

Abstract
Asthma remains an important cause of morbidity and mortality with an incidence that continues to rise. Despite the importance of this disease, the mechanisms by which the host develops allergic airways disease remain poorly understood. The development of allergic airways disease appears to be contingent on activation of both the innate and adaptive immune system, but little is known about the cross-talk between these two systems. The extracellular matrix protein mindin (Spondin 2) has been previously demonstrated to have functional roles in both the innate and adaptive immunological responses. Previous work supports that pulmonary challenge with fungal-associated allergenic proteinase (FAP) induces an innate allergic response. We hypothesized that mindin would modify the biological response to FAP. Saline or FAP was administered by oropharyngeal aspiration to C57BL/6 wild type or mindin-null mice every 4 days for a total of five exposures. FAP exposed C57BL/6 mice developed enhanced airway hyperresponsiveness (AHR) to methacholine challenge and increased neutrophils and eosinophils in the bronchoalveolar lavage as compared to saline exposed controls. These responses were significantly reduced in mindin-null mice exposed to FAP. FAP challenge was associated with a broad induction of cytokines (IL-1β, TNFα, Th1, Th2, and IL-17), chemokines, and growth factors, which were reduced in mindin-null mice exposed to FAP. RNA expression in lung monocytes for representative M1 and M2 activation markers were increased by FAP, but were independent of mindin. Our observations support that challenge with FAP results in activation of both innate and adaptive immune signaling pathways in a manner partially dependent on mindin. These findings suggest a potential role for the extracellular matrix protein mindin in cross-talk between the innate and adaptive immune systems.
Keywords: Environment, Asthma, Reactive airways disease, Extracellular matrix, Allergy, Aspergillus
Introduction
Asthma remains a significant cause of morbidity and a frequent presenting complaint for acute/emergency care and hospitalizations. It is a particular burden in industrialized nations for reasons which remain unclear. Asthma prevalence continues to increase in the United States with the most recent CDC analysis revealing a rise in the incidence from 7.3% to 8.2% from 2001 to 2009 [1]. Asthma is a chronic respiratory syndrome characterized by inflammation, mucus production, airflow limitation, and AHR. Understanding the mechanisms that contribute to the development, progression, and exacerbation of allergic asthma remain an area of high clinical interest.
Previous studies clearly identify a complex interaction between the innate immune system and the adaptive immune system in the development of allergic airways disease. The most common animal model system of allergic airways disease utilizes ovalbumin (OVA) as an allergen, which requires the use of a strong adjuvant such as alum for sensitization. In the absence of a robust adjuvant, low level exposure to the inert antigen OVA induces immunological tolerance [2,3]. We now recognize that the adjuvant alum activates innate immunity through uric acid-dependent activation of the inflammasome [4-6]. Alternatively, airway sensitization to OVA can be facilitated through activation of toll-like receptors with ligands such as LPS [7]. Therefore, the OVA model of allergic airways disease appears to require activation of innate immunity to achieve sensitization. It is additionally recognized that after established antigen sensitization activation of innate immune pathways can either enhance allergic inflammation or exacerbate airways disease through neutrophil-predominant inflammation [7].
Despite the utility of the OVA model system for understanding the mechanisms of allergic inflammation, the OVA model will not necessarily elucidate the mechanisms required for response to some clinically relevant and commonly encountered allergens. In contrast to the OVA model, pollen and fungal-derived allergens, such as those derived from Aspergillus, can initiate allergic inflammation on direct airway challenge without need for additional adjuvants [8-10]. Previous work supports that these allergens are an abundant source of exogenous proteinases; and protease activity is required for development of allergic airways disease [11-13]. Studies utilizing protease-dependent allergens will provide novel insight into the pathogenesis of allergic airways disease in a manner independent of a requirement for use of robust adjuvant to achieve sensitization.
Fungal antigens appear to induce innate allergic inflammation. A single airway challenge to FAP is sufficient to induce both eosinophilic airway inflammation and lung IL-4 production [14]. Interestingly, the response to FAP is independent of both classical cells of the adaptive immune system and genes of innate immunity including: TLR4, MyD88, and STAT6. However, the innate allergic response to FAP was dependent on intact proteinase activity. These observations suggest that FAP induces allergic inflammation through a unique and potentially unrecognized innate immune signaling mechanism.
Mindin is a member of the mindin/F-spondin family of secreted extracellular matrix (ECM) proteins. Mindin has two structural domains including an F-spondin (FS) domain and a thrombospondin type 1 repeat (TSR). FS1 and FS2 are the only conserved sequence motifs of the f-spondin domain in mindin, when compared to F-spondin. It is now recognized that the mindin FS domain function as an integrin ligand and that mindin recognizes LPS through the TSR domain [15,16]. Mindin was original discovered in zebrafish where it was found in the basal lamina [17]. Previous work identified this ECM protein as a pattern-recognition molecule, which directly binds to bacteria and functions as an opsonin to direct macrophage phagocytosis [18]. This work highlighted the critical role of mindin to the innate immune response to bacterial pathogens. More recently mindin was demonstrated to be important in the intranasal clearance of viruses suggesting that its functions are not limited to bacterial pathogens [19]. In addition to the role in innate immunity, mindin has a role in the development of adaptive immune responses as it is important for both efficient dendritic cell priming of T cells and eosinophil trafficking into the airspace [20,21]. Interestingly, mindin contributes to the airway response following inhalation exposure to endotoxin and ambient ozone [22]. The host response to ozone is believed to be dependent on genes of innate immunity [23-25]. Together these findings support that mindin has critical roles in both innate and adaptive immune system activation. However, the role of mindin in fungal antigen challenge has remained unexplored. Therefore, we hypothesized that the extracellular matrix mindin would be critical to the development of fungal allergen-induced allergic airways disease.
In the present study, we evaluated the role of mindin on the development of allergic airways disease after FAP exposure. We identified that mindin is required for the development of AHR and recruitment of granulocytes into the airspace after FAP exposure. We further identified that mindin deficiency result in broad reduction of pro-inflammatory secreted factors. These data highlight a role of mindin in the development of airways disease in context of fungal allergen challenge.
Methods
Animals
C57BL/6 mice were purchased from Jackson Laboratory. Mindin−/− mice were generated by Y-W He as previously reported and backcrossed onto C57BL/6 [18]. Experimental groups consisted of 6-8 week old female mice for all experiments. Animal experiments were conducted in accordance with National Institute of Health guidelines and protocols approved by the Animal Care and Use Committee at Duke University.
Exposure protocol
Following administration of isofluorane for anesthesia, C57BL/6 and Mindin −/− mice were administered either saline or FAP by oropharyngeal aspiration. FAP was composed of Aspergillus oryzae proteinase (1 mg/ml; Sigma, St. Louis, MO, USA) and ovalbumin (0.5 mg/ml; Sigma, St. Louis, MO, USA) in a volume of 50 μL. This dosing was repeated every four days for a total of five challenges per a previously published protocol [11,26]. The mice were characterized for physiologic measurement and BAL cytokine and cell count analysis 24-hours after the last saline/FAP challenge.
Airway physiology
Direct measurements of respiratory mechanics in response to methacholine were made using the flexiVent system (SCIREQ, Montreal, Canada) and reported as total resistance (RT) cmH2O/mL/s. Anesthesia was achieved with 60 mg/kg pentobarbital sodium injected i.p. Mice were then given a neuromuscular blockade (0.8 mg/kg pancuronium bromide). Mice were ventilated with a computer-controlled ventilator (flexiVent, SCIREQ) with a tidal volume of 7.5 mL/kg and a positive end-expiratory pressure of 3 cmH2O. Measurements of respiratory mechanics were made by the forced oscillation technique. Response to aerosolized methacholine (0 mg/mL, 10 mg/mL, 25 mg/mL, and 100 mg/mL) was determined by resistance measurements every 30 s for 5 min, ensuring the parameters calculated had peaked. Total lung capacity breaths were administered after each dose of methacholine to maintain patent airways and return the measurements back to baseline. The resistance measurements were then averaged at each dose and graphed (RT cmH2O/mL/s) along with the initial baseline measurement.
Bronchoalveolar lavage fluid (BALF)
Immediately after pulmonary function measurements, mice were euthanized by Nembutal (100mg/kg) overdose. The chest was opened, the trachea was exposed, and bronchoalveolar lavage (BAL) was performed by inserting a catheter into the mouse trachea with PE-90 tubing and instilling saline until the lung reached total lung capacity. This was repeated three times. The total volume returned was the lavage return volume. Cells from the BALF were isolated using centrifugation (1500 rpm, 15 min) and the supernatant was stored at −80°C for assessment of cytokines.
Cytokine measurements
Cytokines and chemokines were analyzed using a MILLIPLEX™ MAP Mouse Cytokine/Chemokine 22 Plex assay kit (Millipore, Billerica, MA, USA) per the manufacturer’s protocol. This is a bead-based suspension array using the Luminex technology (Bio-Rad, Hercules, CA, USA) in which fluorescent-coded beads, known as microspheres, have cytokine capture antibodies on the bead surface to bind the proteins. Serum total IgE levels were determined by ELISA (Biolegend, San Diego, CA, USA).
Lung digestion and mononuclear cell enrichment
After airway physiology measurements and BAL the lungs were removed from the mouse and placed in a tissue culture dish. They were minced and digested for 40 minutes at 37°C in HBSS with 1 mg/mL collagenase A (Roche, Indianapolis, IN, USA) and 0.2 mg/mL of DNase1 (Sigma, St. Louis, MO, USA). The digestion solution was passed through a 70-μm mesh strainer and centrifuged at 535 × g at room temperature over an 18% nycodenz (Accurate Chemical Co, Westbury, NY, USA) cushion. Low density cells were collected, underwent red blood cell lysis and then washed × 2. The cell pellet was then processed for RNA analysis.
Real time PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen Inc., Valencia, CA, USA), according to the manufacturer’s protocol. Samples are treated with DNase treatment and removal reagents to remove DNA contamination (Catalogue #AM1906, Ambion Inc., Austin, TX, USA). RNA samples were then reverse-transcribed into cDNA using Super-Script II RT (Invitrogen Corp., Carlsbad, CA, USA) following the manufacturer’s instruction. All real-time quantitative PCRs were performed using ABI SDS 7500 and SYBR Green reagent (Applied Biosystems, Foster City, CA, USA). Primers were designed using ABI software and were produced by IDT, Inc. (Coralville, IA, USA). The sequences are as follows: STAT1 forward, GACCACGCCTTTGGGAAGTA; STAT1 reverse, TCGCTTAGGGTCGTCAAGCT; STAT6 forward, GGTGGTCATCGTGCATGGTA; STAT6 reverse, GGCATTGTCCCATAGGATGGT; Arginase-1 forward, CATGGGCAACCTGTGTCCTT; Arginase-1 reverse, TCCTGGTACATCTGGGAACTTTC; NOS-2 forward, GGCAGCCTGTGAGACCTTTG; NOS-2 reverse, TGAAGCGTTTCGGGATCTG; Mannose Receptor C1 forward, CCCAAGGGCTCTTCTAAAGCA; Mannose Receptor C1 reverse, CGCCGGCACCTATCACA. PCR amplification was performed by the following program: 50°C, 2 min; 95°C, 10 min; 95°C 15s, 60°C 1 min, for 40 cycles. Gene expression values were normalized to the housekeeping gene 18s. The data were presented as a fold change compared to 18s.
Statistics
Data are expressed as mean ± standard error. The statistical difference between groups was assessed by Student’s t test. A two-tailed p value of < 0.05 was considered statistically significant.
Results
Sub-acute exposure to FAP enhanced AHR to methacholine challenge and was dependent on mindin
We were interested to understand the role that mindin would have on the development of allergic airways disease after exposure to FAP. To do this we utilized an established model of repeated exposure to saline control or FAP that was initiated on day 1, and repeated every 4th day for a total of five exposures as previously described [11]. The mice received the final dose of either saline or FAP and then were phenotyped 24-hours after the last challenge. Mice underwent assessment of AHR by flexiVent after increasing doses of methacholine. Basal measurement of C57BL/6 and mindin−/− mice exposed to saline demonstrated no difference between the groups in terms of AHR (Figure 1). C57BL/6 mice exposed to FAP had a significant increase in AHR at the 25 and 100mg/mL dose of methacholine as compared to saline exposed C57BL/6 mice (Figure 1, C57BL/6 saline shaded box, C57BL/6 FAP closed box). This response was almost completely abrogated in the mindin−/− mice (Figure 1 open box). These data strongly support that AHR develops in response to repeated allergic challenge of the lower respiratory tract with FAP, a result that is dependent on the extracellular matrix protein mindin.
Figure 1. Airway physiology measurements in C57BL/6 and Mindin −/− mice after saline or FAP.

C57BL/6 and Mindin−/− mice were exposed to saline or FAP every 4 days for a total of five exposures. The mice were then assessed for airway hyperresponsiveness to escalating doses of methacholine. Saline exposed C57BL/6 (dotted box) and Mindin−/− (shaded box) mice demonstrated no difference in response to methacholine. C57BL/6 mice exposed to FAP (closed box) had a significant increase in AHR as compared to saline controls. This effect was reduced in the Mindin −/− mice exposed to FAP (open box). (*P<0.05 between C57BL/6 saline and FAP exposed and C57BL/6 and Mindin−/− FAP exposed mice, N=6, representative of two experiments).
FAP exposure enhances total cell counts, neutrophils and eosinophils in the airspace which was partially dependent on mindin
To expand upon the interaction between exposure to FAP and mindin in the development of allergic airways disease, we collected and analyzed BAL fluid for cell counts and differentials from C57BL/6 and mindin−/− mice after exposure to saline and FAP. There was no difference in the total cell counts of C57BL/6 and mindin−/− exposed to saline (Figure 2A). FAP exposed C57BL/6 mice had a significant increase in total cells as compared to saline exposed C57BL/6 mice (Figure 2A). Interestingly, on cell differential analysis there was an increase in both neutrophils (Figure 2B) and eosinophils in the FAP exposed C57BL/6 mice (Figure 2C). FAP exposed Mindin−/− mice also had an increase in total cells, neutrophils and eosinophils as compared to mindin−/− saline exposed mice (Figure 2A-C). However these were all significantly reduced as compared to FAP exposed C57BL/6 mice. We did not observe mindin-dependent differences in the absolute number of macrophages. The results highlight that FAP results in both neutrophil and eosinophil influx onto airway and alveolar surfaces and that this response is partially dependent on mindin.
Figure 2. Cell counts and differentials in C57BL/6 and Mindin −/− mice after saline or FAP.
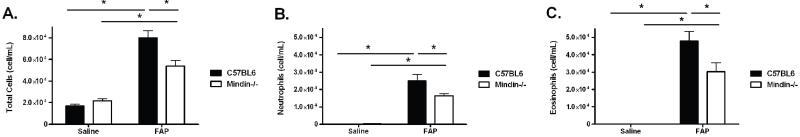
After physiologic measurements, bronchoalveolar lavage was performed for total cell count and differentials for C57BL/6 wild type (closed box) and Mindin −/− (open box) after either saline or FAP. (A) Total cell count, (B) neutrophils, and (C) eosinophils are all elevated in wild type mice after FAP exposure when compared to saline or mindin−/− mice exposed to FAP. (*P<0.05 between C57BL/6 saline and FAP, Mindin−/− saline and FAP and C57BL/6 and Mindin−/− FAP exposed mice, N=6, representative of two experiments).
FAP enhances levels of innate and adaptive pro-inflammatory factors in a manner partially dependent on mindin
To help elucidate factors influential to the cellular influx, we analyzed a variety of cytokines in the bronchoalveolar lavage fluid from WT and mindin−/− exposed to saline or FAP. WT and mindin−/− saline exposed mice expressed low levels of cytokines and there were no differences in the activity levels of cytokines between the two groups. FAP exposure resulted in enhanced expression of inflammatory cytokines IL-1β and TNF-α (Figure 3A, closed box), Th1 cytokines IL-12p70 and IFN-γ (Figure 3B), Th2 cytokines IL-13 and IL-4 (Figure 3C), the Th17 cytokine IL-17 (Figure 3D), a variety of chemokines including CCL2, CCL4, CXCL1 and CXCL10 (Figure 3E), eotaxin (Figure 3F) and the growth factors GM-CSF and G-CSF (data not shown). In FAP exposed mindin−/− mice these cytokine elevations were all reduced (Figure 3A-E, open box). We analyzed serum levels of total IgE in order to determine whether there was evidence of altered allergic sensitization in a manner dependent on mindin. While FAP did cause increased total serum IgE, there was no difference in the level of total serum IgE between WT and mindin −/−. This suggested that allergic sensitization to FAP appeared independent of mindin (Supplemental Figure 1). Based on the observation of differences in both Th1 and Th2 cytokine production, we were interested to determine if the effect of FAP and mindin was related to polarization of macrophages into either M1 or M2 phenotypes. We analyzed the mRNA expression from whole lung digests which were enriched for mononuclear cells. FAP exposure caused increased expression of both traditional M1 (STAT1; Supplemental Figure 2A) and M2 (Arginase-1, Mannose Receptor, and STAT6; Supplemental Figure 2B) markers. There also was a trend to increase mRNA expression of NOS-2 (Supplemental Figure 2A) which did not reach statistical significance. Though we identified a broad increase in macrophage expression in response to FAP, we did not identify differences in the mRNA expression between WT and mindin −/− mice (Supplemental Figure 2A, B). This supports that the effect of FAP on macrophage mRNA expression of these M1 and M2 markers appeared to be independent of mindin. These data highlight that FAP causes a broad array of Th1, Th2, Th17, and cytokine/chemokine stimulation, which is associated with enhanced expression of markers of both M1 and M2 activation. While the profound effects of mindin do not appear associated with classic markers of M1/M2 activation, our observations do support a broad impact of mindin on the production of pro-inflammatory factors associated with activation of both the innate and adaptive immunological response.
Figure 3. BAL cytokine analysis in C57BL/6 and Mindin −/− mice.

Using the cell-free supernatants from the BAL in the C57BL/6 saline (shaded box), Mindin−/− saline (checkered box), C57BL/6 FAP (closed box) and Mindin−/− FAP (open box) a multiplex was performed to assess protein levels reflecting activation of either innate or adaptive immunity including: (A) IL-1β and TNF-α; (B) Th1 cytokines IL-12p70 and IFN-γ; (C) Th2 cytokines IL-13 and IL-4; (D) Th17 cytokine IL-17; (E) chemokines CCL2, CCL4, CXCL1 and CXCL10; and (F) eotaxin. (*P<0.05 between C57BL/6 saline and FAP exposure and C57BL/6 and Mindin−/− FAP exposure, N=6).
Discussion
Our results demonstrate that mindin, a component of the extracellular matrix, modifies the severity of allergic airways disease after sub-acute exposure to FAP. Mindin contributes to several cardinal features of allergic airways disease including; the development of AHR, eosinophil recruitment into the airways, and the production of pro-inflammatory Th2 cytokines. We did not observe mindin-dependent differences in sensitization to antigen as supported by similar levels of serum total IgE, though this observation was limited by a small sample size. Interestingly, FAP additionally enhanced neutrophil recruitment into the airspaces and induced a broad array of inflammatory, Th1, and Th17 cytokines. The cytokine expression and neutrophil recruitment was partially dependent on the presence of mindin. These data suggest that blockade of mindin could impact both innate and adaptive immunological response and may represent a novel therapeutic approach for allergic airways disease in context of fungal antigen exposures.
Direct airway challenge to fungal allergens represents a clinically relevant environmental exposure that can result in allergic airway disease. Aspergillus fungal allergens have been recognized to cause an innate-allergic inflammatory response in the lungs [27,28]. They appear to cause broad induction of cytokines as demonstrated by the ability of Aspergillus fumigatus exposure to activate T cells and produce Th1, Th2 and Th17 responses [29]. The allergic response of fungal allergens appears to be dependent on proteinase activity, which has lead to interest in understanding the mechanisms of fungal-derived proteinase allergic inflammation. Murine exposure to FAP has many features similar to human asthma resulting in allergic airways disease and induced a strong Th2 response [11]. The response to FAP is dependent on proteinase activity and does not require use of a secondary adjuvant for sensitization [30,31]. FAP causes neutrophil and eosinophil influx and broad stimulation of cytokines, chemokines and growth factors traditionally associated with both the innate and adaptive immune systems. This suggests that FAP may activate both innate and adaptive immune system pathways. This does not occur in a manner typical of other allergic responses as the allergic response to FAP can occur in a manner independent of T cells, B-cells, and STAT6. These cells and pathways are traditionally believed required for the development of allergic airways disease and for activation of the adaptive immune system [14,32]. Instead, Kiss et al. proposed that FAP acted via an innate immune mechanism [14], which appear independent of the potential low level endotoxin contamination of reagents. While work identifies that toll-like receptor 4 contributes to surface recognition of Aspergillus [33-35], the innate-allergic response to FAP appears independent of either toll-like receptor 4 or the down-stream adaptor protein MyD88 [14]. These data suggest that FAP function through a previously unrecognized innate immune pathway for fungal recognition.
We therefore considered the possible role of extracellular matrix, mindin, in the response to FAP. Mindin has previously been recognized to modify both innate and adaptive immunological responses. Prior work supports that mindin is required for the development of both allergic [20,21] and non-allergic airways disease [22] and is required for the innate immune response to live pathogens [18,19]. We now identify after challenge to FAP that mindin is partially required for the influx of both neutrophils and eosinophils into the air space. Mindin has previously been recognized to contribute to both neutrophil [36] and eosinophil chemotaxis [21] in a manner dependent on integrin binding. However, we additionally identify that the broad induction of pro-inflammatory cytokines/chemokines by FAP is partially dependent on mindin. The specific mechanism by which mindin contributes to the global defect in Th1, Th2 and Th17 cytokines remains unknown. However, the mindin-dependent defect in response to FAP further supports a central role of this extracellular matrix protein in both innate and adaptive immunity.
Based on published results and our new observations, we suggest that mindin may function in a role bridging innate and adaptive immune responses. There is evidence that both neutrophils and (recently reviewed in [37,38]). The role of mindin may be related to known defects in the trafficking of these innate and cells [21,36]. Another possibility for the cytokine and inflammatory cell defects could be as a consequence of defective lymphocyte function resulting in reduced Th1, Th2 and Th17 cytokine production. Mindin is known to be required to direct DC priming of T lymphocytes [20]. Mindin−/− mice have defective CD4+ T cell priming and humoral response to antigens as a consequence of inefficient engagement with T cells. While this remains a possibility, the response to FAP is independent of T cells as demonstrated by the finding effects were present in RAG−/− mice [14]. Previous work supports that mindin contributes to macrophage-derived cytokines [18]. We consider that mindin could regulate sub-populations of lung macrophages response to FAP. Interestingly, FAP challenge induced markers of both M1 and M2 macrophage polarization. However, the changes to be independent of mindin. A limitation of these experiments is small sample size of the RNA analysis. It remains unclear from work whether mindin has other effects on macrophages subpopulations or non-traditional polarization. This pattern of reduced allergic airway disease with normal macrophage differentiation and normal responses has previously been observed in several animal For example, both the LFA-1 (CD18/CD11a) deficient and the arrestin2 deficient mice maintain Th2 differentiation, but have a defect in Th2 homing [39,40]. While we do not know the specific mechanism, by which, mindin regulates the response to fungal antigen, our support a central role of the extracellular matrix in this mouse of innate allergic airways disease.
In summary, our observations demonstrate that mindin the response to airway exposure to the FAP including; AHR, influx both neutrophils and eosinophils into the airspace, and production cytokines/chemokines/growth factors. Mindin-dependent in allergic airways response to FAP appear independent of defects antigen sensitization. Because challenge to FAP results in an innate-allergic response, our observations suggest that mindin may contribute to the bridge between the innate and adaptive arms of the system. Together, current evidence supports that the extracellular matrix protein mindin contributes to both innate and immunologic responses. Therefore, blockade of mindin could an important novel therapeutic target in airways disease.
Supplementary Material
Acknowledgement
This work was supported by National Institutes of Health Grant ES016126 and ES020426 (to J.W.H.) and HL77291 (to P.W.N.). We appreciate the critical comments and infrastructure support provided by W. Michael Foster PhD.
References
- 1.Centers for Disease Control and Prevention (CDC) Vital signs: asthma prevalence, disease characteristics, and self-management education: United States, 2001--2009. MMWR Morb Mortal Wkly Rep. 2011;60:547–552. [PubMed] [Google Scholar]
- 2.Tsitoura DC, DeKruyff RH, Lamb JR, Umetsu DT. Intranasal exposure to protein antigen induces immunological tolerance mediated by functionally disabled CD4+ T cells. J Immunol. 1999;163:2592–2600. [PubMed] [Google Scholar]
- 3.Sedgwick JD, Holt PG. Induction of IgE-isotype specific tolerance by passive antigenic stimulation of the respiratory mucosa. Immunology. 1983;50:625–630. [PMC free article] [PubMed] [Google Scholar]
- 4.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 5.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–882. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, et al. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, et al. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci U S A. 1997;94:1344–1349. doi: 10.1073/pnas.94.4.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grunig G, Corry DB, Leach MW, Seymour BW, Kurup VP, et al. Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J Exp Med. 1997;185:1089–1099. doi: 10.1084/jem.185.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corry DB, Grunig G, Hadeiba H, Kurup VP, Warnock ML, et al. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 11.Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, et al. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol. 2002;169:5904–5911. doi: 10.4049/jimmunol.169.10.5904. [DOI] [PubMed] [Google Scholar]
- 12.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 13.Lim KC, Sun E, Bahgat M, Bucks D, Guy R, et al. Blockage of skin invasion by schistosome cercariae by serine protease inhibitors. Am J Trop Med Hyg. 1999;60:487–492. doi: 10.4269/ajtmh.1999.60.487. [DOI] [PubMed] [Google Scholar]
- 14.Kiss A, Montes M, Susarla S, Jaensson EA, Drouin SM, et al. A new mechanism regulating the initiation of allergic airway inflammation. J Allergy Clin Immunol. 2007;120:334–342. doi: 10.1016/j.jaci.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Cao C, Jia W, Yu L, Mo M, et al. Structure of the F-spondin domain of mindin, an integrin ligand and pattern recognition molecule. EMBO J. 2009;28:286–297. doi: 10.1038/emboj.2008.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan K, Lawler J. The structure of the Ca(2)+-binding, glycosylated F-spondin domain of F-spondin - A C2-domain variant in an extracellular matrix protein. BMC Struct Biol. 2011;11:22. doi: 10.1186/1472-6807-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higashijima S, Nose A, Eguchi G, Hotta Y, Okamoto H. Mindin/F-spondin family: novel ECM proteins expressed in the zebrafish embryonic axis. Dev Biol. 1997;192:211–227. doi: 10.1006/dbio.1997.8760. [DOI] [PubMed] [Google Scholar]
- 18.He YW, Li H, Zhang J, Hsu CL, Lin E, et al. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat Immunol. 2004;5:88–97. doi: 10.1038/ni1021. [DOI] [PubMed] [Google Scholar]
- 19.Jia W, Li H, He YW. Pattern recognition molecule mindin promotes intranasal clearance of influenza viruses. J Immunol. 2008;180:6255–6261. doi: 10.4049/jimmunol.180.9.6255. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Oliver T, Jia W, He YW. Efficient dendritic cell priming of T lymphocytes depends on the extracellular matrix protein mindin. EMBO J. 2006;25:4097–4107. doi: 10.1038/sj.emboj.7601289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Garantziotis S, Jia W, Potts EN, Lalani S, et al. The extracellular matrix protein mindin regulates trafficking of murine eosinophils into the airspace. J Leukoc Biol. 2009;85:124–131. doi: 10.1189/jlb.0208135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 22.Frush S, Li Z, Potts EN, Du W, Eu JP, et al. The Role of the Extracellular Matrix Protein Mindin in Airway Response to Environmental Airways Injury. Environ Health Perspect. 2011 doi: 10.1289/ehp.1003339. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Hollingsworth JW, 2nd, Cook DN, Brass DM, Walker JK, Morgan DL, et al. The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med. 2004;170:126–132. doi: 10.1164/rccm.200311-1499OC. [DOI] [PubMed] [Google Scholar]
- 24.Garantziotis S, Li Z, Potts EN, Lindsey JY, Stober VP, et al. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J Respir Crit Care Med. 2010;181:666–675. doi: 10.1164/rccm.200903-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 25.Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE. Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. Am J Respir Cell Mol Biol. 2000;22:620–627. doi: 10.1165/ajrcmb.22.5.3912. [DOI] [PubMed] [Google Scholar]
- 26.Goswami S, Angkasekwinai P, Shan M, Greenlee KJ, Barranco WT, et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Driscoll BR, Hopkinson LC, Denning DW. Mold sensitization is common amongst patients with severe asthma requiring multiple hospital admissions. BMC Pulm Med. 2005;5:4. doi: 10.1186/1471-2466-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulimood TB, Corden JM, Bryden C, Sharples L, Nasser SM. Epidemic asthma and the role of the fungal mold Alternaria alternata. J Allergy Clin Immunol. 2007;120:610–617. doi: 10.1016/j.jaci.2007.04.045. [DOI] [PubMed] [Google Scholar]
- 29.Murdock BJ, Shreiner AB, McDonald RA, Osterholzer JJ, White ES, et al. Coevolution of TH1, TH2, and TH17 responses during repeated pulmonary exposure to Aspergillus fumigatus conidia. Infect Immun. 2011;79:125–135. doi: 10.1128/IAI.00508-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9:310–318. doi: 10.1038/ni1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh M, Lee SH, Porter P, Xu C, Ohno A, et al. Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125:1369–1378. e1362. doi: 10.1016/j.jaci.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 33.Wang JE, Warris A, Ellingsen EA, Jorgensen PF, Flo TH, et al. Involvement of CD14 and toll-like receptors in activation of human monocytes by Aspergillus fumigatus hyphae. Infect Immun. 2001;69:2402–2406. doi: 10.1128/IAI.69.4.2402-2406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mambula SS, Sau K, Henneke P, Golenbock DT, Levitz SM. Toll-like receptor (TLR) signaling in response to Aspergillus fumigatus. J Biol Chem. 2002;277:39320–39326. doi: 10.1074/jbc.M201683200. [DOI] [PubMed] [Google Scholar]
- 35.Netea MG, Warris A, Van der Meer JW, Fenton MJ, Verver-Janssen TJ, et al. Aspergillus fumigatus evades immune recognition during germination through loss of toll-like receptor-4-mediated signal transduction. J Infect Dis. 2003;188:320–326. doi: 10.1086/376456. [DOI] [PubMed] [Google Scholar]
- 36.Jia W, Li H, He YW. The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood. 2005;106:3854–3859. doi: 10.1182/blood-2005-04-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10:1325–1334. doi: 10.1016/j.intimp.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Shamri R, Xenakis JJ, Spencer LA. Eosinophils in innate immunity: an evolving story. Cell Tissue Res. 2011;343:57–83. doi: 10.1007/s00441-010-1049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SH, Prince JE, Rais M, Kheradmand F, Shardonofsky F, et al. Differential requirement for CD18 in T-helper effector homing. Nat Med. 2003;9:1281–1286. doi: 10.1038/nm932. [DOI] [PubMed] [Google Scholar]
- 40.Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, et al. Beta-arrestin-2 regulates the development of allergic asthma. J Clin Invest. 2003;112:566–574. doi: 10.1172/JCI17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.