

Abstract
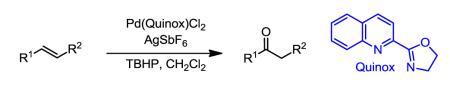
The Pd-catalyzed TBHP-mediated Wacker-type oxidation of internal alkenes is reported. The reaction uses 2-(4,5-dihydro-2-oxazolyl)quinoline (Quinox) as ligand and TBHP(aq) as oxidant to deliver single ketone constitutional isomer products in a predictable fashion from electronically-biased olefins. This methodology is showcased through its application on an advanced intermediate in the total synthesis of the anti-malarial drug, artemisinin.
The conversion of terminal olefins to methyl ketones using the Tsuji-Wacker oxidation has found extensive application in the synthesis of natural products, pharmaceuticals, and commodity chemicals.1 In contrast, the use of the Tsuji-Wacker reaction for the selective in the case of unbiased olefins.2 Additionally, the reaction of internal alkenes is slow relative to terminal olefins due to the increased congestion of the substrate. The sluggish rate likely arises from turnover-limiting addition of the nucleophile to the activated Pd-alkene complex.1a There have been several notable reports demonstrating the potential utility of internal olefin oxidation for select substrate classes.2 For example, Tsuji and co-workers have shown the regioselective oxidation of α,β-unsaturated ester 1 to give β-keto ester 2 (Figure 1).3 The authors suggest the high regioselectivity observed results from coordination of the neighboring oxygen group to the catalyst.3a In addition, Feringa and co-workers have reported the oxidation of allylic phthalimide 3 to selectively deliver the methyl ketone 4.4 Kaneda and co-workers have demonstrated a copper-free oxidation of nitrile 5 to produce ketone 6.5 These examples illustrate that electronic bias is often required to achieve high selectivity in the Wacker oxidation as unfunctionalized internal alkenes lead to multiple isomeric products.
Figure 1.

Previously reported Wacker oxidations using electronically biased internal alkenes.
Due to the potential utility of internal alkene oxidation to provide access to more highly substituted ketone products, we wanted to explore the versatility of our previously reported tert-butylhydroperoxide (TBHP)-mediated Wacker oxidation using Pd(Quinox) as the catalyst.6 For a wide range of terminal olefin substrates, the use of this catalyst system results in excellent selectivity for formation of the methyl ketone product (Markovnikov addition). This has been proposed to arise from the use of a bidentate ligand and precoordination of TBHP leaving only a single coordination site for alkene binding prior to a well-defined syn-peroxypalladation step6,7 and precluding interactions with remote functional groups in contrast to the proposal by Tsuji.3a Additionally, this method generally requires shorter reaction times and reduced catalyst loadings as compared to the traditional Tsuji-Wacker oxidation.6 Herein, we report the results of this investigation highlighted by a Wacker-type oxidation of a complex internal alkene en route to the synthesis of the antimalarial natural product, artemisinin.
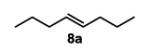
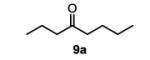
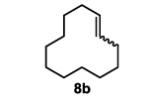
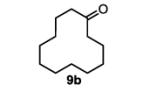
To initiate our studies, the symmetrical alkene, 4-octene, was submitted to our previously reported optimized Pd(Quinox)-TBHP reaction conditions, which afforded a 71% yield of 4-octanone, with no appreciable amount of other ketone isomers observed (Table 1, entry 1). Notably, when anhydrous TBHP was used, significant isomerization was observed, which led to a complex mixture of ketone isomers. Next, a symmetric cyclic alkene, cyclododecene as a 70:30 mixture of trans:cis isomers (8b), was submitted to the reaction conditions, which gave a 76% yield of the ketone product 9b (Table 1, entry 2). Submission of an unsymmetrical, non-biased alkene, 2-octene, resulted in a ~1:1 mixture of 2- and 3-octanone. This result is consistent with previous observations supporting the hypothesis that electronic bias of the substrate is required for high selectivity.3-5 Therefore, we sought to evaluate the type of functional groups necessary to achieve a highly regioselective alkene oxidation.
Table 1.
Scope of the TBHP-Mediated Wacker Oxidation 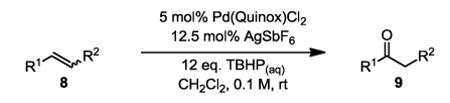
| Entry | substrate | Product | % Yield |
|---|---|---|---|
| 1 |
|
|
71 |
| 2 |
|
|
76 |
| 3 |
|
|
48 |
| 4 |
|
|
68 |
| 5 |
|
|
61 |
| 6 |
|
|
93 |
| 7 |
|
|
57 |
| 8 |
|
|
66 |
| 9 |
|
|
79 |
| 10 |
|
|
31(59)a |
| 11 |
|
|
<1 |
| 12 |
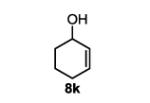
|
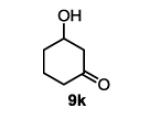
|
<1 |
All yields represent an average of two experiments on at least 0.5 mmol scale.
10 mol% Pd(Quinox)Cl2 and 25 mol% AgSbF6.
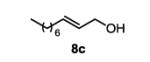
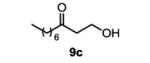
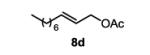
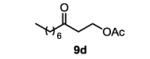
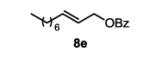
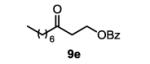
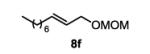
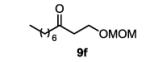
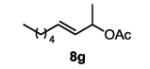
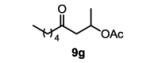
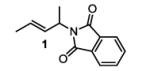
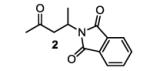
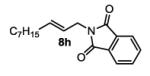
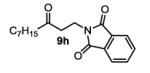
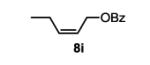
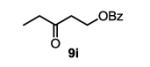
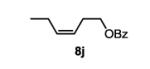
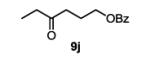
The first functionalized substrate class to be evaluated was allylic alcohols. When simple allylic alcohol 8c was subjected to the standard reaction conditions, a highly selective oxidation occurred at the alkene carbon distal to the alcohol, albeit in a modest 48% yield (Table 1, entry 3). The starting material was entirely consumed but no major byproducts were detected. To determine if a protected variant would improve the yield, three common alcohol protecting groups were evaluated (Table 1, entries 4-6). Both acetate 8d (Table 1, entry 4) and benzoate 8e (Table 1, entry 5) delivered the corresponding ketone products in improved yields and as single isomers (>20:1 by 1H NMR) in 68% and 61% yields, respectively. Interestingly, allylic methoxy ether (MOM) 8f proved superior by producing ketone 9f in 93% yield as a single regioisomer (Table 1, entry 6). When secondary allylic acetate 8g was subjected to the reaction conditions, the ketone product was isolated in 57% yield (Table 1, entry 7). In addition to allylic alcohol derivatives, allylic phthalimides were investigated based on our previous success with such substrates.6d While both the secondary (1) and primary (8h) allylic phthalimides produced single ketone products (>20:1 by 1H NMR) distal to the phthalimide, substrate 1 delivered ketone 2 in 66% yield (Table 1, entry 8) and phthalimide 8h provided ketone 9h in 79% yield (Table 1, entry 9). Finally, cis-alkene 8i was submitted to the reaction conditions resulting in 31% yield (Table 1, entry 10). Increasing the catalyst loading to 10 mol% did enhance the yield of the process to 59%. In both cases, the remainder of the mass balance was unreacted starting material. Further efforts using cis-alkenes were unsuccessful (Table 1, entries 11-12). It remains unclear why cis-alkenes are less effective in the TBHP-mediated Wacker oxidation.
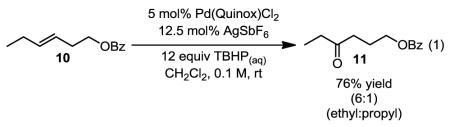
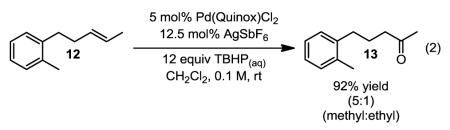
To test whether the selectivity arises from an electronic bias, a protected homoallylic alcohol substrate was evaluated. In the reaction, oxidation of homoallylic benzoate 10 (eq. 1) delivered a 6:1 mixture of the ethyl and propyl ketones in 76% yield. In this case, the mixture could be effectively separated by column chromatography. This result is consistent with the selectivity being dependent on the alkene’s proximity to the directing group and suggests as the group is farther away from the alkene that poorer differentiation would result. Interestingly, an aryl group might facilitate directed oxidation as observed in the transformation of 12 to 13, which results in 92% yield of a 5:1 mixture of methyl:ethyl ketone products (eq. 2).
 |
 |
After demonstrating the requirement of a functional group to bias oxypalladation, we next challenged the methodology by subjecting an advanced precursor in the synthesis of anti-malarial drug, artemisinin, to our conditions. Ortho ester 14 has been previously subjected to an unusual variant of the Wacker oxidation (PdCl2 and excess H2O2) for 4 days yielding the desired methyl ketone as a 2:1 mixture with the isomeric ethyl ketone in an overall 92% yield.8 Using our variant, a similar result is observed after 24 hours using this electronically unbiased alkene, furnishing product 15 in an overall 55% yield as a 1.6:1 mixture of methyl:ethyl ketones (Scheme 1).
Scheme 1.
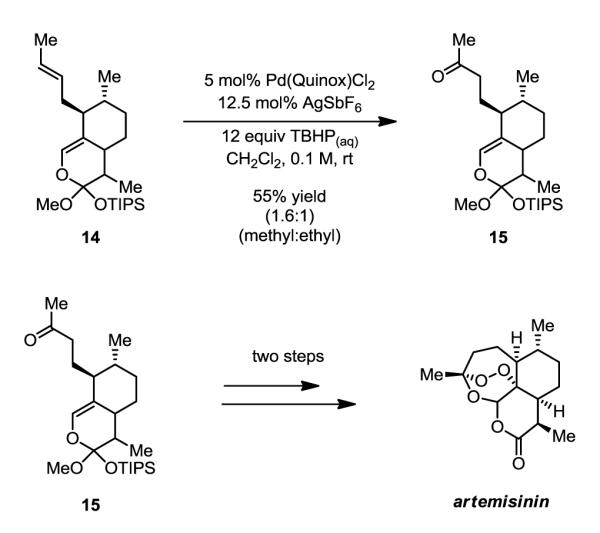
The TBHP-Mediated Wacker Oxidation Provides Access to 15, an Intermediate in the Synthesis of Natural Product Artemisinin.
In summary, the palladium-catalyzed TBHP-mediated Wacker oxidation of internal olefins is a useful complement to the traditional Tsuji-Wacker system.2a The TBHP-mediated Wacker oxidation effectively differentiates the two internal alkene carbons using electronically biased substrates delivering single ketone product constitutional isomers. This system is highlighted by its high predictability, and its attractive ease of set up and purification.
EXPERIMENTAL SECTION
A procedure for a one-pot synthesis of Quinox is outlined in a recent publication.6d ALTHOUGH NO PROBLEMS OCCURRED DURING THESE STUDIES, HIGHLY CONCENTRATED SOLUTIONS OF TBHP IN THE PRESENCE OF TRANSITION METALS CAN BE EXPLOSIVE.
General Procedure for the Pd(Quinox)Cl2–TBHP Oxidation
In the dark, AgSbF6 (43 mg, 0.125 mmol, 0.125 equiv), Pd(Quinox)Cl2 complex (19 mg, 0.05 mmol, 0.05 equiv), and a magnetic stir bar were added to a 25-mL round-bottom flask. CH2Cl2 (3.3 mL) was charged to the flask and the mixture was stirred for 10 min, after which aqueous 70% wt/wt TBHP (1.7 mL, 12 mmol, 12 equiv) and remaining CH2Cl2 (5.0 mL) were charged to the flask. The mixture, which turned a deep orange, was stirred for 10 min before the substrate (1.0 mmol, 1 equiv) was added. The reaction was monitored by TLC and upon complete consumption of starting material (or 24 hours), the reaction was cooled to 0 °C and quenched with Na2SO3 (15 mL) to consume excess TBHP. The mixture was transferred to a separatory funnel and diluted with EtOAc (25 mL). The aqueous layer was back extracted with ethyl acetate (25 mL). The combined organics were washed with H 2O (4 × 10 mL) and brine (25 mL) and then dried over MgSO4. After filtration and concentrating under reduced pressure, the crude material was purified by silica gel flash chromatography and the product containing fractions were combined and concentrated under reduced pressure.
General Procedure for Benzoyl Protection
Into a dry 250 mL round-bottom flask equipped with a stirbar were added the alcohol (10.0 mmol, 1.0 equiv), 244 mg of DMAP (2.0 mmol, 0.20 equiv), 1.8 mL of Et3N (13.0 mmol, 1.3 equiv) and 50 mL of DCM. The flask was equipped with a septum and connected to a N2 line. The resulting mixture was cooled to 0 °C, and 3.40 g of benzoic anhydride (15.0 mmol, 1.5 equiv) was dissolved in 5 mL of DCM and added dropwise. The mixture was allowed to warm to room temperature and was stirred overnight. The solution was then washed with sat. aq. NH4Cl (50 mL) and H2O (50 mL). The combined aqueous layers were extracted with DCM (50 mL). The combined organic layers were then washed with brine (30 mL), dried over Na2SO4, decanted, and concentrated in vacuo. The product was purified by silica gel flash column chromatography.
General Procedure for the Mitsunobu Reaction
Into a dry 100 mL round-bottom flask equipped with a stirbar were added the corresponding allylic alcohol (10.0 mmol, 1.0 equiv) and 40 mL of dry THF. Next, 4.02 g of triphenylphosphine (15.0 mmol, 1.5 equiv) and 1.47 g of phthalimide (10.0 mmol, 1.0 equiv) were added to the reaction flask. The flask was equipped with a septum and connected to a N2 line. The resulting mixture was cooled to 0 °C, and 2.9 mL of diispropyl azodicarboxylate (DIAD) (15.0 mmol, 1.5 equiv) was added dropwise. The mixture was allowed to warm to room temperature and was stirred overnight. The crude reaction mixture was concentrated in vacuo and the product was purified by silica gel flash column chromatography.
(E)-oct-4-ene (8a)
This commercially available compound was purchased and used without further purification.
cyclododecene (8b)
This commercially available compound was purchased and used without further purification. The ratio of cis:trans was 30:70.
(E)-dec-2-en-1-ol (8c)
This commercially available compound was prepared according to the literature procedure.9 Analytical data are consistent with the literature.10
(E)-dec-2-en-1-yl acetate (8d)
This commercially available compound was prepared according to the literature procedure.11 Analytical data are consistent with the literature.12
(E)-dec-2-en-1-yl benzoate (8e)
This compound was prepared with the general procedure for benzoyl protection using 1.56 g of (E)-dec-2-en-1-ol. The product was purified by silica gel flash chromatography eluting with 5% diethyl ether in hexanes to give the product as a colorless oil in 93% yield (2.42 g). Rf = 0.5 (10% Et2O in hexanes) 1H NMR (400 MHz, CDCl3): δ 0.88 (t, J = 6.6 Hz, 3H), 1.18-1.47 (m, 10H), 2.08 (q, J = 6.8 Hz, 2H), 4.76 (d, J = 6.4 Hz, 2H), 5.68 (dt, J = 15.6, 6.4 Hz, 1H), 5.86 (dt, J = 15.2, 6.6 Hz, 1H),_7.43 (t, J = 7.6 Hz, 2H), 7.55 (t, J = 7.4 Hz, 1H), 8.05 (d, J = 7.2 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 14.3, 22.9, 29.1, 29.3(2), 32.0, 32.5, 66.0, 123.9, 128.5, 129.5, 130.6, 133.1, 137.0, 166.7. IR (neat): 2924, 2853, 1717, 1450, 1377, 1265, 1107, 1068, 1025, 968, 935, 707, 686 cm−1. HRMS (ESI-TOF) m/z calculated for C17H24O2Na [M+Na]+ : 283.1674, obsvd. 283.1669.
(E)-1-(methoxymethoxy)dec-2-ene (8f)
This compound was prepared according to the literature procedure.13 Analytical data are consistent with the literature.13
(E)-non-3-en-2-yl acetate (8g)
This compound was prepared with the general procedure for benzoyl protection with the modification that 1.56 g of (E)-dec-2-en-1-ol was protected using 1.4 mL of acetic anhydride instead of benzoic anhydride. The product was purified by silica gel flash chromatography eluting with DCM to give the product as a colorless oil in 56% yield (1.03 g). Rf = 0.7 (DCM) 1H NMR (500 MHz, CDCl3): δ 0.88 (t, J = 7.0 Hz, 3H), 1.23-1.41 (m, 11H), 2.03 (s, 3H), 5.31 (quint, J = 6.6 Hz, 1H), 5.45 (dd, J = 15.3, 6.8 Hz, 1H), 5.70 (dt, J = 15.3, 6.7 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 14.2, 20.6, 21.7, 22.7, 28.8, 31.6, 32.3, 71.4, 129.5, 133.7, 170.6. IR (neat): 2957, 2927, 2857, 1735, 1456, 1369, 1235, 1135, 1041, 1014, 967, 947, 841, 668, 609 cm−1. HRMS (ESI-TOF) m/z calculated for C11H20O2Na [M+Na]+: 207.1361, obsvd. 207.1364.
(E)-2-(pent-3-en-2-yl)isoindoline-1,3-dione (1)
This compound was prepared with the general procedure for the Mitsunobu reaction.3 Analytical data are consistent with the literature.3
(E)-2-(dec-2-en-1-yl)isoindoline-1,3-dione (8h)
This compound was prepared with the general procedure for the Mitsunobu reaction using 1.56 g of (E)-dec-2-en-1-ol. The product was purified by silica gel flash chromatography eluting with 5% ethyl acetate in hexanes to give the product as a colorless oil in 98% yield (2.80 g). Rf = 0.5 (5% EtOAc in hexanes) 1H NMR (500 MHz, CDCl3): δ 0.85 (t, J = 7.0 Hz, 3H), 1.18-1.37 (m, 10H), 1.99 (q, J = 7.2 Hz 2H), 4.23 (d, J = 6.0 Hz, 2H), 5.50 (dt, J = 15.5, 6.6 Hz, 1H), 5.74 (dt, J = 15.5, 7.1 Hz, 1H), 7.69-7.72 (m, 2H) 7.82-7.86 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 14.3, 22.8, 29.1, 29.3, 32.0, 32.3, 39.8, 71.4, 123.2, 123.4, 132.4, 134.0, 135.6, 168.2. IR (neat): 2923, 2853, 1771, 1709, 1466, 1390, 1354, 1186, 1099, 966, 716 cm−1. HRMS (AP-TOF) m/z calculated for C18H24NO2[M+H]+: 286.1807, obsvd. 286.1802.
(Z)-pent-2-en-1-yl benzoate (8i)
This compound was prepared with the general procedure for benzoyl protection using 861 mg of (Z)-pent-2-en-1-ol. The product was purified by silica gel flash chromatography eluting with 5% ethyl acetate in hexanes to give the product as a colorless oil in 90% yield (1.71 g). Analytical data are consistent with the literature.14
(Z)-hex-3-en-1-yl benzoate (8j)
This compound was prepared with the general procedure for benzoyl protection using 1.00 g of (Z)-hex-3-en-1-ol. The product was purified by silica gel flash chromatography eluting with 5% ethyl acetate in hexanes to give the product as a colorless oil in 86% yield (1.76 g). Analytical data are consistent with the literature.15
(E)-hex-3-en-1-yl benzoate (10)
This compound was prepared with the general procedure for benzoyl protection using 1.56 g of (E)-dec-2-en-1-ol. The product was purified by silica gel flash chromatography eluting with 10% ethyl acetate in hexanes to give the product as a colorless oil in 95% yield (1.94 g). Analytical data are consistent with the literature.15
(((7R,8S)-8-((E)-but-2-en-1-yl)-3-methoxy-4,7-dimethyl-4,4a,5,6,7,8-hexahydro-3H-isochromen-3-yl)oxy)triisopropylsilane (14)
This compound was prepared according to the literature procedure.8 Analytical data are consistent with the literature.8
octan-4-one (table 1, entry 1) (9a)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 112 mg of (E)-oct-4-ene (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 5% ethyl acetate in hexanes to afford the product as a colorless oil in 71% yield (91 mg). The spectral data were in accordance with the known commercially available compound.
cyclododecanone (table 1, entry 2) (9b)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 166 mg of cyclododecene (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 5% ethyl acetate in hexanes to afford the product as a white solid in 76% yield (138 mg). The spectral data were in accordance with the known commercially available compound.
1-hydroxydecan-3-one (table 1, entry 3) (9c)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 78 mg of (E)-dec-2-en-1-ol (0.5 mmol). The crude mixture was purified by flash chromatography eluting with 40% ethyl acetate in hexanes to afford the product as a colorless oil in 32% yield (55 mg). Rf = 0.5 (40% EtOAc in hexanes) 1H NMR (500 MHz, CDCl3): δ 0.88 (t, J = 7.0 Hz, 3H), 1.22-1.34 (m, 8H), 1.59 (quint, J = 7.3 Hz, 2H), 2.44 (t, J = 7.5 Hz, 2H), 2.67 (t, J = 5.3 Hz, 2H), 3.85 (t, J = 5.3 Hz, 2H) 13C NMR (100 MHz, CDCl3): δ 14.3, 22.8, 23.9, 29.3, 29.4, 31.9, 43.6, 44.5, 58.2, 212.3. IR (neat): 3420, 2924, 2854, 1701, 1457, 1375, 1047, 668 cm−1. HRMS (ESI-TOF) m/z calculated for C10H20O2Na [M+Na]+: 195.1361, obsvd. 195.1361.
3-oxodecyl acetate (table 1, entry 4) (9d)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 198 mg of (E)-dec-2-en-1-yl acetate (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 10% ethyl acetate in hexanes to afford the product as a colorless oil in 68% yield (146 mg). Rf = 0.3 (10% EtOAc in hexanes) 1H NMR (300 MHz, CDCl3): δ 0.88 (t, J = 6.8 Hz, 3H), 1.21-1.33 (m, 8H), 1.54-1.62 (m, 2H), 2.04 (s, 3H), 2.44 (t, J = 7.4 Hz, 2H), 2.74 (t, J = 6.3 Hz, 2H), 4.34 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 14.2, 21.0, 22.7, 23.7, 29.2, 29.3, 31.8, 41.3, 43.4, 59.6, 171.0, 208.2. IR (neat): 2926, 2855, 1739, 1715, 1366, 1233, 1036, 668 cm−1. HRMS (ESI-TOF) m/z calculated for C12+H22O3Na [M+Na]+: 237.1467, obsvd. 237.1469.
3-oxodecyl benzoate (table 1, entry 5) (9e)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 130 mg of (E)-dec-2-en-1-yl benzoate (0.5 mmol). The crude mixture was purified by flash chromatography eluting with 20% diethyl ether in hexanes to afford the product as a colorless oil in 61% yield (84 mg). Rf = 0.5 (20% Et2O in hexanes) 1H NMR (400 MHz, CDCl3): δ 0.87 (t, J = 6.6 Hz, 3H), 1.20-1.32 (m, 8H), 1.57-1.64 (m, 2H), 2.47 (t, J = 7.4 Hz, 2H), 2.87 (t, J = 6.3 Hz, 2H), 4.60 (t, J = 6.3 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.55 (t, J = 7.6 Hz, 1H), 8.00 (d, J = 5.8 Hz, 2H). 13C_NMR (100 MHz, CDCl3): δ 14.2, 22.7, 23.8, 29.2(2), 31.8, 41.5, 43.4, 60.1, 128.4, 129.7, 130.1, 133.1, 166.5, 208.1. IR (neat): 2925, 2854, 1717, 1602, 1451, 1271, 1110, 964, 708, 686 cm−1. HRMS (ESI-TOF) m/z calculated for C17H24O3Na [M+Na]+ : 299.1623, obsvd. 299.1622.
1-(methoxymethoxy)decan-3-one (table 1, entry 6) (9f)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 100 mg of (E)-1-(methoxymethoxy)dec-2-ene (0.5 mmol). The crude mixture was purified by flash chromatography eluting with 10% diethyl ether in hexanes to afford the product as a colorless oil in 93% yield (100 mg). Rf = 0.2 (10% Et2O in hexanes) 1H NMR (400 MHz, CDCl3): δ 0.88 (t, J = 6.5 Hz, 3H), 1.21-1.34 (m, 8H), 1.55-1.62 (m, 2H), 2.45 (t, J = 7.4 Hz, 2H), 2.68 (t, J = 6.1 Hz, 2H), 3.37 (s, 3H), 3.80 (t, J = 6.0 Hz, 2H) 4.60 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 14.3, 22.8, 23.8, 29.2, 29.3, 31.9, 42.8, 43.6, 55.4, 62.9, 96.7, 209.4. IR (neat): 3743, 2925, 2855, 1715, 1457, 1379, 1213, 1149, 1110, 1041, 918, 668 cm−1. HRMS (ESI-TOF) m/z calculated for C12+H24O3Na [M+Na]+: 239.1623, obsvd. 239.1624.
4-oxononan-2-yl acetate (table 1, entry 7) (9g)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 92 mg of (E)-non-3-en-2-yl acetate (0.5 mmol). The crude mixture was purified by flash chromatography eluting with dichloromethane to afford the product as a colorless oil in 57% yield (57 mg). Rf = 0.3 (DCM) 1H NMR (500 MHz, CDCl3): δ 0.88 (t, J = 6.8 Hz, 3H), 1.20-1.34 (m, 7H), 1.52-1.60 (m, 2H), 2.00 (s, 3H), 2.40 (t, J = 7.3 Hz, 2H), 2.52 (dd, J = 16.2, 5.8 Hz, 1H), 2.76 (dd, J = 16.3, 7.0 Hz, 1H), 5.27 (sext., J = 6.3 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 14.1, 20.3, 21.4, 22.6, 23.5, 31.5, 43.6, 48.7, 67.4, 170.5, 208.1. IR (neat): 2956, 2932, 2872, 1736, 1714, 1457, 1370, 1236, 1141, 1037, 956, 668, 607cm−1. HRMS (ESI-TOF) m/z calculated for C11H20O3Na [M+Na]+: 223.1310, obsvd. 223.1305.
2-(4-oxopentan-2-yl)isoindoline-1,3-dione (table 1, entry 8) (2)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 430 mg of (E)-2-(pent-3-en-2-yl)isoindoline-1,3-dione (2.0 mmol). The crude mixture was purified by flash chromatography eluting with 15% ethyl acetate in hexanes to afford the product as a colorless oil in 66% yield (305 mg). The spectral data were in accordance with the previous report.3
2-(3-oxodecyl)isoindoline-1,3-dione (table 1, entry 9) (9h)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 258 mg of (E)-2-(dec-2-en-1-yl)isoindoline-1,3-dione (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 10% ethyl acetate in hexanes to afford the product as a white solid in 79% yield (237 mg). Rf = 0.3 (10% EtOAc in hexanes). M. P. = 88-90 °C. 1H NMR (400 MHz, CDCl3): δ 0.87 (t, J = 6.9 Hz, 3H), 1.19-1.31 (m, 8H), 1.56 (quint, J = 7.3, 2H), 2.41 (t, J = 7.6 Hz, 2H), 2.84 (t, J = 7.6 Hz, 2H), 3.96 (t, J = 7.4 Hz, 2H), 7.71 (dd, J = 5.5, 3.0 Hz, 2H), 7.83 (dd, J = 5.3, 3.1 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 14.3, 22.8, 23.9, 29.3, 29.4, 31.9, 33.3, 40.8, 43.2, 123.5, 132.3, 134.2, 168.4, 208.5. IR (neat): 2923, 2853, 1704, 1652, 1436, 1360, 1221, 1091, 721, 668 cm−1. HRMS (ESI-TOF) m/z calculated for C18H23NO3Na [M+Na]+ : 324.1576, obsvd. 324.1574.
3-oxopentyl benzoate (table 1, entry 10) (9i)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 190 mg of (Z)-pent-2-en-1-yl benzoate (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 10% ethyl acetate in hexanes to afford the product as a colorless oil in 31% yield (64 mg). Rf = 0.3 (10% EtOAc in hexanes). 1H NMR (500 MHz, CDCl3): δ 1.09 (t, J = 7.3 Hz, 3H), 2.50 (q, J = 7.3 Hz, 2H), 2.88 (t, J = 6.5, 2H), 4.59 (t, J = 6.3 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.55 (t, J = 7.6 Hz, 1H), 8.00 (d, J = 8.1 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 7.8, 36.6, 41.3, 60.2, 128.5, 129.8, 130.2, 133.2, 166.6, 208.5. IR (neat): 2976, 1711, 1601, 1451, 1314, 1369, 1111, 1070, 1025, 973, 708, 617 cm−1. HRMS (ESI-TOF) m/z calculated for C12H14O3Na [M+Na]+: 229.0841, obsvd. 229.0837.
4-oxohexyl benzoate (11)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 204 mg of (E)-hex-3-en-1-yl benzoate (1.0 mmol). The crude mixture was purified by flash chromatography eluting with 10% ethyl acetate in hexanes to afford the product as a colorless oil in 65% yield (143 mg). The spectral data were in accordance with the previous report.16
5-(o-tolyl)pentan-2-one (13)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 200 mg of (E)-1-methyl-2-(pent-3-en-1-yl)benzene (1.25 mmol). The crude mixture was purified by flash chromatography eluting with 5% ethyl acetate in hexanes to afford the product as a colorless oil in 92% yield (202 mg). The spectral data were in accordance with the previous report.17
4-((7R,8S)-3-methoxy-4,7-dimethyl-3-((triisopropylsilyl)oxy)-4,4a,5,6,7,8-hexahydro-3H-isochromen-8-yl)butan-2-one (15)
The general procedure for the TBHP-mediated Wacker oxidation was followed using 48 mg of (((7R,8S)-8-((E)-but-2-en-1-yl)-3-methoxy-4,7-dimethyl-4,4a,5,6,7,8-hexahydro-3H-isochromen-3-yl)oxy)triisopropylsilane (14) (0.11 mmol). The crude mixture was purified by flash chromatography eluting with 10% ethyl acetate in hexanes to afford the products as a colorless oil in 55% yield (27 mg) as a 1.6:1 mixture. The spectral data were in accordance with the previous report.8
Supplementary Material
Acknowledgment
This work was supported by the National Institutes of Health (NIGMS RO1 GM3540).
Footnotes
Supporting Information. Copies of NMR Spectra. This information is available free of charge via the Internet at http://pubs.acs.org
References
- (1) (a).Keith JA, Henry PM. Angew. Chem. Int. Ed. 2009;48:9038. doi: 10.1002/anie.200902194. [DOI] [PubMed] [Google Scholar]; (b) Cornell CN, Sigman MS. Inorg. Chem. 2007;46:1903. doi: 10.1021/ic061858d. [DOI] [PubMed] [Google Scholar]; (c) Takacs JM, Jiang X.-t. Curr. Org. Chem. 2003;7:369. [Google Scholar]; (d) Tsuji J. Synthesis. 1984;369 [Google Scholar]
- (2) (a).Morandi B, Wickens ZK, Grubbs RH. Angew. Chem., Int. Ed. 2013 doi: 10.1002/anie.201303587. DOI: 10.1002/anie.201209541. [DOI] [PubMed] [Google Scholar]; (b) Gaunt MJ, Yu J, Spencer JB. Chem. Commun. 2001;1844 doi: 10.1039/b103066n. [DOI] [PubMed] [Google Scholar]; (c) Nelson DJ, Li R, Brammer C. J. Am. Chem. Soc. 2001;123:1564. doi: 10.1021/ja002190j. [DOI] [PubMed] [Google Scholar]; (d) Betzemeier B, Lhermitte F. d. r., Knochel P. Tetrahedron Lett. 1998;39:6667. [Google Scholar]; (e) Okota T, Fujibayashi S, Nishiyama Y, Sakaguchi S, Ishii Y. J. Mol. Catal. A: Chem. 1996;114:113. [Google Scholar]; (f) Kang S-K, Jung K-Y, Chung J-U, Namkoong E-Y, Kim T-H. J. Org. Chem. 1995;60:4678. [Google Scholar]; (g) Sommovigo M, Alper H. J. Mol. Catal. 1994;88:151. [Google Scholar]; (h) Hosokawa T, Nakahira T, Takano M, Murahashi S-I. J. Mol. Catal. 1992;74:489. [Google Scholar]; (i) Miller DG, Wayner DDM. Can. J. Chem. 1992;70:2485. [Google Scholar]; (j) Kulkarni MG, Sebastian MT. Synth. Commun. 1991;21:581. [Google Scholar]; (k) Miller DG, Wayner DDM. J. Org. Chem. 1990;55:2924. [Google Scholar]; (l) Takehira K, Hayakawa T, Orita H, Shimizu M. J. Mol. Catal. 1989;53:15. [Google Scholar]; (m) Grattan TJ, Whitehurst JS. J. Chem. Soc., Chem. Commun. 1988;43 [Google Scholar]; (n) Akermark B, Soederberg BC, Hall SS. Organometallics. 1987;6:2608. [Google Scholar]; (o) Takehira K, Orita H, Oh IH, Leobardo CO, Martinez GC, Shimidzu M, Hayakawa T, Ishikawa T. J. Mol. Catal. 1987;42:247. [Google Scholar]; (p) Tsuji J, Minato M. Tetrahedron Lett. 1987;28:3683. [Google Scholar]; (q) Keinan E, Seth KK, Lamed R. J. Am. Chem. Soc. 1986;108:3474. [Google Scholar]; (r) Zahalka HA, Januszkiewicz K, Alper H. J. Mol. Catal. 1986;35:249. [Google Scholar]; (s) Alper H, Januszkiewicz K, Smith DJH. Tetrahedron Lett. 1985;26:2263. [Google Scholar]; (t) Takehira K, Hayakawa T, Orita H. Chem. Lett. 1985;14:1835. [Google Scholar]; (u) Ogawa H, Fujinami H, Taya K, Teratani S. Bull. Chem. Soc. Jpn. 1984;57:1908. [Google Scholar]
- (3) (a).Tsuji J, Nagashima H, Hori K. Tetrahedron Lett. 1982;23:2679. [Google Scholar]; (b) Tsuji J, Nagashima H, Hori K. Chem. Lett. 1980;9:257. [Google Scholar]
- (4).Weiner B, Baeza A, Jerphagnon T, Feringa BL. J. Am. Chem. Soc. 2009;131:9473. doi: 10.1021/ja902591g. [DOI] [PubMed] [Google Scholar]
- (5).Mitsudome T, Mizumoto K, Mizugaki T, Jitsukawa K, Kaneda K. Angew. Chem. Int. Ed. 2010;49:1238. doi: 10.1002/anie.200905184. [DOI] [PubMed] [Google Scholar]
- (6) (a).Michel BW, Sigman MS. Aldrichimica Acta. 2011;44:55. [PMC free article] [PubMed] [Google Scholar]; (b) McCombs JR, Michel BW, Sigman MS. J. Org. Chem. 2011;76:3609. doi: 10.1021/jo200462a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Michel BW, Steffens LD, Sigman MS. J. Am. Chem. Soc. 2011;133:8317. doi: 10.1021/ja2017043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Michel BW, McCombs JR, Winkler A, Sigman MS. Angew. Chem., Int. Ed. 2010;49:7312. doi: 10.1002/anie.201004156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Anderson BJ, Keith JA, Sigman MS. J. Am. Chem. Soc. 2010;132:11872. doi: 10.1021/ja1057218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Michel BW, Camelio AM, Cornell CN, Sigman MS. J. Am. Chem. Soc. 2009;131:6076. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Mimoun H. Angew. Chem. 1982;94:750. [Google Scholar]; (b) Mimoun H. J. Mol. Catal. 1980;7:1. [Google Scholar]; (c) Mimoun H, Charpentier R, Mitschler A, Fischer J, Weiss R. J. Am. Chem. Soc. 1980;102:1047. [Google Scholar]; (d) Roussel M, Mimoun H. J. Org. Chem. 1980;45:5387. [Google Scholar]; (e) Halfpenny J, Small RWH. J. Chem. Soc., Chem. Commun. 1979;879 [Google Scholar]
- (8).Zhu C, Cook SP. J. Am. Chem. Soc. 2012;134:13577. doi: 10.1021/ja3061479. [DOI] [PubMed] [Google Scholar]
- (9).Nutaitis CF, Bernardo JE. J. Org. Chem. 1989;54:5629. [Google Scholar]
- (10).Mayer SF, Steinreiber A, Orru RVA, Faber K. J. Org. Chem. 2002;67:9115. doi: 10.1021/jo020073w. [DOI] [PubMed] [Google Scholar]
- (11).Babler JH, White NA, Kowalski E, Jast JR. Tetrahedron Lett. 2011;52:745. [Google Scholar]
- (12).Campbell AN, White PB, Guzei IA, Stahl SS. J. Am. Chem. Soc. 2010;132:15116. doi: 10.1021/ja105829t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Han JH, Kwon YE, Sohn J-H, Ryu DH. Tetrahedron. 2010;66:1673. [Google Scholar]
- (14).Heller ST, Fu T, Sarpong R. Org. Lett. 2012;14:1970. doi: 10.1021/ol300339q. [DOI] [PubMed] [Google Scholar]
- (15).Tokuyasu T, Kunikawa S, McCullough KJ, Masuyama A, Nojima M. J. Org. Chem. 2004;70:251. doi: 10.1021/jo048359j. [DOI] [PubMed] [Google Scholar]
- (16).Zhao Y, Yim W-L, Tan CK, Yeung Y-Y. Org. Lett. 2011;13:4308. doi: 10.1021/ol2016466. [DOI] [PubMed] [Google Scholar]
- (17).Kise N, Suzumoto T, Shono T. J. Org. Chem. 1994;59:1407. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.