

Abstract
α-Synuclein is the major component of filamentous inclusions that constitute the defining characteristic of neurodegenerative α-synucleinopathies. However, the molecular mechanisms underlying α-synuclein accumulation and spread are unclear. Here we show that intracerebral injections of sarkosyl-insoluble α-synuclein from brains of patients with dementia with Lewy bodies induced hyperphosphorylated α-synuclein pathology in wild-type mice. Furthermore, injection of fibrils of recombinant human and mouse α-synuclein efficiently induced similar α-synuclein pathologies in wild-type mice. C57BL/6J mice injected with α-synuclein fibrils developed abundant Lewy body/Lewy neurite-like pathology, whereas mice injected with soluble α-synuclein did not. Immunoblot analysis demonstrated that endogenous mouse α-synuclein started to accumulate 3 months after inoculation, while injected human α-synuclein fibrils disappeared in about a week. These results indicate that α-synuclein fibrils have prion-like properties and inoculation into wild-type brain induces α-synuclein pathology in vivo. This is a new mouse model of sporadic α-synucleinopathy and should be useful for elucidating progression mechanisms and evaluating disease-modifying therapy.
Keywords: α-synuclein, Lewy bodies, Parkinson’s disease, propagation
Introduction
Filamentous inclusions composed of α-synuclein in nerve cells or glial cells are the defining neuropathological feature of a group of neurodegenerative diseases including Parkinson’s disease, dementia with Lewy bodies, and multiple-system atrophy (Goedert, 2001). In these so-called α-synucleinopathies, α-synuclein is deposited in a hyperphosphorylated form with β-sheet-rich, fibrillar structure (Spillantini et al., 1997, 1998; Baba et al., 1998; Wakabayashi et al., 1998; Fujiwara et al., 2002). Missense mutations (A30P, E46K and A53T) in the α-synuclein gene (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004) and multiplications of the region (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibanez et al., 2004,) have been identified in familial forms of Parkinson’s disease and dementia with Lewy bodies, indicating that abnormalities of α-synuclein cause these diseases. Neuropathologically, α-synuclein lesions are believed to spread progressively throughout the brain and their spread correlates to the staging of clinical symptoms (Muller et al., 2005), as in the case of tau pathology in Alzheimer’s disease (Braak and Braak, 1991). Kordower et al. (2008) and Li et al. (2008) reported that embryonic neurons transplanted into the striatum of an individual with Parkinson’s disease developed Lewy body-like pathologies, suggesting that pathological α-synuclein may be transmissible from diseased neurons to healthy neurons. Recent studies have also shown that exogenous α-synuclein fibrils induced Lewy body pathology in cultured neurons (Desplats et al., 2009; Emmanouilidou et al., 2010; Nonaka et al., 2010; Volpicelli-Daley et al., 2011), transgenic mouse brain (Mougenot et al., 2012; Luk et al., 2012b) and wild-type mouse brain (Luk et al., 2012a). In addition, a growing body of evidence indicates that self-propagating protein aggregates play central roles in many neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease (Clavaguera et al., 2009; Mougenot et al., 2012; Luk et al., 2012b; Stohr et al., 2012). In this work, we have tested whether inoculation of insoluble α-synuclein from brains with dementia with Lewy bodies and synthetic mouse and human α-synuclein fibrils can induce α-synuclein pathology in wild-type mice. As a result, we have established a new mouse model of sporadic α-synucleinopathy using wild-type mice.
Materials and methods
Preparation of recombinant α-synuclein monomer and fibrils
Human and mouse α-synuclein were expressed in E. coli BL21 (DE3) cells, as described (Masuda et al., 2006b). To avoid the production of α-synuclein dimers induced by misexpression of cysteine-containing α-synuclein, the Y136-TAT construct was used (Masuda et al., 2006a). α-Synuclein was purified by boiling, Q-Sepharose® ion exchange chromatography and ammonium sulphate precipitation, before dialysis against 30 mM Tris–HCl, pH 7.5. Recombinant proteins were centrifuged at 113 000g for 20 min at 4°C to remove insoluble materials and used as α-synuclein monomer. Protein concentrations were determined as described (Yonetani et al., 2009). Purified human and mouse α-synuclein (7 mg/ml) were incubated at 37°C in a shaking incubator (200 rpm) in 30 mM Tris–HCl, pH 7.5, containing 0.1% NaN3, for 72 h. α-Synuclein fibrils were pelleted by spinning the assembly mixtures at 113 000g for 20 min.
Preparation of the insoluble fraction of dementia with Lewy bodies brain
Fresh frozen brain tissue from a patient with dementia with Lewy bodies (phosphorylated α-synuclein pathology is shown in Supplementary Fig. 9) was homogenized in 18 volumes (w/v) of Buffer A (10 mM Tris-HCl, pH 7.4, 0.8 M NaCl, 1 mM EGTA, and 10% sucrose), and sarkosyl was added to the homogenate at a concentration of 2%. The mixture was incubated for 30 min at 37°C, sonicated and spun at 9100g for 10 min at 25°C. The supernatant was further centrifuged at 113 000g for 20 min at 25°C, and the sarkosyl-insoluble pellet was washed with Buffer A. The pellet was taken up in saline, sonicated and centrifuged at 800 g for 5 min. The supernatant was used for stereotaxic injection.
Stereotaxic surgery
Four- to six-month-old female C57BL/6 J mice (CLEA Japan, Inc.) anaesthetized with 50 mg/kg pentobarbital sodium were injected with 10 µg of recombinant α-synuclein monomer, fibrils or 5 µl of insoluble fraction of dementia with Lewy bodies brain into substantia nigra (anterior-posterior: −3.0 mm; medial-lateral: −1.3 mm; dorsal-ventral: −4.7 mm from the bregma and dura) using a 10 -µl Hamilton syringe. Mice were anaesthetized with isoflurane and killed by decapitation. For immunohistochemistry, brains were fixed in 10% formalin neutral buffer solution (Wako), and for biochemical analysis, brains were snap-frozen on dry ice and stored at −80°C. All experimental protocols were approved by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Medical Science.
Immunohistochemistry
Brains fixed in 10% formalin were cut on a vibratome (Leica) at 50 µm thickness. The free-floating sections were treated with 0.5% H2O2 in methanol for 30 min to inactivate peroxidase and blocked with 10% calf serum in PBS. Sections were immunostained with appropriate antibodies. Antibodies used in this study are summarized in Supplementary Table 1. After incubation with the biotinylated-secondary antibody (Vector), labelling was detected using the ABC staining kit (Vector).
Confocal microscopy
For double-label immunofluorescence for phosphorylated α-synuclein and ubiquitin or phosphorylated α-synuclein and p62, brain sections were incubated overnight at 4°C in a cocktail of 1175 and anti-ubiquitin or anti-p62 antibody. The sections were then washed and incubated in a cocktail of Alexa Fluor® 568-conjugated goat anti mouse IgG (Molecular Probes) and Alexa Fluor® 488-conjugated goat anti mouse IgG (Molecular Probes). After further washing, sections were stained with TO-PRO®-3, coverslipped with VECTASHIELD® (Vector) and observed with a laser-scanning confocal fluorescence microscope (LSM5 PASCAL, Carl Zeiss).
Biochemical analysis
Mouse brains were homogenized in 20 volumes (w/v) of Buffer A, then spun at 100 000g for 30 min at 4°C, and the supernatant was retained as buffer-soluble fraction. The pellet was homogenized in 20 volumes of Buffer A containing 1% Triton™ X-100 and incubated for 30 min at 37°C. After centrifugation at 100 000g, the Triton™-insoluble pellet was further homogenized in Buffer A containing 1% sarkosyl and incubated at 37°C for 30 min. Samples were spun at 100 000g for 30 min. The sarkosyl-pellet was sonicated in 30 mM Tris-HCl, pH 7.4, and used for immunoblotting as sarkosyl-insoluble fraction. The samples were subjected to SDS-PAGE on 12.5% polyacrylamide gel and proteins were electrotransferred onto a polyvinylidene difluoride membrane, probed with appropriate antibodies and detected as described (Nonaka et al., 2009).
Behavioural tests
Open field test
Each mouse was placed in the centre of the open field apparatus (25-cm diameter). Activity was measured by SUPERMEX system (Muromachi Kikai) over 90-min period and analysed by CompACT AMS software ver.3 (Muromachi Kikai). Total activity was measured by counting the number of photobeam interruptions over every 5-min period.
Wire hang test
Neuromuscular strength was tested with a wire hang test. The mouse was placed on a wire mesh, waved gently so that the mouse gripped the wire and then inverted. Latency to fall was recorded with a 300-s cut-off time.
Rotarod test
The Rotarod test, using an accelerating Rotarod (Muromachi Kikai), was performed by placing mice on 9-cm diameter rods and measuring the time each animal was able to maintain its balance on the rod. We used 9-cm rods to make the test more sensitive to motor skill learning (Shiotsuki et al., 2010). The speed of the rotarod accelerated from 0 to 40 rpm over a 5-min period.
Y-maze test
Y-maze apparatus (Muromachi Kikai) consisted of three arms (40 cm × 3 cm) made of grey plastic joined in the middle to form a Y shape. Mice were placed into one of the arms of the maze and allowed to explore freely the maze for an 8-min session. The alternation between arms was recorded.
Intranasal administration of abnormal α-synuclein fibrils
Twenty micrograms of recombinant α-synuclein monomer or preformed fibrils, or 10 μl of insoluble fraction of dementia with Lewy bodies brain, was administered intranasally once a week for 1 month to 10-week-old female C57BL/6J mice (soluble mouse α-synuclein, soluble human α-synuclein, mouse α-synuclein fibrils, human α-synuclein fibrils and dementia with Lewy bodies extracts, n = 5 per group). At 21 months after the last administration, mice were anaesthetized with pentobarbital sodium and killed by perfusion with phosphate buffer (pH 7.4) and 4% paraformaldehyde in 0.1% phosphate buffer. Brains were cryosectioned and immunostained as described above.
Results
To investigate whether insoluble α-synuclein fibrils can propagate in vivo, we injected recombinant human α-synuclein fibrils into the substantia nigra in the right cerebral hemisphere of C57BL/6J mice. α-Synuclein fibrils were prepared using highly purified recombinant α-synuclein (Supplementary Fig. 1A) by incubation with shaking. Formation of the fibrils was confirmed by electron microscopy (Supplementary Fig. 1B) and thioflavin S assay (data not shown). The fibrils were then collected by ultracentrifugation, sonicated and used for injection. Abnormal phosphorylated α-synuclein-positive structures were observed in the brains of mice injected with human α-synuclein fibrils at 15 months after inoculation (Fig. 1). Phosphorylated α-synuclein pathology was distributed throughout the brain including substantia nigra, amygdala, dentate gyrus, hippocampal CA1-3, molecular layer of hippocampus, fimbria, stria terminalis, hypothalamus, somatosensory area, visual cortex, cingulate cortex and corpus callosum (Fig. 1). Phosphorylated α-synuclein-positive structures were also positive for anti-ubiquitin and p62 antibodies (Fig. 2A). Co-localization was confirmed by confocal microscopy (Fig. 2B and C), indicating that these structures have the same immunoreactive properties as Lewy bodies (Kuusisto et al., 2001). By contrast, no phosphorylated α-synuclein, ubiquitin or p62-positive pathology was observed in the brains of mice injected with soluble human α-synuclein (Supplementary Fig. 2). Remarkably, despite the unilateral injection of α-synuclein fibrils, phosphorylated α-synuclein-positive pathology appeared bilaterally (Fig. 3A). In the right hemisphere (injected side), phosphorylated α-synuclein pathology was seen abundantly in dentate gyrus and amygdala, whereas in the left hemisphere no pathology was seen in amygdala and only sparsely in dentate gyrus (Fig. 3A). These results strongly suggest that α-synuclein pathology propagates throughout the brain from the injection site. To understand the spreading pathway of phosphorylated α-synuclein pathology, we investigated in detail the distribution in four coronal sections at 15 months after inoculation (Fig. 3B). Near the injection level (bregma −3.40 mm), abundant phosphorylated α-synuclein pathology was present in substantia nigra, hippocampus, external capsule and entorhinal cortex in right hemisphere, whereas in the left hemisphere, sparser pathology was detected in hippocampus and external capsule (Fig. 3B). By contrast, at the level of 0.02 mm from bregma (3 mm anterior to the injection level), phosphorylated α-synuclein pathology was concentrated in stria terminalis, septal nucleus and cingulate, motor and somatosensory cortex in the right hemisphere. In the left hemisphere, phosphorylated α-synuclein pathology was detected only in septal nucleus (Fig. 3B). These results suggest that phosphorylated α-synuclein pathology does not spread by simple diffusion and the propensity to accumulate phosphorylated α-synuclein seems to differ among brain regions. The time course of spreading of phosphorylated α-synuclein pathology was analysed by immunohistochemistry and summarized in Table 1. Table 1 clearly indicates that induction of phosphorylated α-synuclein pathology in wild-type mice is time- and brain region-dependent. No signs of astrogliosis and inflammation were observed in human α-synuclein fibril-injected mice compared with soluble-human α-synuclein-injected mice at 15 months after injection (Supplementary Fig. 3).
Figure 1.

Induction of phosphorylated α-synuclein pathology in wild-type mouse brain injected with human α-synuclein fibrils, observed at 15 months after injection. Sections were immunostained with anti-phosphorylated α-synuclein antibody, 1175. The shapes of phosphorylated α-synuclein-positive structures differed among brain areas. Ring-like and Lewy neurite-like structures were observed in substantia nigra, hippocampus, hypothalamus, somatosensory area, visual cortex, cingulate cortex and corpus callosum, whereas Lewy body- and Lewy neurite-like structures were observed in amygdala and stria terminalis.
Figure 2.
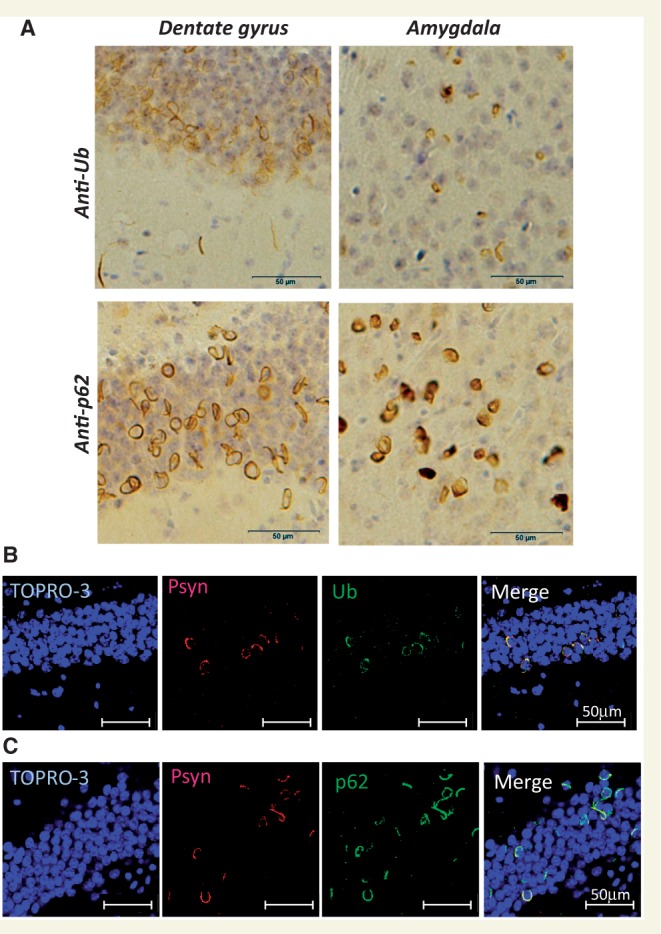
α-Synuclein pathology in fibril-injected mice brain was immunoreactive for ubiquitin (Ub) and p62. (A) Staining of dentate gyrus and amygdala of fibril-injected mice at 15 months after injection, using anti-ubiquitin (upper) and p62 (lower) antibodies. Abundant ubiquitin- and p62-positive pathology can be seen. (B and C) Double-labelled immunofluorescence of dentate gyrus for phosphorylated α-synuclein (Psyn) and ubiquitin (B) or p62 (C). Phosphorylated α-synuclein-positive structures were co-localized with ubiquitin and p62.
Figure 3.

(A) Spreading of phosphorylated α-synuclein pathology on the contralateral side of mouse brain injected with α-synuclein fibrils. Staining of dentate gyrus and amygdala in the right hemisphere (injection side) and in the left hemisphere (non-injected side) with anti-phosphorylated α-synuclein (psyn) antibody, 1175, at 15 months after injection. (B) Distribution of phosphorylated α-synuclein pathology in human α-synuclein fibril-injected mouse brain at 15 months after injection (n = 24). Four coronal sections were stained with phosphorylated α-synuclein antibody, 1175. Red dots indicates Lewy bodies- and Lewy neurites-like pathology. Near the injection level (bregma -3.40 mm), abundant phosphorylated α-synuclein pathology was present in substantia nigra, hippocampus, external capsule, and entorhinal cortex in right hemisphere, whereas in the left hemisphere, sparser pathology was detected in hippocampus and external capsule. At the level of -1.94 mm from bregma, severe phosphorylated α-synuclein pathology was present in hippocampus, amygdala, corpus callosum, hypothalamus and motor, visual, somatosensory, auditory and piriform cortex in the right hemisphere, whereas moderate phosphorylated α-synuclein pathology was observed in corpus callosum, hippocampus, external capsule and motor, somasosensory and auditory cortex in the left hemisphere. At the level of -0.58 mm from bregma, phosphorylated α-synuclein pathology was detected in amygdala, corpus callosum, fimbria, fornix, hypothalamus, striatum and somatosensory and piriform cortex in the right hemisphere, whereas in the left hemisphere, the pathology was present in corpus callosum, fimbria, fornix, hypothalamus and striatum. At the level of 0.02 mm from bregma, phosphorylated α-synuclein pathology was concentrated in stria terminalis, septal nucleus and cingulate, motor and somatosensory cortex in the right hemisphere. In the left hemisphere, phosphorylated α-synuclein pathology was detected only in septal nucleus. Dashed box indicates substantia nigra (injection site). L = left hemisphere of brain; R = right hemisphere.
Table 1.
Semi-quantitative grading of α-synuclein pathology in mice injected with human α-synuclein fibrils
| Non-injection side | Injection side | |||||||
|---|---|---|---|---|---|---|---|---|
| (left hemisphere) |
(right hemisphere) |
|||||||
| Time from injection (days) | Time from injection (days) | |||||||
| 90 | 180 | 450 | 90 | 180 | 450 | |||
| Bregma | 0.02 mm | Stria terminalis | − | − | − | − | ++ | +++ |
| Striatum | − | + | + | + | ++ | ++ | ||
| Cigular cortex | − | − | − | − | + | + | ||
| Septal nucleus | − | − | − | − | + | + | ||
| Bregma | −0.58 mm | Corpus callosum | − | − | + | − | − | ++ |
| Fornix | − | + | ++ | − | + | ++ | ||
| Hippocampal commissure | − | + | ++ | − | + | ++ | ||
| Amygdala | − | − | − | + | +++ | +++ | ||
| Globus pallidus | − | + | + | − | + | ++ | ||
| Striatum | − | − | + | + | + | + | ||
| Somatosensory area | − | − | + | − | + | + | ||
| Insular cortex | − | − | − | + | + | + | ||
| Bregma | −1.94 mm | Corpus callosum | − | − | ++ | − | − | ++ |
| Hippocampus | − | + | +++ | + | ++ | +++ | ||
| Habenular nucleus | − | − | + | − | − | +++ | ||
| Fimbria | − | + | +++ | − | + | +++ | ||
| Amygdala | − | − | − | ++ | +++ | +++ | ||
| Hypothalamus | − | − | + | + | + | ++ | ||
| Thalamus | − | − | − | − | − | + | ||
| Visual cortex | − | − | + | − | + | ++ | ||
| Somatosensory area | − | + | + | − | + | ++ | ||
| Auditory cortex | − | − | + | + | + | ++ | ||
| Piriform cortex | − | − | + | + | + | ++ | ||
| External cupsule | − | − | + | − | − | ++ | ||
| Bregma | −3.40 mm | Substantia nigra | − | − | − | + | + | + |
| Hippocampus | − | + | ++ | + | ++ | ++ | ||
| Superior colliculus | − | + | + | − | + | ++ | ||
| External cupsule | − | − | + | − | − | + | ||
| Visual cortex | − | − | − | + | + | + | ||
| Auditory cortex | + | + | + | + | ++ | ++ | ||
| Entorhinal cortex | − | + | + | + | ++ | ++ | ||
Four coronal sections were stained with anti-phosphorylated α-synuclein antibody at 90, 180 or 450 days after injection. Grading of α-synuclein pathology was performed as follows: −, none; +, slight; ++, moderate; +++, severe. At 90 days after injection, small amounts of phosphorylated α-synuclein-positive structures were observed in substantia nigra, amygdala, striatum, hypothalamus, hippocampus, and stria terminalis in the right hemisphere of brain (injected side), but very few Lewy neurites were detected in cortex in the left hemisphere. At 180 days post-injection, the amount of phosphorylated α-synuclein-positive pathology was increased and was more widely spread in the right hemisphere, while in the left hemisphere, little phosphorylated α-synuclein pathology was apparent in hypothalamus, hippocampus, striatum or globus pallidus. At 450 days (15 months) after injection, phosphorylated α-synuclein pathology had spread throughout the right hemisphere and the left hemisphere.
To clarify which α-synuclein species accumulated in the mice, and when, we performed immunoblot analysis with LB509 and anti-mouse synuclein antibodies, which specifically recognize human α-synuclein and mouse α-synuclein, respectively. The antibody specificities are shown in Supplementary Fig. 1B. At a few hours after injection (Day 0), injected recombinant human α-synuclein fibrils were detected in the sarkosyl-insoluble fraction of the right and left hemispheres by LB509 antibody, suggesting that injected human α-synuclein fibrils in the extracellular space spread quickly throughout the brain. However, at 7 days after injection, the human α-synuclein immunoreactivities had disappeared, and did not reappear at 30 or 90 days after injection (Fig. 4). At 90 days after injection, anti-phosphorylated α-synuclein-positive 15, 20, 30 and 35 kDa bands were detected in the sarkosyl-insoluble fractions. This band pattern is indistinguishable from that of pathological α-synuclein in dementia with Lewy bodies brain (Fig. 4). The 15, 20, 30 and 35 kDa bands correspond to α-synuclein monomer, mono-ubiquitinated α-synuclein, dimer and ubiquitinated dimer, respectively. Most interestingly, anti-mouse α-synuclein strongly labelled the sarkosyl-insoluble phosphorylated α-synuclein-positive bands at Day 90, but these were not immunostained with LB509. These results clearly show that endogenous mouse α-synuclein is accumulated as phosphorylated and ubiquitinated forms. Immunohistochemical analysis with anti-tyrosine hydroxylase suggested that dopaminergic neurons are retained in substantia nigra of human α-synuclein fibril-injected mice at 6 months after injection (Fig. 5A and B). However, dramatic loss of the neurotransmitter enkephalin was observed in globus pallidus and amygdala central nucleus, where abundant phosphorylated α-synuclein-positive structures are detected (Fig. 5C and D). These data suggest that neuronal dysfunction occurs without apparent neuronal loss. We also performed behavioural analyses of mice injected with soluble human α-synuclein monomers or human α-synuclein fibrils. However, significant differences were not observed in open field test, wire hang test, rotarod test and Y-maze test (Supplementary Fig. 4) at 6 months after injection.
Figure 4.

Endogenous mouse α-synuclein was aggregated in wild-type mouse brain injected with human α-synuclein (hsyn) fibrils. The brain was divided into two parts at the longitudinal fissure of the cerebrum. Sarkosyl-insoluble fractions were obtained from the right and left hemispheres, and analysed by immunoblotting with #64, LB509 or anti-mouse α-synuclein (msyn) antibodies. Representative images are shown (n = 14). Sarkosyl-insoluble phosphorylated α-synuclein (psyn) started to accumulate, predominantly in the right hemisphere, at 90 days after injection. It was composed of endogenous mouse α-synuclein, not exogenous human α-synuclein.
Figure 5.
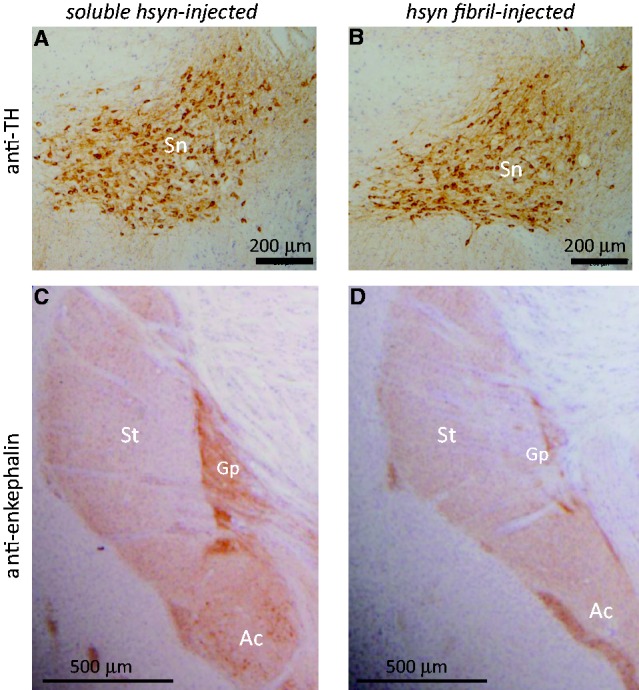
Fibril-injected mice show apparent reduction of a neurotransmitter enkephalin in amygdala central nucleus and globus pallidus at 15 months after injection. Brain sections were stained with anti-tyrosine hydroxylase (TH) (A and B) and anti-enkephalin (C and D) antibodies. Ac = amygdala central nucleus; Gp = globus pallidus; Sn = substantia nigra; St = striatum.
Next, we tested whether fibrils composed of recombinant mouse α-synuclein can induce α-synuclein pathology more efficiently than those composed of human α-synuclein, because the sequences of human and mouse α-synuclein are slightly different (Supplementary Fig. 5), and there could be a species difference. Mouse α-synuclein complementary DNA was cloned, and the protein was expressed in Escherichia coli and purified. Fibrils or soluble mouse α-synuclein were inoculated into substantia nigra of wild-type mouse brains and the pathology was evaluated. Strikingly, all the mice injected with mouse α-synuclein fibrils developed phosphorylated α-synuclein pathology in the injected side of the brain, whereas no pathology was detected in mice injected with soluble mouse α-synuclein (Table 2). The phosphorylated α-synuclein pathologies were basically the same as those of mice injected with human α-synuclein fibrils (data not shown). The efficiency of the induction of phosphorylated α-synuclein pathology by human α-synuclein fibrils was ∼90% (Table 2), which is quite high, but slightly lower than that with mouse α-synuclein fibrils, suggesting that there may be a small species difference between mouse and human α-synuclein.
Table 2.
Comparison of propagation efficiency in mice at 15 months after injection
| Injection samples | anti-psyn | Right hemisphere (injection side) anti-ubiquitin | anti-p62 | Left hemisphere (non-injected side) anti-psyn | |
|---|---|---|---|---|---|
| Soluble human α-syn | (n = 8) | 0/8 (0%) | 0/8 (0%) | 0/8 (0%) | 0/8 (0%) |
| Insoluble human α-syn fibril | (n = 24) | 22/24 (91.6%) | 21/24 (87.5%) | 22/24 (91.6%) | 19/24 (79.2%) |
| Soluble mouse α-syn | (n = 4) | 0/4 (0%) | 0/4 (0%) | 0/4 (0%) | 0/4 (0%) |
| Insoluble mouse α-syn fibril | (n = 8) | 8/8 (100%) | 7/8 (87.5%) | 8/8 (100%) | 8/8 (100%) |
| DLB brain extracts | (n = 14) | 7/14 (50%) | 0/14 (0%) | 5/14 (35.7%) | 1/14 (7.1%) |
In the right hemisphere, mice showing immunopositive structures for anti-phosphorylated α-synuclein (psyn), ubiquitin (Ub) or p62 were counted. In the left hemisphere, mice showing immunopositive structures for anti-phosphorylated α-synuclein were counted. Values show number of immunopositive mice/total mice, with percentage of immunopositive mice. DLB = dementia with Lewy bodies.
Finally, we tested whether pathological α-synuclein deposited in the brains of patients has similar prion-like properties in brains of wild-type mice. Surprisingly, pathological α-synuclein-enriched fractions also induced phosphorylated α-synuclein-positive pathologies in various areas of brain, including the substantia nigra, amygdala, hippocampus, striatum, hypothalamus, somatosensory area, motor cortex, piriform cortex and superior colliculus (Fig. 6). In brains of these mice, the phosphorylated α-synuclein-positive pathologies mostly resembled Lewy neurite-like structures. Lewy body-like pathology was detected only in amygdala and piriform cortex. The percentage of mice that developed phosphorylated α-synuclein pathology in the injected side of the brains was 50% in the group injected with insoluble phosphorylated α-synuclein of dementia with Lewy bodies brains, which is less than that in mice injected with recombinant α-synuclein fibrils (Table 2). Thus, these results demonstrate that inoculation of either pure synthetic recombinant α-synuclein fibrils or dementia with Lewy bodies brain extracts into wild-type mice can induce Lewy body/neurite-like phosphorylated α-synuclein pathology efficiently and reproducibly. Our results raise an important question, i.e. whether or not α-synuclein fibrils are transmissible among individuals. To test this possibility, we intranasally administered at high concentrarion of abnormal α-synuclein fibrils (preformed recombinant human or mouse α-synuclein fibrils) or the insoluble fraction from dementia with Lewy bodies brain to normal mice. However, no pS129-positive abnormal structures were detected in the brain at 21 months after the final administration (Supplementary Fig. 6), even with highly sensitive immunohistochemical staining, suggesting that the abnormal α-synuclein cannot pass through the nasal mucosa.
Figure 6.

α-Synuclein pathology in wild-type mice brain injected with dementia with Lewy bodies-insoluble fraction observed at 15 months after injection. Sections were immunostained with anti-phosphorylated α-synuclein antibody, 1175.
Discussion
In this study, we have shown that the inoculation of α-synuclein fibrils made of recombinant α-synuclein or dementia with Lewy bodies brain extracts into wild-type mouse brain is sufficient to cause the appearance of Lewy body/neurite-like α-synuclein pathology in vivo. Similar work was recently published by Luk et al. (2012a) but there are important differences between our study and theirs. Luk et al. (2012a) showed that only inoculation of synthetic mouse α-synuclein fibrils into wild-type mouse brain induced synuclein pathology. In our present study, we inoculated not only fibrils made of recombinant mouse α-synuclein but also ones from human α-synuclein fibrils, and importantly also insoluble α-synuclein from dementia with Lewy bodies brains, into wild-type mouse brain. This is the first report showing efficient induction of α-synuclein pathology by inoculation of material from human brain. Furthermore, our biochemical analyses clearly demonstrate that endogenous mouse α-synuclein is converted into abnormal form and deposited in neurons of the brain through a prion-like mechanism or by seed-dependent aggregation by crossing the species barrier (Fig. 4). Since soluble α-synuclein never induced such pathology (Supplementary Fig. 2), we can conclude that the structural difference between soluble and filamentous forms of α-synuclein, i.e. cross-β structure in the α-synuclein fibrils (Serpell et al., 2000) is critical for the pathogenesis. It has been reported that recombinant α-synuclein fibrils enhance the initiation and progression of α-synuclein pathology in transgenic mice overexpressing mutant α-synuclein (Mougenot et al., 2012; Luk et al., 2012b) and wild-type mice (Luk et al., 2012a). In those models, α-synuclein pathology appeared at 90 days after inoculation. In our mouse model, abnormal phosphorylated α-synuclein pathology was also detected at 90 days after injection (Fig. 4 and Table 1), suggesting that it takes about this length of time for the formation of abnormal phosphorylated α-synuclein pathology in vivo after the seeding procedure. Despite a diffusion of injected exogenous α-synuclein fibrils to the bilateral sides of brain within a few hours after injection (Fig. 4), phosphorylated α-synuclein pathology seems to be initiated in the injected side and to spread from the injected side to the non-injected side in a time-dependent manner (Table 1). Thus, it is reasonable to speculate that exogenous fibrils enter neurons at the injection site as a result of infusion pressure, a temporary high concentration, or some other mechanism, and then the pathological process starts to develop from these cells.
Propagation patterns of pathology in the inoculated mice were basically identical regardless of the species of injected seeds (i.e. recombinant human α-synuclein fibrils, mouse α-synuclein fibrils or dementia with Lewy bodies brain extracts), but extracts of brains with dementia with Lewy bodies showed lower propagation efficiency than recombinant fibrils (Table 2). This relatively low efficiency may be explained by the lesser amount of abnormal α-synuclein contained in the dementia with Lewy bodies brain extracts. Comparison of human α-synuclein fibrils and mouse α-synuclein fibrils indicated that mouse α-synuclein fibrils showed slightly higher efficiency (Table 2). In vitro experiments also indicated that mouse α-synuclein fibrils promote fibrillization of the soluble mouse α-synuclein monomer faster than human α-synuclein fibrils (Supplementary Fig. 7). It is well known that prion propagation can cross the species barrier (Prusiner, 1993) and the efficiency of propagation depends on the amino acid sequences of prion proteins. In the case of α-synuclein, mouse α-synuclein and human α-synuclein share 95% amino acid sequence homology (Supplementary Fig. 5), and this may be the reason why endogenous mouse α-synuclein is capable of aggregation by inoculation of human α-synuclein fibrils. Another factor may be that mouse α-synuclein protein has a threonine residue at amino acid position 53 (Supplementary Fig. 5), which is known as an aggregation-prone mutation in familial Parkinson’s disease (Polymeropoulos et al., 1997).
Time course analyses of the pathology in these mice (Table 1) showed that at 90 days after injection, phosphorylated α-synuclein pathology was mainly observed near the injection level, but also seen in striatum, amygdala, stria terminalis and dentate gyrus: areas far from the injection site had developed pathology. The striatum and the amygdala central nucleus have projections from substantia nigra, and the stria terminalis serves as a major output pathway of the amygdala (Supplementary Fig. 8). Although the dentate gyrus does not have direct projection to substantia nigra, regions connecting with dentate gyrus (i.e., hippocampal CA1, CA3, entorhinal cortex, fimbria, fornix and hippocampal commissure) also showed moderate pathology (Table 1). These results may indicate that α-synuclein pathology propagates unidirectionally through the neural circuit (Supplementary Fig. 8). Spread of pathology from the right hemisphere to the left hemisphere might occur via the corpus callosum, hippocampal commissure, etc., connecting with the contralateral side of the brain (Fig. 3B and Table 1). Phosphorylated α-synuclein pathology in our mouse model was mainly observed in neurons and was hardly detected in glial cells, while the band pattern of sarkosyl-insoluble phosphorylated α-synuclein in mice was indistinguishable from that of dementia with Lewy bodies brains (Fig. 4), where phosphorylated α-synuclein pathology was mainly seen in neurons. Although the mechanism remains to be clarified, exogenous α-synuclein fibrils may enter cells through a selective mechanism(s), such as neuron-specific receptors. Alternatively, differences in expression levels of endogenous α-synuclein or cellular environments may also be important for formation of the pathology, even if abnormal α-synuclein has already entered the cells.
Luk et al. (2012a) reported dopaminergic neuronal loss and motor dysfunction (by Rotarod test and wire hang test) in wild-type mice injected with mouse α-synuclein fibrils at 6 months after inoculation into striatum. In contrast, our human α-synuclein or mouse α-synuclein fibril-injected mice did not show any motor and cognitive deficits at 6 months after inoculation and dopaminergic degeneration even after 15 months, a dramatic reduction of enkephalin was observed in the amygdala central nucleus and globus pallidus, with severe pathology, at 6 months after injection (Fig. 5 and Supplementary Fig. 4). The different phenotypes of these mice might be explained by differences in the injection sites [striatum in Luk et al. (2012a) and substantia nigra in our study]. Nonetheless, the spreading pattern of the pathological α-synuclein is different between our study and theirs. Differential vulnerability of neurons to these abnormal proteins may also affect phenotypes of these mice.
In summary, we have shown that intracerebral injection of insoluble α-synuclein fibrils can induce aggregation of endogenous mouse α-synuclein through a prion-like propagation mechanism. Our data suggest that phosphorylated α-synuclein pathologies do not induce acute neuronal loss but induce a slow neurodegeneration by disrupting neuronal function. These models should be useful not only for elucidating the molecular mechanisms of propagation of intracellular abnormal proteins, but also for development and evaluation of disease-modifying therapy.
Funding
This work was supported by MEXT KAKENHI Grant Numbers 12937622, 12901980 (to M.H.), JSPS KAKENHI Grant Number 11024780 (to M.M-S.) and MHLW Grant Number 12946221 (to M.H.).
Supplementary material
Supplementary material is available at Brain online.
References
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–4. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–71. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–6. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–90. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012a;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M, Dohmae N, Nonaka T, Oikawa T, Hisanaga S, Goedert M, et al. Cysteine misincorporation in bacterially expressed human alpha-synuclein. FEBS Lett. 2006a;580:1775–9. doi: 10.1016/j.febslet.2006.02.032. [DOI] [PubMed] [Google Scholar]
- Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, et al. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006b;45:6085–94. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchere J, Lakhdar L, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–8. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Muller CM, de Vos RA, Maurage CA, Thal DR, Tolnay M, Braak H. Staging of sporadic Parkinson disease-related alpha-synuclein pathology: inter- and intra-rater reliability. J Neuropathol Exp Neurol. 2005;64:623–8. doi: 10.1097/01.jnen.0000171652.40083.15. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18:3353–64. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–98. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Genetic and infectious prion diseases. Arch Neurol. 1993;50:1129–53. doi: 10.1001/archneur.1993.00540110011002. [DOI] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci USA. 2000;97:4897–902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotsuki H, Yoshimi K, Shimo Y, Funayama M, Takamatsu Y, Ikeda K, et al. A rotarod test for evaluation of motor skill learning. J Neurosci Methods. 2010;189:180–5. doi: 10.1016/j.jneumeth.2010.03.026. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–73. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, Dearmond SJ, et al. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci USA. 2012;109:11025–30. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180–2. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- Yonetani M, Nonaka T, Masuda M, Inukai Y, Oikawa T, Hisanaga S, et al. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–50. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.