate and migrate toward the genital ridge. Then, DNA methylation is globally decreased in both males (blue line) and females (red line) with erasure of methylation marks of imprinted genes, X-linked genes (only in females), and some germline-specific genes (see Fig. 4) through TET-catalyzed oxidation (Hackett et al. 2013). During this migration, whole-genome CpG methylation levels are relatively higher in males than in females (see Fig. 2). Following gonadal sex determination, new DNA methylation patterns are established in each germ cell in a sex-specific manner. In the male embryo, de novo CpG and non-CpG methylation occurs in mitotically arrested gonocytes (see Fig. 5). Establishment of the paternal methylation imprints (e.g., H19) is completed before birth (meiosis), and these imprints are maintained during subsequent spermatogenesis and throughout meiosis; however, the presence of non-CpG methylation is rarely observed in the mature spermatozoon. In the female embryo, PGCs enter meiosis as primary oocytes and arrest in the prophase of the first meiotic division; the oocyte genome remains globally hypomethylated, but parts of maternal ICRs (e.g., Peg10, Mest, Peg3, Snrpn) exhibit partial methylation (see Fig. 4). DNA methylation marks are established after birth during the growth phase of the oocyte. At puberty, fully grown oocytes are still arrested at meiotic prophase, a stage known as the germinal vesicle (GV) stage. The GV oocyte genome exhibits global hypermethylation at transcribed regions, but the whole-genome CpG methylation level of oocytes is less than half that of spermatozoa (Kobayashi et al. 2012). When GV oocytes resume the first meiotic division, they undergo GV breakdown, extrude a first polar body, and develop to metaphase of the second meiotic division (MII). MII oocytes complete meiosis only with fertilization. In the zygote, striking asymmetric DNA demethylation between the two parental genomes is observed within the zygote's cytoplasm. The paternal genome is actively demethylated before the first mitotic division through the involvement of TET protein-mediated 5-methylcytosine oxidation (conversion to 5-hydroxymethylcytosine) (Gu et al. 2011; Wossidlo et al. 2011). The maternal genome resists hydroxylation (Nakamura et al. 2012) and instead undergoes passive DNA replication-dependent demethylation. Multiple maternal and zygotic DNA-binding factors specifically recognize ICRs and protect them from these post-fertilization demethylation events (Nakamura et al. 2007; Li et al. 2008; Messerschmidt et al. 2012), resulting in epigenetic allelic asymmetries that affect associated imprinted genes. Following blastocyst implantation, the embryo undergoes a wave of de novo methylation (black line) that establishes a new DNA methylation landscape, and this process is associated with cellular differentiation.
Historically, most CGIs were thought to be unmethylated in various tissues; however, a substantial proportion of CGIs were reported to undergo methylation during genomic imprinting, X-chromosome inactivation (one of the two copies of the X chromosome present in females is inactivated), carcinogenesis, and even in normal tissues (Illingworth and Bird 2009). Moreover, the methylation status of CGIs in promoter regions is highly correlated with gene expression. CGIs have been studied by multiple array-based and sequencing-based global methylation assessments, and several imprinted, cancer-specific, and tissue-specific DMRs have been identified (Bock et al. 2010; Laird 2010). CGI methylation profiling revealed increasing mgPMRs, and significant differences were observed in autosomal chromosomes during mitotic arrest but not in chromosome X, where chromosome-wide de novo methylation occurred (Fig. 2). Our findings indicated that CGIs (mostly associated with X-linked genes) on chromosome X were resistant to de novo methylation during fetal spermatogenesis. This is reminiscent of previous methylome studies that showed an absence of sperm-specific methylated DMRs (compared between fully grown oocytes and sperm) on chromosome X (Smallwood et al. 2011; Kobayashi et al. 2012). The mechanism that protects the genome from de novo methylation pressure is unknown, but DNA-binding factors may maintain the unmethylated status of X-linked genes (Illingworth et al. 2010). Meanwhile, among mgPMRs at E16.5, all three known paternal ICRs were re-identified. This result was consistent with previous studies, which revealed that methylation of paternal ICRs is established during PGC mitotic cell division before birth (Li et al. 2004; Hiura et al. 2007; Kato et al. 2007). In contrast, the number of fgPMRs decreased during PGC progression and was quite low at E16.5. Interestingly, although some of these fgPMRs were also shown to be maternally methylated imprinted regions (i.e., Peg10, Mest, Peg3, Snrpn, Kcnq1, Slc38a4, and Impact), previous studies have shown that increasing expression of Dnmts and establishment of maternal methylation imprints occurred during oocyte growth at postnatal stages (Lucifero et al. 2004; Hiura et al. 2006). Of the maternal ICRs, Peg10, Peg3, and Impact ICRs were identified as fgPMRs at E13.5. These regions may be partially resistant to global demethylation, similar to some retrotransposons. Previously, a maternal–zygotic effect gene, Zfp57, was shown to be required not only for the establishment of DNA methylation in the female germline, but also for methylation re-acquisition, specifically at the maternally derived allele in the Snrpn imprinted region (Li et al. 2008), indicating that the parental alleles were not equivalent and retained their identity in the absence of Snrpn methylation. A type of DNA-independent epigenetic memory may exist in these regions to permit maternal methylation, even after global demethylation in female PGCs. Meanwhile, complete establishment of germline methylation at imprinted Mest DMR (also known as Peg1 DMR) is slower than the other maternally methylated imprinted loci (Lucifero et al. 2004; Hiura et al. 2006). Further DNA methylome analysis in growing oocytes will help us to understand a detailed mechanism that determines the timing of maternal methylation imprinting.
In this study, we identified more than 200 fgPMRs in E10.5 PGCs, most of which were located on chromosome X (X-DMRs). This may reflect the CGI methylation of X-linked genes by X-chromosome inactivation in females. These X-DMRs became hypomethylated at E13.5, similar to genomic imprints; this is consistent with re-activation of X-linked genes during PGC development (Sugimoto and Abe 2007). In addition, demethylation during PGC migration may activate some germline-specific genes, including those specifically expressed in PGCs. Thus, our results showed that CGI methylation on X-linked genes, germline-specific genes, and imprinted genes was erased by global DNA demethylation during entry of PGCs into embryonic gonads. This idea is also supported by other, more recent reports based on WGSBS or methyl-DNA immunoprecipitation (MeDIP) studies (Seisenberger et al. 2012; Hackett et al. 2013). Conversely, we showed that the global DNA methylation levels were slightly higher in male PGCs than in female PGCs, even before gonadal sex determination at E10.5. This result is consistent with studies demonstrating that male ES cells and PGCs have higher methylation levels than female ES cells and PGCs at E13.5 (Zvetkova et al. 2005; Popp et al. 2010). A potential explanation for the observed sex-based difference was the global reduction in methylation due to the presence of two active X chromosomes in females; however, this might be unlikely because a sex-based difference was observed in all autosomal chromosomes. Thus, the reason for sex-based differences in the global DNA methylation of PGCs is still unknown. Interestingly, recent observations using low-coverage bisulfite sequencing have indicated that the cytidine deaminase AICDA (also known as AID) partially contributed to demethylation in PGCs and could explain sex-specific differences in DNA methylation in E13.5 PGCs (Popp et al. 2010). This also supports the hypothesis that the base excision repair pathway is involved in DNA demethylation during PGC migration and that other pathways exist in the demethylation process, such as ten–eleven translocation (TET) protein-mediated oxidation of 5-methylcytosine (Hajkova et al. 2010). In fact, recent reports revealed that hydroxylation of 5-methylcytosines mediated by TET proteins is involved in active DNA demethylation of the zygotic paternal genome after fertilization and both active and passive demethylation of the PGC genome during expansion and migration (Fig. 6; Supplemental Fig. 14; Gu et al. 2011; Wossidlo et al. 2011; Hackett et al. 2013). TET1 knockout mice were reported to be viable and fertile and exhibit normal gametogenesis (Dawlaty et al. 2011); however, in a more recent report, significant reduction of oocyte numbers and fertility was observed in other TET1 knockout mice (Yamaguchi et al. 2012). Moreover, bisulfite-based techniques to detect 5-hydroxymethylcytosines have been reported by two groups (Booth et al. 2012; Yu et al. 2012). Thus, further investigations using new technologies may elucidate DNA demethylation mechanisms in males and females during PGC formation.
Our study of global DNA demethylation revealed that most genomic CpG sites (and CGIs) were hypomethylated at E13.5, but L1 LINE, ERVK LTR, and ERV1 LTR retrotransposons were resistant to demethylation at relatively high CpG densities. In addition, our previous study showed that methylation of LINE/LTR sequences was retained in Dnmt3l-deficient oocytes (Kobayashi et al. 2012). Furthermore, separate studies have reported that the intracisternal A-particle (IAP), a member of the ERVK LTR retrotransposons, generally appears to be resistant to demethylation processes in gametogenesis and embryogenesis (Hajkova et al. 2002; Lane et al. 2003; Seisenberger et al. 2012). Recently, Guibert et al. (2012) revealed that ERV1 (with a higher CpG richness than the bulk of ERV1 sequences) is incompletely demethylated in PGCs. These results suggest the existence of a mechanism for preferentially maintaining cytosine methylation at evolutionarily young and potentially active transposable elements, which may be necessary to prevent the deleterious effects of their activation during epigenetic reprogramming. As mentioned above, Peg10, Peg3, and Impact ICRs were also partially protected from PGC demethylation. Furthermore, two recent studies also showed that demethylation rates of Peg10 and Peg3 ICRs were slower than that of the other ICRs or the predicted rates of passive demethylation (in a similar fashion to that of IAP retrotransposons) (Hackett et al. 2013; Kagiwada et al. 2013). While the mechanisms that allow resistance to global demethylation are unclear, it may involve DNA binding factors that specifically protect some sequences from demethylation, as shown with multiple maternal and/or zygotic DNA-binding factors, including DPPA3 (also known as PGC7/STELLA), ZFP57, and TRIM28 (also known as KAP-1), all of which protect ICRs from demethylation in pre-implantation embryos (Supplemental Fig. 14; Nakamura et al. 2007; Li et al. 2008; Messerschmidt et al. 2012). Conversely, X-linked genes may be protected by de novo methylation during spermatogenesis. Thus, it is important to realize that these mechanisms protect DNA specifically via a wave of demethylation or de novo methylation.
Following gonadal sex determination, new differential DNA methylation patterns are established during spermatogenesis and oogenesis, resulting in distinct DNA methylation profiles of mature spermatozoon and oocytes. In this study, we identified increasing cytosine methylation at both CpG and non-CpG dinucleotides only in male gonocytes (PGCs at E16.5). In contrast, previous studies based on bisulfite sequencing revealed that non-CpG methylation occurred in fully grown oocytes, but rarely in mature spermatozoa (Tomizawa et al. 2011; Kobayashi et al. 2012; Smith et al. 2012). It is possible that such non-CpG methylation in gonocytes may be lost during spermatogonial mitotic proliferation after birth; however, the reason remains unknown. In fact, Ichiyanagi et al. (2013) found non-CpG methylation at a subfamily of SINEs in male gonocytes and prospermatogonia at prepubertal stages but not in mature sperm cells. Several murine studies have demonstrated the presence of non-CpG methylation in ES cells and early embryos (Ramsahoye et al. 2000; Haines et al. 2001), but this modification is completely absent in most adult somatic tissues. Recently, non-CpG methylation was observed in ES cells and induced pluripotent cells, with loss of methylation in differentiated cells (Lister et al. 2009); however, how these methylation modifications can be gained and maintained in daughter pluripotent stem cells after mitosis is unknown. Furthermore, it has been suggested that the expression of Dnmts may be responsible for this modification (Ramsahoye et al. 2000; Arand et al. 2012). Up-regulation of Dnmts and de novo methylation in the male germline is initiated in mitotically arrested prospermatogonia before birth and the onset of meiosis; however, de novo methylation in the female germline occurs in postnatal meiotic prophase I oocytes (Lucifero et al. 2004; Sakai et al. 2004). These data indicate that non-CpG methylation may be a result of abundant or sustained Dnmt protein expression, particularly in nondividing cells. Comparisons of experimental data and examined methylation profiles during gametogenesis or stem cell formation (including embryogenesis) may be useful in discovering the underlying mechanisms responsible for each biological process.
Methods
Isolation of PGCs from the mouse fetus
Mouse PGCs were isolated from E10.5, E13.5, and E16.5 embryos (dissected in the evening of embryonic days 10, 13, and 16) of female C57BL/6N mice (Clea Japan, Tokyo, Japan) mated with male Pou5f1-ΔPE-GFP mice (Yoshimizu et al. 1999). Embryonic sex was distinguished by PCR of genomic DNA from the embryonic head by using specific primers (Zfy: forward primer, 5′-CCTATTGCATGGACTGCAGCTTATG-3′ and reverse primer, 5′-GACTAGACATGTCTTAACATCTGTCC-3′; Xist: forward primer, 5′-AGGATAATCCTTCATTATCGCGC-3′ and reverse primer, 5′-AAACGAGCAAACATGGCTGGAG-3′) for E10.5 samples or by assessing gonad morphology for E13.5 and E16.5 samples. Dorsal mesenteries and fetal gonads were isolated from Pou5f1-ΔPE-GFP E10.5–E16.5 mouse embryos, and digestion with collagenase and trypsin was performed as described (Hiura et al. 2007). GFP-positive PGCs were isolated on a FACSAria II cell sorter (BD Biosciences), sorting for green fluorescence; the isolated cell populations were >97% pure.
PBAT library preparation
To construct the PBAT library, 1000 or 2000 PGCs were lysed by incubation with 20 μL of DNase-free water containing 0.1% SDS, 50 ng/μL carrier RNA (QIAGEN), and 1 μg/μL proteinase K (Invitrogen) for 60 min at 37°C. Sample DNA lysates containing 0.1 ng of unmethylated lambda phage DNA (Invitrogen) were subjected to bisulfite treatment with the Methylcode Bisulfite Conversion Kit (Invitrogen) according to the manufacturer's instructions. PBAT libraries were then synthesized as previously described (Miura et al. 2012). Briefly, bisulfite-treated DNA was re-annealed to double-stranded DNA using Klenow fragments (3′–5′ exo−; New England Biolabs) with random primers containing 5′ biotin tags and an Illumina adaptor (BioPEA2N4: 5′-biotin-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNN-3′). The biotinylated molecules (first strand) were captured using Dynabeads M280 Streptavidin (Invitrogen) and re-annealed to double-stranded DNA again using Klenow fragments (3′–5′ exo−) with random primers containing Illumina adaptors (PE-reverse-N4 for SR sequencing: 5′-CAAGCAGAAGACGGCATACGAGATNNNN-3′ and Primer4-N15 for PE sequencing: 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTNNNNNNNNNNNNNNN-3′). Finally, template DNA strands were synthesized as cDNA with a second strand (where unmethylated C's were converted to T's) using Phusion Hot Start High-Fidelity DNA Polymerase (New England Biolabs) with the Illumina primer PE 1.0 (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′). Concentrations of PBAT libraries were determined by qPCR using Illumina PE-primers (PE-forward: 5′-AATGATACGGCGACCACCGAGATCTACAC-3′ and PE-reverse: 5′-CAAGCAGAAGACGGCATACGAGAT-3′) (Miura et al. 2012). The PhiX v2 Control Kit (Illumina) was used as a standard for quantification.
Illumina sequencing
Based on the qPCR quantification, 4 × 108 to 10 × 108 copies of dsDNA from the PBAT library was sequenced per lane on a HiSeq 2000 (Illumina) as described (Miura et al. 2012). Cluster generation and sequencing were performed with ∼102-nt SR and PE methods using the TruSeq SR Cluster Kit v3 -cBot -HS (Illumina) and the TruSeq SBS Kit v3 -HS (Illumina) according to the manufacturer's protocols.
Mapping of reads
Sequenced PBAT reads were processed using the Illumina standard base-calling pipeline (v1.8.0–1.8.2). Before read-mapping, the first 4 bases (or 15 bases of read 2 of PE sequences) and last 1 base of all SR/PE sequences were trimmed due to derivation from random primers and/or low quality. Generated sequence tags were mapped onto the mouse genome using the Burrows-Wheeler Aligner (BWA) tool and default parameter settings. Mapping and filtering of PBAT tags was performed by following procedures described in previous studies, with an original customized Perl program (Kobayashi et al. 2012). Briefly, any guanines in all SR tags and PE read 1 (forward) tags were replaced with adenines, and any cytosines in all PE read 2 (forward) tags were replaced with thymines. Next, these tags were aligned to two in silico–converted mouse genome reference sequences (mm9, UCSC Genome Browser, July 2007, Build 37.1) and the lambda DNA sequence (accession no. V00636), with cytosines in the first strand converted to thymine (“Watson” strand) and guanines in the second strand converted to adenines (“Crick” strand). Finally, all tags that were mapped uniquely without any mismatches (>32 nt of perfectly matched tags) to both “Watson” and “Crick” strands were used for further analyses.
Methylation analysis
The percentage of individual cytosines methylated at all CpG and non-CpG sites covered by at least one, three, or five reads was calculated as 100 × [number of aligned cytosines (methylated cytosines)]/[total number of aligned cytosines and thymines (originally unmethylated cytosines)]. All genomic CpG methylation data are available on our website (http://www.nodai-genome.org/mouse_en.html). Bisulfite conversion rates were calculated by read C:T ratios from lambda DNA mapping data. The conversion rates are shown in Supplemental Table 1. Locations of transposable elements in the mouse genome (mm9) were obtained from the UCSC Genome Browser, and the average methylation levels of the whole genome and each transposable element were recalculated from the ratio of the aligned cytosines and thymines in each sequence. Lists of a total of 23,021 CGIs were obtained from a previous report (Illingworth et al. 2010). These computational analyses were performed using a custom Perl script and R statistical package.
Conventional bisulfite sequencing
Genomic DNA was extracted from FACS-purified PGCs (about 10,000 cells) using the QIAamp DNA Micro Kit (QIAGEN) and treated with sodium bisulfite using the Methylcode Bisulfite Conversion Kit (Invitrogen). The bisulfite-treated DNA was PCR-amplified using a reaction mix containing 0.5 μM each primer set, 200 μM dNTPs, 1× PCR buffer, and 1.25 units of EpiTaq HS DNA polymerase (Takara Bio) in a total volume of 20 μL. The primers were Snrpn DMR (5′-ATTGGTGAGTTAATTTTTTGGA-3′ and 5′-ACAAAACTCCTACATCCTAAAA-3′) and Mest DMR (5′-TTTTAGATTTTGAGGGTTTTAGGTTG-3′ and 5′-AATCCCTTAAAAATCATCTTTCACAC-3′). The following PCR program was used for Snrpn and Mest DMRs: 1 min of denaturation at 94°C followed by 35 cycles of 30 sec at 94°C, 30 sec at 60°C, and 30 sec at 72°C, and final extension for 5 min at 72°C. Subcloning and sequencing analyses were performed as described earlier (Kobayashi et al. 2009).
Data access
All WGSBS data in this study have been deposited in the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (http://trace.ddbj.nig.ac.jp/dra/index_e.shtml) under accession number DRA000607.
Acknowledgments
We are grateful to Professor Hiroyuki Sasaki and Dr. Tomoko Ichiyanagi (Kyushu University, Japan) for advice on improving our assessment of non-CpG methylation and communicating their results prior to publication. We are also grateful to Professor Tom Moore (University College Cork, Ireland) for his helpful suggestions. We also thank Terumi Horiuchi (University of Tokyo, Japan) and Satoshi Sano (Tokyo University of Agriculture, Japan) for their assistance with analysis of the Illumina data. T.K. was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (Grant No. 222228004), including the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (Grant No. S0801025), and was partially supported by Grants-in-Aid for Scientific Research on Innovative Areas “Genome Research” from MEXT (Grant No. 221S0002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.148023.112.
Freely available online through the Genome Research Open Access option.
References
- Abe M, Tsai SY, Jin SG, Pfeifer GP, Szabó PE 2011. Sex-specific dynamics of global chromatin changes in fetal mouse germ cells. PLoS ONE 6: e23848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, et al. 2012. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet 8: e1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S 2012. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336: 934–937 [DOI] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. 2011. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9: 166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JR, O'Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. 2010. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 20: 972–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg M, Snow MH, McLaren A 1990. Primordial germ cells in the mouse embryo during gastrulation. Development 110: 521–528 [DOI] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. 2011. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477: 606–610 [DOI] [PubMed] [Google Scholar]
- Guibert S, Forné T, Weber M 2012. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res 22: 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA 2013. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339: 448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines TR, Rodenhiser DI, Ainsworth PJ 2001. Allele-specific non-CpG methylation of the Nf1 gene during early mouse development. Dev Biol 240: 585–598 [DOI] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 117: 15–23 [DOI] [PubMed] [Google Scholar]
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA 2008. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452: 877–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA 2010. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 329: 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckel A, Chebli K, Kota SK, Arnaud P, Feil R 2012. Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J 31: 606–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura H, Obata Y, Komiyama J, Shirai M, Kono T 2006. Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells 11: 353–361 [DOI] [PubMed] [Google Scholar]
- Hiura H, Komiyama J, Shirai M, Obata Y, Ogawa H, Kono T 2007. DNA methylation imprints on the IG-DMR of the Dlk1-Gtl2 domain in mouse male germline. FEBS Lett 581: 1255–1260 [DOI] [PubMed] [Google Scholar]
- Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H 2013. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res 41: 738–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP 2009. CpG islands–‘a rough guide.’ FEBS Lett 583: 1713–1720 [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP 2010. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 6: e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M 2013. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J 32: 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903 [DOI] [PubMed] [Google Scholar]
- Kaneda M, Hirasawa R, Chiba H, Okano M, Li E, Sasaki H 2010. Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells 15: 169–179 [DOI] [PubMed] [Google Scholar]
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H 2007. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16: 2272–2280 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Yamada K, Morita S, Hiura H, Fukuda A, Kagami M, Ogata T, Hata K, Sotomaru Y, Kono T 2009. Identification of the mouse paternally expressed imprinted gene Zdbf2 on chromosome 1 and its imprinted human homolog ZDBF2 on chromosome 2. Genomics 93: 461–472 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. 2012. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8: e1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW 2010. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11: 191–203 [DOI] [PubMed] [Google Scholar]
- Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, Reik W 2003. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 35: 88–93 [DOI] [PubMed] [Google Scholar]
- Li JY, Lees-Murdock DJ, Xu GL, Walsh CP 2004. Timing of establishment of paternal methylation imprints in the mouse. Genomics 84: 952–960 [DOI] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC 2008. A maternal–zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15: 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifero D, Mann MR, Bartolomei MS, Trasler JM 2004. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet 13: 839–849 [DOI] [PubMed] [Google Scholar]
- Maatouk DM, Kellam LD, Mann MR, Lei H, Li E, Bartolomei MS, Resnick JL 2006. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 133: 3411–3418 [DOI] [PubMed] [Google Scholar]
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB 2012. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335: 1499–1502 [DOI] [PubMed] [Google Scholar]
- Miura F, Enomoto Y, Dairiki R, Ito T 2012. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res 40: e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Matsui Y 2010. Epigenetic profiles in primordial germ cells: Global modulation and fine tuning of the epigenome for acquisition of totipotency. Dev Growth Differ 52: 517–525 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, et al. 2007. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol 9: 64–71 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T 2012. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 486: 415–419 [DOI] [PubMed] [Google Scholar]
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W 2010. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463: 1101–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R 2000. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci 97: 5237–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabour D, Arauzo-Bravo MJ, Hubner K, Ko K, Greber B, Gentile L, Stehling M, Scholer HR 2011. Identification of genes specific to mouse primordial germ cells through dynamic global gene expression. Hum Mol Genet 20: 115–125 [DOI] [PubMed] [Google Scholar]
- Saga Y 2008. Mouse germ cell development during embryogenesis. Curr Opin Genet Dev 18: 337–341 [DOI] [PubMed] [Google Scholar]
- Sakai Y, Suetake I, Shinozaki F, Yamashina S, Tajima S 2004. Co-expression of de novo DNA methyltransferases Dnmt3a2 and Dnmt3L in gonocytes of mouse embryos. Gene Expr Patterns 5: 231–237 [DOI] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48: 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y, Hayashi K, Itoh K, Mizugaki M, Saitou M, Matsui Y 2005. Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Dev Biol 278: 440–458 [DOI] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Abe K 2007. X chromosome reactivation initiates in nascent primordial germ cells in mice. PLoS Genet 3: e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, Sasaki H 2011. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 138: 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2: 241. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, Zhang K, Zhang Y 2012. Tet1 controls meiosis by regulating meiotic gene expression. Nature 492: 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Low EW, Marikawa Y, Iwahashi K, Bartolomei MS, McCarrey JR, Yanagimachi R 2005. Adult mice cloned from migrating primordial germ cells. Proc Natl Acad Sci 102: 11361–11366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T, Sugiyama N, De Felice M, Yeom YI, Ohbo K, Masuko K, Obinata M, Abe K, Scholer HR, Matsui Y 1999. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Dev Growth Differ 41: 675–684 [DOI] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. 2011. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7: e1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvetkova I, Apedaile A, Ramsahoye B, Mermoud JE, Crompton LA, John R, Feil R, Brockdorff N 2005. Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet 37: 1274–1279 [DOI] [PubMed] [Google Scholar]