

Abstract
Mutations of the retinoblastoma tumour suppressor gene (RB1) or components regulating the RB pathway have been identified in almost every human malignancy. The E2F transcription factors function in cell cycle control and are intimately regulated by RB. Studies of model organisms have revealed conserved functions for E2Fs during development, suggesting that the cancer-related proliferative roles of E2F family members represent a recent evolutionary adaptation. However, given that some human tumours have concurrent RB1 inactivation and E2F amplification and overexpression, we propose that there are alternative tumour-promoting activities for the E2F family, which are independent of cell cycle regulation.
Two decades of experimentation link E2F activity to cell cycle control1–4. The E2Fs are a large family of transcription factors containing one or more conserved DNA binding domains (DBDs) that bind target promoters and regulate their expression1,2. The RB tumour suppressor and the RB-related pocket proteins p107 and p130 directly associate with E2Fs and can be co-recruited to E2F-responsive promoters to modulate gene expression5. Functional inactivation of RB1 in various human cancers and transgenic animal models leads to deregulated E2F activity, which has been correlated with aberrant cell proliferation and in some instances cell death6–8. Consistent with these observations, global gene expression profiling and promoter occupancy (chromatin immunoprecipitation (ChIP)-on-chip and ChIP–sequencing) arrays have confirmed that many genes that are crucial for proper cell cycle progression are bona fide E2F targets9–15, therefore establishing a direct role for E2Fs in governing cell proliferation. In recent years, more members and isoforms of the E2F family have been described and their functions explored in different physiological settings unrelated to the control of cell proliferation1,2,8,10,16–21. These studies have yielded results inconsistent with the central dogma of the RB–E2F pathway that has been defined by in vitro and cell culture assays. The aim of this Review is to present emerging data from mouse models of development and tumorigenesis that challenge the principles of RB–E2F function in coordinating cell cycle progression. We present the view that in vivo E2F functions extend beyond the control of cell proliferation, and discuss how these functions may be implicated in the pathogenesis of multiple human cancers independent of cell cycle control.
E2Fs: the current paradigm
E2F function is regulated by cyclin-dependent kinases (CDKs) and RB
It was discovered nearly two decades ago that infection by DNA tumour viruses caused excessive host cell proliferation, which was associated with the binding of specific cellular factors – later named E2Fs – to viral gene promoters22,23. These initial observations paved the way for the identification of an intricate signalling pathway that has been shown to culminate in the control of E2F transcription activity (BOX 1). Since the identification of the founding E2F family member E2F1 (Refs 24,25) two distinct genes in Drosophila melanogaster and seven additional genes in mammals have been found to encode the signature DBDs that give these transcription factors their DNA binding specificity1,2,8 (FIG. 1; see Supplementary information S1 (box)). The key finding that overexpression of E2F activators could trigger quiescent cells to enter G1 phase, independent of growth factor stimulation, linked this transcriptional module to cell cycle control26 and fuelled the initial identification of important E2F target genes that are involved in DNA replication. Thereafter, numerous studies focusing on how E2Fs contribute to cell proliferation elucidated a molecular signalling network that converged on the post-translational modifications of RB and RB-related proteins (which are mediated by cyclin–CDK complexes) and the subsequent waves of E2F-dependent transcription activation and repression that guarantee the timely movement of cells through all four phases of the cell cycle27s–30 (FIG. 2). It should be emphasized that the simple classification of E2Fs as activators and repressors is mostly based on the analysis of cells cultured in vitro and lacks in vivo validation.
Box 1. Alterations of the CDK–RB–E2F pathway in human cancer.
Uncontrolled cell growth is an invariable characteristic of human cancer138. The proliferation of cancer cells is sustained in the absence of growth factors and is insensitive to growth-inhibitory signals138. Members of the human epidermal growth factor receptor family of receptor tyrosine kinases are deregulated in many types of human cancer139. Overexpression or mutation of these receptors leads to the constitutive activation of downstream signalling pathways140,141.
On mitogenic stimulation, intracellular levels of D-type cyclins (D1, D2 and D3) increase, resulting in the formation and nuclear localization of cyclin D–cyclin-dependent kinase 4 (CDK4) and cyclin D–CDK6 complexes that phosphorylate RB early in G1 phase142. Subsequently, levels of mitogen-independent E-type cyclins accumulate and these cyclins associate with CDK2 to further phosphorylate RB in preparation for progression into S phase142. Genetic alterations of G1/S regulators have been extensively documented in human cancers (see the figure)29,59,143. The G1 cyclins, in particular D1 and E1, and their catalytic partners CDK4 and CDK6, are overexpressed, mutated or their genomic loci amplified in a wide variety of tumours29,59,143. The activity of CDKs is regulated by two families of CDK inhibitors (CDKIs), with the first family comprised of the INK4 proteins (INK4A, INK4B, INK4C and INK4D). The second family consists of p21, p27 and p57. Decreased expression of these negative G1/S regulators in human cancer has been attributed to genetic and epigenetic changes3–5,29,59,143. In short, deregulation of factors that control the G1/S transition, which ultimately relies on the engagement of E2F activity in the current view of cell cycle regulation, seems to be a universal theme in the process of neoplastic transformation. LOH, loss of heterozygosity.
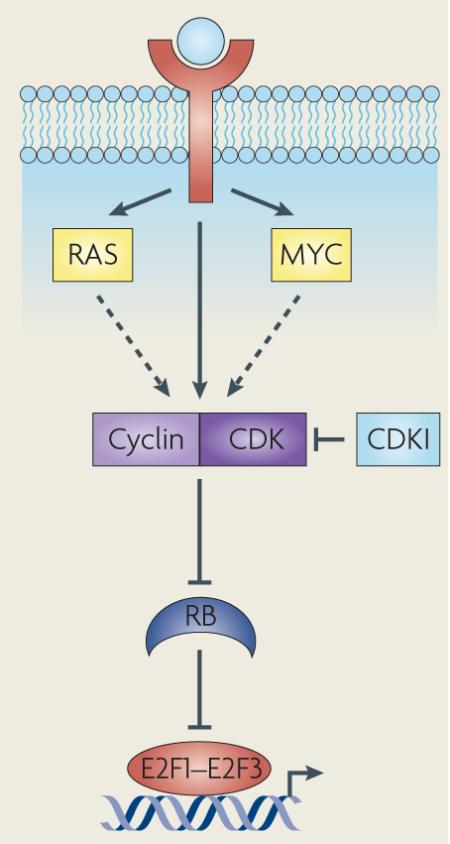
Figure 1. The mammalian E2F family of transcription factors.

This family is defined by their signature winged-helix DNA binding domain (DBD), and all members are expressed from eight chromosomal loci that encode nine distinct gene products. Based predominantly on results from in vitro studies, the E2F family has been traditionally divided into activator (E2F1–E2F3) and repressor (E2F4–E2F8) subclasses. Most E2F family members (E2F1–E2F6) bind DNA as heterodimers with one of three dimerization partner (DP) proteins, TFDP1, TFDP2 and TFDP3, and heterodimerization is mediated by the leuzine zipper (LZ) and marked box (MB) domains. RB binds within the transactivation domain (RB) of E2F1–E2F3. Alternative promoters at the E2F3 locus drive the expression of two highly related isoforms, E2F3a and E2F3b51. Unlike the activating E2F1–E2F3, E2F4 associates with all three pocket protein family members and E2F5 associates predominantly with p130 (REFS 1,2,8). Ectopically expressed E2F activators are localized to the nucleus owing to their amino-terminal nuclear localization signal (NLS) sequence143,144, which is adjacent to the cyclin A-binding site (CycA). E2F4 and E2F5 have bipartite nuclear export signals (NES) that mediate their export to the cytoplasm145,146. Repressors E2F6–E2F8 do not possess the canonical carboxy-terminal features of E2F4 and E2F5 and so are presumed to repress E2F-responsive genes independently of RB and related pocket proteins40,41,147,148. Indeed, E2F6 can repress E2F target expression when overexpressed in cell culture147–149. The structurally unique and most recently identified E2Fs, E2F7 and E2F8, comprise a separate and highly evolutionarily conserved repressor arm in the E2F family. Unlike the E2F3 isoforms that are transcribed from distinct promoters, E2F7a and E2F7b isoforms are produced by alternative splicing of the primary transcript41.
Figure 2. The expression and activity of mammalian E2F family members during the cell cycle.
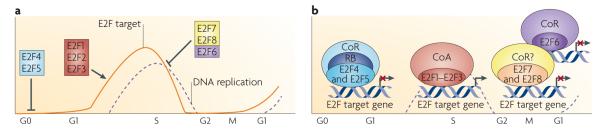
a| In quiescent (G0) cells, the ubiquitously expressed E2F4 and E2F5 associate with pocket proteins and other co-repressors to maintain repression of E2F-responsive genes (line labelled E2F target) that promote entry into the G1 phase of the cell cycle. On mitogenic stimulation, the sequential phosphorylation of RB by activated cyclin-dependent kinases results in the loss of RB function, release of E2F repressors142 and the accumulation of newly synthesized free E2F1, E2F2 and E2F3 late in G1 phase. Together, these events initiate a transcriptional programme driving cells into S phase150 (dashed line labelled DNA replication). This G1/S-specific transcriptome is then attenuated on completion of S phase in G2 by the action of the repressors E2F6, E2F7 and E2F8, which are thought to function independently of RB and RB-related proteins40,41,151. E2F7 and E2F8 can also specifically mediate the repression of E2F activators such as E2f1 (REFS 17,41). Heterodimeric or homodimeric E2F7 and E2F8 complexes directly bind the E2f1 promoter and repress its expression throughout S phase to restrain activator E2F function. Without this brake, E2F1 proteins would persist inappropriately and continue the activation of genes encoding components of the transcriptional machinery, resulting in ectopic DNA replication. b| In quiescent or differentiated cells, pocket protein-bound E2F4 and E2F5 have been found to associate with various co-repressors (CoRs), such as histone deacetylases, the DNA methyltransferase DNMT1 and C-terminal binding protein (CtBP), leading to chromatin compaction and transcription inhibition152–155. Conversely, during cell proliferation, when RB is hyperphosphorylated, E2F activators recruit the basal transcription factor TFIID and other co-activators (CoA), such as histone acetyltransferases, p300 and CBP, GCN5 and TIP60, to specific gene promoters156–158, leading to an open chromatin configuration and transcription initiation. The more recently identified repressors E2F6–E2F8 mediate repression of E2F-responsive genes independent of pocket protein binding. Although E2F6, through interaction with the polycomb complex149, still requires dimerization with a dimerization partner protein to function in transcription repression, E2F7 and E2F8 are unique in that they form homodimers (E2F7–E2F7 and E2F8–E2F8) or heterodimers (E2F7–E2F8) to repress transcription of cell cycle-related genes17,40,41. The co-repressors that associate with E2F7 and E2F8 are currently unknown.
Redundancy, antagonism and feedback: the complexities of E2F function
Three obstacles have precluded a thorough analysis of the role of E2Fs in vivo. First, the high degree of functional redundancy among activators (E2F1, E2F2, E2F3a and E2F3b)1,2,6–8,31,32 and repressors (E2F4, E2F5, E2F6, E2F7 and E2F8)17,33 has made the study of these proteins technically challenging and experimental results difficult to interpret. The systematic analyses of mice with single and compound E2F mutations initially revealed the redundant nature of E2F activity. Moreover, recent gene knock-in strategies in mice have demonstrated that expression from the endogenous E2f3a locus of the highly related E2f3b isoform or the more distantly related E2f1 family member suppressed most of the early postnatal developmental phenotypes associated with the inactivation of E2f3a31.
The second obstacle impeding a complete understanding of E2F functions in vivo is the apparent functional antagonism between E2F-mediated activation and repression in the control of normal cell proliferation. Results from initial studies in D. melanogaster seemed to validate the straightforward model of E2F function as an activity that promoted cell cycle progression. Unlike in mammals, the D. melanogaster E2F family consists of a single transcription activator (E2F) and repressor (E2F2). Both proteins require dimerization with the transcription factor DP, which is encoded by a single gene. Loss-of-function mutations in E2f lead to a decrease in the expression of classic E2F-regulated genes, DNA synthesis and larval survival34,35. Surprisingly, loss of E2f2 restored cell proliferation in E2f-mutant larvae and significantly delayed lethality until mid or late pupal stages35. These results suggest that the net effect of E2Fs on cell proliferation is dependent on the balanced interplay between activation and repression on a shared set of E2F-regulated genes. However, subsequent analysis of gene expression in S2 cells lacking E2f, E2f2 or Dp (the latter of which abrogates total E2F activity in D. melanogaster) revealed surprisingly little overlap in genes regulated by E2f and E2f2 (REF. 11). Consistent with this gene expression data, a recent genetic mosaic screen found that the cell cycle defects induced by loss of E2F could be rescued independently of functionally inactivating E2f2 and abrogating E2F2-mediated repression36. Therefore, in addition to regulating a common set of cell cycle genes, E2F and E2F2 seem to contribute independently to the normal proliferative potential of a cell.
The third reason why the study of E2Fs remains a challenge is their ability to regulate the expression of other members of the family, forming complex feedback loops to ensure a balanced level of activators and repressors in each phase of the cell cycle. A clear example of this was revealed by the recent molecular analysis of E2f7−/−;E2f8−/− mouse embryos17. E2F7 and E2F8 directly repress E2f1 expression and double-knockout embryos have increased levels of E2F1 and widespread apoptosis. The removal of E2f1 from these mutant embryos significantly reduced the level of apoptosis. E2F7 and E2F8 are the most recently discovered E2F family members and represent a structurally distinct arm of the repressor subclass that interestingly is not unique to mammals. The model plant Arabidopsis thaliana has three such ‘atypical’ E2Fs, E2FE, E2FD and E2FF37 (Supplementary information S1 (box)). The general functions of these plant E2F repressors involve the control of endocycle onset and progression, as well as the regulation of cell size38,39. Similar to their plant counterparts, the expression of mammalian E2F7 and E2F8 is E2F-regulated – in silico analysis of their promoters identified consensus E2F binding motifs to which E2F1, E2F3, E2F4 and E2F7 were shown to bind using ChIP assays40,41.
RB–E2F developmental mouse models support the canonical pathway
A considerable number of developmental studies using RB and RB–E2F compoundmutant mice seem to reinforce the role of E2Fs in cell proliferation and apoptosis that was originally identified in vitro42–44. Although homozygous inactivation of Rb1 in mice leads to embryonic lethality that is accompanied by massive ectopic proliferation and apoptosis in multiple tissues45–47, the deletion of E2F activators has been shown to partially rescue the proliferative and/or apoptotic phenotypes. Specifically, the loss of E2f1 or E2f3a most robustly suppressed Rb1-mutant phenotypes and extended the viability of Rb1−/− embryos to late stages of embryogenesis42,48. The loss of E2f3b, which encodes an E2F3 isoform thought to repress gene expression, also alleviated a subset of phenotypes that are associated with the complete loss of Rb1 (REF.48–51). The observation that loss of the repressor E2f4 or E2f5 did not rescue Rb1-mutant phenotypes in the mouse embryo is consistent with their role as binding partners of RB that mediate repression44. Taken together, these observations generally support the canonical role of E2F1–E2F3 activators and E2F4 and E2F5 repressors as effectors of RB that control cell proliferation and apoptosis.
In summary, the prevailing view has been that phenotypes associated with the inactivation of specific E2F family members result from the loss of E2F-mediated activation or repression of genes directly involved in DNA synthesis, mitotic progression and apoptosis. Although the above analyses of RB–E2F compoundmutant mice support the function of E2F activators in regulating proliferation and apoptosis, the detailed analysis of E2F-knock-in mice31 suggests that members of this gene family have overlapping functions in the regulation of many essential processes in addition to the cell cycle. Evidence from the model organism D. melanogaster also contradicts the simple antagonistic nature of E2F-mediated activation and repression, and recent knockout models in mice illustrate the existence of an extensive E2F cross-regulatory network. Clearly, these exciting new findings reveal that we are only beginning to understand the immense complexity of how the E2F family participates in the control of cell proliferation and in other, as yet uncharted, biological processes.
Regulation of E2F target genes
E2F family members regulate overlapping and distinct subsets of genes. The specificity of E2F-dependent activation and repression is dictated by interactions between individual family members and various cofactors. For instance, in vitro pull-down assays showed that E2Fs can associate with different histonemodifying enzymes that direct specific chromatin configurations13,52 (FIG. 2b).
As changes in gene expression reflect direct and indirect functions of transcription factors, several laboratories have expended considerable effort on ChIP-based technologies to identify direct E2F-regulated targets10,12–15,20,53. This approach has led to the identification of many E2F-responsive genes beyond those involved in proliferation and apoptosis, including genes that participate in biological processes as diverse as cell differentiation, metabolism and animal development9–12,54. Furthermore, these studies have distinguished targets that are uniquely regulated by individual E2F family members from targets that are co-regulated by multiple members. Work from the Farnham laboratory has also demonstrated the recruitment of E2F1 to an unprecedented number of target promoters that are not always restricted to consensus E2F binding sites15,20. This level of promiscuity in E2F–chromatin associations could be due to the recruitment of E2Fs by other transcription factors, such as nuclear factor-κβ (NF-κβ)55, MYC56 and CAAT enhancer/binding protein-α (CEBPα)57,58, or by components of the transcriptional machinery. Although they are undoubtedly intriguing, most of these results were obtained using cell culture systems. Whether the newly identified transcriptional networks involving E2Fs reflect their true physiological roles in vivo (such as the regulation of inflammatory response together with NF-κβ55) remains to be determined.
Revealing the full range of E2F functions will require the identification of direct targets by two synergistic approaches, global gene (including microRNA gene) expression profiling and ChIP–sequencing. The first approach is unbiased and identifies all genes that have deregulated expression in the absence of E2F family members. The second approach validates these genes as direct E2F targets. The fact that both types of analysis can be carried out with materials extracted from control and E2F knockout mouse tissues allows for a rigorous examination of tissue-specific compared with general effects of E2F functions throughout multiple stages of metazoan development. The ability to carry out these analyses in normal tissue and tumour tissue ranging from pre-malignant to metastatic will also distinguish physiological E2F functions from those that may have evolved during tumour development. For example, a requirement for different CDKs in normal and oncogenic cell proliferation has been reported59. This idea is supported by elegant work from the Sicinski group that clearly demonstrated that Cdk4−/− mice develop normal mammary glands but are resistant to Erbb2- or Hras-driven mammary tumorigenesis60,61.
Complexity of E2F functions in vivo
Studies using germline Rb1+/− and chimeric Rb1−/− mice that develop various neuroendocrine tumours have produced unexpected phenotypes when E2Fs are inactivated. The logic behind these studies was based on developmental studies showing that the inactivation of Rb1 led to the deregulated function of activator E2Fs and that the subsequent removal of these E2Fs resulted in a reversion of Rb1 mutant phenotypes. Although the results from these studies do not dismiss a role for E2Fs in cell proliferation and apoptosis, they have not truly furthered our understanding of the mechanisms, molecular partners and cellular contexts that contribute to E2F-mediated activation and repression. In brief, the following studies in mice contradict a uniform role for E2Fs in cancer.
The role of E2F activators in cancer, as defined in the context of Rb1 mutation
Unlike humans inheriting one mutated or deleted RB1 allele who develop retinoblastomas, germline Rb1+/− mice develop intermediate lobe pituitary and C cell thyroid tumours62,63. The initial studies by Jacks and colleagues exploring the role of E2F activators in driving tumour initiation showed that ablation of E2f1 in Rb1+/− mice significantly reduced the incidence of both pituitary and thyroid tumours, prolonging the tumour-free lifespan of Rb1+/− mice64. This was the first genetic evidence to support an oncogenic role for an E2F family member in the context of Rb1 inactivation. The interpretation that E2F1 has a uniform oncogenic role, however, is clouded by the observation that E2f1−/− mice develop lymphoma or occasionally tumours of mesenchymal origin (TABLE 1). The fact that the loss of Rb1 in T cells does not result in lymphomagenesis65 suggests that the tumour-suppressive role of E2f1 in this context may be RB independent. The role of E2f3 as an effector of RB action seems to be slightly more complex. Loss of E2f3 in germline Rb1+/− mice, similar to the loss of E2f1, suppressed the development of pituitary tumours66. This is consistent with E2f3 functioning as an oncogene downstream of Rb1. However, its loss also promoted the development and metastasis of medullary thyroid tumours in Rb1+/− mice, suggesting that E2f3, like E2f1, may function as a tumour suppressor66. It is possible that E2F3 might promote proliferation in the pituitary but have additional RB-independent roles in the thyroid. As the direct functions of E2F1 and E2F3 have not been extensively explored in the pituitary and thyroid, the precise molecular basis for their apparent pro-tumorigenic and anti-tumorigenic actions in these tissues remains unclear.
Table 1.
Developmental defects in E2F-knockout mouse models
| Genotype | Phenotype | Refs |
|---|---|---|
| E2f1 −/− | Defective thymic-negative selection, testicular atrophy and increased susceptibility to tumorigenesis in different tissues |
85,86 |
| E2f2 −/− | Development of autoimmune disease owing to combined hematopoietic defect | 88 |
| E2f1−/−;E2f2−/− | Development of exocrine pancreatic insufficiency and diabetes | 159,160 |
| E2f3 −/− | Embryonic lethal in pure FVB (E12.5) or 129Sv genetic backgrounds, reduced viability in mixed (C57BL/6 × 129Sv) genetic background |
31,87,161 |
| E2f3a −/− | Reduced WAT deposits with age in pure genetic background; no specific defects observed in mixed genetic background |
31,32 |
| E2f3b −/− | None observed | 31,32 |
| E2f1−/−;E2f3a−/− | Death by ~1 month of age, runted growth with under-developed sexual organs, reduced WAT deposits, reduced number of pancreatic exocrine cells and multiple other tissue-specific developmental defects |
31,32 |
| E2f1−/−;E2f3b−/− | Viable and fertile | 31,32 |
| E2f1−/−;E2f2−/−;E2f3a−/− | Neonatal lethality (~E19.5) in mixed genetic background | 31 |
| E2f1−/−;E2f2−/−;E2f3b−/− | Runted growth with under-developed sexual organs in mixed genetic background | 31 |
| E2f1−/−;E2f2−/−;E2f3−/− | Embryonic lethal (E10.5) with no obvious disturbances in cell proliferation | 214,215 |
| E2f4 −/− | Neonatal lethality owing to increased susceptibility to opportunistic infections resulting from craniofacial defects. Maturation defects in multiple hematopoietic lineages and the gut epithelium, macrocytic anaemia owing to impaired erythroid proliferation |
89,90,162 |
| E2f5 −/− | Shortened lifespan owing to development of hydrocephalus resulting from overproduction of cerebrospinal fluid by the choroid plexus |
91 |
| E2f6 −/− | Normal lifespan with homeotic transformations of the axial skeleton | 92 |
| E2f7 −/− | None observed | 17 |
| E2f8 −/− | None observed | 17 |
| E2f7−/−;E2f8−/− | Embryonic lethal (E12.5) with widespread apoptosis, vascular dilation and haemorrhage |
17 |
E, embryonic day; WAT, white adipose tissue.
Further attempts to inactivate E2F function in chimeric Rb1−/− mice produced intriguing but inconclusive results. Rb1−/− chimeras were previously shown to be fully viable and develop pituitary tumours at a young age67,68. In more recent studies by Parisi and colleagues, thyroid tumours and pulmonary neuroendocrine cell (PNEC) hyperplasia were also observed in Rb1−/− chimeras69. Ablation of E2f3 in Rb1−/− chimeras suppressed PNEC, which is thought to precede the development of small cell lung carcinoma (SCLC), but failed to suppress the onset or development of either pituitary or thyroid tumours69. Why deletion of E2f3 can mediate different pituitary and thyroid tumour outcomes in Rb1+/− compared with Rb1−/− chimeric mice is not clear. It is entirely possible that the role of E2F3 in these contexts is tumour cell non-autonomous and that loss of E2F3 in these two different microenvironments leads to different or even opposing outcomes. Given that several cell non-autonomous functions have been described for RB and E2F in Caenorhabditis elegans and mice70–73, it would be prudent to caution against the over-interpretation of the above analyses until more definitive molecular mechanisms have been provided.
Finally, loss of E2f2, but not E2f1 or E2f3, in mice increased Myc-induced T cell lymphomagenesis, and the reintroduction of E2F2 into these tumours stimulated apoptosis of the tumour cells74. This result implicates E2F2 as a tumour suppressor. By contrast, loss-of-function models have also revealed that E2f2 seems to behave as an oncogene in simian virus 40 (SV40) large T antigen-induced intestinal hyperplasia, as its ablation reduced epithelial cell proliferation75. These findings provide another example of an E2F activator, functioning as an oncogene or as a tumour suppressor in a tissue-specific manner. Consistent with this, the disruption of RB and components of the RB pathway has been shown to affect gastrointestinal but not haematopoietic malignancies, suggesting that the role of E2F2 in T cells may be RB independent. It should be noted that the inability to identify the exact site of gene action is a confounding characteristic of the analysis of germline knockout mice and may underlie the observed discrepancies that mitigate a unified view of how E2F1–E2F3 activity influences cancer.
The role of E2F repressors in cancer, as defined in the context of Rb1 mutation
The analysis of E2f4 inactivation and its effects on tumour formation in germline Rb1+/− mice revealed tissue-specific roles for this abundantly expressed E2F repressor. The loss of E2F4, a classic repressor known to physically bind all pocket protein family members, extended the lifespan of Rb1+/− mice by suppressing pituitary and thyroid tumorigenesis76, suggesting an oncogenic role for E2F4 in these tissues. This result was partially recapitulated in chimeric Rb−/−;E2f4−/− mice, which had a reduced incidence and delayed development of pituitary tumours77. Interestingly, as in compound Rb1−/−;E2f3−/− chimeras, the Rb1−/−;E2f4−/− chimeras also had reduced PNEC hyperplasia. It is intriguing that the loss of an activator (for example, E2F3) or repressor (for example, E2F4) leads to similar outcomes in Rb1-deficient tumours. To reconcile this paradox, the authors suggested that in the absence of E2F4, the E2F1 and E2F3 activators, which normally do not bind p107 and p130, could now be recruited into a complex with these pocket proteins. The consequence of this pocket protein–E2F rearrangement is the sequestration of E2F activators into a protein complex that has a repressor function, raising the possibility that activators could compensate for repressors and vice versa in a context-dependent manner. By contrast, the development of ganglionic neuroendocrine neoplasms and urothelial transitional carcinomas in Rb1−/−;E2f4−/− double chimeras, which was not observed in chimeric Rb1−/− or E2f4−/− mice, suggests that E2F4 has tumour suppressive properties. It therefore seems that by regulating alternative sets of genes in different tissues E2F4 can function as a tumour suppressor or an oncogene. A full understanding of how this potential switch in target gene specificity might occur in multiple cellular contexts is lacking.
Given the lack of clearcut in vivo data to support the classification of E2Fs as repressors or activators, it is conceivable that these two subclasses function similarly in the animal. Therefore, a simple explanation for why E2F3 and E2F4 can function as either oncogenes or tumour suppressors is that both may behave as transcription activators or repressors in vivo. Additional supporting evidence of this has been published in several recent studies. Work from the Zubiaga group has shown that E2F2 suppresses cell cycle gene expression in T cells and that the targeted disruption of E2f2 accelerated S phase entry and cell division78. E2F3b, which is constitutively expressed in all phases of the cell cycle, was initially shown to bind to the Arf promoter and to directly repress Arf transcription49. Knockin and E2f3 isoform-specific knockout studies in mice have revealed overlapping functions for E2f3a and E2f3b in early mammalian development31,32 and indicate that both isoforms could function as activators or repressors.
Currently, the role of the new repressor arm of the E2F family, E2F7 and E2F8, in cancer is virtually unknown. However, given their crucial role in the control of proliferation and apoptosis in embryonic development, it would not be surprising to find that these E2Fs are involved in cancer. The expression of these E2Fs needs to be evaluated in clinical samples and in the context of tumour models.
Challenges to the current RB–E2F paradigm
CDKs and E2Fs: a role in normal cell proliferation?
A seminal series of mouse knockout studies recently revealed that most cells in the animal continue to proliferate in the absence of CDK2, CDK4 or CDK6 (REFS 79,80). Mice individually lacking Cdk2, Cdk4 or Cdk6 are fully viable and have developmental phenotypes that pinpoint specific roles for these Cdks in the control of homeostatic proliferation of specialized cell types59. Strikingly, Cdk2−/−;Cdk4−/−;Cdk6−/− triple-knockout mouse embryos develop to mid gestation and die around embryonic day 12.5 (E12.5) from defective haematopoiesis, without obvious disturbances in cell proliferation81. By contrast, Cdk1−/− embryos fail to develop beyond the two-cell stage. These observations led the authors to conclude that the mitotic CDK1 is the only CDK required for normal cell proliferation81 and that interphase CDKs cannot compensate for the activity of CDK1 (REF. 82). On the basis of these findings, it would seem that factors positively regulating cell cycle progression are not as crucial for normal cell proliferation in mammals as studies in yeast had originally implied. It has been suggested that oncogene-stimulated proliferation may result in the need for a specific CDK family member, as seems to occur with CDK8 in colorectal cancer83. Given that the human CDK family consists of 13 members, it will be interesting to determine the extent of redundancy in development and cancer among members of this diverse family of kinases. Even though the combinations of CDKs involved in cell proliferation may change in different cell types, it is thought that E2Fs are the crucial downstream effectors of CDK activity that ultimately drive cell cycle progression. Although the loss of E2f1–E2f3 leads to the complete arrest of mouse embryonic fibroblasts (mEFs) in cell culture and therefore supports this idea84, this hypothesis remains to be validated in vivo.
The idea that E2F function is indispensable for the control of cell proliferation has dominated several decades of experimentation. Incompatible with this view is the fact that mice deficient for individual or a combination of E2F genes did not have widespread defects in cell proliferation17,85–92 (TABLE 1). Recent work has shown that the combined ablation of the entire subset of E2F activators (E2f1–E2f3) resulted in mid gestational lethality of mouse embryos with only a negligible effect on cell proliferation in most of the tissues examined214,215. These results seem to oppose the finding that mEFs lacking E2f1–E2f3 are profoundly arrested at G1/S84. However, the inactivation of the p53 pathway was sufficient to stimulate quiescent triple-knockout MEFs to re-enter the cell cycle and be transformed by MYC and Ras93,94, indicating that conditions permissive for the proliferation of triple-knockout cells exist in a cell type- or oncogene-dependent manner. Collectively, these observations suggest that the redundant role of E2F activators in normal cell proliferation, as revealed by the recent E2F-knockin studies in mice, may be restricted to a fairly narrow period in the development of mammals and that compensatory factors exist to permit cell growth and division in the absence of E2F activators. Similar to CDKs, many studies have also suggested an acquired preference for specific E2F activators in oncogenic proliferation. For instance, E2F transgenic mice develop various tumours (TABLE 2), and overexpression and/or amplification of E2F1 and E2F3 has been observed in different human cancers (TABLE 3). In addition, E2F activators were shown to be necessary for the unscheduled proliferation of cells mutant for either Rb1 or components of the Hippo tumour suppressor pathway42,43,48,95. Taken together, these results suggest a differential requirement for E2Fs in the control of cell proliferation in normal compared with oncogenic environments. Confirmation of this hypothesis will require a thorough evaluation of E2F function in both normal tissues and associated tumours.
Table 2.
E2F-transgenic mouse models
| Promoter | Tissue with E2F expression | Phenotype | Refs |
|---|---|---|---|
| E2f1 | |||
| Gfap | Brain | Development of medulloblastoma, choroid plexus carcinoma and primary neuroectodermal tumour in young mice; development of malignant gliomas in aged mice |
163 |
| Alb | Liver | Altered lipid metabolism and early development of preneoplastic lesions, with 100% of these progressing to adenomas |
164 |
| αA-crystallin | Retina (post-mitotic lens fibre cell) | Cell cycle re-entry and subsequent apoptosis of lens fibre cells | 165 |
| Pf4 | Megakaryocyte | Impaired terminal differentiation and apoptosis of megakaryocytes resulting in thrombocytopenia |
166 |
| Hmga | Testis | Testicular atrophy and sterility | 167 |
| K5 | Basal epidermal layer and squamous epithelial tissues | Hyperplasia and p53-dependent apoptosis of epithelial tissue, development of spontaneous skin tumours by ~1 year of age and reduced response to two-step carcinogenesis protocol |
168, 169 |
| E2f2 | |||
| Eμ and Pim1 | Thymic epithelium | Development of cortical thymomas | 170 |
| αA-crystallin | Retina (post-mitotic lens fibre cell) | Cell cycle re-entry and subsequent apoptosis of lens fibre cells | 165 |
| E2f3a | |||
| K5 | Basal epidermal layer and squamous epithelial tissues | Development of spontaneous skin tumours and increased response to two-step skin carcinogenesis protocol |
171 |
| αA-crystallin | Retina (post-mitotic lens fibre cell) | Cell cycle re-entry and subsequent apoptosis resulting in microphthalmia |
172 |
| E2f4 | |||
| K5 | Basal epidermal layer and squamous epithelial tissues | Increased response to two-step skin carcinogenesis protocol | 173 |
| αA-crystallin | Retina (post-mitotic lens fibre cell) | Moderate level of cell cycle re-entry with normal-size lens | 172 |
| E2f5 | |||
| αA-crystallin | Retina (post-mitotic lens fibre cell) | Inhibition of E2F1- or E2F3a-induced ectopic cell cycle entry together with p130 overexpression |
174 |
Alb, albumin; Eμ, immunoglobulin heavy chain gene enhancer; Gfap, glial fibrillary acidic protein; K5, keratin 5; Hmga, hydroxy-methyl-glutaryl-Coenzyme A reductase; Pf4, platelet factor 4.
Table 3.
Genetic alterations of E2F family members in human cancers
| Gene (chromosome) | Genetic alteration | Human cancer | Notes |
|---|---|---|---|
| E2F1 (20q11.2) | Amplification | HCC96-98, oesophageal SCC175, NSCLC176 and cancer177–179 |
None |
| Increased expression | NSCLC111,135,179,180, SCLC112,181, glioblastoma109, oesophageal SCC181,182, HCC183,184, pancreatic ductal carcinoma185, and GI stromal186, breast187 and ovarian cancer130,131,188 |
None | |
| Decreased expression | Gastric adenocarcinoma189, oral SCC190, and colon191 and bladder cancer192 |
None | |
| E2F2 (1p36) | Amplification | SCLC193, alveolar rhabdomyosarcoma194 and osteosarcoma195 |
Detection of 1p32–1p36 amplicon in SCLC |
| Deletion | Neuroblastoma196, pheochromocytoma197 and breast cancer198 |
None | |
| Increased expression | Ovarian cancer130,131 | None | |
| E2F3 (6p22) | Amplification | Retinoblastoma105,106,133, uveal melanoma199, and breast200,201 and bladder cancer103,104 |
Detection of 6p21.2 amplicon in breast cancer; complete RB1 inactivation through LOH in retinoblastoma |
| Increased expression | NSCLC111, SCLC111, and bladder103,104, breast118, ovarian188 and prostate cancer202 |
Decreased RB1 expression in breast cancer | |
| E2F4 (16q21–q22) | Amplification | Bladder cancer101 | CGH analysis carried out on 12 transitional cell carcinoma lines |
| Deletion | HCC203 and breast cancer117,204,205 | None | |
| Increased expression | Breast117 and colon cancer206 | None | |
| Mutation (AGC repeat) |
GI cancer207–209 | Expanded or reduced polyserine stretch and LOH frequently observed |
|
| E2F5 (8q21.2) | Amplification | Osteosarcoma195, and bladder102, colon210 and breast cancer128,200, |
Minimal common region of 8q21.3–8q23 in osteosarcoma; MOS and/or MYC amplification in breast cancer |
| Increased expression | Ovarian115,188 and breast cancer200 | None | |
| E2F6 (2p25.1) | Amplification | Neuroblastoma211 and ganglioneuroblastoma212 | Detection of 2p25 amplicon in ganglioneurob- lastoma; MYCN amplification in neuroblastoma |
| E2F7 (12q21.2) | Increased expression | Cutaneous SCC132 | None |
| Decreased expression | Ovarian cancer115 | None | |
| E2F8 (11p15.1) | Deletion | Medulloblastoma213 | LOH of subchromosomal region 11p13–11p15.1 |
| Increased expression | Ovarian cancer114 | None |
CGH, comparative genomic hybridization; GI, gastrointestinal; HCC, hepatocellular carcinoma; LOH, loss of heterozygosity; NSCLC, non-small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer.
E2Fs linked to human cancer
In the current RB–E2F paradigm (FIG. 3) it is thought that genetic alterations resulting in the loss of RB function cause cancer by unleashing E2F activity and deregulating cell proliferation. From this traditional view of how E2F factors control cell proliferation, clear predictions of their involvement in human cancer can be made. As a whole, E2F repressors may be expected to behave as tumour suppressors, and activators as oncogenes. Therefore, oncogenic alterations of repressor E2F genes would include loss-of-function mutations, chromosomal deletions and/or epigenetic silencing. Conversely, one would expect to observe gain-of-function mutations, amplification and/or overexpression of the activator E2F genes.
Figure 3. The paradigm of RB–E2F function in human cancer.

a | In the nuclei of non-proliferating cells, RB remains hypophosphorylated and forms RB–E2F repressor complexes to inhibit the expression of genes that promote S phase entry. Hypophosphorylated RB also directly binds to and inhibits the activity of E2F activators (E2F1–E2F3). RB is inactivated in most types of human cancer and this occurs through the direct mutation of RB1, located on the long arm of chromosome 13 (13q14.3), or through a disruption in the regulatory components of the RB–E2F pathway. For instance, CCND1 is overexpressed or amplified in cancer143, an event that leads to deregulated E2F function by activating cyclin-dependent kinase 4 (CDK4) and CDK6 and by stimulating RB hyperphosphorylation. CDK4 and CDK6 are also overexpressed, amplified and mutated in cancers29,59,143, leading to the loss of RB function. Many types of tumours exhibit decreased expression or mutations of CDK inhibitors, such as INK4A, p21 and p27, which normally antagonize RB phosphorylation29,59,143. In human cancer, it is the prevailing view that the inactivation of RB leads to the disassembly of RB–E2F co-repressor complexes, lifting the repression of genes that are necessary for progression through the cell cycle. Furthermore, the dissociation of hyperphosphorylated RB from E2F activators leads to the inappropriate accumulation of free E2F1, E2F2 and E2F3 with unmasked transactivation domains, resulting in additional transactivation of these genes. b | Amplification of the genomic locus 6p22 harbouring E2F3 is detected in more than 50% of cases of retinoblastoma105,106,133, as well as in bladder tumours that have complete loss of RB function100–104. RB1 is mutated in more than 90% of small-cell lung cancer cases, many of which also exhibit overexpression of E2F1 or E2F3 (REF. 110). The inactivation of RB in these cancers leads to increased proliferation of tumour cells as a result of deregulated E2F function and increased expression of classic E2F target genes. The additional increase in the level of E2F activators might contribute to the activation of genes important for tumour initiation or progression that function independently of cell cycle control. CoA, co-activator; CoR, co-repressor.
At least in humans, a clear theme regarding the oncogenic role of E2F1–E2F3 in human cancer has emerged. Several reports have described the amplification of the E2F1 or E2F3 gene locus as a frequent genetic event observed in hepatocellular carcinoma96–99, bladder cancer100–104, retinoblastoma105,106, liposarcoma107,108 and many other malignancies. Overexpression of E2F1 or E2F3 has also been detected in glioblastoma109 and lung110–113, ovarian114,115, breast116–118, gastric119 and colon cancer120,121. In some cases of neuroblastoma122,123, thyroid cancer122 and pancreatic cancer124,125, large chromosomal deletions of regions including the E2F1, E2F2 or E2F3 genes have been detected. Generally, however, the levels of E2F activators are increased in most cancer types and are presumed to mediate the uncontrolled proliferation of cancer cells.
Inconsistent with the prediction of an involvement in tumour suppression, E2F repressors have not frequently been found to be mutated, deleted or silenced in human cancers. Instead, current data support a role for E2F repressors in tumour promotion. For example, the E2F4–p130 complex was shown to mediate the repression of the DNA repair genes RAD51 and BRCA1 in response to hypoxic stress in vitro126,127. Additionally, an increased gene copy number of E2F5 was detected in two independent cohorts of patients with breast cancer128,129. In one of these studies, there was a positive association of E2F5 amplification with a basal-like phenotype (an oestrogen receptor-, progesterone receptor- and ERBB2-negative phenotype) and worse clinical outcome129. Finally, the evaluation of E2F7 in human cancer has yielded conflicting results130–132 and further investigation will be required to understand its function in cancer.
In summary, deregulated expression of E2F family members is common in human cancer, but whether this contributes to the genesis of these cancers has not been unequivocally established. Future studies will need to determine whether their potential contribution to the tumorigenic process involves deregulation of cell cycle progression or extends beyond the framework of RB and cell cycle control.
E2Fs: beyond proliferation
Generally, cell transformation results from the accumulation of stochastic mutations or ‘hits’ that confer a selection advantage over neighbouring cells, and so mutations affecting multiple components in the same pathway that yield a similar outcome are rarely found in cancer. However, a close examination of the genetics of RB1-mutant human cancers reveals a pervasive association between RB and E2F expression that is inconsistent with the current view of how deregulation of the RB–E2F pathway leads to cancer.
RB1 is mutated in essentially all patients with hereditary or sporadic retinoblastoma. Amplification of 6p22, which contains the E2F3 locus, has been reported in more than 50% of cases of retinoblastoma and leads to increased expression of E2F3 mRNA and protein105,106,133. This trend has also been observed in other types of cancers, including bladder cancer. Loss of heterozygosity (LOH) at the RB1 locus is found in 29% of patients with bladder cancer and is associated with 6p22 amplification, E2F3 over expression and advanced tumour stage100–104. In human SCLC, RB1 is mutated in more than 90% of cases110,134, and most SCLCs also have E2F1 or E2F3 overexpression112–113. A similar but less pronounced relationship between RB1 and E2F1 was reported for non-small-cell lung cancer111,113,135. Finally, the Tlsty laboratory recently unmasked a strict inverse correlation between RB1 and E2F3 expression in the basal-like subtype of human breast cancer118. In their study, global gene expression profiling revealed low expression of either CCND1 or RB1, consistent with their mutually exclusive association in many cancers, but high levels of E2F3. This overlap between RB1 inactivation and E2F3 hyperactivity in retinoblastoma and bladder, lung and breast cancer is not readily explained by current paradigms of RB–E2F action.
Why do RB1 inactivation and E2F amplification and/or overexpression coexist in tumour cells? In several knockout-mouse models the inactivation of Rb1 resulted in unrestrained E2F3 activity and cell proliferation, and the concomitant loss of Rb1 and E2f3 suppressed most of this ectopic cell proliferation43,48. This demonstrates that the levels of E2F3 activity achieved by RB inactivation are sufficient to promote uninhibited cell proliferation. We propose, therefore, that in cancers such as retinoblastoma and bladder cancer the deregulation of E2F3 activity (as might occur through the loss of RB function) and the further increase in E2F3 expression (as might occur through the amplification of 6p22) may represent separate molecular events with different physiological outcomes. In this context, RB1 mutation would provide a proliferative advantage to tumour cells, whereas E2F3 hyperactivity would provide functions that extend beyond the traditional control of the cell cycle. A preliminary example of such a function is evident in mice in which the targeted ablation of E2f3 from tumour cells overexpressing the oncogene Erbb2 is associated with an infiltration of innate and adaptive immune cells in the tumour microenvironment, leading to tumour regression (G.L., unpublished observations). Several developmental studies in mice, flies and worms also support a role for E2Fs that is independent of cell cycle control. For instance, loss-of-function studies have identified roles for lin-35 (an RB1 orthologue) and efl-1 (an E2F orthologue) in epidermal growth factor-mediated cell fate determination in C. elegans70,71 and for mammalian E2Fs in angiogenesis136, adipogenesis31,137 and cell migration21. On the basis of these examples, it can be argued that the role of E2Fs in cell cycle control might be a recent evolutionary adaptation and that the role of E2Fs in cancer might also include more evolutionarily ancient functions.
The observations outlined above suggest that the tight control of E2F1, and E2F3 in particular, is crucial for tissue homeostasis and that disruption of their expression may have a causative relationship with cancer development. A thorough molecular analysis of mice that either lack or have misregulated E2F1 and E2F3 should begin to clarify the mechanisms underlying their link with cancer. Genetic and gene expression profiling studies of human tumour samples, together with the analysis of mice deficient for multiple E2Fs, have begun to reveal functions of E2Fs beyond the control of the cell cycle and apoptosis. The extent to which these poorly defined functions contribute to the cancer phenotype will need to be evaluated in the coming years, with the promise that they might represent events that could be targeted by therapeutic regimens.
Conclusions and future perspectives
The prevailing view is that E2F repressors and activators cooperate to orchestrate proper cell cycle progression and that disruption of this carefully coordinated network contributes to cancer. However, recent clinical and mouse studies have begun to challenge this view. We suggest that the simple paradigm of proliferation control by E2F repressors and activators does not correlate with the complexity of E2F function observed during development and tumorigenesis.
As factors that control the cell cycle, apoptosis, differentiation and stress responses, the E2F family of transcription factors has earned the status of master regulators of cell proliferation. We have learned that many signals can regulate the activity of E2F family members and that E2Fs can in turn regulate many different targets that affect a wide range of biological processes that are intertwined with the control of cell proliferation. Given that E2Fs have the capacity to determine whether cells proliferate or not, what more could be learnt about E2Fs other than the finer details of their work ethic in specific contexts and cell types? Quite a bit we think, and we could begin by asking whether E2Fs are necessary for the proliferation of all cell types in mammals and other organisms. If not, then identifying the crucial accomplices that function in concert with E2Fs, and how their activities are coordinated to commit cells to proliferation, would be important. It might be that E2F functions are not required in some cell types. Second, are the biological roles of E2Fs strictly dependent on their activation or repression functions and when are such activities relevant during the developmental life of a cell? We should further address why different levels of E2F-dependent activation and repression induce such complex outcomes in mammalian cells. The combined use of new genetic mouse models and molecular techniques such as ChIP–sequencing, gene expression arrays and proteomics could help us map and elucidate the mechanism of E2F action in intact tissues. From a pessimistic standpoint, the complexity of E2F activities might simply represent an imperfect evolutionary adaptation to perform the vital task of cell proliferation in multicellular organisms. Once these normal functions of E2Fs have been fully understood, we could then ask whether the classic cell proliferation functions long ascribed to E2Fs are relevant to the development of cancer. If so, for which cancers and at what point in their malignant evolution might they be especially crucial? If not, then what additional pro-oncogenic or anti-oncogenic roles might they have?
So, despite xs~3,500 scientific publications focusing on E2Fs since the first family member was discovered two decades ago, we know embarrassingly little about what E2Fs do in vivo or how they do it. We have certainly garnered volumes of data relating to how E2F activities can be controlled and which targets and biological processes E2Fs can in turn potentially regulate. But from this bulk of information, we have yet to identify a single physiological circumstance in which a defined signal elicits the E2F programme to exert its powers on a specific biological outcome. Perhaps by studying these factors in whole animals, we may finally have the opportunity to discover the extent to which E2Fs function in specific capacities or as versatile modulators poised at biological crossroads that can be commandeered to execute diverse processes in health and disease.
Supplementary Material
At a glance.
A long-standing paradigm has been that E2F activity is tightly regulated by the RB tumour suppressor and that the disruption of this regulation leads to unscheduled progression through the cell cycle.
Based on structure–function studies in vitro, the mammalian E2F family of transcription factors has been artificially subdivided into activators (E2F1–E2F3) and repressors (E2F4–E2F8).
E2F1–E2F3 activators are highly redundant during development.
Tumour models using RB–E2F compound-mutant mice and E2F-transgenic mice show dual roles for E2Fs in tumour promotion and suppression. These results suggest tissue-specific functions and argue against a uniform role for E2Fs in cancer.
Mice lacking E2F1, E2F2 or E2F3 survive to mid gestation without global defects in the cell cycle, suggesting that the activators are not essential for normal mammalian cell proliferation. We propose that under normal conditions E2Fs do not substantially contribute to the proliferative potential of a cell.
Deregulated expression or activity of most members of the E2F family has been detected in many human cancers. We propose that the requirement for certain E2F family members in proliferation under oncogenic conditions represents a recent evolutionary adaptation.
RB inactivation and E2F amplification coexist in cancer. RB inactivation leads to inappropriate cell cycle progression through the deregulation of E2F function. We propose that the additional increase in E2F activity caused by amplification has cell proliferation-independent functions in cancer.
Acknowledgement
This work was funded by National Institutes of Health grants to G.L. (R01CA85619, R01CA82259, R01HD04470 and P01CA097189).
Glossary
- Endocycle
A specialized type of cell cycle, consisting of alternating S and G phases, that is widely used by both plants and animals. For example, mammalian cells using the endocycle include trophoblast giant cells, hepatocytes and megakaryocytes. Cells achieve greater than 2N genomes by uncoupling DNA replication from mitosis.
- Chimera
An animal produced by mixing wild-type cells with mutant cells at the pre-implantation stages of embryonic development to overcome embryonic lethal mutations and facilitate the identification of the primary site of action or the function of a gene in later lineages.
Footnotes
DATABASES Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.
fcgi?db=gene
CDK8 ∣ E2F1 ∣ Erbb2 ∣ Hras
UniProtKB: http://www.uniprot.org
CDK1 ∣ CDK2 ∣ CDK4 ∣ CDK6 ∣ CEBPα ∣ E2F2 ∣ E2F4 ∣ E2F5 ∣ E2F6 ∣ E2F7 ∣ E2F8 ∣ MYC ∣ p107 ∣ p130 ∣ RB
FURTHER INFORMATION Gustavo Leone’s homepage: http://www.cancergenetics.med.ohio-state.edu/2848.cfm
SUPPLEMENTARY INFORMATION See online article: S1 (box)
References
- 1.Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nature Rev. Mol. Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 3.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 4.Nevins JR. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 5.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nature Rev. Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 2006;6:739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 7.Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 2007;19:649–657. doi: 10.1016/j.ceb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 9.Ren B, et al. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cam H, et al. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell. 2004;16:399–411. doi: 10.1016/j.molcel.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 11.Dimova DK, Stevaux O, Frolov MV, Dyson NJ. Cell cycle-dependent and cell cycle-independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev. 2003;17:2308–2320. doi: 10.1101/gad.1116703. By coupling RNA interference with gene expression arrays, this study shows that E2F family members can control the transcription of genes other than those associated with the cell cycle.
- 12.Wells J, Graveel CR, Bartley SM, Madore SJ, Farnham PJ. The identification of E2F1-specific target genes. Proc. Natl Acad. Sci. USA. 2002;99:3890–3895. doi: 10.1073/pnas.062047499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 14.Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol. Cell. Biol. 2000;20:5797–5807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin VX, Rabinovich A, Squazzo SL, Green R, Farnham PJ. A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data — a case study using E2F1. Genome Res. 2006;16:1585–1595. doi: 10.1101/gr.5520206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zalmas LP, et al. DNA-damage response control of E2F7 and E2F8. EMBO Rep. 2008;9:252–259. doi: 10.1038/sj.embor.7401158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev. Cell. 2008;14:62–75. doi: 10.1016/j.devcel.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buttitta LA, Edgar BA. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr. Opin. Cell Biol. 2007;19:697–704. doi: 10.1016/j.ceb.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeGregori J. E2F and cell survival: context really is key. Dev. Cell. 2005;9:442–444. doi: 10.1016/j.devcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClellan KA, Slack RS. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle. 2007;6:2917–2927. doi: 10.4161/cc.6.23.4997. [DOI] [PubMed] [Google Scholar]
- 22.Kovesdi I, Reichel R, Nevins JR. Identification of a cellular transcription factor involved in E1A transactivation. Cell. 1986;45:219–228. doi: 10.1016/0092-8674(86)90386-7. [DOI] [PubMed] [Google Scholar]
- 23.Yee AS, Reichel R, Kovesdi I, Nevins JR. Promoter interaction of the E1A-inducible factor E2F and its potential role in the formation of a multi-component complex. EMBO J. 1987;6:2061–2068. doi: 10.1002/j.1460-2075.1987.tb02471.x. References 22 and 23 describe the identification of E2F (from the nuclear extracts of adenovirus-infected cells) as the cellular factor responsible for binding and activating the adenovirus early E2 promoter, which drives viral DNA replication.
- 24.Helin K, et al. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- 25.Kaelin WG, et al. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- 26.Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 27.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 28.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 29.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7:892–898. doi: 10.4161/cc.7.7.5637. [DOI] [PubMed] [Google Scholar]
- 31.Tsai S-Y, et al. Mouse development with a single E2F activator. Nature. 2008;454:1137–1141. doi: 10.1038/nature07066. This study shows in vivo that the activators E2F1, E2F3a and E2F3b have interchangeable roles in the control of mammalian development and provides the first genetic and molecular evidence of their functional plasticity.
- 32.Danielian PS, et al. E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene. 2008;27:6561–6570. doi: 10.1038/onc.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaubatz S, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 34.Duronio RJ, O’Farrell PH, Xie JE, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- 35.Frolov MV, et al. Functional antagonism between E2F family members. Genes Dev. 2001;15:2146–2160. doi: 10.1101/gad.903901. References 34 and 35 are classic studies in D. melanogaster that have contributed to the understanding of fundamental principles of E2F-mediated cell cycle control.
- 36.Ambrus A, Nicolay B, Rasheva V, Suckling R, Frolov M. dE2F2-independent rescue of proliferation in cells lacking an activator dE2F1. Mol. Cell. Biol. 2007;27:8561–8570. doi: 10.1128/MCB.01068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lammens T, Li J, Leone G, De Veylder L. Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 2009;3:111–118. doi: 10.1016/j.tcb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lammens T, et al. Atypical E2F activity restrains APC/CCCS52A2 function obligatory for endocycle onset. Proc. Natl Acad. Sci. USA. 2008;105:14721–14726. doi: 10.1073/pnas.0806510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramirez-Parra E, et al. Role of an atypical E2F transcription factor in the control of Arabidopsis cell growth and differentiation. Plant Cell. 2004;16:2350–2363. doi: 10.1105/tpc.104.023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen J, et al. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005;33:5458–5470. doi: 10.1093/nar/gki855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003;22:6289–6298. doi: 10.1093/emboj/cdg613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai KY, et al. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- 43.Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001;15:386–391. doi: 10.1101/gad.858801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wenzel PL, et al. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007;21:85–97. doi: 10.1101/gad.1485307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee EY, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 46.Jacks T, et al. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 47.Clarke AR, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 48.Chong J-L, et al. E2f3a and E2f3b contribute to the control of cell proliferation and mouse development. Mol. Cell. Biol. 2009;2:414–424. doi: 10.1128/MCB.01161-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev. 2004;12:1413–1422. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miki J, Fujimura Y, Koseki H, Kamijo T. Polycomb complexes regulate cellular senescence by repression of ARF in cooperation with E2F3. Genes Cells. 2007;12:1371–1382. doi: 10.1111/j.1365-2443.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- 51.Leone G, et al. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol. Cell. Biol. 2000;10:3626–3632. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu X, et al. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young AP, Nagarajan R, Longmore GD. Mechanisms of transcriptional regulation by Rb-E2F segregate by biological pathway. Oncogene. 2003;22:7209–7217. doi: 10.1038/sj.onc.1206804. [DOI] [PubMed] [Google Scholar]
- 55.Lim CA, et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 56.Leung JY, Ehmann GL, Giangrande PH, Nevins JR. A role for Myc in facilitating transcription activation by E2F1. Oncogene. 2008;27:4172–4179. doi: 10.1038/onc.2008.55. [DOI] [PubMed] [Google Scholar]
- 57.Timchenko NA, Wilde M, Darlington GJ. C/EBPα regulates formation of S-phase-specific E2F–p107 complexes in livers of newborn mice. Mol. Cell. Biol. 1999;19:2936–2945. doi: 10.1128/mcb.19.4.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iakova P, Awad SS, Timchenko NA. Aging reduces proliferative capacities of liver by switching pathways of C/EBPα growth arrest. Cell. 2003;113:495–506. doi: 10.1016/s0092-8674(03)00318-0. [DOI] [PubMed] [Google Scholar]
- 59.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 60.Yu Q, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 61.Reddy HK, et al. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–10178. doi: 10.1158/0008-5472.CAN-05-2639. [DOI] [PubMed] [Google Scholar]
- 62.Williams BO, et al. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature Genet. 1994;7:480–484. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- 63.Hu N, et al. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–1027. [PubMed] [Google Scholar]
- 64.Yamasaki L, et al. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1+/− mice. Nature Genet. 1998;18:360–364. doi: 10.1038/ng0498-360. [DOI] [PubMed] [Google Scholar]
- 65.Opavsky R, et al. CpG island methylation in a mouse model of lymphoma is driven by the genetic configuration of tumor cells. PLoS Genet. 2007;3:1757–1769. doi: 10.1371/journal.pgen.0030167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol. Cell. Biol. 2003;23:6542–6552. doi: 10.1128/MCB.23.18.6542-6552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maandag EC, et al. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams BO, et al. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parisi T, et al. Selective requirements for E2f3 in the development and tumorigenicity of Rb-deficient chimeric tissues. Mol. Cell. Biol. 2007;6:2283–2293. doi: 10.1128/MCB.01854-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ceol CJ, Horvits HR. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell. 2001;7:461–473. doi: 10.1016/s1097-2765(01)00194-0. [DOI] [PubMed] [Google Scholar]
- 71.Myers TR, Greenwald I. lin-35 Rb acts in the major hypodermis to oppose ras-mediated vulval induction in C. elegans. Dev. Cell. 2005;8:117–123. doi: 10.1016/j.devcel.2004.11.015. References 70 and 71 represent just two in a series of elegant studies demonstrating a role for RB–E2F in the regulation of cell fate specification in C. elegans.
- 72.de Bruin A, et al. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc. Natl Acad. Sci. USA. 2003;100:6546–6551. doi: 10.1073/pnas.1031853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu L, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 74.Opavsky R, et al. Specific tumor suppressor function for E2F2 in Myc-induced T cell lymphomagenesis. Proc. Natl Acad. Sci. USA. 2007;104:15400–15405. doi: 10.1073/pnas.0706307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saenz-Robles MT, et al. Intestinal hyperplasia induced by simian virus 40 large tumor antigen requires E2F2. J. Virol. 2007;81:13191–13199. doi: 10.1128/JVI.01658-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee EY, et al. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell. 2002;2:463–472. doi: 10.1016/s1535-6108(02)00207-6. [DOI] [PubMed] [Google Scholar]
- 77.Parisi T, Bronson RT, Lees JA. Inhibition of pituitary tumors in Rb mutant chimeras through E2f4 loss reveals a key suppressive role for the pRB/E2F pathway in urothelium and ganglionic carcinogenesis. Oncogene. 2009;28:500–508. doi: 10.1038/onc.2008.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Infante A, et al. E2F2 represses cell cycle regulators to maintain quiescence. Cell Cycle. 2008;7:3915–3927. doi: 10.4161/cc.7.24.7379. [DOI] [PubMed] [Google Scholar]
- 79.Pagano M, Jackson PK. Wagging the dogma; tissue-specific cell cycle control in the mouse embryo. Cell. 2004;118:535–538. doi: 10.1016/j.cell.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 80.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 81.Santamaria D, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. This landmark study shows that interphase CDKs — CDK2, CDK4 and CDK6 — are not essential for cell division in the mammal as their combined genetic ablation does not alter the cell cycle of most cell types.
- 82.Satyanarayana A, et al. Genetic substitution of Cdk1 by Cdk2 leads to embryonic lethality and loss of meiotic function of Cdk2. Development. 2008;135:3389–3400. doi: 10.1242/dev.024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Firestein R, et al. CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu L, et al. The E2F1–3 transcription factors are essential for cellular proliferation. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- 85.Yamasaki L, et al. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- 86.Field SJ, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. Contrary to expectations, references 85 and 86 provide in vivo evidence of a role for E2F1 in the suppression of cell proliferation and tumorigenesis.
- 87.Humbert PO, et al. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- 88.Murga M, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity. 2001;15:959–970. doi: 10.1016/s1074-7613(01)00254-0. [DOI] [PubMed] [Google Scholar]
- 89.Humbert PO, et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell. 2000;6:281–291. doi: 10.1016/s1097-2765(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 90.Rempel RE, et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol. Cell. 2000;6:293–306. doi: 10.1016/s1097-2765(00)00030-7. [DOI] [PubMed] [Google Scholar]
- 91.Lindeman GJ, et al. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998;12:1092–1098. doi: 10.1101/gad.12.8.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pohlers M, et al. A role for E2F6 in the restriction of male-germ-cell-specific gene expression. Curr. Biol. 2005;15:1051–1057. doi: 10.1016/j.cub.2005.04.060. [DOI] [PubMed] [Google Scholar]
- 93.Timmers C, et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol. Cell. Biol. 2007;27:65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma N, et al. Control of the p53-p21CIP1 axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and cellular transformation. J. Biol. Chem. 2006;281:36124–36131. doi: 10.1074/jbc.M604152200. [DOI] [PubMed] [Google Scholar]
- 95.Nicolay BN, Frolov MV. Context-dependent requirement for dE2F during oncogenic proliferation. PLoS Genet. 2008;4:e1000205. doi: 10.1371/journal.pgen.1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Midorikawa Y, Makuuchi M, Tang W, Aburatani H. Microarray-based analysis for hepatocellular carcinoma: from gene expression profiling to new challenges. World J. Gastroenterol. 2007;13:1487–1492. doi: 10.3748/wjg.v13.i10.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Midorikawa Y, et al. Distinct chromosomal bias of gene expression signatures in the progression of hepatocellular carcinoma. Cancer Res. 2004;64:7263–7270. doi: 10.1158/0008-5472.CAN-04-1275. [DOI] [PubMed] [Google Scholar]
- 98.Zondervan PE, et al. Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: analysis of 26 carcinomas and 12 concurrent dysplasias. J. Pathol. 2000;192:207–215. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH690>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 99.Wang G, et al. Genetic aberration in primary hepatocellular carcinoma: correlation between p53 gene mutation and loss-of-heterozygosity on chromosome 16q21-q23 and 9p21-p23. Cell Res. 2000;10:311–323. doi: 10.1038/sj.cr.7290058. [DOI] [PubMed] [Google Scholar]
- 100.Hovey RM, et al. Genetic alterations in primary bladder cancers and their metastases. Cancer Res. 1998;58:3555–3560. [PubMed] [Google Scholar]
- 101.Yu DS, Hsieh DS, Chang SY. Detection of chromosomal alterations in bladder cancer by comparative genomic hybridization. BJU Int. 2001;87:889–893. doi: 10.1046/j.1464-410x.2001.02175.x. [DOI] [PubMed] [Google Scholar]
- 102.Veltman JA, et al. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–2880. [PubMed] [Google Scholar]
- 103.Feber A, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004;23:1627–1630. doi: 10.1038/sj.onc.1207274. [DOI] [PubMed] [Google Scholar]
- 104.Hurst CD, Tomlinson DC, Williams SV, Platt FM, Knowles MA. Inactivation of the Rb pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. Oncogene. 2008;27:2716–2727. doi: 10.1038/sj.onc.1210934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grasemann C, et al. Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene. 2005;24:6441–6449. doi: 10.1038/sj.onc.1208792. [DOI] [PubMed] [Google Scholar]
- 106.Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosom. Cancer. 2006;45:72–82. doi: 10.1002/gcc.20263. References 103–106 report the curious observation that amplification and/or overexpression of E2F3 occurs in human tumours with RB inactivation.
- 107.Szymanska J, et al. Gains and losses of DNA sequences in liposarcomas evaluated by comparative genomic hybridization. Genes Chromosom. Cancer. 1996;15:89–94. doi: 10.1002/(SICI)1098-2264(199602)15:2<89::AID-GCC2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 108.Szymanska J, et al. Overrepresentation of 1q21–23 and 12q13–21 in lipoma-like liposarcomas but not in benign lipomas: a comparative genomic hybridization study. Cancer Genet. Cytogenet. 1997;99:14–18. doi: 10.1016/s0165-4608(96)00436-0. [DOI] [PubMed] [Google Scholar]
- 109.Alonso MM, et al. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J. Natl Cancer Inst. 2005;97:1589–1600. doi: 10.1093/jnci/dji340. [DOI] [PubMed] [Google Scholar]
- 110.Wikenheiser-Brokamp KA. Retinoblastoma regulatory pathway in lung cancer. Curr. Mol. Med. 2006;6:783–793. doi: 10.2174/1566524010606070783. [DOI] [PubMed] [Google Scholar]
- 111.Imai MA, Oda Y, Oda M, Nakanishi I, Kawahara E. Overexpression of E2F1 associated with LOH at RB locus and hyperphosphorylation of RB in non-small cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2004;130:320–326. doi: 10.1007/s00432-003-0538-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eymin B, Gazzeri S, Brambilla C, Brambilla E. Distinct pattern of E2F1 expression in human lung tumours: E2F1 is upregulated in small cell lung carcinoma. Oncogene. 2001;20:1678–1687. doi: 10.1038/sj.onc.1204242. [DOI] [PubMed] [Google Scholar]
- 113.Cooper CS, et al. Nuclear overexpression of the E2F3 transcription factor in human lung cancer. Lung Cancer. 2006;54:155–162. doi: 10.1016/j.lungcan.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 114.Reimer D, et al. Expression of the E2F family of transcription factors and its clinical relevance in ovarian cancer. Ann. NY Acad. Sci. 2006;1091:270–281. doi: 10.1196/annals.1378.073. [DOI] [PubMed] [Google Scholar]
- 115.Reimer D, et al. Clinical relevance of E2F family members in ovarian cancer — an evaluation in a training set of 77 patients. Clin. Cancer Res. 2007;13:144–151. doi: 10.1158/1078-0432.CCR-06-0780. [DOI] [PubMed] [Google Scholar]
- 116.Han S, et al. E2F1 expression is related with the poor survival of lymph node-positive breast cancer patients treated with fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res. Treat. 2003;82:11–16. doi: 10.1023/B:BREA.0000003843.53726.63. [DOI] [PubMed] [Google Scholar]
- 117.Rakha EA, Pinder SE, Paish EC, Robertson JF, Ellis IO. Expression of E2F-4 in invasive breast carcinomas is associated with poor prognosis. J. Pathol. 2004;203:754–761. doi: 10.1002/path.1573. [DOI] [PubMed] [Google Scholar]
- 118.Gauthier ML, et al. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12:479–491. doi: 10.1016/j.ccr.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee J, et al. Impact of E2F-1 expression on clinical outcome of gastric adenocarcinoma patients with adjuvant chemoradiation therapy. Clin. Cancer Res. 2008;14:82–88. doi: 10.1158/1078-0432.CCR-07-0612. [DOI] [PubMed] [Google Scholar]
- 120.Bramis J, et al. E2F-1 transcription factor immunoexpression is inversely associated with tumor growth in colon adenocarcinomas. Anticancer Res. 2004;24:3041–3047. [PubMed] [Google Scholar]
- 121.Haller F, et al. Prognostic role of E2F1 and members of the CDKN2A network in gastrointestinal stromal tumors. Clin. Cancer Res. 2005;11:6589–6597. doi: 10.1158/1078-0432.CCR-05-0329. [DOI] [PubMed] [Google Scholar]
- 122.Bagchi A, Mills AA. The quest for the 1p36 tumor suppressor. Cancer Res. 2008;68:2551–2556. doi: 10.1158/0008-5472.CAN-07-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Janoueix-Lerosey I, et al. Gene expression profiling of 1p35–36 genes in neuroblastoma. Oncogene. 2004;23:5912–5922. doi: 10.1038/sj.onc.1207784. [DOI] [PubMed] [Google Scholar]
- 124.Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006;97:1–7. doi: 10.1111/j.1349-7006.2005.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rigaud G, et al. Allelotype of pancreatic acinar cell carcinoma. Int. J. Cancer. 2000;88:772–777. doi: 10.1002/1097-0215(20001201)88:5<772::aid-ijc14>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 126.Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. 2007;26:2048–2057. doi: 10.1038/sj.onc.1210001. [DOI] [PubMed] [Google Scholar]
- 127.Bindra RS, Glazer PM. Basal repression of BRCA1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biol. Ther. 2006;5:1400–1407. doi: 10.4161/cbt.5.10.3454. [DOI] [PubMed] [Google Scholar]
- 128.Polanowska J, et al. Human E2F5 gene is oncogenic in primary rodent cells and is amplified in human breast tumors. Genes Chromosom. Cancer. 2000;28:126–130. [PubMed] [Google Scholar]
- 129.Umemura S, et al. Overexpression of E2F-5 correlates with a pathological basal phenotype and a worse clinical outcome. Br. J. Cancer. 2009;100:764–771. doi: 10.1038/sj.bjc.6604900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reimer D, et al. Expression of the E2F family of transcription factors and its clinical relevance in ovarian cancer. Ann. NY Acad. Sci. 2006;1091:270–281. doi: 10.1196/annals.1378.073. [DOI] [PubMed] [Google Scholar]
- 131.Reimer D, et al. Clinical relevance of E2F family members in ovarian cancer – an evaluation in a training set of 77 patients. Clin. Cancer Res. 2007;13:144–151. doi: 10.1158/1078-0432.CCR-06-0780. [DOI] [PubMed] [Google Scholar]
- 132.Endo-Munoz L, et al. E2F7 can regulate proliferation, differentiation, and apoptotic responses in human keratinocytes: implications for cutaneous squamous cell carcinoma formation. Cancer Res. 2009;69:1800–1808. doi: 10.1158/0008-5472.CAN-08-2725. [DOI] [PubMed] [Google Scholar]
- 133.Chen D, Gallie BL, Squire JA. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet. Cytogenet. 2001;129:57–63. doi: 10.1016/s0165-4608(01)00427-7. [DOI] [PubMed] [Google Scholar]
- 134.Lindblad-Toh K, et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nature Biotechnol. 2000;18:1001–1005. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- 135.Karakaidos P, et al. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability — evidence of E2F-1 transcriptional control over hCdt1. Am. J. Pathol. 2004;165:1351–1365. doi: 10.1016/S0002-9440(10)63393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qin G, et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl Acad. Sci. USA. 2006;103:11015–11020. doi: 10.1073/pnas.0509533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fajas L, et al. E2Fs regulate adipocyte differentiation. Dev. Cell. 2002;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 138.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 139.Muller H, et al. Induction of S-phase entry of E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol. 1997;17:5508–5520. doi: 10.1128/mcb.17.9.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nature Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 141.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nature Rev. Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 142.Harbour JW, et al. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 143.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature Rev. Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 144.Magae J, et al. Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci. 1996;109:1717–1726. doi: 10.1242/jcs.109.7.1717. [DOI] [PubMed] [Google Scholar]
- 145.Verona R, et al. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol. Cell. Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol. Cell. Biol. 2001;21:1384–1392. doi: 10.1128/MCB.21.4.1384-1392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cartwright P, et al. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- 148.Gaubatz S, Wood JG, Livingston DM. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc. Natl Acad. Sci. USA. 1998;95:9190–9195. doi: 10.1073/pnas.95.16.9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Trimarchi JM, Farichild B, Wen J, Lees JA. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc. Natl Acad. Sci. USA. 2001;98:1519–1524. doi: 10.1073/pnas.041597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leone G, et al. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 1998;12:2120–2130. doi: 10.1101/gad.12.14.2120. This paper and reference 87 show that E2F3 has a central role in the control of cell proliferation in vitro by mediating activation of most of the classic E2F target genes in response to mitogenic stimulation.
- 151.Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004;23:4615–4626. doi: 10.1038/sj.emboj.7600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol. Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 153.Ogawa H, Ishiguro K, Gaubatz S, Livingston D, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 154.Rayman JB, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brehm A, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 156.Taubert S, et al. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol. Cell. Biol. 2004;24:4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nicolas E, Roumillac C, Trouche D. Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol. Cell. Biol. 2003;23:1614–1622. doi: 10.1128/MCB.23.5.1614-1622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lang SE, McMahon SB, Cole MD, Hearing P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J. Biol. Chem. 2001;276:32627–32634. doi: 10.1074/jbc.M102067200. [DOI] [PubMed] [Google Scholar]
- 159.Li FX, et al. The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc. Natl Acad. Sci. USA. 2003;100:12935–12940. doi: 10.1073/pnas.2231861100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Iglesias A, et al. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J. Clin. Invest. 2004;113:1398–1407. doi: 10.1172/JCI18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cloud JE, et al. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol. Cell. Biol. 2002;22:2663–2672. doi: 10.1128/MCB.22.8.2663-2672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kinross KM, Clark AJ, Iazzolino RM, Humbert PO. E2f4 regulates fetal erythropoiesis through the promotion of cellular proliferation. Blood. 2006;108:886–895. doi: 10.1182/blood-2005-09-008656. [DOI] [PubMed] [Google Scholar]
- 163.Olson MV, et al. Transgenic E2F1 expression in the mouse brain induces a human-like bimodal pattern of tumors. Cancer Res. 2007;67:4005–4009. doi: 10.1158/0008-5472.CAN-06-2973. [DOI] [PubMed] [Google Scholar]
- 164.Conner EA, et al. Dual functions of E2F-1 in transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19:5054–5062. doi: 10.1038/sj.onc.1203885. [DOI] [PubMed] [Google Scholar]
- 165.Chen Q, Hung FC, Fromm L, Overbeek PA. Induction of cell cycle entry and cell death in postmitotic lens fiber cells by overexpression of E2F1 or E2F2. Invest. Ophthalmol. Vis. Sci. 2000;41:4223–4231. [PubMed] [Google Scholar]
- 166.Guy CT, Zhou W, Kaufman S, Robinson MO. E2F-1 blocks terminal differentiation and causes proliferation in transgenic megakaryocytes. Mol. Cell. Biol. 1996;16:685–693. doi: 10.1128/mcb.16.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Holmberg C, Helin K, Sehested M. Karlstrom, O. E2F-1-induced p53-independent apoptosis in transgenic mice. Oncogene. 1998;17:143–155. doi: 10.1038/sj.onc.1201915. [DOI] [PubMed] [Google Scholar]
- 168.Pierce AM, Fisher SM, Conti CJ, Johnson DG. Deregulated expression of E2F1 induces hyperplasia and cooperates with ras in skin tumor development. Oncogene. 1998;16:1267–1276. doi: 10.1038/sj.onc.1201666. [DOI] [PubMed] [Google Scholar]
- 169.Pierce AM, et al. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol. Cell. Biol. 1999;19:6408–6414. doi: 10.1128/mcb.19.9.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Scheijen B, Bronk M, van der Meer T, De Jong D, Bernards R. High incidence of thymic epithelial tumors in E2F2 transgenic mice. J. Biol. Chem. 2004;279:10476–10483. doi: 10.1074/jbc.M313682200. [DOI] [PubMed] [Google Scholar]
- 171.Paulson QX, McArthur MJ, Johnson DG. E2F3a stimulates proliferation, p53-independent apoptosis and carcinogenesis in a transgenic mouse model. Cell Cycle. 2006;5:184–190. doi: 10.4161/cc.5.2.2307. [DOI] [PubMed] [Google Scholar]
- 172.Chen Q, Liang D, Yang T, Leone G, Overbeek PA. Distinct capacities of individual E2Fs to induce cell cycle re-entry in postmitotic lens fiber cells of transgenic mice. Dev. Neurosci. 2004;26:435–445. doi: 10.1159/000082285. [DOI] [PubMed] [Google Scholar]
- 173.Wang D, Russell JL, Johnson DG. E2F4 and E2F1 have similar proliferative properties but different apoptotic and oncogenic properties in vivo. Mol. Cell. Biol. 2000;20:3417–3424. doi: 10.1128/mcb.20.10.3417-3424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen Q, Liang D, Overbeek PA. Overexpression of E2F5/p130, not E2F5 alone, can inhibit E2F-induced cell cycle entry in transgenic mice. Mol. Vis. 2008;25:602–614. [PMC free article] [PubMed] [Google Scholar]
- 175.Fujita Y, et al. Chromosome arm 20q gains and other genomic alterations in esophageal squamous cell carcinoma, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Hepatogastroenterology. 2003;50:1857–1863. [PubMed] [Google Scholar]
- 176.Tonon G, et al. High-resolution genomic profiles of human lung cancer. Proc. Natl Acad. Sci. USA. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wilting SM, et al. Integrated genomic and transcriptional profiling identifies chromosomal loci with altered gene expression in cervical cancer. Genes Chromosom. Cancer. 2008;47:890–905. doi: 10.1002/gcc.20590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wilting SM, et al. Increased gene copy numbers at chromosome 20q are frequent in both squamous cell carcinomas and adenocarcinomas of the cervix. J. Pathol. 2006;209:220–230. doi: 10.1002/path.1966. [DOI] [PubMed] [Google Scholar]
- 179.Huang CL, et al. E2F1 overexpression correlates with thymidylate synthase and survivin gene expressions and tumor proliferation in non small-cell lung cancer. Clin. Cancer Res. 2007;13:6938–6946. doi: 10.1158/1078-0432.CCR-07-1539. [DOI] [PubMed] [Google Scholar]
- 180.Gorgoulis VG, et al. Transcription factor E2F-1 acts as a growth-promoting factor and is associated with adverse prognosis in non-small cell lung carcinomas. J. Pathol. 2002;198:142–156. doi: 10.1002/path.1121. [DOI] [PubMed] [Google Scholar]
- 181.Mega S, et al. Cyclin D1, E2F1 expression levels are associated with characteristics and prognosis of esophageal squamous cell carcinoma. Dis. Esophagus. 2005;18:109–113. doi: 10.1111/j.1442-2050.2005.00463.x. [DOI] [PubMed] [Google Scholar]
- 182.Ebihara Y, et al. Over-expression of E2F-1 in esophageal squamous cell carcinoma correlates with tumor progression. Dis. Esophagus. 2004;17:150–154. doi: 10.1111/j.1442-2050.2004.00393.x. [DOI] [PubMed] [Google Scholar]
- 183.Xu XR, et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc. Natl Acad. Sci. USA. 2001;98:15089–15094. doi: 10.1073/pnas.241522398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yamazaki K, et al. Expression of transcription factor E2F-1 in pancreatic ductal carcinoma: an immunohistochemical study. Pathol. Res. Pract. 2003;199:23–28. doi: 10.1078/0344-0338-00348. [DOI] [PubMed] [Google Scholar]
- 186.Haller F, et al. Prognostic role of E2F1 and members of the CDKN2A network in gastrointestinal stromal tumors. Clin. Cancer Res. 2005;11:6589–6597. doi: 10.1158/1078-0432.CCR-05-0329. [DOI] [PubMed] [Google Scholar]
- 187.Han S, et al. E2F1 expression is related with the poor survival of lymph node-positive breast cancer patients treated with fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res. Treat. 2003;82:11–16. doi: 10.1023/B:BREA.0000003843.53726.63. [DOI] [PubMed] [Google Scholar]
- 188.De Meyer T, et al. E2Fs mediate a fundamental cell-cycle deregulation in high-grade serous ovarian carcinomas. J. Pathol. 2009;217:14–20. doi: 10.1002/path.2452. [DOI] [PubMed] [Google Scholar]
- 189.Lee J, et al. Impact of E2F-1 expression on clinical outcome of gastric adenocarcinoma patients with adjuvant chemoradiation therapy. Clin. Cancer Res. 2008;14:82–88. doi: 10.1158/1078-0432.CCR-07-0612. [DOI] [PubMed] [Google Scholar]
- 190.Kwong RA, et al. Overexpression of E2F-1 is associated with increased disease-free survival in squamous cell carcinoma of the anterior tongue. Clin. Cancer Res. 2003;9:3705–3711. [PubMed] [Google Scholar]
- 191.Bramis J, et al. E2F-1 transcription factor immunoexpression is inversely associated with tumor growth in colon adenocarcinomas. Anticancer Res. 2004;24:3041–3047. [PubMed] [Google Scholar]
- 192.Rabbani F, et al. Prognostic significance of transcription factor E2F-1 in bladder cancer: genotypic and phenotypic characterization. J. Natl Cancer Inst. 1999;91:874–881. doi: 10.1093/jnci/91.10.874. [DOI] [PubMed] [Google Scholar]
- 193.Ried T, et al. Mapping of multiple DNA gains and losses in primary small cell lung carcinomas by comparative genomic hybridization. Cancer Res. 1994;54:1801–1806. [PubMed] [Google Scholar]
- 194.Weber-Hall S, et al. Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res. 1996;56:3220–3224. [PubMed] [Google Scholar]
- 195.Tarkkanen M, et al. DNA sequence copy number increase at 8q: a potential new prognostic marker in high-grade osteosarcoma. Intl J. Cancer. 1999;84:114–121. doi: 10.1002/(sici)1097-0215(19990420)84:2<114::aid-ijc4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 196.Janoueix-Lerosey I, et al. Gene expression profiling of 1p35–36 genes in neuroblastoma. Oncogene. 2004;23:5912–5922. doi: 10.1038/sj.onc.1207784. [DOI] [PubMed] [Google Scholar]
- 197.Aarts M, et al. Microarray-based CGH of sporadic and syndrome-related pheochromocytomas using a 0.1–02 Mb bacterial artificial chromosome array spanning chromosome arm 1p. Genes Chromosom. Cancer. 2006;45:83–93. doi: 10.1002/gcc.20268. [DOI] [PubMed] [Google Scholar]
- 198.Bieche I, Khodja A, Lidereau R. Deletion mapping of chromosomal region 1p32-pter in primary breast cancer. Genes Chromosom. Cancer. 1999;24:255–263. doi: 10.1002/(sici)1098-2264(199903)24:3<255::aid-gcc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 199.Speicher MR, et al. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54:3817–3823. [PubMed] [Google Scholar]
- 200.Kallioniemi A, et al. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc. Natl Acad. Sci. USA. 1994;91:2156–2160. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nishizaki T, et al. Genetic alterations in primary breast cancers and their metastases: direct comparison using modified comparative genomic hybridization. Genes Chromosom. Cancer. 1997;19:267–272. doi: 10.1002/(sici)1098-2264(199708)19:4<267::aid-gcc9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 202.Foster CS, et al. Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene. 2004;23:5871–5879. doi: 10.1038/sj.onc.1207800. [DOI] [PubMed] [Google Scholar]
- 203.Sakai K, Nagahara H, Abe K, Obata H. Loss of heterozygosity on chromosome 16 in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1992;7:288–292. doi: 10.1111/j.1440-1746.1992.tb00982.x. [DOI] [PubMed] [Google Scholar]
- 204.Cleton-Jansen AM, et al. Loss of heterozygosity mapping at chromosome arm 16q in 712 breast tumors reveals factors that influence delineation of candidate regions. Cancer Res. 2001;61:1171–1177. [PubMed] [Google Scholar]
- 205.Rakha EA, Armour JA, Pinder SE, Paish CE, Ellis IO. High-resolution analysis of 16q22.1 in breast carcinoma using DNA amplifiable probes (multiplex amplifiable probe hybridization technique) and immunohistochemistry. Int. J. Cancer. 2005;114:720–729. doi: 10.1002/ijc.20738. [DOI] [PubMed] [Google Scholar]
- 206.Mady HH, Hasso S, Melhem MF. Expression of E2F-4 gene in colorectal adenocarcinoma and corresponding covering mucosa: an immunohistochemistry, image analysis, and immunoblot study. Appl. Immunohistochem. Mol. Morphol. 2002;10:225–230. doi: 10.1097/00129039-200209000-00007. [DOI] [PubMed] [Google Scholar]
- 207.Ogata S, et al. Microsatellite alterations and target gene mutations in the early stages of multiple gastric cancer. J. Pathol. 2001;194:334–340. doi: 10.1002/path.895. [DOI] [PubMed] [Google Scholar]
- 208.Schwemmle S, Pfeifer GP. Genomic structure and mutation screening of the E2F4 gene in human tumors. Int. J. Cancer. 2000;86:672–677. doi: 10.1002/(sici)1097-0215(20000601)86:5<672::aid-ijc11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 209.Souza RF, et al. Frequent mutation of the E2F-4 cell cycle gene in primary human gastrointestinal tumors. Cancer Res. 1997;57:2350–2353. [PubMed] [Google Scholar]
- 210.Lassmann S, et al. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatelliteunstable sporadic colorectal carcinomas. J. Mol. Med. 2007;85:293–304. doi: 10.1007/s00109-006-0126-5. [DOI] [PubMed] [Google Scholar]
- 211.Fix A, et al. Characterization of amplicons in neuroblastoma: High-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes Chromosom. Cancer. 2008;47:819–834. doi: 10.1002/gcc.20583. [DOI] [PubMed] [Google Scholar]
- 212.Toraman AD, Keser I, Luleci G, Tunali N, Gelen T. Comparative genomic hybridization in ganglioneuroblastomas. Cancer Genet. Cytogenet. 2002;132:36–40. doi: 10.1016/s0165-4608(01)00521-0. [DOI] [PubMed] [Google Scholar]
- 213.Yin XL, et al. Analysis of loss of heterozygosity on chromosomes 10q, 11, and 16 in medulloblastomas. J. Neurosurg. 2001;94:799–805. doi: 10.3171/jns.2001.94.5.0799. [DOI] [PubMed] [Google Scholar]
- 214.Chen D, et al. Division and apoptosis of E2f-deficient retinal progenitors. Nature. doi: 10.1038/nature08544. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chong J-L, et al. A switch of E2F1-3 function from activators in progenitor cells to repressors in differentiating cells. Nature. doi: 10.1038/nature08677. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.