


Abstract
Death domain–associated protein (Daxx) cooperates with X-linked α-thalassaemia retardation syndrome protein (ATRX), a putative member of the sucrose non-fermentable 2 family of ATP-dependent chromatin-remodelling proteins, acting as the core ATPase subunit in this complex, whereas Daxx is the targeting factor, leading to histone deacetylase recruitment, H3.3 deposition and transcriptional repression of cellular promoters. Despite recent findings on the fundamental importance of chromatin modification in host-cell gene regulation, it remains unclear whether adenovirus type 5 (Ad5) transcription is regulated by cellular chromatin remodelling to allow efficient virus gene expression. Here, we focus on the repressive role of the Daxx/ATRX complex during Ad5 replication, which depends on intact protein–protein interaction, as negative regulation could be relieved with a Daxx mutant that is unable to interact with ATRX. To ensure efficient viral replication, Ad5 E1B-55K protein inhibits Daxx and targets ATRX for proteasomal degradation in cooperation with early region 4 open reading frame protein 6 and cellular components of a cullin-dependent E3-ubiquitin ligase. Our studies illustrate the importance and diversity of viral factors antagonizing Daxx/ATRX-mediated repression of viral gene expression and shed new light on the modulation of cellular chromatin remodelling factors by Ad5. We show for the first time that cellular Daxx/ATRX chromatin remodelling complexes play essential roles in Ad gene expression and illustrate the importance of early viral proteins to counteract cellular chromatin remodelling.
INTRODUCTION
For >50 years, adenovirus biology has mainly focused on virus/host interactions and has established that manipulation of host cell homeostasis is required for efficient infection. Despite recent findings on the fundamental importance of chromatin status in host-cell gene regulation, it remains unclear whether adenovirus (Ad) transcription is subject to cellular chromatin remodelling. Recent reports demonstrated that Ad DNA is present in a tightly condensed state in the nucleocapsid, arguing for the requirement of altering chromatin modification early in infection to allow efficient virus gene expression.
We and others reported previously that the transcriptional repressor ‘death domain–associated protein’ (Daxx) is a principal component of ‘promyelocytic leukemia protein (PML) nuclear bodies’ (PML-NBs) and a negative regulator of Ad5 replication during productive infection (1,2). Daxx is mainly found in the nucleus, associated to PML-NBs, or at heterochromatin areas in a complex with ‘X-linked α-thalassaemia retardation syndrome protein’ (ATRX) (3–5). PML-NB association of Daxx was found to alleviate gene repression and activate apoptosis, whereas chromatin-bound Daxx acts in a transcriptionally repressive manner [summarized in Figure 10A; (6–8)]. Daxx association to either PML-NBs or chromatin depends on the status of the host cell and on the interaction of Daxx with other nuclear proteins (e.g. PML, ATRX), which can be regulated by post-translational modifications. Recently, Ishov et al. (9) observed that cell cycle dependent phosphorylation regulates the exit of Daxx from PML-NBs prior to assembly to ATRX and chromatin associated proteins like histone deacetylases, acetylated histone H4 and Dek at condensed chromatin regions (10).
Figure 10.

Model for Ad5-mediated restriction of cellular Daxx/ATRX chromatin remodelling complexes. (A) A schematic representation of known cellular Daxx localizations in human cells. Nuclear Daxx is associated with either PML-NBs or ATRX at heterochromatin foci. Daxx ability to repress transcription is inhibited by its localization to the PML-NB (82). Cytoplasmic Daxx has been reported to be involved in cell death (56,83). (B) Daxx is reported to bind ATRX via two paired amphipathic helices. ATRX acts as the core ATPase subunit in this complex, whereas Daxx is the targeting factor, leading to deacetylation of histone tails (HDAC) and histone variant H3.3 depositioning resulting in transcriptional repression of target promoters. As the Daxx protein has no DNA binding region, ATRX is thought to be the molecular bridge connecting chromatin with Daxx bound to Daxx-interacting sequence-specific transcription factors (yellow sphere). (C) Daxx/ATRX repressive complexes assemble on viral genomes. Efficient transcription of Ad5 gene products necessitates inhibiting Daxx repressive complexes and/or preventing their assembly. Binding of E1B-55K triggers Daxx degradation via a proteasome, whereas ATRX restriction additionally requires the E4orf6 protein. These processes displace the repressive Daxx/ATRX complex from the viral genome, relieving negative regulation of Ad transcription.
So far, the mechanisms of negative transcriptional regulation by Daxx remain only poorly understood, although Daxx association with repressive chromatin remodelling complexes has been proposed [summarized in Figure 10B; (10,11)]. Recently, it was shown that Daxx interacts with ATRX, a large protein of 280 kDa containing a putative ATPase/helicase domain, homologous to members of the ‘Switch/Sucrose non-fermentable’ (SWI/SNF) family of chromatin remodelling proteins (9,12,13). In addition, ATRX contains a ‘plant homeodomain’, similar to the DNA methyltransferase 3 family of proteins (14,15). Daxx interacts with ATRX through its NH2-terminal PAH1 domain (‘paired amphipathic alpha helix’) (9,12,13). Furthermore, Daxx recruits ATRX to the PML-NBs and interferes with ATRX-mediated transcriptional repression [summarized in Figure 10A; (9)]. These results suggest that Daxx regulates ATRX activity by altering its localization in the nucleus. The association between ATRX and PML-NBs also supports the observation that these nuclear bodies regulate diverse cellular processes by modulating transcription.
Others reported that Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres (16). Moreover, ATRX and H3.3 play essential roles in maintaining telomere chromatin (17,18). Further insight into H3.3-specific deposition pathways was gained by identifying the highly conserved N terminus of Daxx as the direct binding partner of H3.3. It was shown that recombinant Daxx assembles H3.3/H4 tetramers on DNA templates, and that deposition and remodelling of H3.3-containing nucleosomes is catalysed by the Daxx/ATRX complex (19). Although originally associated with euchromatic sites of active transcription, H3.3 has recently been found associated with regulatory elements and constitutive heterochromatin (20–22). In summary, Daxx alone or in a complex with ATRX was observed to most presumably repress transcription by effectively assembling H3.3-containing nucleosomes (16).
Ad are non-enveloped viruses with an icosahedral capsid and linear double-stranded DNA (23). The viral genome includes transcription units encoding ∼40 regulatory and structural proteins, and usually two non-coding RNAs (‘virus-associated RNAs’), depending on the serotype. Ad5 proteins are derived from early (E1A, E1B, E2A, E2B, E3 and E4) or late (L1–L5) transcription units. The E1A region is the first transcription unit activated following Ad5 infection (24,25). Mechanistically, E1A proteins stimulate the infected cell to enter S phase and are required to promote the transcription of other early viral genes (26–28). The function of the early region E1B-55K protein is more diverse. As a multifunctional phosphoprotein, it promotes efficient viral replication via a number of different mechanisms. In the early phase of productive Ad5 infection, E1B gene products counteract anti-proliferative processes induced by the host cell (29–31). Additionally, E1B-55K controls efficient late viral protein production by stimulating preferential cytoplasmic accumulation and translation of viral late mRNAs during the late phase of infection (32,33). Multiple functions of E1B-55K require interaction with viral ‘early region 4 open reading frame protein 6’ (E4orf6). Together, both proteins assemble a Skp, Cullin, F-box (SCF)-like E3-ubiquitin ligase complex, initiating proteasomal degradation of cellular targets such as p53, Mre11, DNA ligase IV, integrin α3 subunit and Bloom-helicase (34–38). In contrast, we recently discovered that E1B-55K antagonizes the innate antiviral activities of Daxx by targeting the cellular protein for E4orf6-independent proteasomal degradation (1).
Here, we demonstrate that in addition to Daxx, the nuclear, chromatin remodelling factor ATRX represses Ad5 replication in infected human cells. We observed significantly enhanced viral gene expression after limiting functional Daxx/ATRX chromatin remodelling complexes in the nucleus. These data provide evidence that chromatin modulating proteins play a major role in a host cell intrinsic defense mechanism against adenoviruses. We observed that ATRX protein concentrations are antagonized by proteasomal degradation via the E1B-55K/E4orf6 E3 ubiquitin ligase complex during Ad5 productive infection. Our findings affect future studies on Ad chromatinization and influence further research areas. As Ad-based vectors are frequently used in gene therapy, understanding viral gene expression within the host improves vector efficacy. Considering the fundamental importance of chromatin formation in host-cell gene regulation, investigating whether Ad transcription is regulated by cellular chromatin remodelling will contribute to the identification of new therapeutic targets to limit or prevent virus-mediated diseases and mortality of infected patients.
MATERIALS AND METHODS
Cell culture
Hep reDaxx and Hep Daxx PAH were established using parental lentivirus vector pLKO-shDaxx2 (39) and inserting yellow fluorescent protein (EYFP)-Daxx and EYFP-hDaxx_PAH1 (39). For the Daxx/ATRX knockdown, we constructed a lentivirus for double depletion of Daxx and ATRX (pLKO-shDaxx/ATRX) by replacing the shPML-siRNA sequence with a short hairpin RNA against ATRX (5′-CGACAGAAACTAACCCTGTAA-3′) (40) in a lentivirus designed for simultanous depletion of Daxx and PML (41). A scrambled shRNA-expressing lentivirus was used as negative control. Lentivirus supernatants were prepared by co-transfection of HEK293T cells with the respective pLKO vector, pVSV-G (expressing the vesicular stomatitis virus (VSV) envelope protein) and pCMV.DR8.91 (expressing lentivirus helper functions), as described previously (42). Stable cell lines were selected using puromycin (initially 1 μg/ml and then reduced to 0.5 μg/ml during subsequent passage). H1299 Cullin-5 negative cells and the H1299 control cell line were described previously (37,43). Daxx-depleted U2OS cells (U2OS + shDaxx) were generated by transduction of lentivirus vectors expressing shRNA targeted to the coding strand sequence 5′-GGAGTTGGATCTCTCAGAA-3′ located at nt 626–643 in Daxx (44). HepaRG cell lines (1,39,45), HEK293 cells (46), p53 negative human cell line H1299 (47) and osteosarcoma cell line U2OS (48) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 100 U of penicillin and 100 µg of streptomycin per millilitre in a 5% CO2 atmosphere at 37°C. For HepaRG cells, the media was supplemented with 5 µg/ml of bovine insulin and 0.5 µM of hydrocortisone.
Plasmids, mutagenesis and transient transfections
Ad5 proteins examined in this study were expressed from their respective complementary DNA under the control of the cytomegalo virus (CMV) immediate early promoter, derived from pcDNA3 vector (Invitrogen), to express E1B-55K and E4orf6 (49,50). Luciferase reporter constructs were generated by cloning polymerase chain reaction (PCR)-amplified Ad promoter constructs upstream of the promoterless firefly luciferase gene in the pGl3-basic vector (Promega). The respective Ad promoters were amplified from CAT vectors (kindly provided by Shenk) with specific oligonucleotides, introducing a XhoI/HindIII restriction site (see Table1). For transient transfections, cells were treated as described previously (1). Plasmid pEGFP-ATRX (51) was a kind gift from Picketts.
Table 1.
Oligonucleotide sequences used to amplify specific Ad promoters
| Primer name | Primer sequence |
|---|---|
| E1A fwd | 5′ ATACTCGAGCATCATCAATAATATACCTTA TTTTGGATTGA 3′ |
| E1A rev | 5′ TATAAGCTTGTCGGAGCGGCTCGGAG 3′ |
| E1B fwd | 5′ ATACTCGAGGTGTCTAGAGAATGCAATAGTAG 3′ |
| E1B rev | 5′ ATAAAGCTTTAACCAAGATTAGCCCACGG 3′ |
| E2 early fwd | 5′ ATACTCGAGTAGGATTGCCTGACGAGGCG 3′ |
| E2 early rev | 5′ ATAAAGCTTTACTGCGCGCTGACTCTTAAGG 3′ |
| MLP fwd | 5′ ATACTCGAGCTCACAGATTTGCATTTCCCA 3′ |
| MLP rev | 5′ ATAAAGCTTACAGCGATGCGGAAGAGA 3′ |
Viruses
H5pg4100 served as the wild-type Ad5 parental virus in this study (52). The mutant viruses H5pm4149 and H5pm4154 were generated as described recently (53). Both viruses carry stop codons in the E1B-55K (H5pm4149) or E4orf6 (H5pm4154) open reading frames and do not express the respective viral protein (54). The H5pm4139 was generated carrying amino acid changes in the (elongins B and C) BC1 and BC2 box motifs of E4orf6 resulting in a drastically reduced ability to associate with the Ad5 E3 ubiquitin ligase complex compared with the E4orf6 protein from wild-type virus (53). Viruses were propagated and titrated in HEK293 monolayer cultures. Infections were performed as described previously (55). To measure virus growth, infected cells were harvested 24, 48 and 72 h post-infection and lysed by three cycles of freeze thaw. The cell lysates were serially diluted in Dulbecco’s modified Eagle’s medium for infection of HEK293 cells, and virus yield was determined by quantitative E2A-72K immunofluorescence staining 24 h after infection (52). Viral DNA replication was monitored by quantitative PCR using E1B-specific primers (E1B-fwd 3′-CGCGGGATCCATGGAGCGAAGAAACCCATCTGAGC-5′; E1B-rev 3′-CGGTGTCTGGTCATTAA GC TAAAA-5′) exactly as described previously (50). PCR products were analysed on a 1% agarose gel and quantified using the G-Box system and Gene-Tools software (Syngene). Amount of viral particles was monitored by quantitative PCR. HepaRG cells were infected with wild-type virus H5pg4100, harvested at 48 h post-infection (h. p. i.), total cell extracts were prepared and treated with proteinase K. Quantitative real-time PCR was performed using hexon-specific primers (hexon-qPCR-fw: 5′-CGCT GGACATGACTTTTGAG-3′; hexon-qPCR-rev: 5′-GAACGGTGTGCGCAGGTA-3′). Ad5 H5pg4100 bacmid DNA was used as a control to obtain a standard curve.
Quantitative real-time PCR analysis
Subconfluent cells were infected with wild-type virus and harvested at 48 h. p. i. Total RNA was isolated with ‘Trizol reagent’ (Invitrogen) as described by the manufacturer. The amount of total RNA was measured, and one microgram of RNA was reverse transcribed using the ‘Transcriptor High Fidelity cDNA Synthesis Sample Kit’ from Roche including anchored-oligo(dT)18 primer specific to the poly(A)+RNA. Quantitative real-time (qRT) PCR was performed with a first strand method in a Rotor-Gene 6000 (Corbett Life Sciences, Sydney, Australia) in 0.5 ml of reaction tubes containing a 1/100 dilution of the cDNA template, 10 pmol/µl of each synthetic oligonucleotide primer, 12.5 µl/sample ‘Power SYBR Green PCR Master Mix’ (Applied Biosystems). The PCR conditions were as follows: 10 min at 95°C, 55 cycles of 30 s at 95°C, 30 s at 55°C–62°C (depending on the primer set) and 30 s at 72°C. The average Ct value was determined from triplicate reactions, and levels of viral mRNA relative to cellular 18S rRNA were calculated as described recently (56). The identities of the products obtained were confirmed by melting curve analysis.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay (ChIP) analysis was performed as described previously with some modifications (57,58). Proteins were cross-linked to DNA with 1% formaldehyde in phosphate buffered saline for 10 min at RT. The reaction was quenched, cells were washed with phosphate buffered saline and harvested by scraping off the dish. Nuclei were isolated by incubating cross-linked cells with 500 µl of buffer I [50 mM Hepes-KOH, 140 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 0.5% NP-40, 0.25% Triton X-100] for 10 min on ice and pelleted by centrifugation. Nuclei were subsequently washed with 500 µl of buffer II (10 mM Tris–HCl, 200 mM NaCl, 1 mM EDTA, 0.5 mM (EGTA) ethylene glycol tetraacetic acid), pelleted again and resuspended in 500 µl of buffer III [1% sodium dodecyl sulphate (SDS), 10 mM EDTA, 50 mM Tris–HCl]. Chromatin was fragmented by sonication using a BioruptorTM (Diagenode) to an average length of 100–300 bp. After addition of 10% Triton X-100, cell debris was pelleted by centrifugation and supernatants were collected. Chromatin was diluted with buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl, 167 mM NaCl). To reduce non-specific background, chromatin was pre-incubated with salmon-sperm DNA protein-A agarose beads (Upstate). Antibodies (Ab) were added and incubated for 16 h at 4°C. The 50 µl of agarose beads were added to precipitate the chromatin immunocomplexes for 4 h at 4°C. Beads were washed once with low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 150 mM NaCl), once with high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 500 mM NaCl), once with LiCl-wash buffer (0.25 M LiCl, 1% Nonidet P-40, 1% Na-deoxycholate, 1 mM EDTA, 10 mM Tris–HCl) and twice with TE buffer. Chromatin was eluted from the beads in elution-buffer (50 mM Tris–HCl (pH 8.0), 10 mM EDTA, 1% SDS) for 10 min at 95°C. ProteinaseK was added for protein degradation, and samples were incubated for 1 h at 55°C. To prepare input controls, samples were treated like IP samples except that non-specific Abs were used. qRT PCR analysis was performed using a Rotor Gene 6000 (Corbett Life Sciences) in 0.5 ml of reaction tubes containing 1/100 dilution of the precipitated chromatin, 10 pmol/µl of each synthetic oligonucleotide primer (see Table 2), 5 µl/sample SYBR Green PCR Master Mix (Applied Biosystems). The PCR conditions used: 7 min at 95°C, 45 cycles of 12 s at 95°C, 40 s at 60°C and 15 s at 72°C. The average Ct-value was determined from triplicate reactions and normalized against non-specific immunoglobulin G (IgG) controls (sc-2027; Santa Cruz Biotechnology) with standard curves for each primer pair. The identities of the products obtained were confirmed by melting curve analysis.
Table 2.
Oligonucleotide sequences used to amplify Ad DNA in ChIP assays
| Primer name | Primer sequence |
|---|---|
| E1A fwd | 5′ TCCGCGTTCCGGGTCAAAGT 3′ |
| E1A rev | 5′ GTCGGAGCGGCTCGGAG 3′ |
| E1B fwd | 5′ GGTGAGATAATGTTTAACTTGC 3′ |
| E1B rev | 5′ TAACCAAGATTAGCCCACGG 3′ |
| E2 early fwd | 5′ TACTGCGCGCTGACTCTTAAGG 3′ |
| E2 early rev | 5′ ATGGCGCTGACAACAGGTGCT 3′ |
| MLP fwd | 5′ TGATTGGTTTGTAGGTGTAGG 3′ |
| MLP rev | 5′ ACAGCGATGCGGAAGAGA 3′ |
Luciferase reporter assays
For dual luciferase assays, subconfluent cells were transfected as described previously (55,59), using 0.5 μg of reporter and 0.5 µg of pRL-TK (Promega), which expresses Renilla luciferase under the control of the herpes simplex virus thymidine kinase promoter. As described by the manufacturer (Promega) for dual luciferase assays, total cell extracts were prepared 48 h after transfection, and retinal ganglion cells (RGC) firefly luciferase activity was assayed with 5 µl of lysed extract in an automated luminometer (Berthold Technologies). All samples were normalized for transfection efficiency by measuring Renilla luciferase activity. All experiments shown were performed as triplicates, and data are presented as mean values.
Micrococcal nuclease nuclease accesibility assay
Cell pellets were lysed in a hypotonic buffer (10 mM Tris–HCl (pH 7.4), 10 mM KCl, 15 mM MgCl2) on ice for 10 min. Nuclei were pelleted by centrifugation and resuspended in Micrococcal nuclease (MNase) digestion buffer [0.32 M sucrose, 50 mM Tris–HCl (pH 7.5), 4 mM MgCl2, 1 mM CaCl25, 0.1 mM phenylmethylsulfonyl fluoride (PMSF)] supplemented with 20 U MNase and incubated at 37°C for varying periods. The MNase reaction was stopped by the addition of 10 mM EDTA followed by centrifugation. The pellet was then resuspensed in MNase digestion buffer supplemented with 10 mM EDTA and RNase and incubated at 37°C for 30 min. The DNA was extracted and purified by standard procedures. Digested chromatin was analysed on a 1.4% agarose gel using the G-Box system and Gene-Tools software (Syngene). Band intensities were quantified with ImageJ and analysed with GraphPad Prism software.
Protein analysis and Ab
Cells were resuspended in radioimmunoprecipitation buffer (RIPA) buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol (DTT), 0.1% SDS, 1% NP-40, 0.1% Triton X-100, 0.5% sodium deoxycholate) containing 1% (v/v) PMSF, 0.1% (v/v) aprotinin, 1 µg/ml of leupeptin, 1 µg/ml of pepstatin, 1 mM DTT, 25 mM iodacetamide and 25 mM N-ethylmaleimide. After 1 h on ice, the lysates were sonicated, and insoluble debris was pelleted at 15 000×g/4°C. Fractionation of soluble and chromatin-rich fractions was reported recently (60). For immunoprecipitation and immunoblotting, protein lysates were treated as described recently (61). Primary Ab specific for Ad proteins used in this study included E1A mouse monoclonal antibody (MAb) M73 (62), E1B-55K mouse MAb 2A6 (63), E2A-72K mouse MAb B6-8 (64), E4orf6 MAb RSA3 (65) and Ad5 rabbit polyclonal serum L133 (66). Primary Ab specific for cellular proteins included ATRX rabbit Ab H-300 (Santa Cruz Biotechnology), ATRX mouse MAb clone 39F (kindly provided by Gibbons), Daxx rabbit polyclonal Ab 07-471 (Upstate/Millipore), anti-histone H3.3 rabbit polyclonal Ab ab62642 (Abcam), PML rabbit poyclonal Ab H-238 (Novus Biologicals) and ß-actin mouse MAb AC-15 (Sigma-Aldrich). EYFP epitopes were detected with anti-green fluorescent protein (GFP) rabbit Ab ab290 (Abcam). Secondary Ab conjugated to horseradish peroxidase to detect proteins by immunoblotting were anti-rabbit IgG and anti-mouse IgG (Jackson/Dianova).
RESULTS
Efficient Ad5 replication is restricted by Daxx and ATRX
Ad5 replication centres establish in the vicinity of nuclear PML-NBs, as demonstrated by recruitment of viral and host proteins required for DNA replication, transcription and gene expression to these nuclear foci (67). We previously showed that the transcriptional repressor and PML-NB resident factor Daxx suppresses Ad5 replication early in infection (1). Daxx is linked to transcriptional repression via interaction with ATRX in a functional, ATP-dependent, SWI/SNF chromatin remodelling complex, recruiting histone deacetylase (HDACs) to sites of active transcription and depositing histone variant H3.3. To analyse the role of Daxx-associated ATRX during Ad5 infection, we performed experiments in a non-transformed human hepatocyte cell line, HepaRG, expressing shRNAs targeting either Daxx, ATRX or both (1,39). Before our analysis, we investigated cell proliferation in Daxx- and ATRX knockdown cells and observed no significant difference. Moreover, immunofluorescence analysis showed that neither Daxx nor ATRX depletion affected formation of PML-NB structures in the nucleus (data not shown). Previously, we found that depletion of transcription factor Daxx correlated with enhanced Ad5 early gene expression. Consistent with this, Daxx knockdown increased production of Ad5 particles ∼4-fold, 72 h post infection [h. p. i.; Figure 1A; (1)]. As expected, the repressive effect of Daxx was partially rescued by expressing of a EYFP-Daxx fusion protein in the Daxx-depleted cells (1,39). Reconstituted Daxx substantially reduced Ad5 virus growth in these Hep reDaxx cells [Figure 1A; (1)]. To investigate the effect of ATRX on Ad5 progeny production, we determined virus growth in ATRX-depleted cells (Hep shATRX; Figure 1A). As with Hep shDaxx cells, ATRX depletion enhanced progeny virus production 2 - (48 h. p. i.) to 4-fold (72 h. p. i.) compared with control cells (Hep parental/Hep neg; Figure 1A). To reveal whether Daxx and ATRX clearly act together on the repression of viral progeny production rather than in a parallel manner, we generated a double knockdown cell line depleting Daxx and ATRX simultaneously (Hep shDaxx/shATRX). We observed an increased production of Ad5 progeny ∼2- (48 h. p. i.) to 4-fold (72 h. p. i.) in comparison with the cell lines expressing a functional Daxx/ATRX chromatin remodelling complex (Hep parental, Hep neg; Figure 1A). These results suggest that both Daxx and ATRX mediate repressive effects regarding Ad5 progeny production. To further determine whether Daxx/ATRX nuclear localization might affect Daxx/ATRX-dependent transcriptional repression of viral gene expression, we also analysed Ad gene expression in HepaRG cells depleted for endogenous Daxx and expressing an EYFP-Daxx fusion protein with a functional mutation in the sumo interacting motif (SIM), completely abrogating Daxx association with PML-NBs. Our analysis showed that Daxx SIM mutant cells support efficient repression of Ad5 gene expression compared with control cells (data not shown). Our findings strongly suggest that repressive functions are achieved, despite inhibition of Daxx colocalization with PML-NBs in the SIM mutation. As recent reports show that Daxx is degraded during late adenovirus infection via an E1B-55K-dependent, proteasomal pathway (1), we next analysed the expression levels of Daxx and ATRX during Ad5 infection (Figure 1B). Our data show stable expression of shRNAs depleting either Daxx, ATRX or both (Hep shDaxx; Hep shDaxx/shATRX) and/or ATRX (Hep shATRX; Hep shDaxx/shATRX). Moreover, we confirmed Daxx reduction in Ad5 wild-type infected cells, highly dependent on E1B-55K concentrations (Hep parental; Hep shATRX; Hep reDaxx; Hep neg; Supplementary Figure S1A). Hence, by monitoring ATRX levels in infected cells, we observed reproducibly declined concentrations (Hep parental; Hep shDaxx; Hep reDaxx; Hep neg; Supplementary Figure S1A). Therefore, we are tempted to speculate that efficient Ad gene expression requires Daxx/ATRX elimination during infection or at least limitation of the protein concentrations to counteract the repressive activity determined by both transcription factors. We further determined quantitative PCR analysis to precisely quantify virus concentration using hexon-specific primers (Supplementary Figure S1B). We observed reduced amount of Ad5 viral DNA copies in cells depleted for Daxx and/or ATRX (Hep shDaxx, Hep shATRX, Hep shDaxx/shATRX; Supplementary Figure S1B).The results from these assays are consistent with the data obtained by virus yield and stengthen the hypothesis that Daxx and ATRX act together, rather than in a parallel manner to repress viral titer and gene expression. Similar effects were observed monitoring viral DNA synthesis in infected cells (Figure 1B). As with Ad5 progeny production, DNA synthesis was more efficient in Daxx- and/or ATRX-depleted cells (Figure 1B). These results suggest that both cellular transcription factors mediate repressive effects during Ad5 infectious cycle. To further test this hypothesis, expression of viral early E1A (Supplementary Figure S2A), E1B-55K (Supplementary Figure S2B), E2A/DBP (Supplementary Figure S2C) and late proteins hexon and fiber (Supplementary Figure S2D) was monitored at different time points after Ad5 wild-type infection. Expression of E1A, E1B and E2A/DBP was substantially increased in cells with reduced amount of Daxx and/or ATRX (Hep shDaxx, Hep shATRX, Hep shDaxx/shATRX; Supplementary Figure S1B) and in comparison with the control cells (Hep par, Hep neg, Hep reDaxx; Supplementary Figure S1B). Consistent with this observation, we show an increase in protein concentration of late hexon and fiber proteins in infected Daxx- and/or ATRX-depleted cells (Supplementary Figure S2D).
Figure 1.
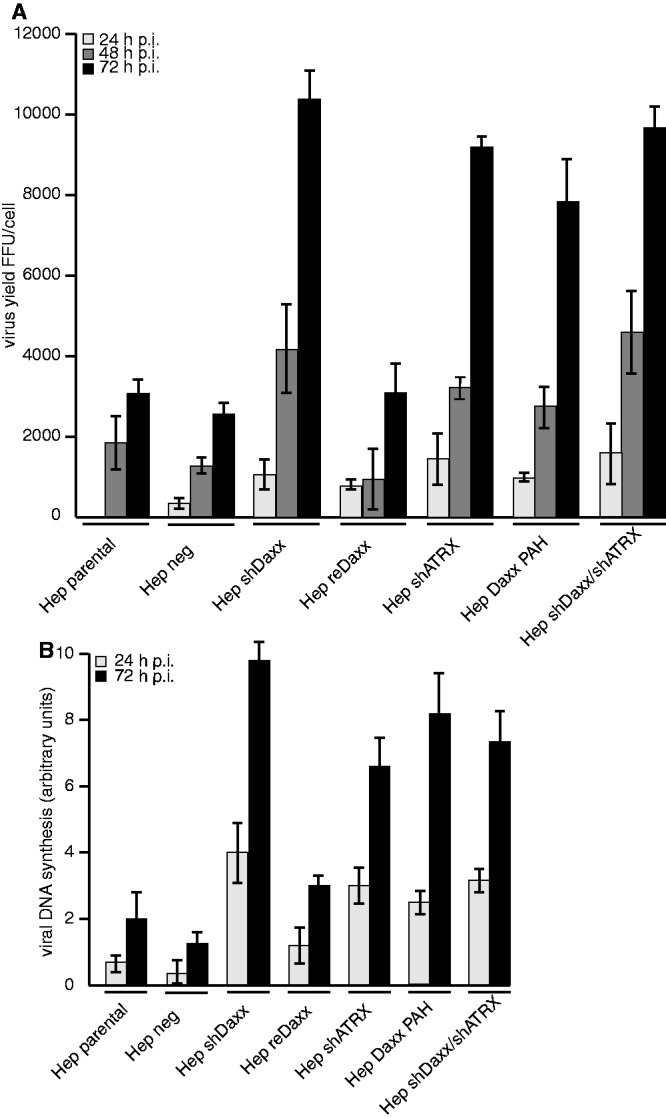
Functional inhibition of Daxx/ATRX repressive complex efficiently stimulates Ad5 viral gene expression. Hepa RG cells were infected with wild-type H5pg4100 at moi of 50 FFU per cell. (A) Viral particles were harvested 24, 48 and 72 h. p. i., and virus yield was determined by quantitative E2A-72K immunofluorescence staining on HEK293 cells. Shown are averages from three independent experiments. Error bars indicate the standard error of the mean. (B) Total cell extracts were prepared and treated with proteinase K. PCR was performed, and identicle amounts of PCR product were separated on analytic agarose gels (1%) and quantified with Gene Snap Software (Syngene). The results shown represent the averages from two independent experiments.
Daxx/ATRX functional complex formation is essential to repress Ad5 gene expression
So far, it was unclear whether Daxx and ATRX affect Ad gene expression individually or impair Ad transcription by assembling a functional SWI/SNF chromatin remodelling complex. To test whether functional Daxx/ATRX complexes affect Ad5 replication, we monitored virus growth in a stable cell line depleted for endogenous Daxx, expressing an EYFP-Daxx fusion protein lacking 73 amino acids spanning its ATRX-interacting PAH1 domain (Hep Daxx PAH). These cells express the same amounts of Daxx and ATRX proteins but lack functional cooperation and consequently Daxx/ATRX-mediated transcriptional repression [Supplementary Figure 1A; see (39)]. We found that Ad5 progeny production is enhanced 2- (48 h. p. i.) to 5-fold (72 h. p. i.) in infected cells lacking functional Daxx/ATRX complex formation (Hep Daxx PAH), compared with parental cells (Hep par), cells transduced with scrambled shRNA (Hep neg) or cells expressing an EYFP Daxx fusion protein re-introduced into the Daxx-depleted cell line (Hep reDaxx; Figure 1A). This effect is not due to altered cell proliferation in the Hep Daxx PAH line compared with control cells or any altered localization of PML, Daxx or ATRX in uninfected or Ad5-infected cells (data not shown). Similar results were obtained from studies monitoring viral DNA synthesis (Figure 1B; Supplementary Figure S1B) and expression of viral proteins (Supplementary Figure S2). Taken together, our findings indicate that although both cellular proteins are present, Ad5 repression depends on a functional Daxx/ATRX interaction and most likely occurs at the transcriptional level.
Functional Daxx/ATRX complex is sufficient to repress Ad5 promotor activity and mRNA synthesis
Next, we investigated whether Ad5 early promoters are targeted by Daxx and/or ATRX-mediated repression and whether this can be reversed by functionally inhibiting the repressive SWI/SNF complex. First, we analysed whether early and late Ad5 mRNA levels are affected by Daxx and/or ATRX in Ad5 wild-type virus-infected cells. Viral early E1A, E1B, E2A/DBP and hexon mRNA production was lower in parental cells and in cells expressing reintroduced EYFP-Daxx [Hep reDaxx; (1)], compared with cells lacking a functional Daxx/ATRX complex formation (Hep Daxx PAH, Figure 2). Similar results were obtained for hexon mRNA expression, suggesting either an impact of enhanced synthesis of early gene products or direct repression of the late promoter (Figure 2). Next, we constructed luciferase expression vectors, controlled by adenoviral early promoters (E1A, E1B, E2 early and MLP) and measured luciferase expression in different HepaRG cell lines (Figure 3A).
Figure 2.
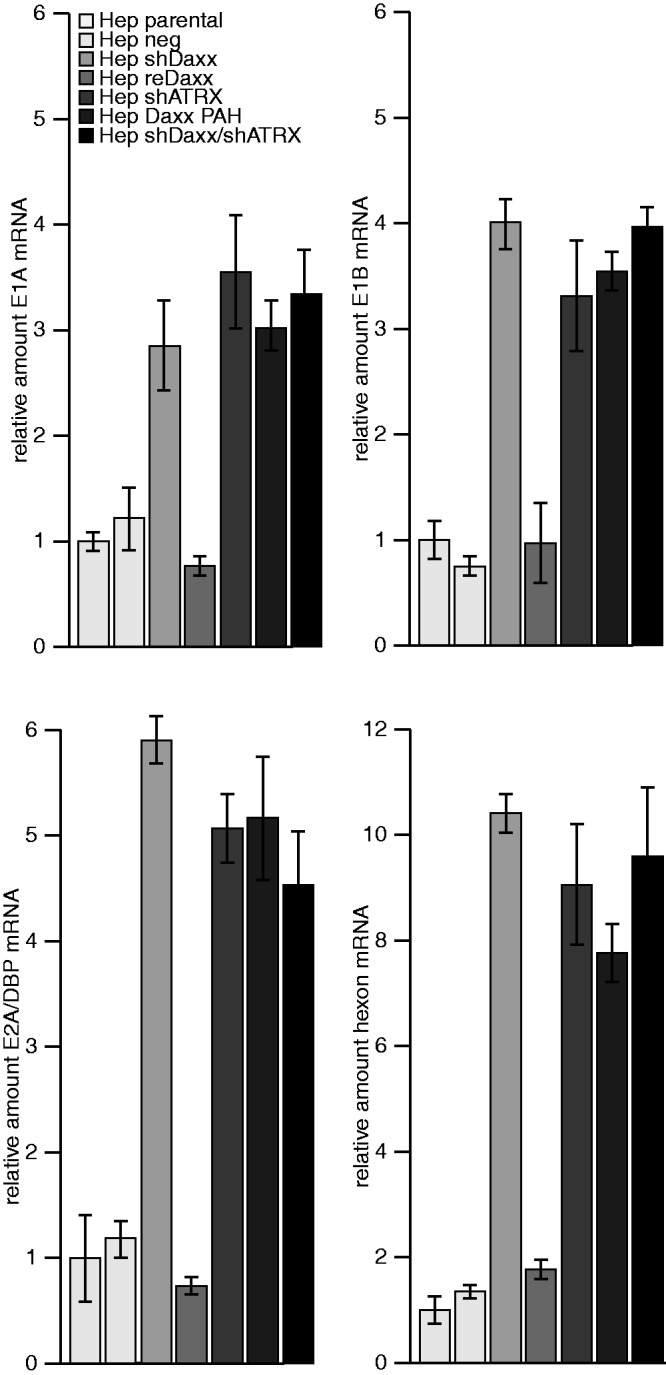
Daxx/ATRX complex represses Ad5 mRNA synthesis. HepaRG cells were infected with H5pg4100 Ad5 wild-type at moi of 50 FFU/cell. Then, 24 h. p. i., total RNA was extracted, reverse transcribed and quantified by RT-PCR using primers specific for E1A (E1A fwd: 5′-GTGCCCCATTAAACCAGTTG-3′; E1A rev: 5′-GGCGTTTACAGCTCAAGTCC-3′), E1B-55K (E1B fwd: 5′-GAGGGTAACTCCAGGG TGCG-3′; E1B rev: 5′-TTTCACTAGCATGAaGCAACCACA-3′, E2A/DBP and hexon (hexon rev: 5′-GAACGGTGTGCGCAGGTA-3′; hexon fwd 5′-CGCTGGACATGACTTTTG AG-3′. Data were normalized to 18S rRNA levels (18S rRNA fwd: 5′-cggctaccacatccaaggaa-3′; 18S rRNA rev: 5′-GCTGGAATTACCGCGGCT-3′). Values correspond to the mean of triplicates, and error bars indicate the standard error of the mean.
Figure 3.

Daxx/ATRX complex mediates Ad transcriptional repression. (A) Hepa RG cells were transfected with luciferase reporter plasmids under Ad promoter control (E1A, E1B, E2early, MLP). Then, 48 h after transfection, samples were lysed, absolute luciferase activity was measured. All samples were normalized for transfection efficiency by measuring Renilla luciferase activity. Promoter activity of E1A, E1B, E2early and MLP promoter in Hep parental cells was normalized to 1. Mean and STD are from three independent experiments. (B) HepaRG cells were infected with H5pg4100 Ad5 wild-ype at moi of 50 FFU/cell. Then, 24 h. p. i. cells were fixed with formaldehyde and analysed by ChIP assays (see ‘Material and Methods’ section). The average Ct-value was determined from triplicate reactions and normalized against non-specific IgG controls with standard curves for each primer pair (Table 2). The y-axis indicates the percentage of immunoprecipitated signal from the input (100%). The white dotted line highlights values >1% of input, commonly stated as significant chromatin/protein binding.
Compared with parental control cells and cells expressing an EYFP-Daxx fusion protein [Hep reDaxx; (1)], the absence of either Daxx and/or ATRX stimulated ∼5-fold expression from E1A and E1B promoters, ∼8-fold from the E2 early promoter and ∼4-fold from the MLP promoter (Figure 3A).
As Daxx contains no DNA binding region, it is supposed that ATRX is the molecular bridge connecting the complex to chromatin. To show direct association of ATRX with adenovirus promoters, we performed ChIP in virus-infected HepaRG cells, using ATRX Ab, unrelated IgG control Ab and Ad promoter-specific primers (Table 2; Figure 3B). The results show that ATRX is associated with Ad promoters in infected Hep parental cells 24 h after infection (Figure 3B). To support the model that ATRX recruits Daxx to Ad promoters, we inclued Hep shATRX cells to our ChIP analysis. As anticipated, we did not observe ATRX chromatin binding to Ad promoter sequences in the shATRX cells, excluding unspecific background of the ATRX-specific Ab. To further elucidate whether ATRX bound to adenoviral promoters is Daxx independent, we carried out additional ChIP experiments in Daxx-depleted (Hep shDaxx) cells and in cells without a functional Daxx/ATRX complex (Hep Daxx PAH). We observe that ATRX is targeted to Ad5 promoters independent of Daxx, as we were still able to detect ATRX recruitment (Figure 3B). Conclusively, we clarified the interdependence of Daxx and ATRX when targeting viral promoters illustrating an ATRX-dependet process, independent of the complex formation with Daxx. Taken together, these results indicate that ATRX relocalizes Daxx to Ad promoter regions, and that both transcription factors mediate repression of Ad5 productive replication.
ATRX expression in ATRX knockout cells is sufficient to repress Ad5 gene expression
Next, we analysed Ad5 replication in U2OS cells, which lack ATRX protein expression (Figure 4A; 39,64,68). First, we monitored Daxx protein levels and observed significant reduction in the presence of wild-type virus (Figure 4A). Consistent with our observations in infected HepaRG cells (Supplementary Figure S1A), Daxx was still degraded in the absence of ATRX. This result confirms that fact that inhibition of both repressors might occur via independent mechanisms. Next, we tested whether Daxx depletion (Figure 4A) affects adenovirus production and gene expression in U2OS cells. We observed no effect on adenovirus progeny production in the absence of Daxx (U2OS + shDaxx; Figure 4B). When ATRX was provided in trans, expressed from a pEGFP construct with 41% efficiency of transfection (data not shown), virus growth was 3-fold less than in control cells, where Ad5 was not repressed (Figure 4B). The same result was obtained when we quantified the amount of viral DNA copies (Figure 4C) and viral DNA synthesis. (Figure 4D). Taken together, depletion of Daxx in U2OS cells, lacking ATRX gene expression, did not show supportive effects on adenovirus gene repression. Moreover, as with HepaRG cells (Figure 1B; Supplementary Figure S1B), amount of Ad5 DNA copies (Figure 4C) and viral DNA synthesis (Figure 4D) was reduced in U2OS cells when transfected with the ATRX-encoding construct before infection. We note that induction of Ad5 gene expression is more pronounced in ATRX knockout (U2OS) than ATRX knockdown (Hep shATRX) cells, likely owing to diverging ATRX concentrations. Next, we tested expression of viral early E1A, E1B-55K, E4orf6 and late proteins (Supplementary Figure S3) at different time points after Ad5 wild-type infection in the different U2OS cells. Expression of all viral proteins monitored was substantially reduced in cells expressing ATRX in trans (Supplementary Figure S3, lanes 7–9). Consistent with this observation, we showed no efficient mRNA synthesis in infected U2OS cells stably expressing the ATRX transcription factor (Figure 5; U2OS + EGFP-ATRX) in comparison with various control cells (U2OS control, U2OS + EGFP-empty, U2OS + shDaxx). These results indicate that both PML-NB components, Daxx and ATRX, are negative regulators of Ad5 productive replication.
Figure 4.

Daxx/ATRX functional complex modulates productive Ad replication. U2OS cells were transfected with either empty vector or pEGFP-ATRX plasmid expressing human ATRX 24 h before infection with wild-type H5pg4100 virus or E1B-55K minus virus mutant H5pm4149 at moi of 50 FFU per cell. (A) U2OS cells were harvested at 48 h. p. i., total cell extracts were prepared and proteins were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and subjected to immunoblotting using mouse ATRX-specific mouse Mab or Daxx-specific rabbit Ab. Corresponding β-actin was included as a loading control. (B) Viral particles were harvested 24, 48 and 72 h. p. i., and virus yield was determined by quantitative E2A-72K immunofluorescence staining on HEK293 cells. Averages from three independent experiments are shown. Error bars indicate the standard error of the mean. (C) U2OS cells were harvested at 48 h. p. i., total cell extracts were prepared and treated with proteinase K. Quantitative real-time PCR was performed using hexon-specific primers. Ad5 H5pg4100 bacmid was used to obtain a standard curve. The results represent the averages from three independent experiments. Error bars indicate the standard error of the mean. (D) Total cell extracts were prepared and treated with proteinase K. PCR was performed and identicle amounts of PCR product were separated on an analytic agarose gels (1%) and quantified with Gene Snap Software (Syngene).
Figure 5.

Daxx/ATRX functional complex modulates productive Ad5 mRNA synthesis. U2OS cells were transfected with either empty vector or pEGFP-ATRX plasmid-expressing human ATRX 24 h before infection with wild-type H5pg4100 virus or E1B-55K minus virus mutant H5pm4149 at moi of 50 FFU per cell. Then, 24 h. p. i., total RNA was extracted, reverse transcribed and quantified by RT-PCR using primers specific for E1A, E1B-55K, E4orf6 (E4orf6 rev: 5′-CCCTCATAAACACGCTGGAC-3′; E4orf6 fwd: 5′-GCTGGTTTAGGATGGTGGTG-3′) and hexon. Data were normalized to 18S rRNA levels. Values correspond to the mean of triplicates, and error bars indicate the standard error of the mean.
ATRX protein levels are reduced during lytic Ad infection in human cells
So far, various reports clearly demonstrate that Ad disrupts PML-NBs in the early phase of infection via relocalization into track-like structures. Our findings show that the Daxx/ATRX complex can repress transcription independent of PML-NB association. Therefore, Ad-mediated track-like formation is most likely not interfering with Daxx/ATRX repressive attributes. Consequently, we proposed that efficient Ad gene expression requires Daxx/ATRX inhibition during infection or at least limitation of repressive activity by post-translational modifications. We therefore monitored ATRX protein levels in Ad-infected cells and observed that ATRX concentrations are reduced during wild-type (wt) (H5pg4100) lytic infection (Figure 6A). In contrast, we observed no ATRX reduction in infected cells lacking either E1B-55K (H5pm4149) or E4orf6 (H5pm4154; Figure 6A). E1B-55K is likely the substrate recognition unit, whereas E4orf6 assembles the cellular components into Ad5 dependent E3 ubiquitin ligase. To determine whether ATRX is a novel binding partner of E1B-55K, we performed binding assays in Ad5 wild-type (H5pg4100), E1B-55K (H5pm4149) and E4orf6 (H5pm4154) mutant virus-infected cells lacking the respective viral protein. Co-immunoprecipitation of E1B-55K with ATRX-specific MAb revealed an interaction between both factors during lytic Ad5 infection (Figure 6A, lanes 2 and 4), whereas no signal was detected in the corresponding controls (Figure 6A, lanes 1 and 3). We detected nearly identical amounts of co-precipitated E1B-55K protein, although the ATRX input levels were greatly altered, dependent on proteasomal degradation. This might be due to limiting conditions of Ab-coupled protein A-sepharose in these IP experiments, which ensures complete saturation of the beads with ATRX Ab. To control co-immunoprecipitation of E1B-55K with ATRX-specific MAb, we repeated the experiment in the Hep shATRX cells in the absence of ATRX and did not observe any non-specific band (data not shown). The decrease in ATRX seen in wild-type infected cells was abolished when we treated the cells with proteasome inhibitor MG 132 (Figure 6B, lanes 2 and 6). These data demonstrate that in contrast to E1B-55K-dependent degradation of Daxx (1), ATRX is targeted via a proteasomal pathway, which requires E1B-55K in cooperation with E4orf6. To test our hypothesis, we first analysed the levels of ATRX protein in plasmid-transfected cells in the absence of a viral background (Figure 6C).
Figure 6.

Proteasomal degradation of ATRX in infected and transfected cells. H1299 cells were infected with wild-type (H5pg4100) and mutant viruses (H5pm4149, H5pm4154) at moi of 50 FFU per cell. (A) Total cell extracts were prepared 48 h. p. i., and proteins were separated by SDS–PAGE and subjected to immunoblotting using mouse MAb 2A6 (E1B-55K), mouse MAb RSA3 (E4orf6), Daxx specific rabbit Ab and ATRX-specific mouse MAb clone 39F. β-actin was included as a loading control. Co-immunoprecipitation (IP) of E1B-55K was performed with ATRX-specific mouse MAb clone 39F followed detection of co-precipitated E1B-55K with mouse MAb 2A6. (B) Infected cells (as aforementioned) were treated for 6 h with proteasome inhibitor (+MG 132), before total cell extracts were prepared and specific proteins detected as described in A. (C) H1299 cells were transfected with pcDNA3-derived plasmids expressing wild-type E1B-55K, E4orf6 or a combination of both. Cells were harvested 48 h. p. i. Total cell extracts were prepared, and specific proteins were immunoprcipitated and detected as described in (A).
Expression of wild-type E1B-55K or E4orf6 alone had no effect on endogenous ATRX protein concentrations (Figure 9C, lanes 2 and 3), whereas expression of both (Figure 6C, lane 4) diminished ATRX protein levels similarly to wild-type-infected cells (Figure 6A, lane 2).
Figure 9.

MNase nuclease accesibility assay to monitor Daxx/ATRX-mediated modulation of chromatin structure. Hepa RG cells were either untreated or infected with H5pg4100 Ad5 wild-type at moi of 100 FFU/cell for 24 h. Chromatin sensitivity assays were performed using digestion with 20 U of MNase for the indicated periods followed by an RNase treatment. Digested chromatin was analysed on a 1.4% agarose gel using the G-Box system and Gene-Tools software (Syngene). Band intesities were quantified with ImageJ and analysed with GraphPad Prism software. Mono-, di-, tri and poly-nucleosomes are indicated on the right.
ATRX protein is degraded via an Ad5 E1B-55K/E4orf6-dependent E3 ubiquitin ligase complex
Ad5 E1B-55K and E4orf6 assemble an E3 ubiquitin ligase complex containing the Cullin family member Cullin5, Elongins B and C, and the RING protein Rbx1 (69). Recently, it was shown that degradation of p53 is significantly impaired in a Cullin5 knockdown cell line (43). On the basis of these data, we examined ATRX protein levels in infected Cullin5 negative and control H1299 cells [Figure 7A; (37)]. Reduction of Daxx and Mre11 is dependent on Cullin5, as no decrease was observed in Cullin5 negative H1299 cells infected with wild-type (Figure 7A, lane 6). These data demonstrate that similar to known targets of the Ad5 E3 ubiquitin ligase, ATRX is degraded via a Cullin5-dependent proteasomal pathway, which requires E1B-55K and E4orf6. In addition to Cullin5, E4orf6 was previously shown to assemble with cellular proteins Rbx1 and Elongins B/C to form the E3 ubiquitin ligase to degrade cellular factors in presence of E1B-55K (69). Moreover, E4orf6 was shown to bind Elongins B and C via two conserved BC box motifs. Next, we examined ATRX protein levels in H1299 cells, infected with a E4orf6 virus mutant carrying point mutations in this BC Box, abrogating the formation of the Ad5 ubiquitin ligase complex. Our results strongly implicate that activity of the E1B-55K/E4orf6 ligase complex is essential to reduce ATRX protein concentration (Figure 7B, lane 5). Consistent with previous publications we observed, Daxx reduction, but no degradation, of Mre11 during lytic infection with the BC-box virus mutant H5pm4139 (Figure 7B, lane 5). This confirms the fact that formation and ligase activity of the E1B-55K/E4orf6 complex is essential for ATRX degradation via host cell proteasomes.
Figure 7.

ATRX is reduced via the E1B-55K/E4orf6 E3 ubiquitin ligase complex. (A) H1299 control and shCullin5 cells were infected with wild-type (H5pg4100) and mutant viruses (H5pm4149, H5pm4154) at moi of 50 FFU per cell. Total cell extracts were prepared 48 h. p. i., and proteins were separated by SDS–PAGE and subjected to immunoblotting using mouse MAb 2A6 (E1B-55K), mouse MAb RSA3 (E4orf6), Daxx- and Mre11-specific rabbit Ab and ATRX-specific mouse MAb clone 39F. β-actin was included as a loading control. (B) H1299 cells were infected with wild-type (H5pg4100) and mutant viruses (H5pm4149, H5pm4154, H5pm4139) at moi of 50 FFU per cell. (A) Total cell extracts were prepared 48 h. p. i., and proteins were separated by SDS–PAGE and subjected to immunoblotting using mouse MAb 2A6 (E1B-55K), mouse MAb RSA3 (E4orf6), Daxx- and Mre11-specific rabbit Ab and ATRX-specific mouse MAb clone 39F. β-actin was included as a loading control. (C) H1299 cells were infected with wild-type (H5pg4100) and E1B minus mutant virus (H5pm4149) at moi of 50 FFU per cell. Then, 48 h. p. i., total cell extracts were prepared after fractionation of soluble and chromatin fractions as described recently (60). Proteins were separated by SDS–PAGE and subjected to immunoblotting using Mre11-specific rabbit Ab, Daxx-specific rabbit Ab and ATRX-specific mouse MAb clone 39F, mouse MAb 2A6 (E1B-55K) and mouse MAb RSA3 (E4orf6).
To further interconnect our conclusions, we monitored ATRX protein levels in the soluble and chromatin-rich fraction of Ad5-infected cells. Unexpectedly, Daxx and ATRX were reproducibly declined during wt (H5pg4100) lytic infection in the chromatin fraction, although only a minor amount of the viral proteins could be detected in this fraction (Figure 7C). In contrast, we observed no reduction of the cellular chromatin-associated factors in infected cells lacking E1B-55K (H5pm4149; Figure 7C, lanes 3 and 6).
Ad-mediated inactivation of Daxx/ATRX functions affects H3.3 association with Ad promoters and chromatin condensation
Daxx is involved in deposition of cellular histone variant H3.3 on actively transcribed genes. Recent reports showed that Daxx either alone or in complex with ATRX actively assembles H3.3-containing nucleosomes (19,20,70). In line with this, Ad DNA was recently observed to preferentially associate with H3.3 (71), indicating that chromatinization by a replication-independent mechanism may play a so far unknown role during Ad life cycle. To enrich the novelty of our findings and giving the fact that we observed Daxx/ATRX complex mainly degraded in the chromatin fraction of the host cell, we experimentally assessed the effects of Ad5 wt infection on Daxx/ATRX-dependent H3.3 recruitment on viral promoters (Figure 8). ChIP analysis showed that H3.3 was found to be complexed with viral promoters (E1A, E1B) 24 h after infection of Hep parental cells (5 fluorescent forming units (FFU)/cell). Although using infected cells, treated with higher amounts of virus particles (100 FFU/cell), inducing Daxx and ATRX reduction, H3.3 association at viral promoters was significantly lost [promoter binding beyond 1%; (72)]. To determine whether this observation could be linked to efficient Ad-dependent Daxx/ATRX proteasomal degradation, we examined H3.3 loss from Ad promoters in infected HepaRG knockdown and control cells. We observed that the majority of H3.3 was lost from the viral DNA, although cells were treated with a very low multiplicity of infection (moi) of 5 FFU/cell (Hep shDaxx cells; Hep shATRX cells; Figure 8A). We observed the same result in infected Hep Daxx PAH cells in the absence of a functional Daxx/ATRX complex (Figure 8A). To validate functional restriction of the Daxx/ATRX complex via Ad-dependent proteasomal degradation, we monitored Daxx and ATRX protein levels in infected HepaRG cells used for our ChIP analysis. Daxx and ATRX protein concentrations reproducibly declined during Ad wt (H5pg4100) infection with a moi of 100 FFU/cell after 24 h (Figure 8B). As anticipated, detection of Daxx and ATRX proteins also failed within the HepaRG shDaxx cells or HepaRG shATRX cells (5 FFU/cell), illustrating the negative control in these experiments. In contrast, in Hep parental cells, Daxx or ATRX reduction was not yet observed 24 h. p. i. after infection with minor amount of infectious particles (5 FFU/cell; Figure 8B). Consistent with recent reports that Daxx assembles H3.3/H4 tetramers on DNA templates, and that deposition and remodelling of H3.3-containing nucleosomes is catalysed by the Daxx/ATRX complex, we could observe that in cells depleted for Daxx and/or ATRX as well as in cells without the functional complex (Hep Daxx PAH), H3.3 fraction bound to Ad promoter sequences of E1A and E1B transcription units is reduced compared with infected control cells (Hep par; 5 FFU/cell). Ad5 impact on Daxx-dependent chromatinization via H3.3 deposition is consistent with our findings that functional Daxx/ATRX chromatin remodelling complex is inactivated during the course of Ad5 infection. This is further consistent with results from the infection studies, showing that E1B-55K-dependent Daxx degradation (Hep par; 100 FFU/cell) and E1B-55K/E4orf6-dependent ATRX degradation (Hep par; 100 FFU/cell) also results in loss of H3.3 deposition on viral DNA. To gain further insight into the ability of Ad to modulate chromatin structure, we performed MNase chromatin sensitivity assays to directly evaluate any consequence of proteasomal Daxx/ATRX reduction due to E1B-55K and E4orf6 expression on bulk cellular chromatin. We analysed MNase sensitivity of chromatin from Ad-infected HepaRG parental (100 FFU/cell), uninfected HepaRG parental and shDaxx cells (Figure 9) and observed that Daxx reduction in Hep shDaxx and Hep parental (100 FFU/cell) resulted in a greater sensitivity of the chromatin to MNase digestion compared with uninfected control cells (Hep parental mock). This is evident from the increase in mono-, di- and tri-nucleosomes after equivalent lengths of MNase treatment (1, 2, 5 and 7.5 min; Figure 9) and by the reduced amount of undigested chromatin in the absence of Daxx (Hep shDaxx mock and Hep parental, 100 FFU/cell). In contrast to this, presence of Daxx (Hep parental mock) promoted reduced sensitivity of the chromatin to MNase digestion (Figure 9). Therefore, it is likely that Daxx reduction by either siRNA-mediated techniques or productive Ad5 infection results in a less condensed and accessible chromatin architecture. These findings demonstrate that functional Daxx/ATRX cooperation is important for chromatin remodelling, and depletion of these cellular factors results in a less condensed chromatin state and enhanced viral gene expression.
Figure 8.

Inhibition of functional Daxx/ATRX complex modulates chromatin structure and condensation. Hepa RG cells were infected with H5pg4100 Ad5 wild-type at moi of 5 and 100 FFU/cell. 24 h p.i. (A) and 48 h p.i. (B), cells were fixed with formaldehyde and analysed by ChIP assays (see ‘Material and Methods’ section). Isolated chromatin was precipitated with histone variant H3.3-specific polyclonal rabbit Ab. The average Ct-value was determined from triplicate reactions and normalized with standard curves for each primer pair (Table 2). The y-axis indicates the percentage of immunoprecipitated signal from the input (=100%). Values above 1% of input (dotted line) indicate chromatin/protein binding. The term ‘% of input’ is commonly used as y-axis label for ChIP assays and reflects the percentage of signal normalized to the original sample prior to IP. Values between 0% and 0.05% reflect no binding to promoter sequences. (C) Hepa RG cells were infected with H5pg4100 Ad5 wild-type at moi of 5 and 100 FFU/cell. Then, 24 h p.i. cells were harvested and total cell extracts were prepared. Proteins were separated by SDS–PAGE and subjected to immunoblotting using Daxx rabbit polyclonal antibody 07–471, ATRX-specific mouse MAb clone 39F, mouse E1B-55K-specific mouse Mab 2A6 and ß-actin mouse MAb AC-15 as a loading control.
DISCUSSION
Chromatin structure is regulated by multiprotein complexes that either covalently modify histone tails or remodel nucleosomes in an ATP-dependent manner (73). Together, these complexes cooperate to dynamically regulate chromatin structure. To date, considerable progress has been made in studying ATP-dependent chromatin remodelling complexes, mediating essential steps in regulating eukaryotic gene expression. These chromatin-associated complexes are highly conserved during evolution and often recruited to targets by sequence-specific transcription factors binding to certain promoters (74). SWI/SNF complexes use the energy of ATP-hydrolysis to alter histone/DNA contacts, leading to changes in chromatin conformation depending on acetylation of the N-terminal core histone tails (see Figure 6B). Although the basic mechanism is still not fully understood, obviously DNA viruses have acquired various strategies to interfere with chromatin remodelling, modulate histone/DNA contacts and establish lytic or latent infection.
Ad viruses infect a wide range of cell types, promoting cell cycle progression and simultaneously blocking apoptosis and growth arrest (23,75–77). Consequently, manipulation of host cell homeostasis is required for Ad productive infection. Considering the fundamental importance of chromatin formation in regulating gene expression in host cells, it remains unclear whether Ad DNA is chromatinized and whether Ad transcription is regulated by cellular chromatin remodelling complexes.
However, recent findings illustrate that Ad genomes are highly condensed by viral core proteins in the capsid (78–80). This core/DNA complex enters the host cell nucleus and decondenses before early viral gene transcription (72). Although the molecular details are still unclear, dynamic regulation of chromatin condensation suggests involvement of cellular chromatin remodelling complexes. Our findings illustrate that cellular Daxx/ATRX chromatin remodelling complexes associate with Ad genomes and promotes active viral gene transcription during the immediate early phase of Ad infection (Figure 3B).
Daxx is reportedly involved in depositing cellular histone variant H3.3 on actively transcribed genes (19,20,70). In line with this, Ad DNA was recently observed to preferentially associate with H3.3 (71), indicating that chromatinization by a replication-independent mechanism may play a so far unknown role during the Ad life cycle. Interestingly, H3.3 was also found to be complexed with ‘herpes simplex virus type 1’ DNA during the early phase of infection (81). Altogether, H3.3 deposition on incoming genomes of DNA viruses suggests a common mechanism for histone deposition in promoting efficient viral gene expression (71,81). Daxx’s impact on chromatinization via H3.3 deposition is consistent with our findings of functional Daxx-ATRX cooperation within a SWI/SNF chromatin remodelling complex (Figure 9, summarized in Figure 10).
Recent results together with our observations demonstrate the importance of a functional interplay between two types of Daxx chromatin remodelling activities on efficient Ad gene expression. As it is speculated that Ad DNA is chromatinized, epigenetic regulation may be as important for expression of Ad-encoded genes as for expression of host cell genes. Komatsu et al. (72) showed an increase over time in acetylated H3 association at all Ad promoters tested.
In agreement, we show here that Ad promoters are affected by Daxx/ATRX-mediated transcriptional repression. As acetylated histones are associated with actively transcribed genes, this suggests that inhibiting Daxx/ATRX recruitment of HDAC to these promoters changes the epigenetic status for optimal viral gene expression. Thus, E1B-55K/E4orf6-dependent restriction of Daxx/ATRX functional complexes may be relevant for the virus to evade antiviral host cell measures to repress viral gene expression. Indeed, recent reports demonstrate that during Ad vector studies, viral chromatin was preferentially associated with deacetylated histones, markers of transcriptionally inactive chromatin (71,84). These observations clearly illustrate the importance of epigenetic regulation of Ad DNA at the chromatin level by expressing early viral proteins that counteract chromatin remodelling functions of the cell.
To ensure efficient viral gene expression, it seems reasonable that Daxx and ATRX are eliminated during Ad5 infection, or their repressive capacity is limited by E1B-55K interaction, Daxx/ATRX protein degradation, or induction of posttranslational modifications. However, our findings illustrate that Ad5-mediated degradation discriminates between E1B-55K- (Daxx) and E1B-55K/E4orf6 (ATRX)-dependent pathways. Nevertheless, it is still unclear how and why Ad5 differentially regulates Daxx and ATRX proteolysis. Furthermore, it remains elusive whether the proposed mechanism is a kinetic process owing to expression or post-translational modification of viral proteins E1B-55K and E4orf6 early during Ad5 infection.
Chromatin modifying complexes have also been implicated in human cancer development. There is growing evidence for a correlation between chromatin modifiers and tumour suppression, especially demonstrated for SWI/SNF complexes, which comprise several subunits displaying tumour suppressor activity (73). Functional disruption of SWI/SNF complexes may induce a state of epigenetic instability, resulting in altered chromatin structure that affects gene expression and interferes with differentiation processes. These epigenetic changes may be closely linked to genomic instability and predispose to oncogenic transformation. In this context, our recent studies already provide evidence that efficient adenoviral transformation requires E1B-55K-mediated degradation of Daxx (61). In accordance to our current study, we assume that this Ad oncoprotein contributes to cell transformation by modulating Daxx-dependent pathways and consequently through disrupting SWI/SNF chromatin remodelling functions. This is a particularly interesting concept, given the oncogenic capabilities of certain adenoviral oncogenes (e.g. E1A, E1B-55K, E4orf6), which we have partially shown to cooperate with either Daxx and/or ATRX. Therefore, elucidating the mechanisms of these cellular regulators, as subunits of cellular SWI/SNF complexes, will help to define the role of chromatin remodelling in both Ad5 transcriptional regulation and adenoviral transformation of primary cells. These findings might also help to identify novel therapeutic approaches to modern cancer therapy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3.
FUNDING
The Heinrich Pette Institute, Leibniz Institute for Experimental Virology is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit (BMG). Part of this work was supported by the Deutsche Akademische Austauschdienst (DAAD) and the Bundesministerium für Bildung und Forschung (BMBF) with the PROCOPE program, the Peter und Traudl Engelhorn Stiftung, the Erich und Gertrud Roggenbuck Stiftung and the Horst Müggenburg Stiftung. Funding for open access charge: Heinrich Pette Institute.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank V. Lukashchuk, D. Cuchet-Lourenco, R. Gibbons, D. Picketts, P. Gripon, T. Shenk and J. Mymrik for providing reagents. They thank H. Wodrich, A. Grundhoff, H. Will, T. Speiseder, W. Ching and J. Berscheminski for scientific discussion and support.
REFERENCES
- 1.Schreiner S, Wimmer P, Sirma H, Everett RD, Blanchette P, Groitl P, Dobner T. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 2010;84:7029–7038. doi: 10.1128/JVI.00074-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ullman AJ, Hearing P. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 2008;82:7325–7335. doi: 10.1128/JVI.00723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torii S, Egan DA, Evans RA, Reed JC. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs) EMBO J. 1999;18:6037–6049. doi: 10.1093/emboj/18.21.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF, 3rd, Maul GG. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999;147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26:963–977. doi: 10.1002/bies.20089. [DOI] [PubMed] [Google Scholar]
- 6.Xu ZX, Zhao RX, Ding T, Tran TT, Zhang W, Pandolfi PP, Chang KS. Promyelocytic leukemia protein 4 induces apoptosis by inhibition of survivin expression. J. Biol. Chem. 2004;279:1838–1844. doi: 10.1074/jbc.M310987200. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H. PML nuclear bodies and apoptosis. Oncogene. 2004;23:2819–2824. doi: 10.1038/sj.onc.1207533. [DOI] [PubMed] [Google Scholar]
- 8.Gostissa M, Morelli M, Mantovani F, Guida E, Piazza S, Collavin L, Brancolini C, Schneider C, Del Sal G. The transcriptional repressor hDaxx potentiates p53-dependent apoptosis. J. Biol. Chem. 2004;279:48013–48023. doi: 10.1074/jbc.M310801200. [DOI] [PubMed] [Google Scholar]
- 9.Ishov AM, Vladimirova OV, Maul GG. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J. Cell Sci. 2004;117:3807–3820. doi: 10.1242/jcs.01230. [DOI] [PubMed] [Google Scholar]
- 10.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002;115:3319–3330. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Leo C, Zhu J, Wu X, O'Neil J, Park EJ, Chen JD. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 2000;20:1784–1796. doi: 10.1128/mcb.20.5.1784-1796.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl Acad. Sci. USA. 2003;100:10635–10640. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang J, Wu S, Liu H, Stratt R, Barak OG, Shiekhattar R, Picketts DJ, Yang X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004;279:20369–20377. doi: 10.1074/jbc.M401321200. [DOI] [PubMed] [Google Scholar]
- 14.Badens C, Lacoste C, Philip N, Martini N, Courrier S, Giuliano F, Verloes A, Munnich A, Leheup B, Burglen L, et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin. Genet. 2006;70:57–62. doi: 10.1111/j.1399-0004.2006.00641.x. [DOI] [PubMed] [Google Scholar]
- 15.Gibbons RJ, Bachoo S, Picketts DJ, Aftimos S, Asenbauer B, Bergoffen J, Berry SA, Dahl N, Fryer A, Keppler K, et al. Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nat. Genet. 1997;17:146–148. doi: 10.1038/ng1097-146. [DOI] [PubMed] [Google Scholar]
- 16.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl Acad. Sci. USA. 107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2007;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KH. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 20:351–360. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl Acad. Sci. USA. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat. Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, Tam A, Earle E, Anderson MA, Mann J, et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2009;19:404–414. doi: 10.1101/gr.084947.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shenk T. Adenoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Virology. 4th edn. Vol. 2. New York: Lippincott-Raven; 2001. pp. 2265–2300. [Google Scholar]
- 24.Nevins JR. Mechanism of activation of early viral transcription by the adenovirus E1A gene product. Cell. 1981;26:213–220. doi: 10.1016/0092-8674(81)90304-4. [DOI] [PubMed] [Google Scholar]
- 25.Nevins JR. Adenovirus E1A: transcription regulation and alteration of cell growth control. Curr. Top. Microbiol. Immunol. 1995;199(Pt.3):25–32. doi: 10.1007/978-3-642-79586-2_2. [DOI] [PubMed] [Google Scholar]
- 26.Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J. Virol. 2008;82:7252–7263. doi: 10.1128/JVI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nat. Rev. Mol. Cell. Biol. 2002;3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- 28.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 29.Chinnadurai G. Control of apoptosis by human adenovirus genes. Semin. Virol. 1998;8:399–408. [Google Scholar]
- 30.White E. Regulation of apoptosis by adenovirus E1A and E1B oncoproteins. Semin. Virol. 1998;8:505–513. [Google Scholar]
- 31.Williams J, Williams M, Liu C, Telling G. Assessing the role of E1A in the differential oncogenicity of group A and group C human adenoviruses. Curr. Top. Microbiol. Immunol. 1995;199:149–175. doi: 10.1007/978-3-642-79586-2_8. [DOI] [PubMed] [Google Scholar]
- 32.Dobner T, Kzhyshkowska J. Nuclear export of adenovirus RNA. Curr. Top. Microbiol. Immunol. 2001;259:25–54. doi: 10.1007/978-3-642-56597-7_2. [DOI] [PubMed] [Google Scholar]
- 33.Flint SJ, Gonzalez RA. Regulation of mRNA production by the adenoviral E1B 55-kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 2003;272:287–330. doi: 10.1007/978-3-662-05597-7_10. [DOI] [PubMed] [Google Scholar]
- 34.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11 Rad50 NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 36.Baker A, Rohleder KJ, Hanakahi LA, Ketner G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007;81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallaire F, Blanchette P, Groitl P, Dobner T, Branton PE. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 2009;83:5329–5338. doi: 10.1128/JVI.00089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orazio NI, Naeger CM, Karlseder J, Weitzman MD. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 2011;85:1887–1892. doi: 10.1128/JVI.02134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukashchuk V, Everett RD. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 2010;84:4026–4040. doi: 10.1128/JVI.02597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukashchuk V, McFarlane S, Everett RD, Preston CM. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008;82:12543–12554. doi: 10.1128/JVI.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glass M, Everett RD. Components of PML Nuclear Bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 2012;87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006;80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng CY, Blanchette P, Branton PE. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology. 2007;364:36–44. doi: 10.1016/j.virol.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Michaelson JS, Leder P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 2003;116:345–352. doi: 10.1242/jcs.00234. [DOI] [PubMed] [Google Scholar]
- 45.Cuchet-Lourenco D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 2011;7:e1002123. doi: 10.1371/journal.ppat.1002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham FL, Smiley J, Russel WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 47.Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D'Amico D, Bodner HK, Oie HK, Linnoila RI, Mulshine JL, Minna JD, et al. p53 gene mutations in non-small-lung cell cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7:171–180. [PubMed] [Google Scholar]
- 48.Niforou KM, Anagnostopoulos AK, Vougas K, Kittas C, Gorgoulis VG, Tsangaris GT. The proteome profile of the human osteosarcoma U2OS cell line. Cancer Genomics Proteomics. 2008;5:63–78. [PubMed] [Google Scholar]
- 49.Nevels M, Täuber B, Spruss T, Wolf H, Dobner T. "Hit-and-run" transformation by adenovirus oncogenes. J. Virol. 2001;75:3089–3094. doi: 10.1128/JVI.75.7.3089-3094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubenwolf S, Schütt H, Nevels M, Wolf H, Dobner T. Structural analysis of the adenovirus type 5 E1B 55-kilodalton-E4orf6 protein complex. J. Virol. 1997;71:1115–1123. doi: 10.1128/jvi.71.2.1115-1123.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berube NG, Healy J, Medina CF, Wu S, Hodgson T, Jagla M, Picketts DJ. Patient mutations alter ATRX targeting to PML nuclear bodies. Eur. J. Hum. Genet. 2008;16:192–201. doi: 10.1038/sj.ejhg.5201943. [DOI] [PubMed] [Google Scholar]
- 52.Groitl P, Dobner T. Construction of adenovirus type 5 early region 1 and 4 virus mutants. Methods Mol. Med. 2007;130:29–39. doi: 10.1385/1-59745-166-5:29. [DOI] [PubMed] [Google Scholar]
- 53.Blanchette P, Kindsmuller K, Groitl P, Dallaire F, Speiseder T, Branton PE, Dobner T. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 2008;82:2642–2651. doi: 10.1128/JVI.02309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kindsmuller K, Groitl P, Hartl B, Blanchette P, Hauber J, Dobner T. Intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by SUMO1 conjugation. Proc. Natl Acad. Sci. USA. 2007;104:6684–6689. doi: 10.1073/pnas.0702158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kindsmuller K, Schreiner S, Leinenkugel F, Groitl P, Kremmer E, Dobner T. A 49-kilodalton isoform of the adenovirus type 5 early region 1B 55-kilodalton protein is sufficient to support virus replication. J. Virol. 2009;83:9045–9056. doi: 10.1128/JVI.00728-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song JJ, Lee YJ. Role of the ASK1-SEK1-JNK1-HIPK1 signal in Daxx trafficking and ASK1 oligomerization. J. Biol. Chem. 2003;278:47245–47252. doi: 10.1074/jbc.M213201200. [DOI] [PubMed] [Google Scholar]
- 57.Gunther T, Grundhoff A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2011;6:e1000935. doi: 10.1371/journal.ppat.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Si H, Verma SC, Robertson ES. Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J. Virol. 2006;80:9017–9030. doi: 10.1128/JVI.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol. Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 60.Mund A, Schubert T, Staege H, Kinkley S, Reumann K, Kriegs M, Fritsch L, Battisti V, Ait-Si-Ali S, Hoffbeck A, et al. SPOC1 Modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res. 2012;40:11363–11379. doi: 10.1093/nar/gks868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schreiner S, Wimmer P, Groitl P, Chen SY, Blanchette P, Branton PE, Dobner T. Adenovirus type 5 early region 1B 55K oncoprotein-dependent degradation of cellular factor daxx is required for efficient transformation of primary rodent cells. J. Virol. 2011;85:8752–8765. doi: 10.1128/JVI.00440-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harlow E, Franza BR, Jr, Schley C. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 1985;55:533–546. doi: 10.1128/jvi.55.3.533-546.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarnow P, Sullivan CA, Levine AJ. A monoclonal antibody detecting the adenovirus type 5-E1b-58Kd tumor antigen: characterization of the E1b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology. 1982;120:510–517. doi: 10.1016/0042-6822(82)90054-x. [DOI] [PubMed] [Google Scholar]
- 64.Hancock MH, Corcoran JA, Smiley JR. Herpes simplex virus regulatory proteins VP16 and ICP0 counteract an innate intranuclear barrier to viral gene expression. Virology. 2006;352:237–252. doi: 10.1016/j.virol.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 65.Marton MJ, Baim SB, Ornelles DA, Shenk T. The adenovirus E4 17-kilodalton protein complexes with the cellular transcription factor E2F, altering its DNA-binding properties and stimulating E1A-independent accumulation of E2 mRNA. J. Virol. 1990;64:2345–2359. doi: 10.1128/jvi.64.5.2345-2359.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kindsmüller K, Groitl P, Härtl B, Blanchette P, Hauber J, Dobner T. Intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by SUMO1 conjugation. Proc. Natl Acad. Sci. USA. 2007;104:6684–6689. doi: 10.1073/pnas.0702158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maul GG, Ishov AM, Everett RD. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology. 1996;217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 68.McFarlane S, Preston CM. Human cytomegalovirus immediate early gene expression in the osteosarcoma line U2OS is repressed by the cell protein ATRX. Virus Res. 2011;157:47–53. doi: 10.1016/j.virusres.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 69.Schreiner S, Wimmer P, Dobner T. Adenovirus degradation of cellular proteins. Future Microbiol. 2012;7:211–225. doi: 10.2217/fmb.11.153. [DOI] [PubMed] [Google Scholar]
- 70.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ross PJ, Kennedy MA, Christou C, Risco Quiroz M, Poulin KL, Parks RJ. Assembly of helper-dependent adenovirus DNA into chromatin promotes efficient gene expression. J. Virol. 2011;85:3950–3958. doi: 10.1128/JVI.01787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Komatsu T, Haruki H, Nagata K. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res. 2011;39:889–901. doi: 10.1093/nar/gkq783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 74.Hassan AH, Neely KE, Vignali M, Reese JC, Workman JL. Promoter targeting of chromatin-modifying complexes. Front. Biosci. 2001;6:D1054–D1064. doi: 10.2741/hassan. [DOI] [PubMed] [Google Scholar]
- 75.Täuber B, Dobner T. Adenovirus early E4 genes in viral oncogenesis. Oncogene. 2001;20:7847–7854. doi: 10.1038/sj.onc.1204914. [DOI] [PubMed] [Google Scholar]
- 76.Täuber B, Dobner T. Molecular regulation and biological function of adenovirus early genes: the E4 ORFs. Gene. 2001;278:1–23. doi: 10.1016/s0378-1119(01)00722-3. [DOI] [PubMed] [Google Scholar]
- 77.Kosulin K, Haberler C, Hainfellner JA, Amann G, Lang S, Lion T. Investigation of adenovirus occurrence in pediatric tumor entities. J. Virol. 2007;81:7629–7635. doi: 10.1128/JVI.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsumoto K, Nagata K, Ui M, Hanaoka F. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J. Biol. Chem. 1993;268:10582–10587. [PubMed] [Google Scholar]
- 79.Okuwaki M, Nagata K. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J. Biol. Chem. 1998;273:34511–34518. doi: 10.1074/jbc.273.51.34511. [DOI] [PubMed] [Google Scholar]
- 80.Chatterjee PK, Vayda ME, Flint SJ. Adenoviral protein VII packages intracellular viral DNA throughout the early phase of infection. EMBO J. 1986;5:1633–1644. doi: 10.1002/j.1460-2075.1986.tb04406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009;83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salomoni P, Khelifi AF. Daxx: death or survival protein? Trends Cell Biol. 2006;16:97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ, Kim S. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J. Biol. Chem. 2001;276:39103–39106. doi: 10.1074/jbc.M105928200. [DOI] [PubMed] [Google Scholar]
- 84.Parks RJ, Bramson JL, Wan Y, Addison CL, Graham FL. Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J. Virol. 1999;73:8027–8034. doi: 10.1128/jvi.73.10.8027-8034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]