

Abstract
Aims
Sarcoidosis is a granulomatous disease of unknown etiology marked by tremendous clinical heterogeneity. Many patients enter remission with good long-term outcomes. Yet, chronic disease is not uncommon, and this important phenotype remains understudied. Identified alterations in local and circulating cytokines—specifically targeted for study, and often in the acute phase of disease—have informed our growing understanding of the immunopathogenesis of sarcoidosis. Our aim was to evaluate a broad panel of circulating cytokines in patients with chronic sarcoidosis. Among those with chronic disease, pulmonary fibrosis occurs in only a subset. To gain more insight into the determinants of the fibrotic response, we also determined if the phenotypes of fibrotic and non-fibrotic pulmonary sarcoidosis have distinct cytokine profiles.
Results
In patients with sarcoidosis compared to controls, IL-5 was decreased, and IL-7 was increased. Both of these comparisons withstood rigorous statistical correction for multiple comparisons. GM-CSF met a nominal level of significance. We also detected an effect of phenotype, where IL-5 was significantly decreased in non-fibrotic compared to fibrotic pulmonary sarcoidosis, and compared to controls. Compared to controls, there was a trend towards a significant increase in IL-7 in fibrotic, but not in non-fibrotic pulmonary sarcoidosis. In contrast, compared to controls, there was a trend towards a significant increase in GM-CSF in non-fibrotic, but not in fibrotic pulmonary sarcoidosis.
Conclusions
In a comprehensive evaluation of circulating cytokines in sarcoidosis, we found IL-5, IL-7, and GM-CSF to be altered. These findings provide a window into the immunopathogenesis of sarcoidosis. IL-7 is a novel sarcoidosis cytokine and, as a master regulator of lymphocytes, is an attractive target for further studies. By observing an effect of phenotype upon cytokine patterns, we also identify specific immune alterations which may contribute to clinical heterogeneity.
Keywords: Sarcoidosis, Cytokines, Interleukin-5, Interleukin-7, Pulmonary fibrosis
1. Introduction
Sarcoidosis is a clinically heterogeneous, granulomatous condition of unknown etiology. While the lungs are most often affected, multi-organ disease is common, and the heart, skin, eyes, liver, and/or nervous system can be affected [1]. Many patients enter remission after a limited duration of disease, experiencing minimal long-term or permanent organ damage [2,3]. However, chronic disease develops in a subset of patients, often marked by high morbidity and an increased risk of death. Outcomes are particularly poor for those who develop fibrotic pulmonary sarcoidosis. While there is no cure for sarcoidosis, immunosuppression is often employed to control inflammation, although the clinical response is variable [4].
The adjusted annual incidence of sarcoidosis is much higher for African Americans compared to European Americans, and African Americans are more likely to develop chronic disease [5–7]. These observations suggest that clinical phenotypes are mediated by distinct, genetically determined immune responses. Indeed, evolving evidence suggests that fibrotic pulmonary sarcoidosis has immune alterations distinct from non-fibrotic pulmonary disease [8–11].
The initial pathogenesis of sarcoidosis centers around antigen presenting cells interacting with naive CD4+ T cells, and leading to a T helper 1 (Th1)-polarized inflammatory response. Engaged CD4+ T cells release cytokines which drive the accumulation of macrophages, which organize to form granulomas [3]. TNF-α and IFN-γ are fundamental to granulomatous inflammation, and both are increased in sarcoid tissues [12]. While the oligoclonal expansion of CD4+ T cells suggests the presentation of antigen, the inciting antigen remains unidentified. In most patients, the activated immune response down regulates after the acute phase of disease, resulting in clinical remission. The mechanisms of persistent inflammation in a subset of patients, and the determinants of an extensive fibrotic response in an even smaller subset, are not known. Most studies of sarcoidosis have targeted acute disease. In contrast, subjects in our cohort predominantly have chronic disease, where events in the immunopathology may differ from those of acute inflammation.
In addition to TNF-α and IFN-γ, multiple other cytokines also are increased in sarcoidosis. While the full array of clinically important cytokine alterations is not known, the biology of cell signaling is no doubt complex and is seemingly concerted. Identified alterations in local cytokines, often specifically targeted for study, have provided insight into the immunopathogenesis [13–16]. Knowledge about corresponding circulating cytokines is more limited, yet there is increasing interest in the circulating immune system in sarcoidosis, where alterations in circulating monocytes, dendritic cells, T regulatory cells, and natural killer T cells have been demonstrated [17–23]. To better elucidate cytokine alterations in sarcoidosis, we evaluated a broad and relatively unbiased array of circulating cytokines in our cohort.
Combining clinically heterogeneous subjects into a single study cohort may impair detection of immune alterations, which may be phenotype-specific. Phenotyping enhances detection of potentially important alterations and allows comparison among phenotypes to reveal mechanisms of clinical diversity, and has been advocated for studies of sarcoidosis [24–26]. Beyond capturing the phenotype of chronic sarcoidosis, we compared cytokines in those with fibrotic and non-fibrotic pulmonary disease, as the clinical features of these phenotypes suggest that there are important differences in their underlying immunopathologies.
2. Materials and methods
2.1. Subjects
Serum samples were collected from 54 adults with biopsy-established sarcoidosis. Details regarding organ involvement, treatment, and smoking status were available for review. Eighteen controls were matched for age, gender and race. Race was self or clinician designated; while imperfect, it was the only available measure of race that was non-invasive and clinically feasible. Exclusion criteria for cases and controls included pregnancy, active infection, cancer, chronic inflammatory conditions other than sarcoidosis, or a glomerular filtration rate <40 mL/min/body surface area. Immunosuppression included use of ≥10 mg/daily of prednisone or the equivalent corticosteroid dose, or any other non-steroidal immunosuppressive therapy. Without selection bias, cases and controls were recruited consecutively in outpatient clinics via an institutional translational research program, and all patients provided informed consent. This study was approved by the University of Chicago Institutional Review Board.
2.2. Phenotypes
Fibrotic pulmonary sarcoidosis was established by the presence of honeycombing, bronchial distortion, and/or linear fibrosis on computerized tomography imaging. Non-fibrotic pulmonary sarcoidosis was established by non-fibrotic parenchymal or bronchial abnormalities on computerized tomography imaging.
2.3. Multiplex cytokine assays
IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 (p70), IL-13, IL-17, G-CSF, GM-CSF, IFN-γ, MCP-1, MIP-1β, and TNF-α were measured using Bio-Plex human cytokine multiplex kits (Bio-Rad Inc., Hercules, CA) per manufacturer instructions. Normalization was performed to control for variance between plates.
3. Theory/calculation
Four cytokines (IL-1β, IL-2, IL-6, and IL-8) performed poorly, with less than 25% of values registering above background fluorescence, and were excluded from further analysis. GM-CSF performed well, but demonstrated significant background staining, which potentially diminished the signal when background fluorescence was subtracted. The data were non-parametric, and were expressed as medians with interquartile ranges. Differences were initially tested using the non-parametric 2-tailed Mann–Whitney U test. Cytokine data were log transformed for multi-variate logistic regressions, which controlled for potential influences of gender, race, smoking, and immunosuppression, as well as cytokine–cytokine interactions. Correlations were measured with the Spearman’s correlation test.
Pairwise Spearman correlations were calculated for each possible pairing of the 13 analyzed cytokines, resulting in 78 pairwise correlations. We expected to find correlations among cytokines, making a strict Bonferroni correction inappropriate, as the variables were not independent. Thus, we used a modified Bonferroni correction to account for the number of cytokines tested and their degree of correlation with each other as outlined in the following equation, where pcorr is the corrected p-value and pobs is the observed p-value:
This resulted in a p-value of <0.005 to define significance. For phenotype analyses, we controlled the family-wise type I error rate at 0.05 using a Bonferroni correction for the number of comparisons (three), which resulted in a calculated p-value of ≥0.017 to define significance.
4. Results
4.1. Subject and control demographics
Subject characteristics are shown in Table 1. Our cohort was enriched with African Americans and women. Many but not all subjects were on immunosuppression. Nearly all had chronic disease, with documented sarcoidosis activity for at least 2 years at the time of the study [27]. Approximately half of the patients with pulmonary disease had pulmonary fibrosis. There were no significant differences in demographic data between cases and controls except for a trend towards a higher rate of smoking among controls.
Table 1.
Demographic and clinical data for cases and controls.
| Characteristic | Controls (N = 18) | Sarcoidosis subjects (N = 54) | p-Value |
|---|---|---|---|
| Age-yr Mean Range | 52 37-74 | 50 31-75 | 0.523 |
| Gender - no.(%) Female | 13 (72) | 40 (74) | 1.000 |
| Ancestry - no. (%) African American European American Hispanic | 13 (72) 5(27) 0 | 43 (80) 10 (10) 1 (2) | 0.611 |
| Active Smoking - no. (%) Yes No Unknown | 3(17) 13 (72) 2(11) | 3 (5) 50 (93) 1(2) | 0.100 |
| Immunosuppression a - no. (%) Yes No Unknown | - | 31 (57) 22 (41) 1 (2) | - |
| Years since diagnosis - no. (%) <2 years (acute disease) >2 years (chronic disease) | - | 2 (4) 52 (96) | - |
| Sub-phenotype no. (%) Lofgren’s syndrome Fibrotic pulmonary sarcoidosis Non-fibrotic pulmonary sarcoidosis | - | 0(0) 19 (35) 21 (39) | - |
Preclnisone =10 mg/daily, or any other immunosuppressive therapy.
4.1.1. Levels of IL-5, IL-7, and GM-CSF were different for cases and controls
Uni-variate analyses were performed for cytokines with good bioassay performance. We identified IL-5, IL-7, GM-CSF, and G-CSF levels as potentially significantly different (p < 0.05) between cases and controls (Fig. 1). These four cytokines were then entered into a regression model for multi-variate analysis, where we found significant differences between cases and controls for IL-5 (p = 0.0047) and IL-7 (p = 0.0023). IL-5 was decreased, whereas IL-7 was increased in cases compared to controls. GM-CSF and G-CSF did not meet strict criteria for significance, with p-values of 0.068 and 0.082, respectively. To control for the effects of gender, race, and smoking, these variables were then included in the regression model. The p-value for G-CSF increased significantly when these variables were entered into the regression, and G-CSF was therefore not carried forward for further analysis. The p-values for IL-5, IL-7, and GM-CSF were not significantly different in the full regression analysis, suggesting that demographic data and smoking status did not account for the differences in these cytokines between cases and controls. Further, we did not find associations between treatment with immunosuppression and levels of IL-5, IL-7, or GM-CSF. In addition to IL-5 and IL-7, we included GM-CSF for phenotype analysis, with the hypothesis that there could be phenotype associations not evident in combined analysis.
Fig. 1.
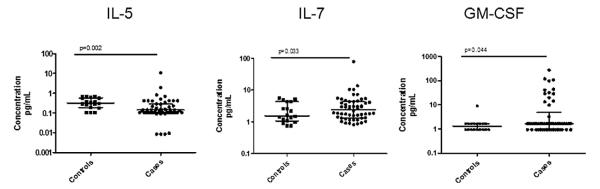
Cytokines in combined analyses. IL-5, IL-7, and GM-CSF in controls and cases are shown. P-values were calculated by Mann–Whitney testing. Central tendencies are represented by the median, with interquartile ranges shown.
4.1.2. Cytokine patterns vary according to phenotype (Fig. 2)
Fig. 2.

Cytokines in phenotype analyses. IL-5, IL-7, and GM-CSF in controls and cases of phenotypes are shown. Patients were stratified by phenotypes defined in the methods. P-values were calculated by Mann–Whitney testing. Central tendencies are represented by the median, with interquartile ranges shown. Pulm = Pulmonary.
IL-5 was significantly decreased in non-fibrotic compared to fibrotic pulmonary sarcoidosis (p = 0.013), and compared to controls (p = 0.0004). Compared to controls, there was a trend towards a significant increase in IL-7 in fibrotic (p = 0.052), but not in non-fibrotic pulmonary sarcoidosis. In contrast, compared to controls, there was a trend towards a significant increase in GM-CSF in non-fibrotic (p = 0.054), but not in fibrotic pulmonary sarcoidosis.
4.1.3. Cytokines were correlated with each other, but cytokine–cytokine interactions did not influence case-control associations
There were many significant correlations among cytokines, with TNF-α and IL-12, TNF-α and IL-17, TNF-α and IFN-γ, IL-5 and IL-13, and GM-CSF and IL-4 demonstrating the highest degree of correlation (Fig. 3a). These correlations were also highly significant. Regression analyses were performed to control for potential cytokine–cytokine interactions which could have influenced the cytokine associations we detected between cases and controls: the p-values of these associations were not significantly different when models were run of IL-5, IL-7, and GM-CSF and their correlated cytokines (data not shown). Interestingly, when correlation matrices were done separately for fibrotic and non-fibrotic pulmonary disease, the patterns of correlations were different according to phenotype, and only four out of 23 correlation pairs were shared (Fig. 3 b and c).
Fig. 3.

Matrix of cytokine correlations. Multiple positive correlations among cytokines are demonstrated in our combined cohort (3a). Distinct patterns of cytokine correlations are observed for fibrotic (3b) and non-fibrotic (3c) pulmonary sarcoidosis. There are no significant negative correlations. Spearman’s Rho value 100× is indicated in each pair box for which a significant correlation was detected. Light shade represents a p-value <0.05–0.0001. Dark shade represents a p-value <0.0001.
4.1.4. Peripheral lymphocyte counts did not correlate with levels of IL-7
IL-7 has a hematopoietic effect, and levels increase during lymphopenic conditions to regulate lymphocyte counts towards normal [28,29]. In this way, IL-7 has been designated by some to be a “master regulator” of lymphocytes [29]. Peripheral lymphopenia is common in chronic sarcoidosis [30]. To evaluate whether alterations in IL-7 in sarcoidosis derive from altered lymphocyte counts, we correlated IL-7 with lymphocyte counts in the 47 subjects with a white blood count differential performed at the time of the serum sample collection. We found no significant correlation between levels of IL-7 and lymphocyte counts (Fig. 4).
Fig. 4.
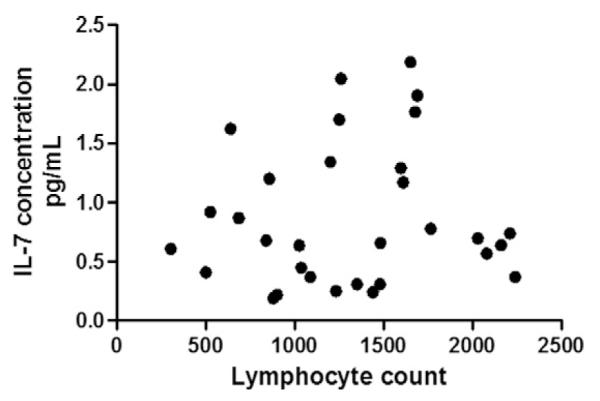
IL-7 levels and lymphocyte counts. IL-7 levels were not correlated with lymphocyte counts (p = 0.566) in the 47 patients with a leukocyte differential performed at the time of the serum sample collection.
5. Discussion
In this study, we found altered levels of circulating IL-5, IL-7, and GM-CSF in patients with chronic sarcoidosis. Circulating IL-5, a T helper 2 (Th2) cytokine, was decreased in sarcoidosis compared to controls. This suggests that circulating cytokines may not merely be due to “spillover” from local sites of active inflammation, but rather may reflect primary alterations, such as suppression of Th2 cells, in the circulating immune system [31]. In contrast to IL-5, IL-7 and GM-CSF were increased in sarcoidosis compared to controls. Historically, IL-7 levels have not been studied in sarcoidosis. However, the up-regulation of IL7 expression in sarcoid lung tissue, alterations in the IL7R gene in patients with sarcoidosis, and increased circulating IL-7 in female patients with severe sarcoidosis have been recently reported, and are of particular interest in light of our current findings [32–35]. IL-7 has a critical role in lymphocyte regulation and homeostasis, and therefore is an attractive target for further studies in sarcoidosis, a lymphocyte-mediated disease. Finally, GM-CSF regulates and activates monocytes, and GM-CSF seems to be up-regulated in sarcoid lung tissue [16]. Previous studies on circulating GM-CSF levels have yielded mixed results, which may relate to different experimental techniques [13,18]. Our result suggests that GM-CSF—an important signal to direct antigen presenting cells, including macrophages—is indeed elevated and potentially important in sarcoidosis. While GM-CSF was not as strong in our analysis, this may have been a function of high background staining.
We are not able to replicate some previously published results of circulating cytokine alterations. Methodological differences may account for our different, yet not contradictory findings: whereas many studies have employed single-cytokine ELISA tests, used plasma or culture supernatant samples, or had cohorts of newly diagnosed disease, we employed a multiplex assay, used serum samples, and had a cohort of chronic disease [18,36]. Our intent was not to detect all possible alterations in circulating cytokines, for which dedicated singleplex assays would likely perform better for a given cytokine. Rather, our aim was to advance our understanding of cell signaling in sarcoidosis through the evaluation of a broad array of cytokines and chemokines, including the study of potentially novel cell signals.
Our findings withstood controlling for variables predicted to be potentially confounding. Clinical and demographic variables did not influence the cytokine associations we observed, and cytokine levels did not correlate with use of immunosuppression. While we found many significant correlations among cytokines, controlling for these correlations did not significantly alter our findings. Finally, IL-7 levels did not correlate with lymphocyte counts, suggesting that increased IL-7 in sarcoidosis is a primary phenomenon.
We found specific cytokine patterns for fibrotic and non-fibrotic pulmonary sarcoidosis. The determinants of pulmonary fibrosis in a subset of patients with chronic disease are not known. However, it is increasingly recognized that pulmonary fibrosis in sarcoidosis likely occurs via specific inflammatory processes which are not shared by all patients, and fibrosis-specific immune and genetic alterations recently have been reported [8–11]. Indeed, we found substantial differences in the patterns of cytokine correlations between these phenotypes. It has been suggested that fibrotic sarcoidosis may result from the transition from a Th1 to a Th2-biased response [37]; our finding of significantly lower levels of IL-5, a Th2 cytokine, in fibrotic compared to non-fibrotic pulmonary sarcoidosis does not support such a transition. Certainly more work in this domain is indicated. Our finding of a trend towards increased IL-7 in fibrotic but not non-fibrotic pulmonary sarcoidosis raises the possibility that this cytokine may have a role in the fibrotic response to sarcoid inflammation.
Most of our subjects were female and African American. The female predominance in our cohort exceeds the higher rate of disease often reported for women [5]. We speculate that women may experience increased disease-associated morbidity compared to men, or for other reasons may more readily seek medical care services for sarcoidosis [38–40]. A predominance of African American patients in our cohort in is line with the ancestral demographics of the catchment area of our medical center. In addition, sarcoidosis can be more severe in African Americans, and disease severity should relate to utilization of tertiary care services, such as those offered by our University Medical Center [5,12,41]. Studies with more gender balanced cohorts and varied ancestral groups will help to generalize our findings. Nonetheless, as we have done here, it is important to study patient groups most likely to be severely affected by sarcoidosis.
Our findings in IL-5, IL-7, and GM-CSF in patients with chronic sarcoidosis provide a window into the immunopathogenesis of disease. They also support the biomarker potential of the circulating immune system in sarcoidosis. Correlating serum and lung cytokines will help to elucidate the similarities and differences between systemic and tissue-specific cytokine alterations, and are important next steps. In addition, repeating our studies in subjects with acute disease will provide insight into the relationship between cytokine alterations and the phase of disease. Finally, by observing an effect of phenotype upon cytokine patterns, we identify specific immune alterations which may contribute to the clinical heterogeneity of sarcoidosis. Further defining the differences between fibrotic and non-fibrotic pulmonary sarcoidosis is important, and will advance our understanding of the fibrotic phenotype, an uncommon yet particularly severe manifestation of disease.
Acknowledgements
Dr. Marissa Alegre, University of Chicago: guidance, Ryan Duggan, University of Chicago: technical support, David Leclerc, University of Chicago: technical support, Nancy Jackson, University of Chicago: graphic assistance.
Funding K.C. Patterson – NIH CTSA Pilot Grant UL1 RR024999.
T.B. Niewold – NIH K08 AI083790, NIH P30 DK42086, NIAID Clinical Research Loan Repayment AI071651, NIH CTSA Core Subsidy Grant and CTSA Pilot Grants from UL1 RR024999, Lupus Research Institute Novel Research Grant, Alliance for Lupus Research Target Identification in Lupus Grant, Arthritis National Research Foundation Eng Tan Scholar Award.
References
- [1].Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391–9. doi: 10.1001/jama.2011.10. Epub 2011/01/27. [DOI] [PubMed] [Google Scholar]
- [2].Nagai S, Handa T, Ito Y, Ohta K, Tamaya M, Izumi T. Outcome of sarcoidosis. Clin Chest Med. 2008;29(3):565–74. x. doi: 10.1016/j.ccm.2008.03.006. Epub 2008/06/10. [DOI] [PubMed] [Google Scholar]
- [3].Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153–65. doi: 10.1056/NEJMra071714. Epub 2007/11/23. [DOI] [PubMed] [Google Scholar]
- [4].Baughman RP, Costabel U, du Bois RM. Treatment of sarcoidosis. Clin Chest Med. 2008;29(3):533–48. ix–x. doi: 10.1016/j.ccm.2008.03.012. Epub 2008/06/10. [DOI] [PubMed] [Google Scholar]
- [5].Rybicki BA, Major M, Popovich J, Jr, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. 1997;145(3):234–41. doi: 10.1093/oxfordjournals.aje.a009096. Epub 1997/02/01. [DOI] [PubMed] [Google Scholar]
- [6].Burke RR, Stone CH, Havstad S, Rybicki BA. Racial differences in sarcoidosis granuloma density. Lung. 2009;187(1):1–7. doi: 10.1007/s00408-008-9111-9. Epub 2008/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rybicki BA, Iannuzzi MC. Epidemiology of sarcoidosis: recent advances and future prospects. Semin Respir Crit Care Med. 2007;28(1):22–35. doi: 10.1055/s-2007-970331. Epub 2007/03/03. [DOI] [PubMed] [Google Scholar]
- [8].Kruit A, Grutters JC, Ruven HJ, van Moorsel CH, Weiskirchen R, Mengsteab S, et al. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest. 2006;129(6):1584–91. doi: 10.1378/chest.129.6.1584. Epub 2006/06/17. [DOI] [PubMed] [Google Scholar]
- [9].Lockstone HE, Sanderson S, Kulakova N, Baban D, Leonard A, Kok WL, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med. 2010;181(12):1367–75. doi: 10.1164/rccm.200912-1855OC. Epub 2010/03/03. [DOI] [PubMed] [Google Scholar]
- [10].Sato H, Williams HR, Spagnolo P, Abdallah A, Ahmad T, Orchard TR, et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur Respir J. 2010;35(2):324–30. doi: 10.1183/09031936.00010209. Epub 2009/08/15. [DOI] [PubMed] [Google Scholar]
- [11].Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. 2010;137(1):89–101. doi: 10.1016/j.clim.2010.06.017. Epub 2010/08/03. [DOI] [PubMed] [Google Scholar]
- [12].Zissel G, Prasse A, Muller-Quernheim J. Sarcoidosis-immunopathogenetic concepts. Semin Respir Crit Care Med. 2007;28(1):3–14. doi: 10.1055/s-2007-970329. Epub 2007/03/03. [DOI] [PubMed] [Google Scholar]
- [13].Beirne P, Pantelidis P, Charles P, Wells AU, Abraham DJ, Denton CP, et al. Multiplex immune serum biomarker profiling in sarcoidosis and systemic sclerosis. Eur Respir J. 2009;34(6):1376–82. doi: 10.1183/09031936.00028209. Epub 2009/06/23. [DOI] [PubMed] [Google Scholar]
- [14].Ziegenhagen MW, Schrum S, Zissel G, Zipfel PF, Schlaak M, Muller-Quernheim J. Increased expression of proinflammatory chemokines in bronchoalveolar lavage cells of patients with progressing idiopathic pulmonary fibrosis and sarcoidosis. J Investig Med. 1998;46(5):223–31. Epub 1998/07/24. [PubMed] [Google Scholar]
- [15].Ziegenhagen MW, Muller-Quernheim J. The cytokine network in sarcoidosis and its clinical relevance. J Intern Med. 2003;253(1):18–30. doi: 10.1046/j.1365-2796.2003.01074.x. Epub 2003/02/18. [DOI] [PubMed] [Google Scholar]
- [16].Itoh A, Yamaguchi E, Furuya K, Kawakami Y. Secretion of GM-CSF by inflammatory cells in the lung of patients with sarcoidosis. Respirology. 1998;3(4):247–51. doi: 10.1111/j.1440-1843.1998.tb00130.x. Epub 1999/04/14. [DOI] [PubMed] [Google Scholar]
- [17].Muller-Quernheim J. Sarcoidosis: immunopathogenetic concepts and their clinical application. Eur Respir J. 1998;12(3):716–38. doi: 10.1183/09031936.98.12030716. Epub 1998/10/08. [DOI] [PubMed] [Google Scholar]
- [18].Prior C, Knight RA, Herold M, Ott G, Spiteri MA. Pulmonary sarcoidosis: patterns of cytokine release in vitro. Eur Respir J. 1996;9(1):47–53. doi: 10.1183/09031936.96.09010047. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- [19].Shigehara K, Shijubo N, Ohmichi M, Kamiguchi K, Takahashi R, Morita-Ichimura S, et al. Increased circulating interleukin-12 (IL-12) p40 in pulmonary sarcoidosis. Clin Exp Immunol. 2003;132(1):152–7. doi: 10.1046/j.1365-2249.2003.02105.x. Epub 2003/03/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mathew S, Bauer KL, Fischoeder A, Bhardwaj N, Oliver SJ. The anergic state in sarcoidosis is associated with diminished dendritic cell function. J Immunol. 2008;181(1):746–55. doi: 10.4049/jimmunol.181.1.746. Epub 2008/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. The immune paradox of sarcoidosis and regulatory T cells. J Exp Med. 2006;203(2):359–70. doi: 10.1084/jem.20050648. Epub 2006/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kobayashi S, Kaneko Y, Seino K, Yamada Y, Motohashi S, Koike J, et al. Impaired IFN-gamma production of V alpha24 NKT cells in non-remitting sarcoidosis. Int Immunol. 2004;16(2):215–22. doi: 10.1093/intimm/dxh020. Epub 2004/01/22. [DOI] [PubMed] [Google Scholar]
- [23].Heron M, Grutters JC, van Velzen-Blad H, Veltkamp M, Claessen AM, van den Bosch JM. Increased expression of CD16, CD69, and very late antigen-1 on blood monocytes in active sarcoidosis. Chest. 2008;134(5):1001–8. doi: 10.1378/chest.08-0443. Epub 2008/07/22. [DOI] [PubMed] [Google Scholar]
- [24].Judson MA, Baughman RP, Teirstein AS, Terrin ML, Yeager H., Jr Defining organ involvement in sarcoidosis: the ACCESS proposed instrument. ACCESS research group. A case control etiologic study of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16(1):75–86. Epub 1999/04/20. [PubMed] [Google Scholar]
- [25].Prasse A, Katic C, Germann M, Buchwald A, Zissel G, Muller-Quernheim J. Phenotyping sarcoidosis from a pulmonary perspective. Am J Respir Crit Care Med. 2008;177(3):330–6. doi: 10.1164/rccm.200705-742OC. Epub 2007/11/03. [DOI] [PubMed] [Google Scholar]
- [26].Salez F, Gosset P, Copin MC, Lamblin Degros C, Tonnel AB, Wallaert B. Transforming growth factor-beta 1 in sarcoidosis. Eur Respir J. 1998;12(4):913–9. doi: 10.1183/09031936.98.12040913. Epub 1998/11/17. [DOI] [PubMed] [Google Scholar]
- [27].Judson MA, Baughman RP, Thompson BW, Teirstein AS, Terrin ML, Rossman MD, et al. Two year prognosis of sarcoidosis: the ACCESS experience. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20(3):204–11. Epub 2003/11/19. [PubMed] [Google Scholar]
- [28].Ponchel F, Cuthbert RJ, Goeb V. IL-7 and lymphopenia. Clin Chim Acta. 2011;412(1-2):7–16. doi: 10.1016/j.cca.2010.09.002. Epub 2010/09/21. [DOI] [PubMed] [Google Scholar]
- [29].Fry TJ, Mackall CL. Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001;22(10):564–71. doi: 10.1016/s1471-4906(01)02028-2. Epub 2001/09/28. [DOI] [PubMed] [Google Scholar]
- [30].Sweiss NJ, Salloum R, Gandhi S, Alegre ML, Sawaqed R, Badaracco M, et al. Significant CD4, CD8, and CD19 lymphopenia in peripheral blood of sarcoidosis patients correlates with severe disease manifestations. PLoS One. 2010;5(2):e9088. doi: 10.1371/journal.pone.0009088. Epub 2010/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Baumer I, Zissel G, Schlaak M, Muller-Quernheim J. Th1/Th2 cell distribution in pulmonary sarcoidosis. Am J Respir Cell Mol Biol. 1997;16(2):171–7. doi: 10.1165/ajrcmb.16.2.9032124. Epub 1997/02/01. [DOI] [PubMed] [Google Scholar]
- [32].Loza MJ, Brodmerkel C, Du Bois RM, Judson MA, Costabel U, Drent M, et al. Inflammatory profile and response to anti-TNF therapy in patients with chronic pulmonary sarcoidosis. Clin Vaccine Immunol. 2011 doi: 10.1128/CVI.00337-10. [Epub 2011/04/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Crouser ED, Culver DA, Knox KS, Julian MW, Shao G, Abraham S, et al. Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2009;179(10):929–38. doi: 10.1164/rccm.200803-490OC. Epub 2009/02/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maver A, Medica I, Peterlin B. Search for sarcoidosis candidate genes by integration of data from genomic, transcriptomic and proteomic studies. Med Sci Monit. 2009;15(12):SR22–8. Epub 2009/12/01. [PubMed] [Google Scholar]
- [35].Heron M, Grutters JC, van Moorsel CH, Ruven HJ, Huizinga TW, van der Helmvan Mil AH, et al. Variation in IL7R predisposes to sarcoid inflammation. Genes Immunol. 2009;10(7):647–53. doi: 10.1038/gene.2009.55. Epub 2009/07/25. [DOI] [PubMed] [Google Scholar]
- [36].Zissel G, Homolka J, Schlaak J, Schlaak M, Muller-Quernheim J. Anti-inflammatory cytokine release by alveolar macrophages in pulmonary sarcoidosis. Am J Respir Crit Care Med. 1996;154(3 Pt 1):713–9. doi: 10.1164/ajrccm.154.3.8810610. Epub 1996/09/01. [DOI] [PubMed] [Google Scholar]
- [37].Muller-Quernheim J, Prasse A, Zissel G. Pathogenesis of sarcoidosis. Presse Med. 2012;41(6 Pt 2):e275–87. doi: 10.1016/j.lpm.2012.03.018. Epub 2012/05/19. [DOI] [PubMed] [Google Scholar]
- [38].Cozier YC, Berman JS, Palmer JR, Boggs DA, Serlin DM, Rosenberg L. Sarcoidosis in black women in the United States: data from the black women’s health study. Chest. 2011;139(1):144–50. doi: 10.1378/chest.10-0413. Epub 2010/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bourbonnais JM, Samavati L. Effect of gender on health related quality of life in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27(2):96–102. Epub 2011/02/16. [PubMed] [Google Scholar]
- [40].Juel K, Christensen K. Are men seeking medical advice too late? Contacts to general practitioners and hospital admissions in Denmark 2005. J Public Health (Oxf) 2008;30(1):111–3. doi: 10.1093/pubmed/fdm072. Epub 2007/11/06. [DOI] [PubMed] [Google Scholar]
- [41].Swigris JJ, Olson AL, Huie TJ, Fernandez-Perez ER, Solomon J, Sprunger D, et al. Sarcoidosis-related mortality in the United States from 1988 to 2007. Am J Respir Crit Care Med. 2011;183(11):1524–30. doi: 10.1164/rccm.201010-1679OC. Epub 2011/02/19. [DOI] [PMC free article] [PubMed] [Google Scholar]