

Abstract
Inflammatory stresses associated with inflammatory bowel diseases up-regulate P2Y2 mRNA receptor expression in the human colon adenocarcinoma cell line Caco-2, the noncancerous IEC-6 cells and in colonic tissues of patient suffering from Crohn’s disease and ulcerative colitis. However, the transcriptional events regulating P2Y2 receptor (P2Y2R) expression are not known. We have identified a putative transcription start site in the P2Y2R gene and demonstrated acetylation of Lys14 on histone H3 and Lys8 on histone H4, thus suggesting that the chromatin associated with the P2Y2 promoter is accessible to transcription factors. We also showed that the transcription factor NF-κB p65 regulates P2Y2R transcription under both proinflammatory and basal conditions. A NF-κB-responsive element was identified at −181 to −172 bp in the promoter region of P2Y2. Hence, activation of P2Y2R by ATP and UTP stimulated cyclooxygenase-2 expression and PGE2 secretion by intestinal epithelial cells. These findings demonstrate that P2Y2R expression is regulated during intestinal inflammation through an NF-κB p65-dependent mechanism and could contribute not only to inflammatory bowel disease but also to other inflammatory diseases by regulating PG release.
Intestinal inflammation is often seen as a pernicious manifestation of lost of homeostasis that can cause significant damage to the host tissue. In fact, inflammation is a key element of mucosal defense. It is aimed at limiting entry of foreign material and microbes in the blood stream and to facilitate the repair of damaged tissues (1). The intestinal epithelium constitutes the first defensive frontline of the mucosal immune system (2, 3). Despite the fact that intestinal epithelial cells (IEC)3 are continually exposed to intraluminal bacteria and their products, the intestinal mucosa maintains a controlled state of inflammation (2, 4). IECs play an active role in cellular responses to inflammatory stimuli by secreting cytokines as well as sending cellular messages to immune cells of the intestinal mucosa and submucosa (5, 6). For many years, cytokines and chemokines and their receptors have been accepted as regulators of cross-talk between the intestinal epithelium, immune cells and the vascular endothelium. Extracellular nucleotides, including ATP, ADP, UTP, and UDP, also have cytokine-like properties (7-10).
Recently, these molecules have been described as endogenous danger signal molecules that display potent innate immune-enhancing activities (7, 10, 11). Hence, inflammation likely promotes the release of nucleotides, such as ATP and UTP or UDP, not only from IECs, as we recently showed (10), but also from other cell types, such as leukocytes, platelets, and smooth muscle cells, as well as from intestinal bacteria (12). Another important source of nucleotides is damaged or dead cells that release nucleotides present at high concentrations in the cytoplasm (7, 10, 11). This increase in the extracellular nucleotide concentration in the environment of inflamed tissues has also been associated with an increased expression of P2Y2 and P2Y6 receptor mRNA in colonic epithelium of patients with Crohn’s disease and ulcerative colitis and in mice with chemical-induced colitis (10). Up-regulation of P2Y2R expression was also observed in a variety of pathophysiological conditions associated with inflammation and/or tissue damage (13-15). This increase in P2Y2R expression and functions has been associated with the aggravation of inflammatory disease symptoms such as in vascular intimal hyperplasia and Sjögren’s syndrome (15-18). However, P2Y2R expression and activity could also be beneficial in tissue repair or wound healing (19, 20). Besides its role in the regulation of Cl− and K+ secretion by IECs (21-23) and its reported overexpression in human colon cancer (24), there is no information on the pathophysiological function of P2Y2R in inflammatory bowel diseases (IBD). More surprisingly, there are no data demonstrating how P2Y2R gene expression is regulated. The characterization of the promoter region of the P2Y2R gene could help to understand the transcriptional regulation of P2Y2R expression in IBD and in other inflammation processes. Among the various transcription factors previously described, the p65 subunit of NF-κB (NF-κB p65) has been shown to regulate the transcription of a broad range of genes related to inflammation in Crohn’s disease and ulcerative colitis (25). The current study describes the cloning and sequence analysis of the proximal promoter region of the P2Y2R gene in which we have detected the presence of three consensus NF-κB-binding sites. Our results also demonstrate that NF-κB p65 stimulates P2Y2R transcription in colonic epithelial cells under proinflammatory conditions. Finally, we demonstrated that up-regulation and activation of the P2Y2R lead to an increase in both cyclooxygenase-2 (COX-2) expression and PGE2 released by IECs.
Materials and Methods
Reagents
DMEM, Eagle’s MEM, F-12 medium, penicillin-streptomycin, HEPES, and FBS were purchased from Wisent. FBS was inactivated by heating at 50°C for 60 min. Optim-MEM and glutamine (GlutaMax) were from Invitrogen. Dextran sulfate sodium (DSS; Mr 36,000–50,000) was obtained from MP Biochemicals. LPS (Escherichia coli O55:B5) and IL-6 were purchased, respectively, from Calbiochem and BioShop Canada. Mouse monoclonal anti-NF-κB p65 Ab was purchased from Santa Cruz Biotechnology. Rabbit anti-acetyl-histone H3 (Lys14) polyclonal Ab, and rabbit anti-acetyl-histone H4 (Lys8) polyclonal Ab were from Millipore. Mouse anti-COX-2 mAb was purchased from Caymen Chemical. PGE2 assay kits were obtained from R&D Systems. The pGL4.10 and pcDNA3.1 vectors were purchased, respectively, from Promega and Invitrogen. The pcDNA3.1/NF-κB p65 construct was kindly provided by Dr. Marc Servant (Université de Montreal, Faculté de Pharmacie, Quebec, Canada).
Cell culture
Human coronary artery endothelial cells (HCAEC; American Type Culture Collection (ATCC) No. CRL-2266) were cultured, as previously described (26). The human colon carcinoma cell line Caco-2 (ATCC No. HTB-37) and the noncancerous rat IEC cell line IEC-6 (ATCC No. CRL-1592) were grown as previously described (10, 27). Cells were incubated in serum free medium for 24 h at 37°C before experiments. When indicated, 0.5% (w/v) DSS, 10 ng/ml IL-6, or 12.5 μg/ml LPS was added to Caco-2 or IEC-6 cells for the specified time period to induce a proinflammatory response, as previously described (10, 28, 29).
Real-time PCR quantification
Total RNA was isolated from Caco-2 and IEC-6 cells with the Totally RNA Kit (Ambion) according to the manufacturer’s instructions. cDNA was then synthesized from 2 μg of purified RNA by reverse transcription using the SuperScript II system (Invitrogen). Five percent of the synthesized cDNA was used as a template for real-time PCR using the QuantiTect SYBR green PCR Kit (Qiagen) and the Stratagene Mx3000P QPCR System. The sequence-specific primers for P2Y2R, COX-2, and TATA-binding protein are listed in supplemental Table I.4
Primer extension reaction
Single-stranded 43-mer anti-sense oligonucleotide (10 pmol) 5′-CTCTCGCCACTGCGCTGCGCTTCTCCTCTCAGGGTGCCGTCGC-3′ (Tm = 75.3°C) corresponding to exon 1 in the P2Y2R gene sequence was designed and chemically synthesized, and end-labeled using polynucleotide kinase from Promega, and [γ-32P]EasyTides ATP was from PerkinElmer. Labeled primers (1 pmol) were hybridized for 20 min with 5 μg of polyadenylate+ RNA isolated from HCAECs, and avian myeloblastosis virus reverse transcriptase was added for 30 min at 41°C to yield the corresponding cDNA. Avian myeloblastosis virus reverse transcriptase was inactivated by incubating all samples at 90°C for 10 min. The resulting products were analyzed on an 8% (w/v) polyacrylamide denaturing gel. Kanamycin-positive mRNA (1.2 kb) from the the Promega kit was used with a corresponding primer as a positive control. The negative control included diethylpyrocarbonate-treated water instead of mRNA in the reaction.
RNA ligase-mediated 5′-RACE (5′-RLM-RACE)
The transcription start site (TSS) of the human P2Y2R gene was determined using the RNA ligase-mediated 5′-RLM-RACE with the FirstChoice RLM-RACE kit following the manufacturer’s instructions (Ambion). Briefly, total RNA was isolated from Caco-2 cells using the RNeasy kit (Qiagen) according to the manufacturer’s instructions, and 10 μg of RNA were treated with alkaline phosphatase to remove phosphate groups on degraded mRNA, rRNA, tRNA, and DNA. RNA was then treated with tobacco acid pyrophosphatase to remove the cap from full-length nascent transcripts. Then, a RACE primer was ligated to phosphorylated uncapped transcripts. Reaction products were reverse transcribed using SuperScript III reverse transcriptase (Invitrogen) and the deoxythymidylate oligonucleotide dT20. Modified cDNA was subjected to PCR analysis using the PCR primer hY2R4 (supplemental Table II) and outer primer detecting the 5′-RACE adapter (Ambion). The generated PCR products were reamplified in a second, nested PCR using the inner primer detecting the 5′-RACE adapter and one of the hY2R1 and hY2R3 primers (supplemental Table II). All PCR amplifications were performed using the Expand High Fidelity PCR system (Roche). PCR products were cloned into pCRII-TOPO using the TOPO cloning kit (Invitrogen). The cloned DNA fragments were identified by automatic DNA sequencing (ABI 3730xl; Applied Biosystems) using the M13 oligonucleotide provided by the TOPO cloning kit.
Human P2Y2R promoter cloning and constructs
Human genomic DNA, isolated from human IECs using the DNeasy Tissue kit (Qiagen), was kindly provided by Dr. Nathalie Rivard (Universiteé de Sherbrooke, Département d’Anatomie et Biologie Cellulaire, Quebec, Canada). The human P2Y2R promoter, −1572 bp to +93 bp relative to the putative TSS, was PCR-amplified from the human genomic DNA using the prP2Y2/NheI forward and prP2Y2/BglII reverse primers (supplemental Table III). The PCR fragment was cloned into the pGL4.10 expression vector (Promega) upstream of the luciferase gene (prP2Y2-luc). Deletion mutants of the P2Y2R promoter were generated by PCR amplification with the following oligonucleotides (supplemental Tables III and IV): prP2Y2Δ-350bp and prP2Y2/BglII primers for deletion of −1572 bp to −351 bp; prP2Y2Δ-784bp and prP2Y2/BglII primers for deletion of −1572 bp to −785 bp; and prP2Y2Δ-1165bp and prP2Y2/BglII primers for deletion of −1572 bp to −1166 bp. The PCR fragments were cloned into the pGL4.10 expression vector (Promega) upstream of the luciferase gene. Deletions within the prP2Y2Δ-350bp mutant were generated by restriction enzyme digests with StuI (−273 bp) (prP2Y2Δ-350bp/StuI), PmlI (−33 bp; prP2Y2Δ-350bp/PmlI) and NheI (−350 bp). Deletion of the putative NF-κB p65-binding motif (NBM) in the prP2Y2Δ-350bp construct was done by overlap extension PCR by replacing those 10 bp by a scrambled nucleotide sequence, as previously described (27). The upstream amplification was performed with the prP2Y2/NheI oligonucleotide primer (supplemental Table III) and the prP2Y2ΔNBM1, prP2Y2ΔNBM2, or prP2Y2ΔNBM3 primer (supplemental Table V). The downstream amplification was performed with the prP2Y2ΔNBM1R, prP2Y2ΔNBM2R, or prP2Y2ΔNBM3R (supplemental Table V) and the prP2Y2/BglII (supplemental Table III) primers. Products resulting from these two PCRs were used as DNA templates for the final PCR using the prP2Y2/NheI and prP2Y2/BglII oligonucleotides. The PCR fragments were then cloned into the pGL4.10 luciferase vector. The presence of the mutations was verified by sequence analysis (McGill University and Genome Quebec Innovation Center).
Transient transfections and luciferase assays
Caco-2 cells, at 80% confluence, were seeded in 24-well plates for 24 h. The next day, 1 h before transfection, complete DMEM was replaced by 300 μl of Opti-MEM (Invitrogen), free of antibiotic and antimycotic. Cells were cotransfected using LipofectAMINE 2000 (Invitrogen) with 0.1 μg of pcDNA3.1/NF-κB p65 and 0.1 μg of the different P2Y2R promoter constructs or 0.1 μg of pGL4.10 (control). After 6 h of transfection, the Opti-MEM was replaced with complete DMEM. Two days after transfection, luciferase activity was measured, as previously described (10). In some experiments, cells were stimulated with DSS at a final concentration of 0.5% (w/v) for 6 h before measuring luciferase activity. Results are expressed as fold induction compared with control (pcDNA3.1/NF-κB p65 plus pGL4.10) values, normalized to Renilla luciferase expression.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using the EZ ChIP assay kit protocol (Upstate). Briefly, 80% confluent Caco-2 cells were cross-linked with 1% (v/v) formaldehyde for 10 min at 37°C. Following cross-linking, chromatin was sheared and immunoprecipitated with 5 μg of mouse anti-NF-κB p65 polyclonal Ab (Santa Cruz Biotechnology) or 5 μg of rabbit anti-acetyl-histone H3 (Lys14) polyclonal Ab or 5 μg of rabbit anti-acetyl-histone H4 (Lys8) polyclonal Ab (Millipore). Five micrograms of normal mouse or normal rabbit IgG (Upstate) were used as a negative control. For re-ChIP assays, a second immunoprecipitation was conducted with 5 μg of rabbit anti-p300 polyclonal Ab, after eluting the immunoprecipitated NF-κB p65-DNA complex from the G protein agarose beads. Input (10% of the lysate before immunoprecipitation) was used to verify the amount of DNA in each immunoprecipitation. Immunoprecipitated DNA was purified and amplified by real-time PCR using the QuantiTect SYBR green PCR Kit (Qiagen) and the Stratagene Mx3000P QPCR System with P2Y2C upstream and downstream oligonucleotide primers amplifying the −221 bp to −105 bp (supplemental Table VI). Data are expressed as fold increase over background (negative control) normalized to input as proposed by SuperArray Biosciences and adapted as described (www.workingthebench.com).
EMSA
Nuclear proteins were obtained as previously described (27). EMSA assays were conducted using 7.5 μg of nuclear protein extracts and 3.5 × 104 cpm of 5′-end-labeled [γ-32P]ATP double-stranded oligonucleotides (supplemental Table VII) in the presence of 50 ng of polydeoxyinosinatepolydeoxycytidylate (Roche) in buffer D (5 mM HEPES, 10% (v/v) glycerol, 0.05 mM EDTA, 0.125 mM PMSF). DNA-protein complexes were separated on a nondenaturing 5% (w/v) polyacrylamide gel run against Tris-glycine buffer, as described (30). In supershift experiments, 3 μg of mouse monoclonal anti-NF-κB p65 Ab were used per sample and added 20 min before the addition of the radiolabeled probes. In competition experiments, 10× or 100× concentrations of the unlabeled probe NBM2 was added to the sample at the same time of labeled probe. Gels were dried and autoradiographed on an Imaging Screen-K (Kodak) for 18 h and imaged using a Bio-Rad Molecular Imager FX apparatus and data were acquired using Quantity One software from Bio-Rad.
Western immunoblotting
After nucleotide stimulation, Caco-2 cells were lysed and processed as previously described (10). Samples were subjected to 7.5% SDS-PAGE, and transferred to polyvinylidene difluoride membranes for protein immunoblotting, as previously described (10). Immunoblotting for human COX-2 was performed using a 1/750 dilution of mouse monoclonal anti- COX-2. Specific protein band on membranes were detected using a 1/10,000 dilution of HRP-conjugated anti-mouse Ab as the secondary Ab and visualized on autoradiographic film using the Millipore chemiluminescence system. Normalization of the signal was realized as previously described (10).
Quantification of PGE2 released by IECs
PGE2 released was quantified using the PGE2 assay kit from R&D Systems. Caco-2 cells were stimulated with 100 μM ATP or UTP for 24 h. Cell media were collected, cleared of debris, and used for PGE2 assays as recommended by the manufacturer.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical significance was determined by performing unpaired t or ANOVA tests as described in the figure legends. The number of replicates for each experiment is presented in the figure legends.
Results
P2Y2R expression is increased in Caco-2 and IEC-6 cells under inflammatory-like conditions
Recently, we showed that P2Y2R mRNA expression was increased in colonic tissues of patients with Crohn’s disease and ulcerative colitis (10). Similar observations were made in the colonic epithelium of mice in which intestinal inflammation was induced by adding DSS to the drinking water (10). Using real-time PCR, we found that addition of DSS for 3–6 h to Caco-2 IECs causes >2-fold increase in level of P2Y2R mRNA (Fig. 1A). In addition, treatment of Caco-2 cells with LPS (12.5 μg/ml) resulted in an 8-fold increase in level of P2Y2R mRNA after 24 h (Fig. 1B), which was also confirmed in a normal IEC cell line, IEC-6, which resulted in a >7-fold increase in the level of P2Y2R mRNA after 12 h (Fig. 1D). Moreover, treatment of IEC-6 cells with IL-6 (10 ng/ml) causes an increase of >2.5-fold in level of P2Y2 mRNA after 18 h (Fig. 1C).
FIGURE 1.
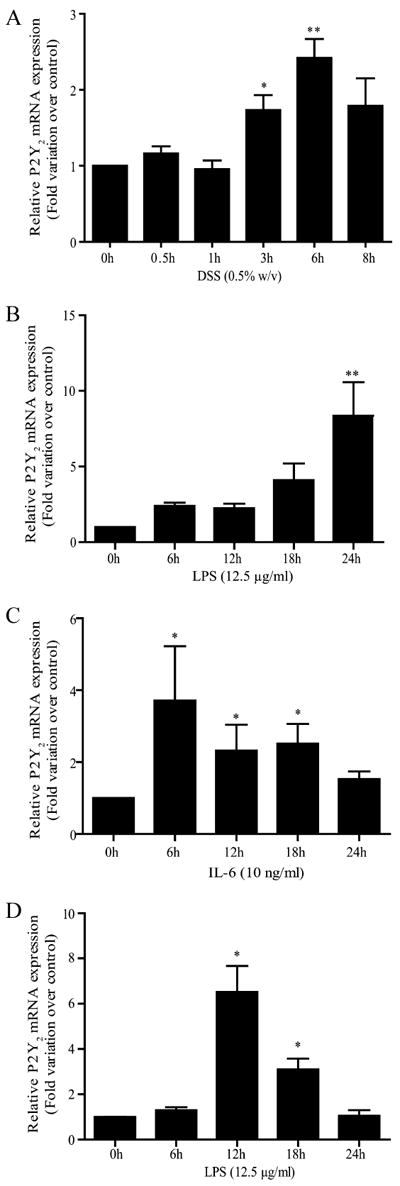
P2Y2R mRNA expression in IECs is enhanced by proinflammatory stimuli. P2Y2R mRNA expression was determined by quantitative RT-PCR in confluent IEC-6 and 3-day-postconfluent Caco-2 cells. A and B, Increases in mRNA expression were significant after a 3- and 6-h incubation with 0.5% (w/v) DSS and 24 h of incubation with 12.5 μg/ml LPS in Caco-2 cells. C and D, Increases in mRNA expression were significant after 12 h of incubation with 12.5 μg/ml LPS and 18 h of treatment with 10 ng/ml IL-6 in IEC-6 cells. Data are expressed as P2Y2R mRNA expression induced by stimuli relative to the untreated control. Results were normalized to the expression of TATA-binding protein mRNA. Values are the means ± SEM of results from three separate experiments done in duplicate. Statistical significance was determined by unpaired t test; *, p < 0.05 and **, p < 0.01, as compared with control.
Localization of the transcription start site (+1) of the human P2Y2R
We used primer extension analysis to identify the potential TSS in the P2Y2R gene using mRNA extracted from HCAECs. The single-stranded 43-mer oligonucleotide used was highly specific for P2Y2R mRNA exon 1. The expected cDNA product size was 134 bp when the P2Y2R TSS conformed to transcripts arrived at for P2Y2R mRNA by in silico analysis (31). The positive control was a 1.2-kb kanamycin-positive in vitro product that was expected to give a product size of 84 bp. The chief cDNA products obtained by primer extension were smaller (57 and 84 bp) and larger (200 bp) than predicted (supplemental Fig. 1A). More than one TSS has been known to be associated with TATA-less promoters (32, 33). However, formation of products due to secondary structures in mRNA in the primer extension reaction cannot be ruled out. To confirm and delineate the TSS, we used RLM-RACE and nested PCR assays with Caco-2 cells (supplemental Fig. 1B). RLMRACE is a more sensitive method than primer extension analysis because it removes uncapped mRNA, DNA, and other non-mRNA molecules by treatment of the RNA sample with calf intestine phosphatase (34, 35). Electrophoretic analysis of the nested PCR amplification products revealed that Caco-2 cells express only variant 2 of the human P2Y2R mRNA and a single TSS, as shown by the presence of a single band for both clones analyzed from the nested PCR assays with hY2R1 and hY2R3 oligonucleotide primers (supplemental Fig. 1B). Sequence alignment of representative clones obtained from the nested PCR with the 5′-untranslatd region sequence of human P2Y2R (variant 2; NM_002564) allowed us to localize the TSS (+1) 72 nucleotides downstream from the first nucleotide of the published P2Y2R gene sequence, as indicated in supplemental Fig. 1C. We also found a mutation at +32 where a thymine (T) was replaced by a cytosine (C) residue, as indicated by the arrowhead in supplemental Fig. 1C. Finally, the translation start point (ATG) was located at position +262, as indicated by the bold letters in supplemental Fig. 1C. We aligned the sequence of the promoter from the two different cell types and identified by computational analysis potential binding sites for Sp1 and NF-κB (supplemental Fig. 2). The P2Y2R promoter of these cell types is 99.2% identical.
Chromatin is decondensed at the P2Y2R promoter region
Modifications of histone tails are known to facilitate or hinder accessibility of transcription factors to their binding sites in promoter regions of genes by promoting condensation or decondensation of the chromatin (36, 37). One of the modifications of significant importance to enhanced transcription of genes is the acetylation of histone H3 and H4 (37). We performed ChIP assays to immunoprecipitate histones H3 and H4 with, respectively, antiacetyl-histone H3 (Lys14) and anti-acetyl-histone H4 (Lys8) Abs followed by real-time PCR amplification to identify those specific acetylations of histones H3 and H4 (supplemental Fig. 3). Acetylation of these two lysine residues is associated with active gene transcription and more particularly with genes that are trans-activated by NF-κB p65 (38). Results indicated a 4.6- and 2.3-fold increase in acetylated histone H3 (on Lys14) and acetylated histone H4 (on Lys8), respectively, over background from the negative control (mouse IgG) normalized to input.
NF-κB p65 regulates the expression of the P2Y2R
To determine whether NF-κB p65 regulated P2Y2R expression, we first verified the presence of potential NF-κB p65 DNA-binding sites in the proximal P2Y2R human promoter using computer analysis (Fig. 2A). We identified multiple putative NF-κB DNA-binding sites by the characteristic GGG(A/G)NN(T/C)(T/C)CC consensus sequence (39). We then cloned −1572 to +93 bp of the proximal human P2Y2R promoter in the luciferase-containing pGL4.10 vector to assess the transcriptional activity in response to NF-κB p65 expression under basal and stress-like conditions (Fig. 2B). P2Y2R promoter transcriptional activity was increased 2-fold under basal conditions, as compared with the absence of promoter (Fig. 2B, p < 0.01) but increased to >7-fold in response to DSS (Fig. 2B, p < 0.05). To delineate the P2Y2R promoter region required for NF-κB p65-dependent induction, we compared the P2Y2R promoter transcriptional activity to the 5′ deletion mutants prP2Y2Δ-1165, prP2Y2Δ-784, and prP2Y2Δ-350. None of the prP2Y2R deletion mutant constructs showed decreased luciferase activity in response to NF-κB p65 expression (Fig. 2C), suggesting that the promoter region and potential NF-κB DNA-binding sites located upstream of nt −350 are not involved in the regulation of the P2Y2R gene expression. In contrast, further deletion downstream of −350-bp site abolished P2Y2R transcriptional activation, as compared with the full-length P2Y2R and P2Y2Δ-350 promoter constructs (Fig. 2D). These data indicate that the promoter region between −273 and −33 nt is essential for NF-κB p65-dependent P2Y2R transcriptional regulation. To prove that NF-κB is involved in the transcriptional regulation of the P2Y2R gene, we specifically mutated three potential NF-κB DNA-binding motifs in the P2Y2R promoter region using the oligonucleotide primers NBM1 (−278 to −269), NBM2 (−181 to −172), and NBM3 (−135 to −126), as shown in Fig. 2A. Mutation of NBM2 significantly reduced the luciferase activity of the minimal P2Y2Δ-350-Luc promoter construct by more than 60%, whereas mutation of NBM1 and NBM3 had no effect on luciferase activity (Fig. 2E), suggesting a role for NBM2 in the transcriptional regulation of P2Y2R expression by NF-κB. We also have identified other potential binding sites for transcription factors in the P2Y2R promoter region, such as SP1 (supplemental Fig. 2), that remain to be characterized.
FIGURE 2.
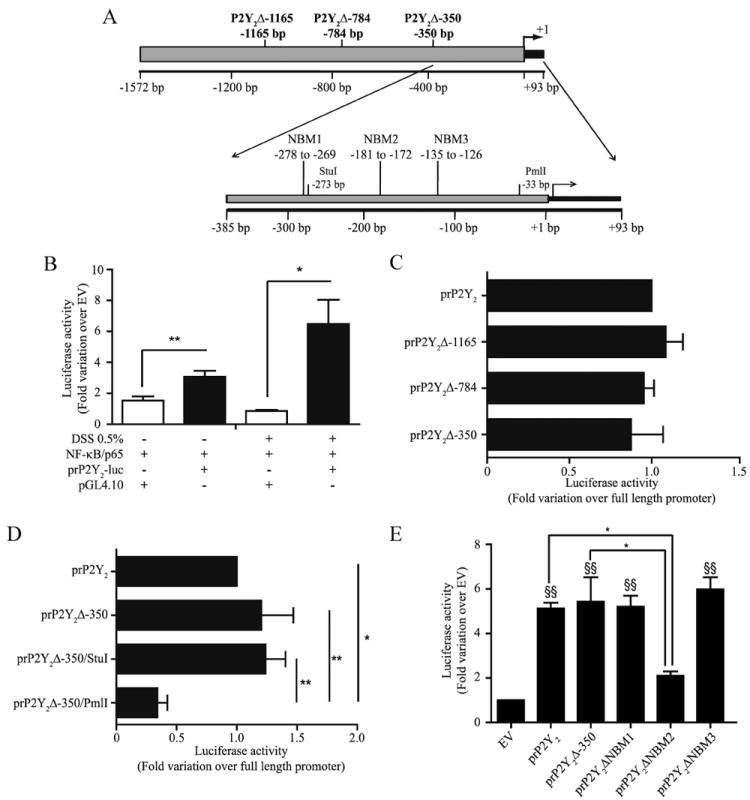
NF-κB p65 trans activates the P2Y2R promoter region under basal conditions and this trans activation is enhanced following DSS-induced stress in Caco-2 cells. A, P2Y2R promoter constructs. B, Subconfluent Caco-2 cells were transiently cotransfected with the P2Y2R promoter-luciferase construct (prP2Y2-luc) and the NF-κB p65-expressing vector or the empty pGL4.10 vector (control). Cells were incubated with or without 0.5% (w/v) DSS for 6 h, and luciferase activity was assayed after 48 h. C, Subconfluent Caco-2 cells were cotransfected with the P2Y2R promoter-deletion luciferase constructs prP2Y2 Δ-350bp, prP2Y2Δ-784bp, or prP2Y2Δ-1165bp and the NF-κB p65-expressing or control vector. D, Subconfluent Caco-2 cells were cotransfected with the P2Y2R promoter-deletion luciferase constructs prP2Y2Δ-350/PmlI or prP2Y2Δ-350/StuI and the NF-κB p65-expressing or control vector. E, prP2Y2ΔNBM1, prP2Y2ΔNBM2, prP2Y2ΔNBM3, P2Y2R full-length promoter constructs or prP2Y2Δ-350bp promoter construct were transiently cotransfected with the NF-κB p65-expressing or control vector. Luciferase activity is expressed as the fold increase relative to the activity of the empty vector cotransfected with the NF-κB p65-expressing vector. Data are the means ± SEM of results from at least four separate experiments done in triplicate. Statistical analysis was performed by an ANOVA test; *, p < 0.05 and **, p< 0.01, as compared with respective controls and indicated on the figures. E, §§, p < 0.01, as compared with empty vector control (EV).
We verified by electrophoretic mobility and supershift assays the ability of labeled NBM1, NBM2, and NMB3 double-stranded oligonucleotides to bind NF-κB p65 in Caco-2 nuclear extracts (Fig. 3A). Of the three NBM sites, the NBM2-protein complex was the major complex supershifted by a NF-κB p65 Ab (Fig. 3A). We also did a competition assay using unlabeled NBM2 probe with NBM2 labeled probe and observed almost complete diminution of the intensity of the labeled protein-DNA complex in the presence of 100× unlabeled probe (Fig. 3B). Thus, NF-κB p65 interacts in vitro with the NBM2−181GCGGCGTCCC−172 sequence. To assess whether NF-κB p65 bound the NBM2 DNA-binding site of the minimal P2Y2R promoter in vivo, we performed a chromatin immunoprecipitation assay with Caco-2 genomic DNA (Fig. 3C) and showed that NF-κB p65 binds the −221 to −105 P2Y2R promoter region encompassing the NBM2 DNA-binding site. Following real-time PCR, we quantified a 2-fold increase in the binding of NF-κB p65 to this region, as compared with the negative control (mouse IgG). We also did a re-ChIP assay (Fig. 3C) in which the second immunoprecipitation was achieved with an Ab against p300. The p300 protein is a coactivator with an histone acetyltransferase (HAT) activity that can bind to and acetylate NF-κB p65 or histones directly or indirectly by the recruitment of other HATs, thereby decondensing chromatin to enhance the transactivation of genes regulated by NF-κB (40, 41). Using real-time PCR amplification we determined that approximately one-half of the immunoprecipitated p65 is bound to p300 (Fig. 3C).
FIGURE 3.
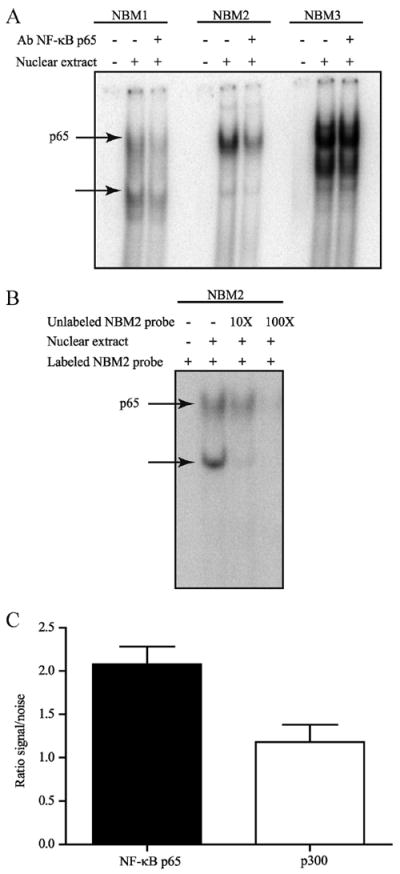
NF-κB p65 binds to the P2Y2R promoter sequence. A, Nuclear extracts from Caco-2 cells were incubated with the putative [γ-32P]ATP-labeled NF-κB p65 DNA-binding site probes NBM1, NBM2, or NBM3 and anti-NF-κB p65 Abs for electrophoretic mobility and supershift assays. DNA-protein complexes were separated from the free probe on a native polyacrylamide gel. Arrow, NF-κB p65 DNAbinding and supershifted complexes. B, Nuclear extracts from Caco-2 cells were incubated with the putative [γ-32P]ATP-labeled NF-κB p65 DNA-binding site probe NBM2 and 10× or 100× unlabeled NBM2 for electrophoretic mobility and competition assays. DNA-protein complexes were separated from the free probe on a native polyacrylamide gel. Results are representative of three independent experiments. NF-κB p65 DNA-binding complex is indicated by the arrow. C, Chromatin was immunoprecipitated with or without rabbit IgG Ab or anti- NF-κB p65 Ab. The re-ChIP assay was performed with anti-p300 Ab following the first immunoprecipitation with anti-p65 Ab. Samples were verified by quantitative RT-PCR analysis with oligonucleotides amplifying the −221 bp to −155 bp region of the P2Y2R promoter and expressed as fold increase over normal rabbit IgG normalized to input. Results are representative of three independent experiments.
Activation of P2Y2R stimulates COX-2 expression and PGE2 released by IECs
In normal intestine, COX-1 is expressed constitutively in epithelial cells where it is associated with intestinal mucosa homeostasis (42). On the opposite, COX-2 is expressed at low basal levels under normal conditions, but its expression is rapidly up-regulated by proinflammatory molecules (43). COX-2-dependent arachidonic acid metabolites, such as PGE2, can modulate the inflammatory response of the intestinal mucosa (43-45). Stimulation of COX-2 expression and PGE2 released through P2Y2R activation have previously been reported in other systems (46-48), but it has never been associated to the increase of P2Y2R expression as observed during intestinal inflammation. In this context, we showed that P2Y2R activation by ATP (Fig. 4A) or UTP (Fig. 4B) significantly increased the expression of COX-2 mRNA after only 30 min and sustained it for up to 60 min. It also increased the expression of COX-2 at the protein level as soon as 3 h following stimulation of P2Y2R (Fig. 4C) in Caco-2 IECs. The P2Y2R-dependent increase of COX-2 expression correlated with PGE2 released by Caco-2 cells in response to ATP and UTP stimulation (Fig. 4D). PGE2 release was stimulated by >2-fold in response to ATP and UTP when compared with nonstimulated Caco-2 cells (Fig. 4D).
FIGURE 4.
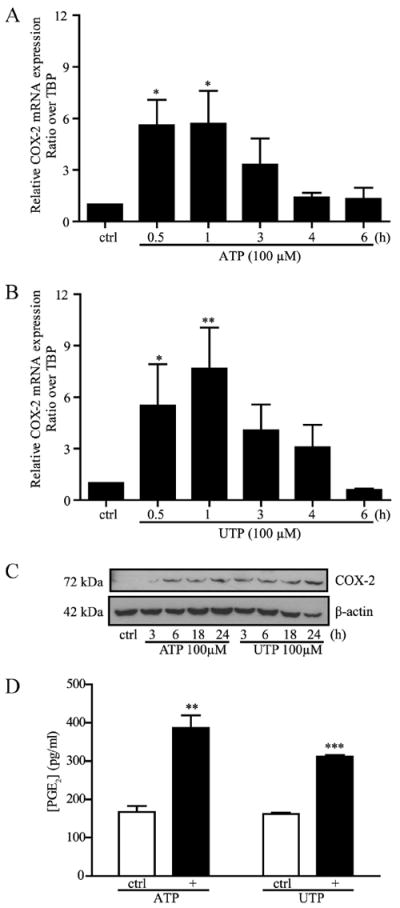
ATP and UTP stimulate COX-2 expression and PGE2 released by Caco-2. Addition of 100 μM ATP (A) or UTP (B) rapidly stimulated the mRNA expression of COX-2 in Caco-2 cells. C, Caco-2 cells were stimulated with 100 μM ATP or UTP for 0 (control; ctrl), 3, 6, 18, and 24 h. COX-2 protein expression was detected by Western analysis. D, PGE2 released in the cell culture medium was determined after a 24-h stimulation of Caco-2 cells with 100 μM ATP or UTP. For A, B, and D, data are the means ± SEM of results from three separate experiments done in duplicate. Statistical analysis was performed by an unpaired t test; *, p < 0.05; **, p < 0.01; and ***, p < 0.001, in comparison with unstimulated cells (ctrl). C, Blot is typical of three separate sets of experiments.
Discussion
We recently reported the up-regulation of P2Y2R mRNA in the colonic tissues isolated from patients suffering from IBDs (10). Similar observations were also made in colon isolated from mice suffering from chemically induced colitis (10). It is well described that immune cells, such as neutrophils and monocytes/macrophages, expressed numerous P2 nucleotide receptors among which the P2Y2R (49). As for many diseases, IBDs are characterized by an increasing number of infiltrating neutrophils and monocytes/macrophages that could have contributed to the observed increased in P2Y2R transcript expression previously described (10). In this study, we demonstrated using the adenocarcinoma-derived Caco-2 cells and the noncancerous IEC-6 cells that the increased expression of the P2Y2R transcript could be attributable in part to the up-regulation of receptor expression by IECs (Fig. 1). This increase in P2Y2R mRNA expression and function has been associated with enhanced tissue damages as reported in a Sjögren’s syndrome-like phenotype in mice (14, 50), chronic pancreatitis (13), and the induction of intimal hyperplasia in an animal model of human atherosclerosis (15, 51). More recently, P2Y2R increased expression and function in rat primary cortical neurons have been associated with the α-secretase-dependent processing of amyloid precursor protein, a well-described molecule involved in neurode-generative disorders (52). In addition, P2Y2R activation in A549 alveolar type II epithelial cells stimulated the expression of COX-2 and PGE2 production that were associated with airway inflammation (46). Despite these numerous reports describing the up-regulation of P2Y2R expression and function in inflammatory diseases and the apparent contribution of a NF-κB-dependent mechanism (52), there are no data describing how P2Y2R gene expression is regulated.
In this report, we identified, cloned and characterized the promoter region of the human P2Y2R gene, from −1572 bp to +93 bp. In addition, we have associated the increase in P2Y2R expression to the stimulation of COX-2 expression and PGE2 released by IECs. NF-κB p65 is one of the prototypic transcription factors associated with inflammation under a number of pathophysiological conditions (25). NF-κB p65 activation is related to the transcriptional regulation of a number of proteins involved in the inflammatory response, such as cytokines and cell adhesion molecules. Thus, it is appropriate that this transcription factor was found to regulate the transcription of the P2Y2R gene under both basal and proinflammatory conditions. We localized a potential NF-κB p65-binding site on the human P2Y2R promoter region and showed that this region is transcriptionally active by measuring histones H3 and H4 acetylation of lysine 14 and 8, respectively (37, 41, 53). Moreover, the specific acetylation of histones H3 and H4 are associated with NF-κB-regulated genes that are transcriptionally active (38). Since we are interested in NF-κB p65 transactivation potential, we also looked at p300, a coactivator with histone acetyltransferase activity that increases transcription factor acetylation and accessibility to the DNA (53). Using re-ChIP assay, we determined that p65 and p300 are associated with the P2Y2R promoter. Accordingly, it was reported that p300 can be associated with the p65 subunit of NF-κB and promote transcription of genes targeted by NF-κB by acetylating histones, recruiting other HATs, or acetylating NF-κB p65 (54) to enhance the transcriptional activity and the DNA-binding of NF-κB p65 (40).
The determination of the transcription start site (TSS) of the P2Y2R promoter by primer extension assays using poly(A)+ RNA isolated from HCAECs and confirmed by 5′RLM-RACE using Caco-2 cells RNA allowed us to focus on a putative promoter region of 1572 bp upstream of the + 1 bp site in the P2Y2R gene. In both assays, regardless of the different nature of the cells, we were able to pinpoint the TSS to the same area of the promoter. The difference of approximately 11 bp in nucleotide localization is due to the nature of the methods used, RLM-RACE being a more sensitive method than primer extension. Many TATA-less promoters contain multiple GC-rich sequences that are recognized by the transcription factor Sp1 (55). Sp1 can recruit the TFIID complex to initiate the assembly of the transcriptional machinery. By computational analysis (supplemental Fig. 2), we identified three potential Sp1-binding sites 50 bp upstream of the P2Y2R TSS. Further studies will be necessary to define the role of these potential binding sites, since multiple Sp1 elements can act independently or synergistically to promote gene transcription (55).
Truncation of the 5′-region of the P2Y2R promoter has allowed us to identify the region between −273 and −33 nt as important for trans activation of P2Y2R gene expression by the transcription factor NF-κB p65. We then conducted computational analysis of this promoter region and indeed found three potential NF-κ-binding sites (NBM1–3). Site-specific mutations of these NBMs in the P2Y2Δ-350bp promoter followed by co-transfection with NF-κB p65 in Caco-2 cells further supported the postulate that NBM2 is a binding site for NF-κB. Actually, mutation of the NBM2 site resulted in a decreased luciferase activity of >50%. This result was confirmed by EMSA experiments which demonstrated that p65 interacts strongly in vitro with NBM2, as compared with NBM1 and NBM3. ChIP assays revealed that p65 binds to the region (−221 bp to −105 bp) that contains the NBM2 motif. trans activation of the P2Y2R promoter by NF-κB p65 occurs under both basal and proinflammatory conditions. Our results suggest that the induction of inflammation in IECs allows nuclear translocation of NF-κB p65 and leads to an increased trans activation of the P2Y2R promoter and subsequently the up-regulation of P2Y2R mRNA. These results are in agreement with recently published data demonstrating that a 24-h stimulation of rat primary cortical neurons with IL-1β-induced P2Y2R mRNA expression possibly through a NF-κB-dependent mechanism (52). To circumvent the cancerous nature of Caco-2 cells, we used the noncancerous IEC-6 cell line and measured an increased in P2Y2R mRNA expression following IL-6 or LPS stimulation. These results thus confirm that P2Y2R transcript expression is up-regulated in IECs under an inflammatory insult.
We have linked the increase in P2Y2R expression and function to the stimulation of COX-2 expression and PGE2 released by IECs (Fig. 4). In the intestine, the role of COX-2-dependent generation of arachidonic acid metabolites, such as the intestine-predominant PGE2, is controversial. On one hand, PGE2 have proinflammatory effects in the acute phase of IBD (44, 56). On the other hand, this molecule could be beneficial because PGE2 production in the intestine appears to have a protective effect on the integrity of the epithelial intestinal wall, presumably through the enhancement of IEC survival and regeneration (44, 56), while having a dampening effect on granulocyte infiltration (57).
In this study, we propose for the first time a promoter sequence for P2Y2R that is euchromatin accessible to transcription factors. We also show that NF-κB p65 regulates P2Y2R gene expression under both basal and proinflammatory conditions. In addition, we are proposing that P2Y2R on IECs may serve two purposes. First, in the acute phase of inflammation, P2Y2R-dependent activation of COX-2 and PGE2 production could stimulate the inflammatory response of the intestinal mucosa, thus resulting in increasing tissue damages but also at limiting entry of foreign molecules to the systemic circulation, as well as facilitating the repair of damage tissues. Second, in the resolution phase following inflammation, these same molecules could enhance epithelium repair by stimulating IEC proliferation and increasing the barrier integrity of the IECs (44, 56).
Supplementary Material
Acknowledgments
We thank Naomie Turgeon and Isabelle Fréchette for their technical help with the ChIP and EMSA studies as well as Dr. Cheikh I. Seye and Sophie Tousignant for critical and careful reading of the manuscript.
Footnotes
This research was supported by the Crohn’s and Colitis Foundation of Canada Grant in Aid of Research, the Canadian Institutes of Health Research (CIHR, NMD-94729), and an establishment grant from the Fonds de la Recherche en Santé du Québec (FRSQ) (to F.P.G.); by grants from the CIHR (IMH-67520 and MOP-68957) and The Arthritis Society (to J.S.); and by National Institutes of Health Grants AG18357, DE07389, and DE17591 (to G.A.W.). F.P.G. is a scholar from the FRSQ and a member of the FRSQ-funded Centre de Recherche Clinique Étienne-Le Bel. D.M.G. is a recipient of a scholarship from the Natural Sciences and Engineering Research Council of Canada. J.S. was a recipient of a new investigator award from the CIHR, and E.G.L. received a scholarship from the FRSQ.
Abbreviations used in this paper: IEC, intestinal epithelial cell; COX-2, cyclooxygenase-2; DSS, dextran sulfate sodium; HCAEC, human coronary artery endothelial cells; IBD, inflammatory bowel diseases; NBM, NF-κB-binding motif; TSS, transcription starting site; ChIP, chromatin immunoprecipitation; RLM, RNA ligase mediated.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Wallace JL, Devchand PR. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br J Pharmacol. 2005;145:275–282. doi: 10.1038/sj.bjp.0706201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKay DM. Good bug, bad bug: in the case of enteric inflammatory disease does the epithelium decide? Mem Inst Oswaldo Cruz. 2005;100(Suppl. 1):205–210. doi: 10.1590/s0074-02762005000900035. [DOI] [PubMed] [Google Scholar]
- 3.Muller CA, Autenrieth IB, Peschel A. Innate defenses of the intestinal epithelial barrier. Cell Mol Life Sci. 2005;62:1297–1307. doi: 10.1007/s00018-005-5034-2. [DOI] [PubMed] [Google Scholar]
- 4.Dunne C. Adaptation of bacteria to the intestinal niche: probiotics and gut disorder. Inflamm Bowel Dis. 2001;7:136–145. doi: 10.1097/00054725-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Dionne S, Ruemmele FM, Seidman EG. Immunopathogenesis of inflammatory bowel disease: role of cytokines and immune cell-enterocyte interactions. Nestle Nutr Workshop Ser Clin Perform Programme. 1999;2:41–61. doi: 10.1159/000061779. [DOI] [PubMed] [Google Scholar]
- 6.Panja A, Siden E, Mayer L. Synthesis and regulation of accessory/proinflammatory cytokines by intestinal epithelial cells. Clin Exp Immunol. 1995;100:298–305. doi: 10.1111/j.1365-2249.1995.tb03668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Brunschweiger A, Muller CE. P2 receptors activated by uracil nucleotides: an update. Curr Med Chem. 2006;13:289–312. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 9.Gendron FP, Newbold NL, Vivas-Mejia PA, Wang M, Neary JT, Sun GY, Gonzalez FA, Weisman GA. Signal transduction pathways for P2Y2 and P2X7 nucleotide receptors that mediate neuroinflammatory responses in astrocytes and microglial cells. Biomed Res. 2003;14:47–61. [Google Scholar]
- 10.Grbic DM, Degagne E, Langlois C, Dupuis AA, Gendron FP. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J Immunol. 2008;180:2659–2668. doi: 10.4049/jimmunol.180.4.2659. [DOI] [PubMed] [Google Scholar]
- 11.Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Ivanova EP, Alexeeva YV, Pham DK, Wright JP, Nicolau DV. ATP level variations in heterotrophic bacteria during attachment on hydrophilic and hydrophobic surfaces. Int Microbiol. 2006;9:37–46. [PubMed] [Google Scholar]
- 13.Kunzli BM, Berberat PO, Giese T, Csizmadia E, Kaczmarek E, Baker C, Halaceli I, Buchler MW, Friess H, Robson SC. Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G223–G230. doi: 10.1152/ajpgi.00259.2006. [DOI] [PubMed] [Google Scholar]
- 14.Schrader AM, Camden JM, Weisman GA. P2Y2 nucleotide receptor up-regulation in submandibular gland cells from the NOD.B10 mouse model of Sjögren’s syndrome. Arch Oral Biol. 2005;50:533–540. doi: 10.1016/j.archoralbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Seye CI, Kong Q, Erb L, Garrad RC, Krugh B, Wang M, Turner JT, Sturek M, Gonzalez FA, Weisman GA. Functional P2Y2 nucleotide receptors mediate uridine 5′-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation. 2002;106:2720–2726. doi: 10.1161/01.cir.0000038111.00518.35. [DOI] [PubMed] [Google Scholar]
- 16.Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, Erb L, Weisman GA. Proinflammatory cytokines tumor necrosis factor-α and interferon-γ alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol. 2008;295:C1191–C1201. doi: 10.1152/ajpcell.00144.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker OJ, Camden JM, Rome DE, Seye CI, Weisman GA. P2Y2 nucleotide receptor activation up-regulates vascular cell adhesion molecule- 1 [corrected] expression and enhances lymphocyte adherence to a human submandibular gland cell line. Mol Immunol. 2008;45:65–75. doi: 10.1016/j.molimm.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seye CI, Yu N, Jain R, Kong Q, Minor T, Newton J, Erb L, Gonzalez FA, Weisman GA. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem. 2003;278:24960–24965. doi: 10.1074/jbc.M301439200. [DOI] [PubMed] [Google Scholar]
- 19.Braun M, Lelieur K, Kietzmann M. Purinergic substances promote murine keratinocyte proliferation and enhance impaired wound healing in mice. Wound Repair Regen. 2006;14:152–161. doi: 10.1111/j.1743-6109.2006.00105.x. [DOI] [PubMed] [Google Scholar]
- 20.Taboubi S, Milanini J, Delamarre E, Parat F, Garrouste F, Pommier G, Takasaki J, Hubaud JC, Kovacic H, Lehmann M. Gα(q/11)-coupled P2Y2 nucleotide receptor inhibits human keratinocyte spreading and migration. FASEB J. 2007;21:4047–4058. doi: 10.1096/fj.06-7476com. [DOI] [PubMed] [Google Scholar]
- 21.Ghanem E, Robaye B, Leal T, Leipziger J, Van Driessche W, Beauwens R, Boeynaems JM. The role of epithelial P2Y2 and P2Y4 receptors in the regulation of intestinal chloride secretion. Br J Pharmacol. 2005;146:364–369. doi: 10.1038/sj.bjp.0706353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerstan D, Gordjani N, Nitschke R, Greger R, Leipziger J. Luminal ATP induces K+ secretion via a P2Y2 receptor in rat distal colonic mucosa. Pflugers Arch. 1998;436:712–716. doi: 10.1007/s004240050693. [DOI] [PubMed] [Google Scholar]
- 23.Matos JE, Robaye B, Boeynaems JM, Beauwens R, Leipziger J. K+ secretion activated by luminal P2Y2 and P2Y4 receptors in mouse colon. J Physiol. 2005;564:269–279. doi: 10.1113/jphysiol.2004.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nylund G, Hultman L, Nordgren S, Delbro DS. P2Y2- and P2Y4- purinergic receptors are over-expressed in human colon cancer. Auton Autacoid Pharmacol. 2007;27:79–84. doi: 10.1111/j.1474-8673.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- 25.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor κB inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1) J Biol Chem. 2004;279:35679–35686. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- 27.Gendron FP, Mongrain S, Laprise P, McMahon S, Dubois CM, Blais M, Asselin C, Rivard N. The CDX2 transcription factor regulates furin expression during intestinal epithelial cell differentiation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G310–G318. doi: 10.1152/ajpgi.00217.2005. [DOI] [PubMed] [Google Scholar]
- 28.Araki Y, Sugihara H, Hattori T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol Rep. 2006;16:1357–1362. [PubMed] [Google Scholar]
- 29.Francoeur C, Escaffit F, Vachon PH, Beaulieu JF. Proinflammatory cytokines TNF-α and IFN-γ alter laminin expression under an apoptosisindependent mechanism in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G592–G598. doi: 10.1152/ajpgi.00535.2003. [DOI] [PubMed] [Google Scholar]
- 30.Laniel MA, Beliveau A, Guerin SL. Electrophoretic mobility shift assays for the analysis of DNA-protein interactions. Methods Mol Biol. 2001;148:13–30. doi: 10.1385/1-59259-208-2:013. [DOI] [PubMed] [Google Scholar]
- 31.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki Y, Taira H, Tsunoda T, Mizushima-Sugano J, Sese J, Hata H, Ota T, Isogai T, Tanaka T, Morishita S, et al. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep. 2001;2:388–393. doi: 10.1093/embo-reports/kve085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. 2007;389:52–65. doi: 10.1016/j.gene.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volloch V, Schweitzer B, Rits S. Ligation-mediated amplification of RNA from murine erythroid cells reveals a novel class of β globin mRNA with an extended 5′-untranslated region. Nucleic Acids Res. 1994;22:2507–2511. doi: 10.1093/nar/22.13.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beilina A, Tassone F, Schwartz PH, Sahota P, Hagerman PJ. Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum Mol Genet. 2004;13:543–549. doi: 10.1093/hmg/ddh053. [DOI] [PubMed] [Google Scholar]
- 36.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 37.Quina AS, Buschbeck M, Di Croce L. Chromatin structure and epigenetics. Biochem Pharmacol. 2006;72:1563–1569. doi: 10.1016/j.bcp.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Miao F, I, Gonzalo G, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279:18091–18097. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]
- 39.Sun Z, Andersson R. NF-κB activation and inhibition: a review. Shock. 2002;18:99–106. doi: 10.1097/00024382-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Chen LF, Greene WC. Regulation of distinct biological activities of the NF-κB transcription factor complex by acetylation. J Mol Med. 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 41.Davie JR, V, Spencer A. Control of histone modifications. J Cell Biochem. 1999;32–33(Suppl):141–148. doi: 10.1002/(sici)1097-4644(1999)75:32+<141::aid-jcb17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 42.Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest. 1997;99:1367–1379. doi: 10.1172/JCI119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology. 1998;115:297–306. doi: 10.1016/s0016-5085(98)70196-9. [DOI] [PubMed] [Google Scholar]
- 44.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23→IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 45.Stenson WF. Prostaglandins and epithelial response to injury. Curr Opin Gastroenterol. 2007;23:107–110. doi: 10.1097/MOG.0b013e3280143cb6. [DOI] [PubMed] [Google Scholar]
- 46.Marcet B, Libert F, Boeynaems JM, Communi D. Extracellular nucleotides induce COX-2 up-regulation and prostaglandin E2 production in human A549 alveolar type II epithelial cells. Eur J Pharmacol. 2007;566:167–171. doi: 10.1016/j.ejphar.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Sun R, Carlson NG, Hemmert AC, Kishore BK. P2Y2 receptormediated release of prostaglandin E2 by IMCD is altered in hydrated and dehydrated rats: relevance to AVP-independent regulation of IMCD function. Am J Physiol Renal Physiol. 2005;289:F585–F592. doi: 10.1152/ajprenal.00050.2005. [DOI] [PubMed] [Google Scholar]
- 48.Welch BD, Carlson NG, Shi H, Myatt L, Kishore BK. P2Y2 receptor-stimulated release of prostaglandin E2 by rat inner medullary collecting duct preparations. Am J Physiol Renal Physiol. 2003;285:F711–F721. doi: 10.1152/ajprenal.00096.2003. [DOI] [PubMed] [Google Scholar]
- 49.Jin J, Dasari VR, Sistare FD, Kunapuli SP. Distribution of P2Y receptor subtypes on haematopoietic cells. Br J Pharmacol. 1998;123:789–794. doi: 10.1038/sj.bjp.0701665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahn JS, Camden JM, Schrader AM, Redman RS, Turner JT. Reversible regulation of P2Y2 nucleotide receptor expression in the duct-ligated rat submandibular gland. Am J Physiol Cell Physiol. 2000;279:C286–C294. doi: 10.1152/ajpcell.2000.279.2.C286. [DOI] [PubMed] [Google Scholar]
- 51.Seye CI, Gadeau AP, Daret D, Dupuch F, Alzieu P, Capron L, Desgranges C. Overexpression of P2Y2 purinoceptor in intimal lesions of the rat aorta. Arterioscler Thromb Vasc Biol. 1997;17:3602–3610. doi: 10.1161/01.atv.17.12.3602. [DOI] [PubMed] [Google Scholar]
- 52.Kong Q, Peterson TS, Baker O, Stanley E, Camden J, Seye CI, Erb L, Simonyi A, Wood WG, Sun GY, Weisman GA. Interleukin-1β enhances nucleotide-induced and α-secretase-dependent amyloid precursor protein processing in rat primary cortical neurons via up-regulation of the P2Y2 receptor. J Neurochem. 2009;109:1300–1310. doi: 10.1111/j.1471-4159.2009.06048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spencer VA, Davie JR. Role of covalent modifications of histones in regulating gene expression. Gene. 1999;240:1–12. doi: 10.1016/s0378-1119(99)00405-9. [DOI] [PubMed] [Google Scholar]
- 54.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 55.Pugh BF, Tjian R. Transcription from a TATA-less promoter requires a multisubunit TFIID complex. Genes Dev. 1991;5:1935–1945. doi: 10.1101/gad.5.11.1935. [DOI] [PubMed] [Google Scholar]
- 56.Wallace JL. COX-2: a pivotal enzyme in mucosal protection and resolution of inflammation. Sci World J. 2006;6:577–588. doi: 10.1100/tsw.2006.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ajuebor MN, Singh A, Wallace JL. Cyclooxygenase-2-derived prostaglandin D2 is an early anti-inflammatory signal in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G238–G244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.