

Abstract
LKB1/STK11 is a tumor suppressor and a negative regulator of mammalian target of rapamycin signaling. It is inactivated in 30% of lung cancer cell lines but only 5–15% of primary lung adenocarcinomas. There is evidence that homozygous deletion (HD) of chromosome 19p at the LKB locus contributes to the inactivation of the gene in primary human lung cancers. Here, we used several complementary genetic approaches to assess the LKB1 locus in primary non-small cell lung cancers (NSCLCs). We first analyzed 124 NSCLC cases for allelic imbalance using eight microsatellite markers on chromosome 19p, which revealed an overall rate of 65% (80 of 124) loss of heterozygosity (LOH). We next used chromogenic in situ hybridization (CISH) to directly examine the chromosomal status of the LKB1 locus. In all, 65 of 124 LOH tested samples were available for CISH and 58 of those (89%) showed either loss of one copy of chromosome 19p (LOH, 40 of 65 cases, 62%) or both copies (HD 18 of 65 cases, 28%). The occurrence of HD was significantly more frequent in Caucasian (35%) than in African-American patients (6%) (P=0.04). A total of 62 of 124 samples with LOH at one or both markers immediately flanking the LKB1 gene were further analyzed by directly sequencing the complete coding region, which identified 7 of 62 (11%) tumors with somatic mutations in the gene. Jointly, our data identified total inactivation of the LKB1 gene by either HD or LOH with somatic mutation in 39% of tested samples, whereas loss of chromosome 19p region by HD or LOH at the LKB1 region occured in 90% of NSCLC.
Keywords: LKB1 gene, LOH, homozygous deletion, mutation, CISH, primary lung cancer
Introduction
The LKB1 gene is a tumor suppressor gene located on chromosome 19p13.3 (Hemminki et al., 1998). It is also known as STK11, has serine–threonine kinase activity and possesses nuclear localization signals in the N-terminal region, central kinase domain and a C-terminal farnesylation region (Alessi et al., 2006). The LKB1 gene is widely expressed at varying levels in all fetal and adult tissues. During development, expression of LKB1 is high in the heart, esophagus, pancreas, kidney, colon, lung, small intestine and stomach (Luukko et al., 1999; Rowan et al., 1999). In adult tissues, expression is high in most epithelium of follicles, including the corpus luteum of the ovary and the seminiferous tubules of the testis, in myocytes from skeletal muscle, and in glia cells (Rowan et al., 1999; Conde et al., 2007).
Functionally, LKB1 regulates cellular energy metabolism and cell polarity by activating AMP-activated protein kinase (AMPK) and other members of the AMPK family (Hawley et al., 2003; Woods et al., 2003; Corradetti et al., 2004; Shaw et al., 2004a, b). Activation of AMPK restores ATP levels by inducing catabolic pathway-like glycolysis and blocking ATP-consuming anabolic pathways—like protein synthesis—thus acting as a sensor of intracellular ATP levels. AMPK controls protein synthesis by regulating the mammalian target of rapamycin through phosphorylation of tuberous sclerosis protein 2 (TSC2), also known as tuberin (Inoki et al., 2003). Deletion of LKB1 results in the loss of AMPK functions and improves glucose uptake (Koh et al., 2006). In the liver, LKB1 knockout causes hyperglycemia due to increased expression of the gluconeogenic gene and hepatic glucose output (Shaw et al., 2005). More recently, genetic and functional studies have demonstrated a critical role of LKB1 in regulating cell polarity and provided an underlying mechanism in colonic polyp development in patients with Peutz–Jeghers syndrome (Zhang et al., 2008; Jansen et al., 2009).
Genetically, mutations in the LKB1 gene are responsible for Peutz–Jeghers syndrome, an autosomal dominant disorder characterized by melanocytic macules of the lips, buccal mucosa, and digits, and multiple gastro-intestinal hamartomatous polyps. Patients with Peutz–Jeghers syndrome have a high risk of gastrointestinal and other cancers (Lim et al., 2003). In addition, cumulative evidence suggests that somatic mutation of LKB1 gene is critically involved in lung and cervical cancers (Sanchez-Cespedes et al., 2002, Wingo et al., 2009). The involvement of LKB1 in lung cancer is several fold. First, chromosome 19p is the second most common region of loss in lung tumors of smokers (Sanchez-Cespedes et al., 2001). Second, inactivation mutations of LKB1 are found in 30% of lung cancer cell lines and in a smaller portion of primary lung adenocarcinomas (Sanchez-Cespedes et al., 2002; Davies et al., 2005). Most somatic mutations involve nonsense or frameshift mutations leading to a truncated protein (Launonen, 2005; Shaw et al., 2005). Third, transgenic animal studies suggest that LKB1 modulates lung cancer differentiation and metastasis (Gurumurthy et al., 2008). Finally, LKB1 regulates lung cancer cell polarity, and deficiency in LKB1 sensitizes mice to carcinogen-induced tumorigenesis (Zhang et al., 2008). In cervical cancer, point mutation and/or deletion of the LKB1 gene was observed in 20% of the primary cervical tumor samples. Deletions were found in about 50% of the cervical cancer cell lines, such as Hela and SiHa. Furthermore, chromosomal loss or mutation of the LKB1 gene is associated with a reduced progression-free survival in patients (Wingo et al., 2009). Taken together, these data further support a critical role of LKB1 in tumorigenesis and progression.
To gain insights into the mechanism of LKB1/STK11 gene alterations in non-small cell lung cancer (NSCLC), we utilized several complementing genetic approaches, including allelic analysis for chromosomal imbalance or loss (loss of heterozygosity (LOH)), sequencing for tumors with LOH, and chromogenic in situ hybridization (CISH) to investigate the genetic status of the gene at a single cell level. Consistent with others, we observed a generally low rate of somatic mutations (11%) at the LKB1 gene. However, we observed that LOH and homozygous deletion (HD) of the LKB1 gene jointly occurred in nearly 90% of the NSCLC tested.
Results
Status of chromosome 19p in NSCLC
We first examined the status of chromosome 19p in 124 lung cancer samples using a panel of eight microsatellite markers (Figure 1a). When considered as a whole, 65% (80 of 124) of the tumors showed allelic imbalance or LOH in two or more markers. When the presence of LOH at each marker was considered, the rate of loss at the marker closest to LKB1/STK11, D19S883, was the lowest (38%, Figure 1b), suggesting a possible mechanism for LKB1 gene inactivation through HD in the region. Tumors from patients with stage I disease (71%) had a higher rate of LOH compared with those with stage II–IV disease (53%) (P=0.04, Table 1).
Figure 1.

Loss of chromosome 19p and the LKB1/STK11 region in primary NSCLC (a) Schematic diagram of LKB1/STK11 locus in relation to CISH probe and tested microsatellite markers. Markers are listed from telomere (left) to centromere (right). The genomic organization of LKB1/SKT11 gene is shown (not to scale) and is 37.8 kb from the most 3′ clone (CTD2315C07) available for CISH. (b) Frequency of LOH by microsatellite analysis. The marker D19S883 is the closest to the LKB1 gene and had the lowest LOH frequency (38.8%) among all tested markers.
Table 1.
Correlation between LKB1/STK11 loss and clinicopathological characteristics of primary lung tumors
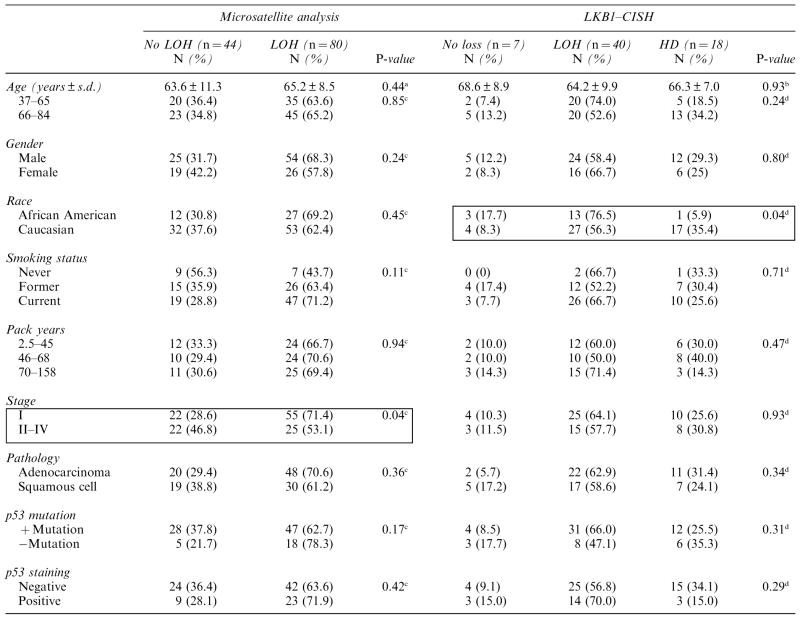
|
Abbreviations: CISH, chromogenic in situ hybridization; HD, homozygous deletion; LOH, loss of heterozygosity.
t-test for unequal variances.
Analysis of variance
χ2-test.
Fisher’s exact test.
The boxes illustrate comparisons with significant P-value.
In situ hybridization analysis of chromosomal status in NSCLC
To directly examine the status of chromosome 19p, we identified a bacterial artificial chromosome (BAC) clone, CDT-2315C07, located near the 3′ end of the LKB1 gene that was uniquely hybridized to the target region (Figure 1a). We developed a CISH method to directly examine the hybridization signals of the BAC clone in tissue samples using the lung tissue microarray (TMA) (examples in Figure 2). In total, 65 of 124 cases analyzed by microsatellite markers were available for analysis by CISH. The distribution of hybridization signals from CISH in cells from the non-tumor lung, adjacent non-involved lung, and lung tumor are summarized in Table 2. Non-tumor lung and adjacent normal lung tissues had a major portion (mode) of signals that were typical of a non-neoplastic tissue, primarily having two signals per cell in 76–86% of nuclei, whereas 3–10% had three to four signals per cell. Among the 65 lung tumors analyzed by CISH, 40 cases (61%) had only one signal per cell, consistent with LOH, whereas 18 cases (28%) had no hybridization signals in most tumor cells, suggesting HD. The remaining seven cases (11%) had approximately 50% of the cells having two signals per nuclei and were considered as no LOH.
Figure 2.
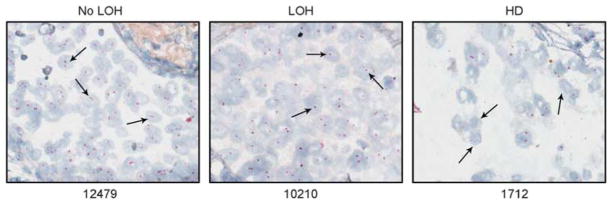
Genomic status of the LKB1/STK11 gene by CISH. Examples of hybridization signals for the LKB1/STK11 gene in normal and tumor lung tissues. Arrows indicate the observed signals inside the cells. The prevailing (mode) distribution of the hybridization signals determines the genetic status of the gene in the tumor as no LOH (#12479), LOH (#10210) or HD (#1712).
Table 2.
CISH signal distribution in all tested normal and tumor lung tissue samples
| Tissue type | Signals/cell (%)
|
||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| Normal | |||||
| Non-tumor (n =9) | 3.6 | 10.9 | 80 | 4.1 | 1.3 |
| Adjacent normal (n = 8) | 4.5 | 20 | 73.5 | 1.9 | 0.1 |
| Tumor | |||||
| No LOH (n = 7) | 9.1 | 24 | 51.9 | 9.8 | 5.3 |
| LOH (n =40) | 19.2 | 46.7 | 28.1 | 4.4 | 1.5 |
| HD (n = 18) | 53 | 33.4 | 12.2 | 0.6 | 0 |
Abbreviations: CISH, chromogenic in situ hybridization; HD, homozygous deletion; LOH, loss of heterozygosity.
The most prevalent mode of chromosome copy at indicated numbers are shown in bold.
We compared the results from CISH with those obtained by microsatellite analysis (Table 3). Of the 56 cases that were informative for D19S883, 21 of 22 cases having LOH had a loss of one signal in a majority of the cells (P<0.0001). In contrast, of the 34 tumors that were considered as ‘normal’ at marker D19S883, 15 (44%) tumors had loss of both alleles by CISH, consistent with having HD, and 13 additional tumors (38%) had a loss in one of the LKB alleles. On the basis of CISH, only 7 of 34 (20%) remaining tumor samples showed no loss and were considered as ‘normal,’ and, of those, 6 also showed no loss by microsatellite marker D19S833. The comparison of LOH data obtained by marker D19S814 with CISH showed a similar trend in that all 26 tumors scored as LOH by microsatellite analysis were confirmed by CISH as either LOH or HD, whereas most of the 25 samples that were ‘normal’ by microsatellite analysis were either LOH (11) or HD (9) by CISH (P=0.003). These results suggest that the CISH method is much more sensitive in detecting chromosome 19p losses when compared with those observed by the standard LOH method. Furthermore, no signal amplification was observed in any of the analyzed tumors further supporting a critical loss of genetic material in this region in NSCLC, and only 10% of the tumors had no loss at the LKB gene locus by both LOH and CISH analysis.
Table 3.
Association of LOH markers with CISH
| Microsatellite analysis | D19S883
|
D19S814
|
Both markers
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| CISH | Normal (n =34) N (%) | LOH (n =22) N (%) | P-value | Normal (n = 25) N (%) | LOH (n = 26) N (%) | P-value | Normal (n = 17) N (%) | LOH (n = 27) N (%) | P-value |
| Normal | 6 (18) | 1 (5) | <0.0001a | 5 (20) | 0 (0) | 0.003a | 5 (29) | 0 (0) | 0.0006a |
| LOH | 13 (38) | 21 (95) | 11 (44) | 22 (85) | 6 (35) | 23 (85) | |||
| HD | 15 (44) | 0 (0) | 9 (36) | 4 (15) | 6 (35) | 4 (15) | |||
Abbreviations: CISH, chromogenic in situ hybridization; HD, homozygous deletion; LOH, loss of heterozygosity.
Fisher’s exact test.
Somatic mutations in the LKB1 gene
Of the 124 tumors analyzed, 62 samples had LOH at either one or both microsatellite markers (D19S814, D19S883) flanking the LKB1/STK11. We sequenced all eight coding exons in these 62 tumors and identified seven tumors (11.3% overall) with a mutation in the LKB1 gene (Table 4). All mutations were detected in the tumors from female patients who were either current or former smokers. Four cases were from African Americans and three were from Caucasians. Histologically, six tumors were adenocarcinomas and one was a squamous carcinoma. The clinical characteristics of all patients and their molecular changes in the mutated LKB1 gene are summarized in Table 4. In total, we identified five cases with point mutations, three predicted to result in amino acid substitutions, two nonsense mutations, and two with single base deletions resulting in a frameshift, leading to premature termination and truncation of the protein. No mutations were found in the paired normal tissues, indicating that these mutations were somatic.
Table 4.
Characteristics of non-small cell lung patients with LKB1 gene mutationsa
| Sampleb | Ethnicity | Gender | Age | Smoking | Pathology | Stage | Family Hx | Mutation | Exon | Nucleotides/amino acid change |
|---|---|---|---|---|---|---|---|---|---|---|
| 1024T | AA | Female | 63 | CS (26PY) | ADC | III | No | Point mutation | 2 | Glu (GAA) 120 stop (TAA) |
| 11669T | C | Female | 67 | FS (105PY) | ADC | I | Yes | Point mutation | 3 | Gln (CAG) 152 stop (TAG) |
| 10152T | AA | Female | 61 | CS (21PY) | ADC | III | No | Point mutation | 4 | Gly (GGG) 180 Val (GTG) |
| 10403T | AA | Female | 55 | CS (84PY) | ADC | I | No | 1-bp Deletion | 5 | Asp (GAC) 237 frameshift (-AC) |
| 1571T | C | Female | 70 | CS (50PY) | SCC | I | Yes | 1-bp Deletion | 6 | Pro (CCG) 281 frameshift (C-G) |
| 11807T | AA | Female | 73 | FS (35PY) | ADC | I | Yes | Point mutation | 7 | Arg (AGG) 297 Ser (AGC) |
| 10272T | C | Female | 70 | FS (33PY) | ADC | IV | Yes | Point mutation | 7 | Arg (CGG) 304 Gly (GGG) |
Abbreviations: AA, African American; ADC, adenocarcinoma; C, Caucasian; CS, current smoker; FS, former smoker; NS, non-smoker; PY, pack/day years; SCC, squamous cell carcinoma.
Reference gene ID: NM 000455.
Known mutation site
Association of LKB1 status in tumors with clinical characteristics of the patient
Overall, CISH analysis identified 89% (58 of 65) of the tested tumors with loss of at least one copy of chromosome 19p. Of those, 18 of 65 (28%) tumors lost both alleles, whereas 40 of 65 (62%) tumors lost one copy of the chromosome. Among the samples included in this study, there was a significant racial difference in loss of LKB1/STK11 with 35% of the Caucasians and 6% of the African Americans having HD, 56% of the Caucasians and 77% of the African Americans having LOH, and 8% of the Caucasians and 17% of the African Americans having no LOH (P=0.04), as shown in Table 1. Overall, 39% of primary NSCLC tumors analyzed had a complete inactivation of the gene through either loss of both copies of the LKB1/STK11 gene or through LOH in one allele and somatic mutation in the remaining allele of the gene. Although not reaching statistical significance, patients with HD and mutations in lung tumors had worsened lung cancer prognosis compared with those whose tumors had only LOH (P=0.06, and data not shown). The survival comparison could not be done between patients whose tumor had mutation and those who did not have LOH due to the small sample numbers in each group.
Discussion
In the present study we used three complementing genetic methods to assess genetic alteration in 19p13.3, and specifically the LKB1/STK11 locus. We documented conclusively the high frequency of loss in this region in NSCLC. Our results support previous work, suggesting a high incidence of cytogenetic abnormalities at 19p in NSCLC (Lukeis et al., 1990) and SCLC (Levin et al., 1995). Sanchez-Cespedes et al. (2002) later showed that a chromosome 19p region, mapped between the telomere and microsatellite marker D19S216, was frequently associated with lung adenocarcinoma with an overall rate of 70% LOH (21 of 30 cases). The frequency of LOH (62%) in our data is consistent with the rate of 58% (15 of 26 cases) LOH observed by Sanchez-Cespedes et al. (2001). Subsequent studies led to the discovery that LKB1 is a genetic target for lung cancer, particularly lung adenocarcinoma (Virmani et al., 1998; Sanchez-Cespedes et al., 2001; Carretero et al., 2004; Matsumoto et al., 2007). However, the rate of inactivating mutations in the gene remains relatively low and a high rate of LOH does not equate to inactivation of the candidate gene in the region. Another candidate tumor suppressor gene, BRG1, located about 10Mb proximal to the gene, can potentiate lung tumor development and is mutated in approximately 20% of the lung cancer cell lines (Glaros et al., 2008; Medina et al., 2008). The high rate of LOH that we observed here and by others strongly suggests that BRG1 could also be a target of inactivation in this region.
We developed the CISH approach to investigate the genetic status of the region immediately adjacent to the LKB1 gene by directly visualizing the chromosomal status in individual tumor cells. Among the 65 tumor samples that were analyzed by both methods, a majority showed loss of either one copy (62%, 40/65) or both copies (28%, 18/65) of the LKB1/STK11 genomic regions by CISH analysis, jointly affecting ~88% of the tested tumors. Homozygous loss of both alleles (HD) for tumor suppressor genes is a result of a second allelic deletion of a chromosomal region that has undergone hemizygous loss in the other allele (Cairns et al., 1995; Hahn et al., 1996; Li et al., 1997; Steck et al., 1997). This high frequency of total loss we observed here (89% total) is consistent with the over 83% LOH in the 19p13.2 region observed in NSCLC cell lines (Virmani et al., 1998), suggesting that the observed loss in NSCLC cell lines most likely originated from their primary tumors in vivo.
Several studies have investigated somatic mutations of the LKB1/STK11 gene in various tumor types (Avizienyte et al., 1998, 1999; Bignell et al., 1998; Wang et al., 1998; Guldberg et al., 1999; Rowan et al., 1999; Zhong et al., 2006; Wingo et al., 2009). Most studies indicate that LKB1 is infrequently mutated in sporadic cancers except in adenocarcinoma of the lung (Carretero et al., 2004) and more recently in cervical cancers of both adeno- and squamous cell pathologies (Wingo et al., 2009). The majority of mutations in the gene result in loss of function. Matsumoto et al. (2007) analyzed 70 lung cancer cell lines, 106 primary adenocarcinomas and 25 brain metastases. In cell lines, point mutations and HD were observed in 13%(9) and 21%(13), respectively. In primary tumors, however, only onemutation was detected in primary adenocarcinomas and three were observed in brain metastases. In the present study, we observed a relatively low frequency of somatic mutations in the LKB1 gene (11%). Among the tumors with a mutation in the LKB1 gene, six were adenocarcinomas and one tumor was squamous cell carcinoma. All mutations were tumor specific and six out of the seven mutations occurred at novel mutation sites, except one nonsense case at codon 120 (E120X). In addition, our data revealed a racial difference in the loss of 19p13.2 with both copies lost in 35% of the Caucasians compared with 6% of the African Americans (P=0.04). Although the sample size is too small in this subset to determine the robustness of the differences between LKB1 gene changes in race and gender, a recent study by Koivunen et al. (2008) suggested strongly that ethnic background of the patient affects LKB1 mutation frequency and spectrum among different patient populations. Cooperation between LKB1 and K-ras was observed in using somatically activated K-ras-driven mouse model of lung cancer (Ji et al., 2007). In this model, LKB1-deficient tumors had a short latency period. More recently, Ding et al. (2008) conducted a large resequencing effort and showed that mutation in LKB1/STK11 gene was among the most common genes frequently mutated in lung adenocarcinoma (Guldberg et al., 1999).
Unique to our study is the finding that a significant fraction of lung tumors harbor HD of the LKB1 gene by direct CISH analyses. Our results demonstrated that the CISH method is highly sensitive in detecting the status of tumor samples for LOH and HD. Another advantage of CISH is that it is not limited by the informativeness of the microsatellite marker and could clearly be resolved under bright field microscopy on the basis of hybridization signals present in the tumor cell nuclei. In addition, the CISH method enabled us to directly assess the chromosomal copy number changes in primary lung cancer samples using TMA samples. In conclusion, our study provides the first direct evidence that 39% of primary NSCLC inactivate the LKB1 gene by either HD or a mutation in the gene, and that nearly 90% of NSCLC has a loss of either single or both copies of the LKB1/SKT11 gene locus, highlighting its critical role in lung tumorigenesis process. Because small deletions below the detection limit of the CISH probe are unlikely to have been detected in our study, the actual frequency of chromosome loss for this region could approach 100%, making LKB1 the highest site of inactivation in lung cancer.
Materials and methods
Tissue samples and DNA extraction
Tumor and matched non-tumor tissues from 124 patients diagnosed with primary lung cancer were obtained after surgical resection at the University of Maryland Medical Center. Samples were collected with informed consent, as approved by the respective institutional review boards. Representative sections from the tumor and paired non-tumor tissues were stained with hematoxylin and eosin and reviewed by a pathologist to determine the tumor histological subtype and to assess tumor content. Fresh frozen tissues from the tumors were grossly dissected to ensure that specimens contained at least 50% tumor cells. Approximately 10 sections of 20 μm thickness were collected from non-tumor and tumor samples and placed in either Trizol reagents (Invitrogen, Carlsbad, CA, USA) for RNA or 1% SDS/proteinase K (10 mg/ml) solution at 58 °C overnight for DNA. For samples placed in Trizol, DNA was extracted from the organic phase after RNA extraction, following the manufacturer’s protocol. All samples were extracted with phenol–chloroform and were ethanol precipitated. Clinical information was available from all 124 patients. The p53 mutation and staining analysis is described in Mechanic et al., (2005).
Microsatellite analysis
We screened for LOH at the short arm of chromosome 19 in 124 paired tumor/non-tumors. Eight fluorescent-labeled microsatellite markers located in chromosome 19p were obtained from Applied Biosystems (Foster City, CA, USA). These markers included D19S814, D19S883, D19S565, D19S1166, D19S424, D19S209, D19S894 and D19S884. For analysis of allelic deletions by LOH, polymerase chain reaction (PCR) was carried out using 6 ng of genomic DNA and 5 pmol of each primer (one of them cyanine 5-labeled). The true allele PCR premix (Applied Biosystems) was used for the PCR amplifications for 32 cycles of 94 °C for 15 s, 55 °C for 15 s, and 72 °C for 30 s, followed by incubation at 72 °C for 30 s. After the amplification, the PCR products were run on an ABI 3100 automatic sequencer together with LIZ 500 molecular weight markers and formamide (Applied Biosystems). Allelic loss was determined when the ratio was <0.75 or >1.25 for the informative alleles between the tumor and the paired non-tumor samples. The sample was considered as having an allelic imbalance when there were at least two markers demonstrating allelic imbalance among all eight chromosome 19p markers tested.
CISH in lung TMA
Identification and labeling of CISH probe
CDT-2315C07, a BAC containing human chromosome regions identified via genetic analysis, was selected for the region around the LBK1/STK11 gene (Invitrogen). This BAC is located at a genomic region 37 807 bp 3′ to the LKB1 gene, as shown in Figure 1a. DNA was isolated, labeled with biotin–UTP, and hybridized with normal blood lymphocyte metaphase-spread slides. The BAC CDT-2315C07 generated a strong and specific hybridization for the target region (data not shown) and was used as a probe for interphase nuclei CISH. For probe labeling and hybridization, conditions were the same as previously described (Gill et al., 2008), and the probe was labeled with digoxigenin–UTP.
CISH hybridization
TMA slides containing tumor and normal samples were baked for 2–4 h at 60 °C before being processed for hybridization with a digoxigenin-labeled probe. Briefly, the tissue was deparaffinized and hydrated through an ethanol series, treated with 0.5mm dithiothreitol for 2 h at room temperature, followed by incubation in 1% sodium thiocyanate overnight at room temperature. After overnight incubation, the slides were incubated at 80 °C for 1 h. The tissues on the slides were further treated with Zymed pretreatment (tissue pretreatment kit; Invitrogen) for 45 min at 95–98 °C, digested with protease solution from the tissue pretreatment kit (Invitrogen) for 14min, washed twice with 2× SSC, and allowed to dry at 37 °C. The tissue was then fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10min at room temperature, washed twice in 2× SSC, and then denatured with 70% formamide and 2× SSC for 10min at 80 °C. Denatured slides were further digested sequentially with collagenase A 10 μg/ml (Roche, Basel, Switzerland) and proteinase K 10 μg/ml (Sigma, Milwaukee, WI, USA) in PBS at 37 °C for 15 and 13min, respectively. The slides were then blocked with avidin-blocking solution (DakoCytomation, Capinteria, CA, USA) for 30min at room temperature and hybridized with preannealed probes for 36 h at 37 °C.
After the hybridization, the slides were washed in 50% formamide and 2× SSC at 45 °C three times for 5min each, 0.5× SSC and 0.1% SDS at 65 °C four times for 5min each, and 2× SSC at room temperature. After washing, they were incubated with casein blocking solution (Invitrogen) containing sheep or goat IgG (1:100 dilution) for 30 min at 37 °C to block nonspecific binding. After blocking, the slides were incubated with alkaline phosphatase-labeled sheep anti-digoxigenin antibody (Invitrogen) for 1 h at 37 °C. The slides were washed in 4× SSC 0.1% Tween-20 solution four times at 45 °C and then washed with 20× SSC twice at room temperature. The slides were developed with fast red substrate (DakoCytomation) for 1 h and then counter stained with Mayer’s hematoxylin.
Microscopy and CISH scoring
TMA slides were scanned by Aperio ScanScope GL System (Aperio, Vista, CA, USA) under an ×40 objective and viewed by Aperio ImageScope software (version 7.3.36; Aperio). Images of hybridization in lung tissue cores were manually scored for number of signals per tumor cell nuclei. Cores with weak signal intensities were analyzed using a Nikon E800 microscope under an ×60 objective. The signals were evaluated by first visually examining the entire core and then individually examining the tumor cell nuclei to determine the number of signals per cell. Nuclei unable to be evaluated due to insufficient hybridization or cell clustering were excluded from scoring. Approximately 100 nuclei were counted for each core. The number of signals per nuclei with the highest percentage (mode) in the sample was used to determine the genetic status of the LKB1 in the sample. A total of 65 samples were also analyzed by LOH study and included for comparisons.
Genomic sequencing of lung tumors with LOH at chromosome
19p. We analyzed 62 of the 124 samples with LOH at markers D19S814 and/or D19S883 by direct sequencing. Genomic DNA from the tumor and matched non-tumor tissues were amplified using eight pairs of primers that amplified the entire coding region of the gene. The PCR reaction was performed using 10 ng of total genomic DNA and all exons of LKB1 were analyzed on an ABI PRISM 3730 (Applied Biosystems) automatic sequencer and subject to mutation analysis using the mutation detector of the Genome-work Bench (Zhang et al., 2008). All exons of LKB1, including intron/exon borders, were directly sequenced in both directions. All alterations were confirmed to be somatic or germ line by sequencing the paired non-tumor samples.
Statistical analysis
We compared the differences in the characteristics of lung cancer patients with or without LOH in 19p and LKB1 mutations by t-test or analysis of variance for continuity, and χ2-tests or Fisher’s exact tests (when 20% of the expected counts were less than 5) for categorical variables as indicated. We also performed comparisons of clinicopathological factors (that is, age at diagnosis, gender, histological type or pathological stage, and smoking status) in no LOH, with LOH and/or HD on the basis of CISH analysis. We used the log-rank test for comparing survival distributions between HD and LOH groups of LKB1/STK11 loss, and we plotted Kaplan–Meier curves for the two groups. All statistical analyses were performed using SAS (SAS version 8.1; SAS Institute, Cary, NC, USA).
Acknowledgments
We thank John Cottrell, Ray Jones, Audrey Salabes and Mike Lipsky at University of Maryland Medical Center for tissue collection and clinical information. We thank the members of Dr. Thomas Reid’s lab for technical support during the course of this study. We thank Ms Stacy Johnson for editorial and graphical/technical assistance. The current addresses are RKG, Laboratory of Receptor Biology and Gene Expression, National Cancer Institute, Bethesda, MD 20892–4258; SHY, Wonkwang University Hospital, Department of Pulmonary and Critical Care Medicine, Iksan, South Korea; HSJ, Laboratory of Biochemistry and Cell Biology, Kyungpook National University, School of Medicine, Deagu, South Korea; SRC, Laboratory of Pathology, National Cancer Institute, Bethesda, MD; AS, Department of Medical Genetics, Tehran University of Medical Sciences, Cancer Institute Hospital, Tehran, Iran; TVD, J. Craig Venter Institute, Rockville, MD; KMH, National Cancer Center, Cancer Cell and Molecular Biology Branch, Goyang, Korea; JF, Toyama University Hospital, Department of Surgical Pathology, Toyama, Japan; and JHZ, St. Jude Children’s Research Hospital, Biotechnology, Memphis, TN. This work was supported by intramural research funds from the Center for Cancer Research at CCR and funds for JJ from the Mayo Cancer Center and Center for Individualized Medicine, Rochester, MN.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Loukola A, Roth S, Hemminki A, Tarkkanen M, Salovaara R, et al. LKB1 somatic mutations in sporadic tumors. Am J Pathol. 1999;154:677–681. doi: 10.1016/S0002-9440(10)65314-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avizienyte E, Roth S, Loukola A, Hemminki A, Lothe RA, Stenwig AE, et al. Somatic mutations in LKB1 are rare in sporadic colorectal and testicular tumors. Cancer Res. 1998;58:2087–2090. [PubMed] [Google Scholar]
- Bignell GR, Barfoot R, Seal S, Collins N, Warren W, Stratton MR. Low frequency of somatic mutations in the LKB1/Peutz-Jeghers syndrome gene in sporadic breast cancer. Cancer Res. 1998;58:1384–1386. [PubMed] [Google Scholar]
- Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11:210–212. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- Carretero J, Medina PP, Pio R, Montuenga LM, Sanchez-Cespedes M. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene. 2004;23:4037–4040. doi: 10.1038/sj.onc.1207502. [DOI] [PubMed] [Google Scholar]
- Conde E, Suarez-Gauthier A, Garcia-Garcia E, Lopez-Rios F, Lopez-Encuentra A, Garcia-Lujan R, et al. Specific pattern of LKB1 and phospho-acetyl-CoA carboxylase protein immunostaining in human normal tissues and lung carcinomas. Hum Pathol. 2007;38:1351–1360. doi: 10.1016/j.humpath.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RK, Vazquez MF, Kramer A, Hames M, Zhang L, Heselmeyer-Haddad K, et al. The use of genetic markers to identify lung cancer in fine needle aspiration samples. Clin Cancer Res. 2008;14:7481–7487. doi: 10.1158/1078-0432.CCR-07-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaros S, Cirrincione GM, Palanca A, Metzger D, Reisman D. Targeted knockout of BRG1 potentiates lung cancer development. Cancer Res. 2008;68:3689–3696. doi: 10.1158/0008-5472.CAN-07-6652. [DOI] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J. Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene. 1999;18:1777–1780. doi: 10.1038/sj.onc.1202486. [DOI] [PubMed] [Google Scholar]
- Gurumurthy S, Hezel AF, Sahin E, Berger JH, Bosenberg MW, Bardeesy N. LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis. Cancer Res. 2008;68:55–63. doi: 10.1158/0008-5472.CAN-07-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89:777–798. doi: 10.1152/physrev.00026.2008. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, et al. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen JP, Kim J, Lee J, Rogers AM, Park JO, Zhao X, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer. 2008;99:245–252. doi: 10.1038/sj.bjc.6604469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launonen V. Mutations in the human LKB1/STK11 gene. Hum Mutat. 2005;26:291–297. doi: 10.1002/humu.20222. [DOI] [PubMed] [Google Scholar]
- Levin NA, Brzoska PM, Warnock ML, Gray JW, Christman MF. Identification of novel regions of altered DNA copy number in small cell lung tumors. Genes Chromosomes Cancer. 1995;13:175–185. doi: 10.1002/gcc.2870130307. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Lim W, Hearle N, Shah B, Murday V, Hodgson SV, Lucassen A, et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer. 2003;89:308–313. doi: 10.1038/sj.bjc.6601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukeis R, Irving L, Garson M, Hasthorpe S. Cytogenetics of non-small cell lung cancer: analysis of consistent non-random abnormalities. Genes Chromosomes Cancer. 1990;2:116–124. doi: 10.1002/gcc.2870020207. [DOI] [PubMed] [Google Scholar]
- Luukko K, Ylikorkala A, Tiainen M, Makela TP. Expression of LKB1 and PTEN tumor suppressor genes during mouse embryonic development. Mech Dev. 1999;83:187–190. doi: 10.1016/s0925-4773(99)00050-7. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911–5918. doi: 10.1038/sj.onc.1210418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechanic LE, Marrogi AJ, Welsh JA, Bowman ED, Khan MA, Enewold L, et al. Polymorphisms in XPD and TP53 and mutation in human lung cancer. Carcinogenesis. 2005;26:597–604. doi: 10.1093/carcin/bgh344. [DOI] [PubMed] [Google Scholar]
- Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–622. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- Rowan A, Bataille V, MacKie R, Healy E, Bicknell D, Bodmer W, et al. Somatic mutations in the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J Invest Dermatol. 1999;112:509–511. doi: 10.1046/j.1523-1747.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, Wu L, et al. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res. 2001;61:1309–1313. [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004a;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004b;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Virmani AK, Fong KM, Kodagoda D, McIntire D, Hung J, Tonk V, et al. Allelotyping demonstrates common and distinct patterns of chromosomal loss in human lung cancer types. Genes Chromosomes Cancer. 1998;21:308–319. doi: 10.1002/(sici)1098-2264(199804)21:4<308::aid-gcc4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Wang ZJ, Taylor F, Churchman M, Norbury G, Tomlinson I. Genetic pathways of colorectal carcinogenesis rarely involve the PTEN and LKB1 genes outside the inherited hamartoma syndromes. Am J Pathol. 1998;153:363–366. doi: 10.1016/S0002-9440(10)65579-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo SN, Gallardo TD, Akbay EA, Liang MC, Contreras CM, Boren T, et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS One. 2009;4:e5137. doi: 10.1371/journal.pone.0005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Zhang S, Schafer-Hales K, Khuri FR, Zhou W, Vertino PM, Marcus AI. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cancer Res. 2008;68:740–748. doi: 10.1158/0008-5472.CAN-07-2989. [DOI] [PubMed] [Google Scholar]
- Zhong D, Guo L, de Aguirre I, Liu X, Lamb N, Sun SY, et al. LKB1 mutation in large cell carcinoma of the lung. Lung Cancer. 2006;53:285–294. doi: 10.1016/j.lungcan.2006.05.018. [DOI] [PubMed] [Google Scholar]