


Abstract
Oligonucleotides containing an immune-stimulatory motif and an immune-regulatory motif act as antagonists of Toll-like receptor (TLR)7 and TLR9. In the present study, we designed and synthesized oligonucleotide-based antagonists of TLR7, 8 and 9 containing a 7-deaza-dG or arabino-G modification in the immune-stimulatory motif and 2′-O-methylribonucleotides as the immune-regulatory motif. We evaluated the biological properties of these novel synthetic oligoribonucleotides as antagonists of TLRs 7, 8 and 9 in murine and human cell-based assays and in vivo in mice and non-human primates. In HEK293, mouse and human cell-based assays, the antagonist compounds inhibited signaling pathways and production of a broad range of cytokines, including tumour necrosis factor alpha (TNF-α), interleukin (IL)-12, IL-6, interferon (IFN)-α, IL-1β and interferon gamma-induced protein (IP)-10, mediated by TLR7, 8 and 9. In vivo in mice, the antagonist compounds inhibited TLR7- and TLR9-mediated cytokine induction in a dose- and time-dependent fashion. Peripheral blood mononuclear cells (PBMCs) obtained from antagonist compound-treated monkeys secreted lower levels of TLR7-, 8- and 9-mediated cytokines than did PBMCs taken before antagonist administration. The antagonist compounds described herein provide novel agents for the potential treatment of autoimmune and inflammatory diseases.
INTRODUCTION
Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns and elicit pathogen-specific innate and adaptive immune responses (1). Of the 11 TLRs identified in humans, TLR3, 7, 8 and 9 are expressed in endolysosomes and recognize pathogen-derived and synthetic nucleic acids (1,2). Several lines of evidence support that TLRs 7, 8 and 9 also recognize endogenous immune complexes containing self-nucleic acids in certain autoimmune disease conditions, including lupus, psoriasis, arthritis and multiple sclerosis, and induce pro-inflammatory cytokines that contribute to the pathogenesis of disease (3–8). Activation of TLRs 7, 8 and 9 by immune complexes leads to expression of interleukin (IL)-12, IL-6, tumour necrosis factor alpha (TNF-α), IL-1β, interferon (IFN)-α and IFN-inducible genes, which is associated with the presence of anti-DNA and anti-RNA autoantibodies in systemic lupus erythematosus (SLE) patients (9,10).
Extensive studies have used TLR7, 8 and 9 knock-out mice to elucidate the role of TLRs in SLE. Lupus disease and disease-associated parameters were abrogated in TLR7 knock-out lupus-prone mice (11). By contrast, lupus disease was exacerbated in TLR9 knock-out mice and these animals had elevated levels of serum IgG and IFN-α (11). Further, lupus disease was abrogated in TLR7 and 9 double–knock-out mice, suggesting that TLR7 plays a key role in lupus disease in mice and TLR9 regulates TLR7 (12). Moreover, TLR8 knock-out mice had elevated levels of nucleic acid autoantibodies and increased incidence of glomerulonephritis associated with increased expression of TLR7. Lupus disease was abrogated in TLR7 and TLR8 double–knock-out mice, however, suggesting that TLR8 controls TLR7 expression and plays a role in the regulation of TLR7 and modulates lupus disease in mice (13). Together these studies suggest that TLRs 7, 8 and 9 play a key role through a cross-talk in lupus and potentially in other autoimmune diseases (13). In humans, a SLE patient who acquired a genetic defect in TLR signaling experienced disease remission with disappearance of anti-DNA antibodies, suggesting further evidence of the role played by TLR signaling in SLE and other autoimmune diseases (14). Together these studies suggest that targeting TLRs 7, 8 and 9 with antagonists may provide a new strategy for treatment of autoimmune diseases, including lupus, psoriasis, arthritis and multiple sclerosis.
The antimalarial agent hydroxychloroquine (HCQ) is commonly used for the treatment of SLE and other autoimmune diseases (15,16). HCQ-treated SLE patient immune cells do not produce IFN-α and TNF-α in response to TLR7 and TLR9 agonist stimulation, suggesting that HCQ inhibits endosomal TLR-mediated immune responses (17). HCQ suppresses TLR-mediated immune responses via neutralization of endosomal acidification (18) and/or by binding to nucleic acids, thereby interfering with interactions between nucleic acids and TLRs without affecting TLR expression (19). However, HCQ causes severe toxicity including retinopathy, neuromyotoxicity and cardiotoxicity (20). Blocking TLR7-, 8- and 9-mediated immune responses with antagonist compounds at the receptor level could be a novel strategy for the treatment of autoimmune diseases while avoiding the toxicities associated with HCQ treatment.
Synthetic oligonucleotides containing poly-dG sequences act as antagonists of TLR9 and/or TLR7 (21–27). Although the mode of action of poly-dG–based compounds is not well understood, treatment of mice with these compounds has had therapeutic effects in mouse models of lupus, arthritis and multiple sclerosis (28–33). Additionally, the potential use of TLR9 inhibitors as possible corticosteroid-sparing agents has been demonstrated in lupus-prone mice (34). Evidence suggests that poly-dG–based compounds interfere with signal transducer and activator of transcription (STAT)1, -3 and -4 and/or other downstream factors proximal to nuclear factor (NF)-κB activation involved in the signaling pathways of TLRs (35,36). The use of poly-dG–based compounds as pharmacotherapies is limited, however, by their tendency to form quadruplex and other higher-order structures and to interact non-specifically with a number of proteins (37,38).
Immune-stimulatory oligonucleotides containing certain cytosine or guanosine modifications in the C or G position, respectively, of a CpG dinucleotide induce TLR9-mediated immune responses (2,39). By contrast, substitution of 2′-O-methyl-C, 2′-O-methyl-5-methyl-C or 5-methyl-dC for C or 2′-O-methyl-G for G leads to loss of immune-stimulatory activity; further, such oligos inhibit TLR7- and TLR9-mediated immune responses (40). Structure-activity relationship studies of immune-stimulatory oligonucleotides have shown that an accessible 5′-end is required for TLR9 activation and that blocking 5′-end with ligands impedes immune-stimulatory activity (2,41–45). In fact, immune-stimulatory oligonucleotides linked through a 3′-3′-attachment and containing two free 5′-ends have greater immune-stimulatory activity than do oligonucleotides containing a single 5′-end (2,41,42,45). Moreover, our studies have identified certain nucleotide and backbone modifications that modulate immune responses when they are incorporated site-specifically in the 5′- or 3′-flanking sequence adjacent to the immune-stimulatory motif (46–49). A systematic study of immune-stimulatory oligonucleotides containing site-specific 2′-O-methyl-ribonucleotide modifications showed that substitutions at the first or second nucleotide position adjacent to the immune-stimulatory motif on the 5′-side abrogated activity, and substitutions three or more nucleotides away in the 5′-flanking sequence enhanced activity (48).
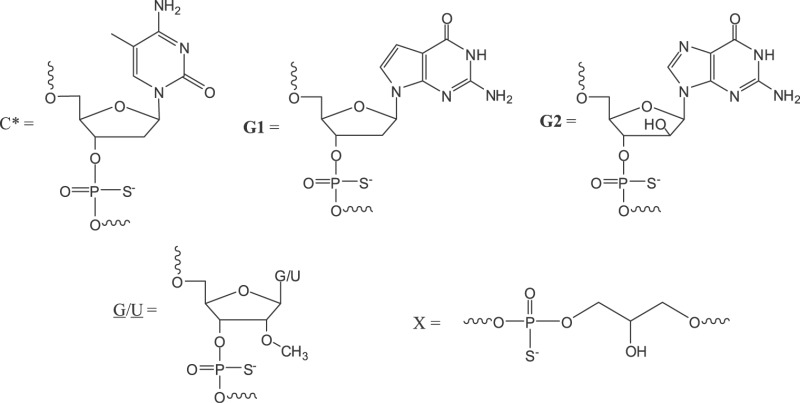
In further studies of oligonucleotides containing two 2′-O-methyl-ribonucleotide substitutions in the 5′-flanking sequence adjacent to the immune-stimulatory motif, we found that these compounds not only lose immune-stimulatory activity but also act as antagonists of TLR7 and TLR9 in vitro and in vivo (50). We referred to such chemical modifications incorporated adjacent to the immune-stimulatory motif, which result in TLR antagonist activity, as immune-regulatory motifs. In the present study, we have designed and synthesized oligonucleotides that contain nucleotide modifications within the CpG immune-stimulatory motif, in which C is replaced with 5-methyl-dC and G is replaced with 7-deaza-dG (G1) or arabino-G (G2), and which have two 2′-O-methylribonucleotides in the immune-regulatory motif (Table 1). Two of the antagonists (1 and 2) contained additional 2′-O-methylribonucleotide substitutions at the 3′-end to increase their stability against 3′-exonucleases (51). We designed antagonist compound 3 with two identical 11-mer sequences attached through their 3′-ends, so that the 3′-end was protected from nuclease degradation and the two free 5′-ends were available for TLR9 binding. We found that these antagonist compounds selectively inhibited TLR7-, 8- and 9-mediated immune responses in mouse and human cell-based assays and in vivo in mice and non-human primates (NHPs).
Table 1.
Antagonists and control compounds used in the study
| Number | Sequence and modificationa | Molecular weightb |
% Purityc |
|||
|---|---|---|---|---|---|---|
| Calculated | Found | IE-HPLC | RP-HPLC | CGE | ||
| 1 | 5′-CTATCTGUC*G1TTCTCTGU-3′ | 5801 | 5803 | 98 | 97 | 96 |
| 2 | 5′-CTATCTGUC*G2TTCTCTGU-3′ | 5818 | 5819 | 99 | 98 | 98 |
| 3 | 5′-CTTGUC*G1TTCT-X-TCTTG1C*UGTTC-5′ | 7266 | 7266 | 98 | 94 | 96 |
| Controls | ||||||
| 4 | 5′-CTATCTCACCTTCTCTGT-3′ | 5624 | 5623 | 98 | 96 | 98 |
| 5 | 5′-TCCATGCTAGGCTTATGT-3′ | 5754 | 5757 | 99 | 95 | 95 |
aAll are phosphorothioate oligodeoxyribonucleotides; G1 and G2 represent 2′-deoxy-7-deazaguanosine and arabinoguanosine, respectively; C* represents 2′-deoxy-5-methyl-cytidine; G and U are 2′-O-methyl-ribonucleotides, X stands for glycerol linker (structures shown below).
bMolecular weight as calculated and determined (found) by MALDI-ToF mass spectrometer.
c% Purity of full-length oligonucleotide as determined by anion-exchange HPLC (IE-HPLC), reverse-phase HPLC (RP-HPLC) and capillary gel electrophoresis (CGE).
MATERIALS AND METHODS
Synthesis and purification of antagonist compounds
All oligonucleotides shown in Table 1 were synthesized on a solid support using automated DNA/RNA synthesizers, deprotected, cleaved from the solid support, purified and analyzed as previously described (45,50). All oligonucleotides were characterized by capillary gel electrophoresis (CGE), high pressure liquid chromatography (HPLC) and Matrix-Assisted Laser Desorption/Ionization-Time of Flight (MALDI-ToF) mass spectrometry (Waters MALDI-ToF mass spectrometer with 337 nm N2 laser) for purity and molecular mass, respectively (Table 1). The purity of full-length oligonucleotides ranged from 95 to 98%, with the remainder lacking one or two nucleotides, as determined by ion-exchange HPLC and CGE. All oligonucleotides were tested for endotoxin levels by the Limulus assay (Bio-Whittaker) and contained <0.075 EU/mg.
Mice
Five- to eight-week-old female C57BL/6 mice were obtained from Charles River Labs (Wilmington, MA, USA) and maintained in the animal facility of Idera Pharmaceuticals under pathogen-free conditions. All the experimental procedures were performed as per the approved protocols and guidelines of the Institutional Animal Care and Use Committee of Idera Pharmaceuticals.
TLR agonists
TLR agonists used in the studies were purchased from the following sources: Poly I:C (TLR3), InvivoGen (San Diego, CA, USA); lipopolysaccharide (LPS, TLR4), Sigma (St. Louis, MO, USA); Flagellin from Salmonella muenchen (TLR5), InvivoGen (San Diego, CA, USA). RNA-based TLR7 agonist (5′-AACUG3ACG3CUU-X-UUCG3CAG3UCAA-5′; G3 stands for 7-deaza-G and X stands for glycerol linker) (52), RNA-based TLR8 agonist (5′-YUGCUGCCUUUG-X-GUUUCCGUCGUY-5′; Y stands for 1,3-propanediol linker) (53) and DNA-based mouse (mTLR9 agonist used in mouse in vitro and in vivo studies; 5′-TCTGACG1TTCT-X-TCTTG1CAGTCT-5′; G1 stands for 7-deaza-dG) (45) and human TLR9 agonist (hTLR9 agonist used in human cell-based assays and in NHPs; 5′-TCTGTCG1TTAG-X-GATTG1CTGTCT-5′) (39) were synthesized at Idera Pharmaceuticals as described above.
Cell culture assays of HEK293 cells expressing TLR3, 4, 7, 8 and 9
Human embryonic kidney (HEK)293 cells stably expressing human TLR3, TLR4/CD14/MD-2 or mTLR9 and HEK293XL cells stably expressing human TLR7 or TLR8 were obtained from Invivogen (San Diego, CA, USA). HEK cells were transiently transfected with reporter gene (SEAP, Invivogen) for 6 h. Appropriate TLR agonists were added to the cultures in the presence or absence of various concentrations of antagonists, and the cultures were continued for 18 h. At the end of the treatment, 20 µl of culture supernatant was taken from each treatment and tested for SEAP activity using 150 µl of Quanti-Blue substrate following the manufacturer’s protocol (Invivogen). The results are expressed as fold change in NF-κB activation over phosphate buffered saline (PBS)-treated cells.
Mouse spleen and J774 cell cultures
Spleen cells from 5- to 8-week-old C57BL/6 mice were cultured in RPMI complete medium as described earlier (39,48). Murine J774 macrophage cells (American Type Culture Collection, Rockville, MD, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics (100 IU/ml of penicillin G/streptomycin). All other culture reagents were purchased from Mediatech (Gaithersburg, MD, USA). Mouse spleen cells were plated in 24-well dishes using 5 × 106 cells/ml. Agonists and antagonists dissolved in TE buffer (10-mM Tris–HCl, pH 7.5, 1-mM ethylenediaminetetraacetic acid) were added alone or in combinations to the cell cultures. The cells were then incubated at 37°C for 24 h and the supernatants were collected for enzyme-linked immunosorbent assay (ELISA) or multiplex assays. The experiments were performed three times in duplicate wells.
The concentrations of cytokines and chemokines in spleen cell culture supernatants were evaluated using a mouse multiplex kit as described below.
Preparation of J774 cell nuclear extracts and electrophoretic mobility shift assay for NF-κB activation
For NF-κB and p38 activation, cells were plated at a density of 4–5 × 106 cells/well in six-well plates and allowed to attach overnight. The next day, the culture medium was changed (DMEM with FBS, no antibiotics) and cells were treated with agonist, antagonist or combinations of both. One hour after addition of the drugs, nuclear extracts were prepared and analyzed by native polyacrylamide gels as described earlier (40,50). Gels were dried and exposed to HyBlot CL autoradiography films at −70°C. Films were scanned, and the images were processed using Adobe imaging software.
Preparation of J774 whole cell lysates and western blotting for p38 activation
J774 cells were treated with agonist, antagonist or combinations as described above, the culture medium was removed, and cells were scraped off the dishes, washed twice with PBS and lysed on ice for 10 min in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). Cells were sonicated briefly and the extracts were centrifuged. Equal amounts of protein were subjected to sodium dodecyl sulphate gel electrophoresis using 10% Ready gels and blotted onto polyvinylidene difluoride (PVDF) membranes (BioRad Laboratories Hercules, CA, USA).We determined the levels of phosphorylated and total p38 mitogen-activated protein kinases (MAPKs) in J774 cells following agonist and/or antagonist treatment by western blotting. All primary antibodies required for the assay were purchased from Cell Signaling Technology, and the secondary antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Healthy human PBMC, mDC and pDC isolation
Peripheral blood mononuclear cells (PBMCs) from freshly drawn healthy human volunteer blood (Research Blood Components, Brighton, MA, USA) were isolated by Ficoll density gradient centrifugation (Ficoll-Paque PLUS, GE Health Care). Plasmacytoid dendritic cells (pDCs) and myeloid dendritic cells (mDCs) were isolated from PBMCs by positive selection using the blood dendritic cell antigen (BDCA)-4 and BDCA-1 cell isolation kits, respectively (Miltenyi Biotec, Auburn, CA, USA), according to the manufacturer’s instructions.
Human B-cell proliferation assay
Human B cells were isolated from PBMCs by positive selection using the CD19 cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. 1 × 105 B cells/0.2 ml were stimulated with different concentrations of agonist and antagonist for 64 h, then pulsed with 0.75 µCi of [3H]-thymidine and harvested 8 h later. The incorporation of [3H]-thymidine was measured using a scintillation counter and the data were shown as proliferation index.
Isolation of PBMCs from lupus patient blood
Whole blood from lupus patient donors was obtained from Bioreclamation Inc. (Westbury, NY, USA) in sodium citrate cell preparation (CP) tubes. On receipt, the entire contents of the tube above the gel layer were transferred into another tube, centrifuged and the pellet consisting of PBMCs was washed twice with PBS. PBMC assays were conducted and cytokine analysis was carried out using a multiplex kit as described below.
Human PBMC, mDC and pDC assays
Human PBMCs (5 × 106/ml), mDCs (1 × 106/ml) and pDCs (1 × 106/ml) were plated into 96-well plates in RPMI medium supplemented with 10% heat-inactivated defined FBS, 1.5 mM glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 50 µM 2-mercaptoethanol and 100 IU/ml penicillin–streptomycin mix. Agonists and antagonists dissolved in PBS were added to the cells at concentrations indicated in specific figure legends. The cells were then incubated at 37°C for 24 h. The levels of cytokines and chemokines in the culture supernatants were measured by using a human 25-plex kit.
Multiplex cytokine assays
Human PBMC, pDC and mDC culture supernatants and selected mouse serum samples from in vivo experiments were assayed using multiplex luminescent beads (human 25-plex and mouse cytokine 20-plex, Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions and analyzed with a Luminex 100 or 200 instrument. Fluorescence intensity was translated into cytokine concentration using StarStation software (Applied Cytometry Systems).
In vivo mouse studies
Female C57BL/6 mice 5–6 weeks old (n = 3) were injected subcutaneously (s.c.) with antagonist compounds at dose levels indicated in individual figure legends followed 24 h later (or as indicated in individual figures) by administration of a TLR agonist. Two hours after agonist administration, blood was collected by retro-orbital bleeding, and the serum cytokine levels were determined by mouse multiplex luminex assay or IL-12 ELISA as described above.
Non-human primate study and blood collection for PBMC assays
Eight healthy young cynomolgus monkeys (Macaca fascicularis) 2–5 years old and weighing ∼3–5 kg were used in the study. All in vivo non-human primate (NHP) studies were conducted at Ricerca (Concord, OH, USA) and were approved by Ricerca’s Institutional Animal Care and Use Committee. Animals were monitored daily. Each antagonist was administered s.c. to four animals (two males and two females) on Day 1 at 1.5 mg/kg. Blood samples (∼6 ml) were collected for PBMC assays pre-administration (Day 1) and at 24 (Day 2), 48 (Day 3) and 168 h (Day 8) after administration of antagonist. The whole blood was then shipped on ice packs to Idera Pharmaceuticals by overnight shipping for PBMC isolation and stimulation with TLR agonists. All animals remained in good health throughout the experiment.
Non-human primate PBMC assay
PBMCs were isolated by Ficoll density gradient method as described above. PBMCs at 1 × 106 cells/0.2 ml/well in 96-well plates were incubated with 50 μg/ml TLR7 agonist, 50 μg/ml TLR8 agonist, 3 μg/ml TLR9 agonist or 0.1 µg/ml TLR4 agonist (LPS) for 24 h. Supernatants were then harvested and stored frozen until cytokine assays.
Cytokine levels in culture supernatants were determined on a Luminex platform using monkey 28-plex magnetic cytokine antibody bead kits (Invitrogen). IFN-α (PBL, Piscataway, NJ, USA) and interferon gamma-induced protein (IP)-10 (R&D Systems, Minneapolis, MN, USA) levels were measured by ELISA.
RESULTS
Antagonist compounds do not induce NF-κB activation in TLR-expressing HEK293 cells
We measured the extent of activation of TLR3, 4, 7, 8 and 9 by antagonist compounds in HEK293 or HEK293XL cells expressing human TLR3, 4, 7, 8 or mouse TLR9. Antagonist compounds 1–3 or control 5 did not activate NF-κB in HEK293 cells expressing TLR3, 4, 7, 8 or 9 (Supplementary Data and Supplementary Figure S1), whereas appropriate agonists of TLR3, 4, 7, 8 and 9 did activate NF-κB in HEK293 cells expressing corresponding TLRs (Supplementary Figure S1). These data demonstrate that the antagonist compounds themselves do not induce TLR-mediated immune responses up to the concentration of 5–10 µg/ml studied in these assays.
Antagonist compounds inhibit TLR7-, 8 - and 9-mediated NF-κB activation in HEK293 cells
HEK293 cells expressing TLR7, 8 or 9 were incubated with their respective TLR agonist and 0–5 µg/ml of antagonist compound 1–3 or control 5. Control 5 did not inhibit TLR7-, 8 - and 9-mediated NF-κB activation (Figure 1). Antagonist compounds 1–3 inhibited TLR7-, 8 - and 9-mediated NF-κB activation in a dose-dependent manner, and almost complete inhibition was observed at the highest dose studied (Figure 1A–C). The IC50 values determined for antagonist compounds 1–3 for the inhibition of TLR7-, TLR8- and TLR9-mediated NF-κB activation are shown in Supplementary Table S1. None of the antagonist compounds significantly inhibited TLR3- or TLR4-agonist-induced NF-κB activation, suggesting that the antagonist compounds 1–3 selectively inhibit endosomal TLRs 7, 8 and 9 (Figure 1D and E).
Figure 1.

Antagonist compounds 1–3, but not control 5, inhibit NF-κB activation in (A) human TLR7-, (B) human TLR8- and (C) mouse TLR9-expressing HEK293 cells. Antagonist compounds do not inhibit NF-κB activation in (D) human TLR3- and (E) human TLR4-expressing HEK293 cells. HEK293 cells expressing human TLR3 and 4 and mouse TLR9 and human TLR7 and 8 XL-HEK293 cells were incubated with 10 µg/ml TLR3 agonist, 0.1 µg/ml TLR4 agonist, 5 µg/ml TLR9 agonist, 50 µg/ml TLR7 agonist and 50 µg/ml TLR8 agonist alone, respectively, and in combination with various concentrations of antagonist compounds 1–3 or control oligo 5. After 18 h of incubation, supernatants were assessed for NF-κB activation using SEAP assays as described in ‘Materials and Methods’ section. The results are expressed as fold change in NF-κB activation compared with PBS control. Each value shown is mean of two wells ± SD. Data shown are representative of three independent experiments.
Antagonist compounds inhibit TLR7- and TLR9-mediated NF-κB and p38 activation in J774 cells
Activation of TLRs by their ligands induces signaling pathways that result in the activation of the transcription factors NF-κB and p38. We evaluated the ability of antagonist compounds to inhibit TLR7- and TLR9-mediated NF-κB activation in mouse macrophage J774 cells. TLR8 is not functional in mice. We selected antagonist compound 1 as a representative example for these studies. As expected, agonists of TLR7 and TLR9, but not antagonist compound 1, induced NF-κB activation in J774 cells (Figure 2A and B). When J774 cells were incubated with antagonist compound 1 for 1 h before the addition of TLR7 or TLR9 agonist and NF-κB activation was measured 1 h after agonist addition, antagonist compound 1 inhibited TLR7- and TLR9-mediated NF-κB activation in a dose-dependent fashion (Figure 2A and B). On the contrary, antagonist compound 1 did not inhibit NF-κB activation induced by a TLR4 agonist (Figure 2C).
Figure 2.
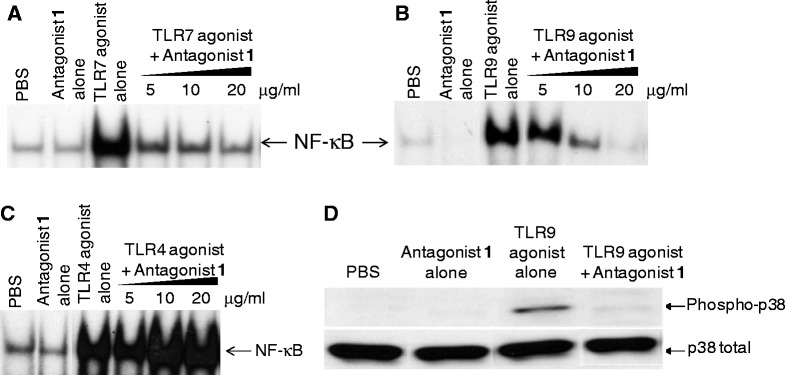
Effect of antagonist compound 1 on (A) TLR7-, (B) TLR9- and (C) TLR4-mediated NF-κB activation in J774 murine macrophage cells. J774 cells were incubated with various concentrations of antagonist compound 1 for an hour, and then further incubated for 1 h after adding 50 µg/ml TLR7 agonist or 1 µg/ml TLR9 agonist. Antagonist compound 1 alone treatment of J774 cells was carried out at 20 µg/ml concentration. Nuclear extracts were then prepared and electrophoretic mobility shift assay was carried out as described in ‘Materials and Methods’ section. (D) Antagonist compound 1 inhibits TLR9 agonist-induced p38 MAPK activation in J774 cells. Cells were incubated with PBS, antagonist compound 1 (10 µg/ml), TLR9 agonist (1 µg/ml) or antagonist compound 1 plus TLR9 agonist. Cells were pre-incubated with antagonist compound 1 for 1 h followed by TLR9 agonist stimulation for 30 min. Whole cell lysates were prepared and analyzed by western blotting as described in ‘Materials and Methods’ section. Data shown are representative of two independent experiments.
We further evaluated the ability of antagonist compound 1 to inhibit TLR9-mediated p38 activation in mouse macrophage J774 cells. Stimulation of J774 cells with a TLR9 agonist induced p38 phosphorylation, whereas antagonist compound 1 alone did not have any affect (Figure 2D). When J774 cells were incubated with the combination of antagonist and TLR9 agonist, the band corresponding to phosphorylated p38 was not observed, suggesting that antagonist compound 1 inhibited TLR9-mediated p38 activation in J774 cells (Figure 2D).
Antagonist compounds inhibit TLR7- and TLR9-mediated cytokine induction in mouse spleen cell cultures
We evaluated the ability of antagonist compounds to inhibit production of TLR7- and TLR9-agonist-induced cytokines in C57BL/6 mouse spleen cell cultures. Both TLR7 and TLR9 agonists induced potent IL-12 and IL-6 production in mouse spleen cell cultures (Figure 3A and B), whereas antagonist compounds alone did not (data not shown and Supplementary Figure S2). When spleen cells were co-incubated with a combination of a TLR agonist and antagonist compound 1, 2 or 3, antagonist compounds inhibited TLR7- and TLR9-mediated IL-12 and IL-6 production in a dose-dependent fashion (Figure 3A and B). Inhibition of additional cytokines by antagonist compound 1 is shown in Supplementary Figure S2. Control 5 did not inhibit TLR7- and TLR9-mediated cytokine production (Figure 3A and B). Antagonist compounds did not inhibit TLR4-mediated IL-12 and IL-6 production (Figure 3C).
Figure 3.

Dose-dependent inhibition of (A) TLR7- and (B) TLR9-mediated induction of IL-12 and IL-6 by antagonist compounds 1, 2 and 3 in mouse spleen cell cultures. Control oligo 5 has no effect on TLR7- and TLR9-mediated cytokine induction. (C) None of the antagonist compounds inhibited TLR4-mediated induction of IL-12 and IL-6. Spleen cells were incubated with TLR7 agonist (50 µg/ml), TLR9 agonist (1 µg/ml) or TLR4 agonist (1 µg/ml) in the absence or presence of various concentrations of antagonist compounds 1–3 or control oligo 5 for 24 h in triplicate wells. Cell culture supernatants were assessed for IL-12 and IL-6 levels by ELISA. Data shown are mean of triplicate wells ± SD and representative of three independent experiments.
Antagonist compounds inhibit TLR7-, TLR8- and TLR9-mediated cytokine production in human cell-based assays
We measured the inhibitory effects of antagonist compounds on TLR-mediated immune responses in cultures of healthy human PBMCs, pDCs, mDCs and B cells. Treatment of human PBMCs with TLR7, TLR8, TLR9 and TLR4 agonists alone resulted in the secretion of a number of cytokines, including TNF-α, IFN-α, IL-12, IL-6, IL-1β and IP-10 (Figure 4A–C and Supplementary Figure S3). The cytokine profile and the levels of cytokines induced varied for each TLR agonist. Co-incubation of human PBMCs with TLR7, TLR8 or TLR9 agonist and an antagonist compound resulted in the inhibition of TLR7-, TLR8- and TLR9-mediated cytokine and chemokine production (Figure 4A–C). All three antagonist compounds produced similar levels of inhibition (Figure 4A–C). At the same concentration, antagonist compounds had minimal effect on TLR4-mediated immune responses in human PBMC cultures (Supplementary Figure S3).
Figure 4.

Antagonist compounds 1, 2 and 3 inhibit (A) TLR7-, (B) TLR8- and (C) TLR9-mediated cytokine induction in human PBMCs. Human PBMCs from healthy volunteer blood were isolated and cultured with 50 µg/ml TLR7 agonist, 50 µg/ml TLR8 agonist or 3 µg/ml TLR9 agonist in the absence or presence of 10 µg/ml antagonist compound 1, 2 or 3 for 24 h. Supernatants were analyzed for cytokine levels by Luminex multiplex assay. Each data point represents one donor, and black line indicates mean of all donors.
Human pDCs express TLR7 and TLR9, and mDCs express TLR8. We therefore evaluated antagonist compounds for their ability to inhibit TLR7- and TLR9-mediated cytokine induction in pDCs and TLR8-mediated cytokine induction in mDCs. In human pDC cultures, both TLR7 and TLR9 agonists potently induced cytokine and chemokine secretion, including TNF-α, IFN-α, IL-12, IL-6 and IP-10 (Figure 5A and B). Antagonist compounds alone did not induce cytokine production (data not shown). Co-incubation of pDCs with TLR7 or TLR9 agonist and antagonist compounds resulted in the inhibition of TLR7- and TLR9-mediated cytokine and chemokine secretion. Figure 5A and B show inhibition of key cytokines and chemokines, TNF-α, IFN-α, IL-12, IL-6 and IP-10, induced by TLR7 and TLR9 agonists in pDCs.
Figure 5.

Antagonist compounds 1, 2 and 3 inhibit (A) TLR7- and (B) TLR9-mediated cytokine induction in human pDCs and (C) TLR8-mediated cytokine induction in human mDCs. Human pDCs from healthy human PBMCs were isolated and cultured with 50 µg/ml TLR7 agonist or 3 µg/ml TLR9 agonist in the absence or presence of 10 µg/ml antagonist compound 1, 2 or 3 for 24 h. Human mDCs from healthy human PBMCs were isolated and cultured with 50 µg/ml TLR8 agonist in the absence or presence of 10 µg/ml antagonist compound 1, 2 or 3 for 24 h. Supernatants were then analyzed for cytokine levels by Luminex multiplex assay. Each data point represents one donor, and black line indicates mean of all donors. (D) Antagonist compound 1 inhibits TLR9-mediated proliferation of B cells in a dose-dependent fashion. Human B cells (CD19+) were isolated from healthy human PBMCs and cultured in the presence of compounds for 72 h. B cell proliferation was determined and expressed as proliferation index as detailed in the ‘Materials and Methods’ section. TLR9 agonist concentration was 5 µg/ml and antagonist alone concentration was 20 µg/ml. Data shown are representative of three independent donors.
In human mDC cultures, TLR8 agonist alone induced production of a number of cytokines and chemokines, including TNF-α, IFN-α, IL-12, IL-6 and IP-10 (Figure 5C). Antagonist compounds alone did not induce cytokine production by mDCs (data not shown). Co-incubation of mDCs with TLR8 agonist and antagonist compounds resulted in the inhibition of TLR8-mediated production of cytokines and chemokines (Figure 5C).
Human B cells express TLR9, and agonists of TLR9 induce B-cell proliferation. Freshly isolated human B cells incubated with TLR9 agonist proliferated at a much higher rate than did PBS-treated cells (Figure 5D). By contrast, antagonist compound 1 alone induced minimal proliferation of human B cells (Figure 5D). A dose-dependent inhibition of TLR9-mediated B-cell proliferation was observed when B cells were co-incubated with TLR9 agonist and various concentrations of antagonist compound 1 (Figure 5D). Control 5 had minimal effects on TLR9-mediated B cell proliferation (data not shown).
Antagonist compounds inhibit TLR-mediated cytokine production by PBMCs from SLE patients
Chronic activation of TLRs by nucleic acid-containing immune complexes is an important component of autoimmune diseases such as SLE. To evaluate the antagonist’s ability to inhibit TLR-mediated immune responses in pathologic conditions, we isolated PBMCs from SLE patient blood, stimulated them with TLR7 and TLR9 agonists in the absence or presence of antagonist compound 1 and measured cytokine levels in the supernatants. Cultures treated with antagonist compound 1 alone produced background levels of cytokines comparable with that of PBS control cultures (Supplementary Figure S4A and B). Both TLR7 and TLR9 agonists alone induced production of several cytokines and chemokines, including IFN-α, TNF-α, IL-6, IP-10, macrophage inflammatory protein (MIP)-1α and MIP-1β (Supplementary Figure S4A and B). The levels of cytokines induced varied for the two TLR agonists (Supplementary Figure S4A and B). PBMCs from SLE patients co-incubated with TLR7 or TLR9 agonist and antagonist compound 1 produced much lower levels of TLR7- and TLR9-mediated cytokines (Supplementary Figure S4A and B).
Antagonist compounds inhibit TLR7- and TLR9-mediated cytokines in vivo in mice
To evaluate if antagonist compounds by themselves induce cytokine responses in vivo in mice, 2 h after s.c. administration of a dose of 15 mg/kg of antagonist compound 1 or 3 alone, blood samples were collected and analyzed for serum cytokine levels by multiplex analysis. TLR7 and TLR9 agonists alone induced production of several cytokines and chemokines, while antagonist compounds 1 and 3 did not (Supplementary Figure S5). Moreover, antagonist compound 1 injected to mice up to 45 mg/kg dose did not induce immune responses (data not shown). These results demonstrate that antagonist compounds by themselves do not induce cytokine responses in mice.
To evaluate the in vivo inhibitory effects of antagonist compounds on TLR7- and TLR9-mediated immune responses, we injected antagonist compounds 1–3 or control 4 (5 mg/kg) s.c. in the left flank and 24 h later TLR7 (10 mg/kg) or TLR9 agonist (0.25 mg/kg) s.c. in the right flank. Two hours after agonist administration, blood was collected and serum IL-12 levels were measured by ELISA. Antagonist compounds inhibited both TLR7- and TLR9-mediated IL-12 production in mice (Figure 6). At the dose level studied, antagonists 1–3 showed 84%, 93% and 78% inhibition of TLR7 agonist-induced IL-12 production, respectively (Figure 6). At the same dose, antagonist compounds 1–3 showed 68%, 69% and 70% inhibition of TLR9 agonist-induced IL-12 production, respectively (Figure 6). Control 4 did not inhibit either TLR7- or TLR9-mediated IL-12 production in mice (Figure 6).
Figure 6.
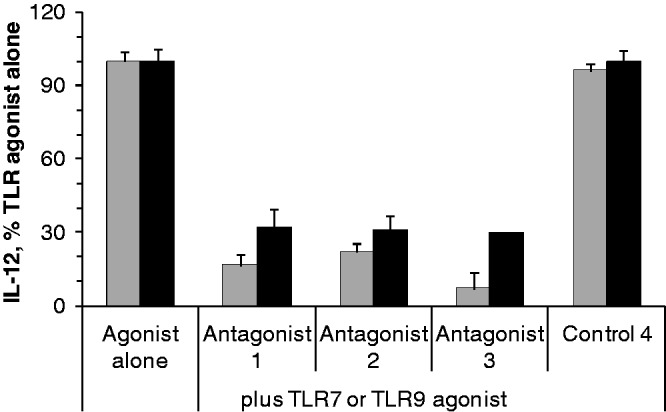
Inhibitory effect of antagonist compounds on TLR7- (grey bars) and TLR9- (black bars) mediated IL-12 induction in mice. Antagonist compound 1, 2, 3 or control oligo 4 was injected s.c. at 5 mg/kg dose at 0 h, and 24 h later, 10 mg/kg TLR7 agonist or 0.25 mg/kg TLR9 agonist was injected to C57BL/6 mice (N = 3). Two hours after agonist administration, blood was collected and serum IL-12 levels were determined by ELISA. Data shown are representative of three independent experiments.
Specificity of antagonist compounds for different TLR agonists in vivo in mice
We further evaluated the impact of antagonists on TLR3- and TLR5-mediated immune responses in vivo in mice. Antagonist compounds 1 and 3 were administered at 5 mg/kg s.c. to mice, and 24 h later an agonist of TLR3 or TLR5 was administered in the opposite flank. Blood was collected 2 h after TLR agonist administration and serum cytokine levels were determined using a Luminex multiplex assay. The agonists of TLR3 and TLR5 alone induced production of various cytokines and chemokines while a PBS control did not (Supplementary Figure S6). Antagonists 1 and 3 had minimal effects on TLR3- and TLR5-mediated cytokine production in mice, suggesting that the inhibitory effects of antagonists are specific to TLR7 and TLR9 in mice (Supplementary Figure S6).
Dose dependency and duration of inhibitory activity of antagonist compound in vivo in mice
We evaluated the effect of the dose of antagonist on the extent and duration of inhibition of TLR7- and TLR9-mediated cytokine production in mice. Mice were injected s.c. with 1-, 5- or 15-mg/kg antagonist compound 1 followed by 10 mg/kg of TLR7 agonist or 0.25 mg/kg of TLR9 agonist at 24 (Day 1), 120 (Day 5), 216 (Day 9) or 336 h (Day 14) after antagonist compound administration, and blood was collected 2 h after agonist administration for serum cytokine analysis by multiplex assay. The extent of inhibition of TLR7- and TLR9-mediated immune responses by the antagonist was dependent on the dose of antagonist compound administered and the type of cytokine induced by the TLR agonist (Figure 7A and B and Supplementary Figures S7 and S8). At the lowest dose of antagonist compound 1 (1 mg/kg), a 30–55% inhibition of various cytokines induced by TLR7 and TLR9 agonists was observed. The extent of inhibition ranged from 55 to 80% at 5-mg/kg and 90 to 100% at 15-mg/kg dose of antagonist compound 1 for various cytokines induced by TLR7 and TLR9 agonists (Figure 7A and B and Supplementary Figures S7 and S8). Maximal inhibition of cytokines was observed at 24–48 h after antagonist administration (Figure 7A and B and Supplementary Figures S7 and S8). The duration of inhibition was dependent on the dose of antagonist compound administered and type of cytokine induced by TLR agonist. A single dose of 5-mg/kg antagonist compound 1 resulted in ≥50% inhibition of most cytokines up to 6–7 days. At 15-mg/kg dose ≥50% inhibition of TLR7- and TLR9-mediated cytokine induction was observed for up to 10–12 days (Figure 7A and B and Supplementary Figures S7 and S8). These results suggest that once-a-week administration of antagonist compound would be sufficient to maintain suppression of TLR7- and TLR9-mediated immune responses.
Figure 7.

Effect of dose of antagonist compound on (A) TLR7- and (B) TLR9-mediated IL-12 induction and duration of activity in mice. C57BL/6 mice (N = 3) were injected s.c. with 1, 5 or 15 mg/kg of antagonist compound 1. After 1, 5, 9, or 14 days of antagonist compound administration, an agonist of TLR7 (10 mg/kg) or TLR9 (0.25 mg/kg) was injected s.c.. Blood was drawn 2 h after TLR agonist administration at each time point and serum cytokines were measured by Luminex multiplex assay. Inhibition of additional cytokines induced by TLR7 and TLR9 agonists is shown in Figures S7 and S8, respectively. Effect of dose of (C) TLR7 and (D) TLR9 agonists on the extent and duration of inhibition by antagonist compound 1 in mice. C57BL/6 mice (N = 3) were injected s.c. with 5 mg/kg antagonist compound 1. After 1, 5, 9 or 14 days of antagonist compound administration, an agonist of TLR7 (5, 10 or 50 mg/kg) or TLR9 (0. 125, 0.25 or 0.5 mg/kg) was injected s.c. Blood was drawn 2 h after TLR agonist administration at each time point and serum cytokines were measured by Luminex multiplex assay. Inhibition of additional cytokines induced by TLR7 and TLR9 agonists is shown in Supplementary Figures S9 and S10, respectively. Data shown are representative of two independent experiments.
Agonist dose dependency and duration of inhibitory activity of antagonist compound in vivo in mice
To study the extent of inhibition of TLR-mediated immune responses by antagonist compounds, we determined the ability of antagonist to inhibit TLR-mediated cytokine induction at different doses of TLR agonists. Mice were injected s.c. with a dose of 5 mg/kg of antagonist compound 1 at 0 h (Day 0) and 0.125, 0.25 or 0.5 mg/kg of TLR9 agonist or 5, 10 or 50 mg/kg TLR7 agonist at 24 (Day 1), 120 (Day 5), 216 (Day 9) or 336 h (Day 14) after antagonist administration. Blood was collected 2 h after agonist administration at each time point and serum cytokines were assessed by multiplex assay. Antagonist compound 1 produced 98, 91 and 83% inhibition of TLR7-mediated IL-12 induction on day 1 at 5, 10 and 50 mg/kg doses of TLR7 agonist, respectively (Figure 7C). Similarly, antagonist compound 1 produced 99, 84 and 58% inhibition of TLR9-mediated IL-12 induction on day 1 at 0.125, 0.25 and 0.5 mg/kg dose of TLR9 agonist, respectively (Figure 7D). The extent and duration of inhibition by the antagonist was dependent on the dose of TLR agonist administered. A single dose of 5 mg/kg of antagonist compound 1 produced ≥50% inhibition of IL-12 for up to 7 and 9 days at 10 and 5-mg/kg TLR7 agonist, respectively (Figure 7C and Supplementary Figure S9), and ≥50% inhibition of IL-12 for up to 8 and 11 days at 0.25- and 0.125-mg/kg TLR9 agonist, respectively (Figure 7D and Supplementary Figure S10). These results demonstrate that antagonist compounds have the potential to inhibit TLR7- and TLR9-mediated immune responses to low levels of agonists for an extended period of time.
Antagonist compounds inhibit TLR7-, TLR8- and TLR9-mediated cytokine induction in vivo in non-human primates
We evaluated the inhibitory effects of antagonist compounds 1 and 3 on TLR7-, TLR8- and TLR9-mediated immune responses in a single-dose study in cynomolgus monkeys at multiple time points during a 1-week period. Monkeys received a single s.c. administration of antagonist compound 1 or 3 at 1.5 mg/kg. Blood was collected at pre-administration and 24, 48 and 168 h after antagonist administration. PBMCs from monkey blood were isolated and stimulated with a TLR7, TLR8, TLR9 or TLR4 agonist for 24 h, and cytokine and chemokine levels in the supernatants were determined. PBMCs from monkeys at the pre-administration time point produced high levels of various cytokines and chemokines in response to the TLR agonists and the cytokine profile was dependent on the type of agonist used for stimulation. A single dose of antagonist compound administered to monkeys resulted in maximal suppression of most cytokines induced by TLR7, 8 and 9 agonists at 48 h. Cytokine levels returned or showed a trend towards returning to pre-administration levels by ∼168 h (Day 8). Data for representative cytokines for duration of inhibition by antagonist compound 1 are shown in Supplementary Figure S11.
Figure 8 shows levels of key cytokines (TNF-α, IL-6, IL-12, IL-1β, IFN-α and IP-10) induced in individual monkey PBMCs by TLR7, TLR8, TLR9 and TLR4 agonists at pre-administration and at 48 h after administration of antagonist compound 1. There was individual variability in cytokine induction by TLR agonists, but cytokine production by PBMCs obtained after administration of the antagonist was greatly reduced. Similar results were observed for antagonist compound 3 (Supplementary Figure S12). Percentage inhibition of various cytokines and chemokines at 48 h compared with pre-administration levels is shown in Table 2 for antagonist compounds 1 and 3. TLR4-mediated cytokine production was affected only minimally (Figures 8D and Supplementary Figure S12D). These results demonstrate that the antagonist compounds inhibit TLR7-, TLR8- and TLR9-, but not TLR4-, mediated cytokine production in NHPs.
Figure 8.

Antagonist compound 1-treated NHP PBMCs show reduced cytokine production in response to (A) TLR7, (B) TLR8 and (C) TLR9, but not (D) TLR4, agonist stimulation. Cynomolgus monkeys (N = 4) were injected s.c. with 1.5 mg/kg antagonist compound 1 and blood was collected at pre-administration and various post-administration time points. PMBCs were isolated and stimulated for 24 h with TLR7 (50 µg/ml), TLR8 (50 µg/ml), TLR9 (3 µg/ml) and TLR4 (0.1 µg/ml) agonists. Cell culture supernatants were analyzed for cytokine levels using a multiplex assay. Pre and Post indicate pre-administration and 48 h post-administration time points. Each individual data point indicates data for one animal, and black line indicates mean of all animals.
Table 2.
Percent inhibition of TLR7, TLR8 and TLR9 agonist-induced immune responses in PBMCs obtained from antagonist-treated NHPs
| % Inhibition to pre-dosea | ||||||
|---|---|---|---|---|---|---|
| Cytokineb | TLR7 agonist |
TLR8 agonist |
TLR9 agonist |
|||
| Antagonist | 1 | 3 | 1 | 3 | 1 | 3 |
| IL-6 | 65.9 | 55.3 | 72.8 | 30.7 | 15.5 | 8.0 |
| IL-12 | 60.2 | 56.8 | 67.5 | 42.0 | 4.0 | 23.3 |
| IP-10 | 15.2 | 36.9 | 46.7 | 57.6 | 64.2 | 84.1 |
| I-TAC | 82.8 | 64.8 | 79.2 | 68.0 | 55.8 | 42.3 |
| MCP-1 | 60.0 | 58.0 | 18.0 | 27.2 | 44.1 | 5.5 |
| IFN-γ | 82.8 | 45.2 | 79.2 | 54.6 | 7.3 | 6.0 |
| IL-1β | 55.7 | 19.0 | 84.1 | 31.0 | 15.2 | |
| IL-1Ra | 51.3 | 38.2 | 26.0 | 19.5 | 21.6 | 28.8 |
| TNF-α | 92.0 | 87.8 | ||||
| IFN-α | 36.7 | 71.2 | 44.3 | 57.2 | 23.7 | |
a% Inhibition of cytokines induced at 48 h post antagonist dosing compared with pre-dose levels. PBMC assays were carried out as described in ‘Materials and Methods’ section. Data shown are mean of four animals. Dotted lines indicate cytokine response was not observed at pre-dose.
bI-TAC, interferon-inducible T cell alpha chemoattractant; MCP-1, monocyte chemotactic protein-1.
DISCUSSION
Our previous studies identified certain nucleotide and backbone modifications that modulate immune responses when incorporated site specifically in the 5′- or 3′-flanking sequence adjacent to the immune-stimulatory motif (46–49). Further studies showed that immune-stimulatory oligonucleotides containing site-specific 2′-O-methyl-ribonucleotide substitutions at the first or second nucleotide position adjacent to the immune-stimulatory motif on the 5′-side abrogate immune-stimulatory activity, and substitutions three or more nucleotides away in the 5′-flanking sequence enhance activity (48). Oligonucleotides containing two 2′-O-methyl-ribonucleotide substitutions in the 5′-flanking sequence adjacent to immune-stimulatory motif do not activate TLR9 but act as antagonists of TLR7 and TLR9 in vitro and in vivo (49). We referred to chemical modifications incorporated adjacent to the immune-stimulatory motif, which result in TLR antagonist activity, as immune-regulatory motifs. In the present study, we designed and synthesized oligonucleotides containing nucleotide modifications within the CpG immune-stimulatory motif, in which C is replaced with 5-methyl-dC and G is replaced with 7-deaza-dG (G1) or arabino-G (G2), and which has two 2′-O-methylribonucleotides in the immune-regulatory motif. Antagonist compound 3 contained the same motifs with two identical 11-mer sequences attached through their 3′-ends.
The new antagonist compounds 1–3 described herein did not induce immune responses in cell-based assays and in vivo in mice compared with PBS and control oligos. Moreover, antagonist compounds inhibited TLR7-, TLR8- and TLR9-mediated NF-κB activation in HEK293 cells in a dose-dependent manner when co-incubated with these TLR agonists. Antagonist compounds inhibited TLR7- and TLR9-mediated NF-κB and TLR9-mediated p38 activation in J774 cells, suggesting that the inhibition of TLR-mediated immune responses occurs upstream, but not downstream, of these transcription factors.
The new antagonist compounds inhibited TLR7-, 8- and 9-mediated cytokine induction, including TNF-α, IFN-α, IP-10, IL-12, IL-6 and IL-1β, in human PBMCs, pDCs and mDCs and TLR9-mediated proliferation of human B cells. Antagonist compounds also inhibited TLR7- and TLR9-mediated cytokine production in cultures of PBMCs from SLE patients, suggesting that the antagonist compounds can inhibit endogenous immune complex-mediated cytokine production in people with autoimmune diseases such as SLE, psoriasis and rheumatoid arthritis.
Antagonist compounds injected systemically at doses of up to 15–45 mg/kg did not induce production of cytokines and chemokines in mice, while TLR7 and TLR9 agonists at the same or lower doses induced robust cytokine and chemokine production. Antagonist compounds administered systemically to mice before TLR agonist administration at a remote site inhibited TLR7- and TLR9-mediated cytokine production. The inhibitory effects were dependent on the dose of antagonist compound as well as that of the TLR agonist administered. The inhibitory effect of antagonist compounds persisted for up to 5–9 days depending on the dose of TLR agonist and antagonist compound administered.
Systemic administration of an antagonist compound to NHPs resulted in reduced TLR7-, TLR8- and TLR9-mediated cytokine responses in PBMCs. A minimal effect was observed on TLR4-mediated cytokine production, suggesting that the effects of antagonist compounds are selective for TLR7, TLR8 and TLR9. The greatest inhibitory effect was observed at 48 h after antagonist administration and the cells regained basal levels of cytokine secretion within 1 week after antagonist compound administration.
Inappropriate or uncontrolled TLR signaling has been implicated in certain autoimmune and inflammatory conditions, including SLE, psoriasis, rheumatoid arthritis and multiple sclerosis (3–8). Endogenous immune complexes containing self-nucleic acids act as ligands for TLR7 and TLR9 and induce pro-inflammatory cytokines, including expression of IL-12, TNF-α, IL-1β, IL-6, IFN-α and IFN-inducible genes, contributing to the pathogenesis of SLE and other autoimmune diseases (9,10). The current treatment options for autoimmune and inflammatory diseases include cytotoxic and immune-modulatory agents. The antimalarial drug HCQ, commonly used for the treatment of autoimmune diseases, inhibits TLR activation through neutralization of endosomal acidification and/or by direct interaction with nucleic acids present within immune complexes (19,20). Monoclonal antibodies directed against cytokines such as TNF-α, IFN-α, IL-6, IL-12/IL-23, IL-17 and IP-10, and factors such as B-cell activating factor (BAFF) that activate B cells are also commonly used as treatments for autoimmune diseases (54–56). These agents act on a single component of the inflammatory response or one type of cell population. The antagonist compounds described herein selectively inhibited multiple cytokines produced after TLR7, 8 and 9 activation in human PBMCs, DCs and B cell proliferation, suggesting that the antagonists can inhibit all sources of inflammation mediated through these receptors implicated in autoimmune diseases. An antagonist compound, referred to as IMO-3100, has shown good safety profile and proof of concept of target engagement of TLR7 and TLR9 in healthy human subjects in phase 1 clinical trials (57) and is currently being evaluated in psoriasis patients in a phase 2 clinical trial. Another antagonist compound IMO-8400, which inhibits TLR7-, 8- and 9-mediated immune responses, is being developed for lupus treatment and is in phase 1 safety studies.
The present studies demonstrated that oligonucleotide-based antagonist compounds containing a (5-methyl-dC)p(7-deaza-dG) or (5-methyl-dC)p(arabino-G) motif and an immune-regulatory 2′-O-methylribonucleotide motif adjacent to the immune-stimulatory motif acted as antagonists of TLR7, TLR8 and TLR9 in various cell-based assays and in mice and NHPs. The two 2′-O-methylribonucleotide modifications at the 3′-end of antagonist compounds 1 and 2 and the 3′-3′-linked structure in antagonist compound 3 contribute to the metabolic stability against 3′-exonucleases (51). These novel antagonist compounds allow specific inhibition of intracellularly expressed TLRs and could be suitable candidates for treating inflammatory and autoimmune diseases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–12.
FUNDING
Funding for open access charge: Idera Pharmaceuticals, Inc.
Conflict of interest statement. All authors are/were employees of Idera Pharmaceuticals, Inc. and hold stock options.
Supplementary Material
REFERENCES
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Kandimalla ER, Agrawal S. Modulation of endosomal Toll-like receptor-mediated immune responses by synthetic oligonucleotides. Adv. Polym. Sci. 2012;249:61–94. [Google Scholar]
- 3.Celhar T, Magalhaes R, Fairhurst AM. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol. Res. 2012;53:58–77. doi: 10.1007/s12026-012-8270-1. [DOI] [PubMed] [Google Scholar]
- 4.Busconi L, Lau CM, Tabor AS, Uccellini MB, Ruhe Z, Akira S, Viglianti GA, Rifkin IR, Marshak-Rothstein A. DNA and RNA autoantigens as autoadjuvants. J. Endotoxin Res. 2006;12:379–384. doi: 10.1179/096805106X118816. [DOI] [PubMed] [Google Scholar]
- 5.Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J. Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 7.Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschnning CJ, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J. Clin. Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alzabin S, Kong P, Medghalchi M, Palfreeman A, Williams R, Sacre S. Investigation of the role of endosomal toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res. Ther. 2012;14:R142. doi: 10.1186/ar3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crow MK. Interferon pathway activation in systemic lupus erythematosus. Curr. Rheumatol. Rep. 2005;7:463–468. doi: 10.1007/s11926-005-0053-4. [DOI] [PubMed] [Google Scholar]
- 10.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. J. Interferon Cytokine Res. 2011;31:887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, Valenzuela DM, Yancopoulos GD, Flavell RA, Alexopoulou L. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Invest. 2010;120:3651–3662. doi: 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visentini M, Conti V, Cagliuso M, Tinti F, Siciliano G, Trombetta AC, Mitterhofer AP, Fiorilli M, Quinti I. Regression of systemic lupus erythematosus after development of an acquired Toll-like receptor signaling defect and antibody deficiency. Arthritis Rheum. 2009;60:2767–2771. doi: 10.1002/art.24760. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012;42:145–153. doi: 10.1007/s12016-010-8243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat. Rev. Nephrol. 2011;7:718–729. doi: 10.1038/nrneph.2011.150. [DOI] [PubMed] [Google Scholar]
- 17.Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production plasmacytoid denderitic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2012;14:R155. doi: 10.1186/ar3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus. 1996;5(Suppl. 1):S4–S10. [PubMed] [Google Scholar]
- 19.Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011;186:4794–4804. doi: 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- 20.Tang C, Godfrey T, Stawell R, Nikpour M. Hydroxychloroquine in lupus: emerging evidence supporting multiple beneficial effects. Intern. Med. J. 2012;42:968–978. doi: 10.1111/j.1445-5994.2012.02886.x. [DOI] [PubMed] [Google Scholar]
- 21.Ashman RF, Goeken JA, Latz E, Lenert P. Optimal oligonucleotide sequences for TLR9 inhibitory activity in human cells: lack of correlation with TLR9 binding. Int. Immunol. 2011;23:203–214. doi: 10.1093/intimm/dxq473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pisetsky DS, Reich CF. Inhibition of murine macrophage IL-12 production by natural and synthetic DNA. Clin. Immunol. 2000;96:198–204. doi: 10.1006/clim.2000.4897. [DOI] [PubMed] [Google Scholar]
- 23.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duramad O, Fearon KL, Chang B, Chan JH, Gregorio J, Coffman RL, Barrat FJ. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J. Immunol. 2005;174:5193–5200. doi: 10.4049/jimmunol.174.9.5193. [DOI] [PubMed] [Google Scholar]
- 25.Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J. Immunol. 2003;171:1393–1400. doi: 10.4049/jimmunol.171.3.1393. [DOI] [PubMed] [Google Scholar]
- 26.Krieg AM, Wu T, Weeratna R, Efler SM, Love-Homan L, Yang L, Yi AK, Short D, Davis HL. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc. Natl. Acad. Sci. USA. 1998;95:12631–12636. doi: 10.1073/pnas.95.21.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenert PS. Classification, mechanisms of action, and therapeutic applications of inhibitory oligonucleotides for Toll-like receptors (TLR) 7 and 9. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/986596. 986596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenert P, Yasuda K, Busconi L, Nelson P, Fleenor C, Ratnabalasuriar RS, Nagy PL, Ashman RF, Rifkin IR, Marshak-Rothstein A. DNA-like class R inhibitory oligonucleotides (INH-ODNs) preferentially block autoantigen-induced B-cell and dendritic cell activation in vitro and autoantibody production in lupus-prone MRL-Fas(lpr/lpr) mice in vivo. Arthritis Res. Ther. 2009;11:R79. doi: 10.1186/ar2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB x NZW mice. Arthritis Rheum. 2005;52:651–658. doi: 10.1002/art.20810. [DOI] [PubMed] [Google Scholar]
- 30.Patole PS, Zecher D, Pawar RD, Gröne HJ, Schlöndorff D, Anders HJ. G-rich DNA suppresses systemic lupus. J. Am. Soc. Nephrol. 2005;16:3273–3280. doi: 10.1681/ASN.2005060658. [DOI] [PubMed] [Google Scholar]
- 31.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol. 2007;37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 32.Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligonucleotides protect against collagen-induced arthritis in mice. Arthritis Rheum. 2004;50:1686–1689. doi: 10.1002/art.20263. [DOI] [PubMed] [Google Scholar]
- 33.Ho PP, Fontoura P, Platten M, Sobel RA, DeVoss JJ, Lee LY, Kidd BA, Tomooka BH, Capers J, Agrawal A, et al. A suppressive oligodeoxynucleotide enhances the efficacy of myelin cocktail/IL-4-tolerizing DNA vaccination and treats autoimmune disease. J. Immunol. 2005;175:6226–6234. doi: 10.4049/jimmunol.175.9.6226. [DOI] [PubMed] [Google Scholar]
- 34.Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, Chan HJ, Wright T, Punaro M, Bolland S, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature. 2010;465:937–941. doi: 10.1038/nature09102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenert P, Stunz L, Yi AK, Krieg AM, Ashman RF. CpG stimulation of primary mouse B cells is blocked by inhibitory oligodeoxyribonucleotides at a site proximal to NF-kappaB activation. Antisense Nucleic Acid Drug Dev. 2001;11:247–256. doi: 10.1089/108729001317022241. [DOI] [PubMed] [Google Scholar]
- 36.Shirota H, Gursel M, Klinman DM. Suppressive oligodeoxynucleotides inhibit Th1 differentiation by blocking IFN-γ- and IL-12-mediated signaling. J. Immunol. 2004;173:5002–5007. doi: 10.4049/jimmunol.173.8.5002. [DOI] [PubMed] [Google Scholar]
- 37.Agrawal S, Iadorola PL, Temsamani J, Zhao Q, Shaw D. Effect of G-rich sequences on the synthesis, purification, hybridization, cell uptake, and hemolytic activity of oligonucleotides. Bioorg. Med. Chem. Lett. 1996;6:2219–2224. [Google Scholar]
- 38.Agrawal S, Tan W, Cai Q, Xie X, Zhang R. In vivo pharmacokinetics of phosphorothioate oligonucleotides containing contiguous guanosines. Antisense Nucleic Acid Drug Dev. 1997;7:245–249. doi: 10.1089/oli.1.1997.7.245. [DOI] [PubMed] [Google Scholar]
- 39.Yu D, Putta MR, Bhagat L, Li Y, Zhu F, Wang D, Tang JX, Kandimalla ER, Agrawal S. Agonists of Toll-like receptor 9 containing synthetic dinucleotide motifs. J. Med. Chem. 2007;50:6411–6418. doi: 10.1021/jm070881l. [DOI] [PubMed] [Google Scholar]
- 40.Yu D, Wang D, Zhu FG, Bhagat L, Dai M, Kandimalla ER, Agrawal S. Modifications incorporated in CpG motifs of oligodeoxynucleotides lead to antagonist activity of toll-like receptors 7 and 9. J. Med. Chem. 2009;52:5108–5114. doi: 10.1021/jm900730r. [DOI] [PubMed] [Google Scholar]
- 41.Yu D, Zhao Q, Kandimalla ER, Agrawal S. Accessible 5′-end of CpG-containing phosphorothioate oligodeoxynucleotides is essential for immunostimulatory activity. Bioorg. Med. Chem. Lett. 2000;10:2585–2588. doi: 10.1016/s0960-894x(00)00537-0. [DOI] [PubMed] [Google Scholar]
- 42.Yu D, Kandimalla ER, Bhagat L, Tang JY, Cong Y, Tang J, Agrawal S. ‘Immunomers’-novel 3′-3′-linked CpG oligodeoxyribonucleotides as potent immunomodulatory agents. Nucleic Acids Res. 2002;30:4460–4469. doi: 10.1093/nar/gkf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kandimalla ER, Bhagat L, Yu D, Cong Y, Tang J, Agrawal S. Conjugation of ligands at the 5′-end of CpG DNA affects immunostimulatory activity. Bioconjug. Chem. 2002;13:966–974. doi: 10.1021/bc0200374. [DOI] [PubMed] [Google Scholar]
- 44.Putta MR, Zhu FG, Wang D, Bhagat L, Dai M, Kandimalla ER, Agrawal S. Peptide conjugation at the 5′-end of oligodeoxynucleotides abrogates Toll-like receptor 9-mediated immune stimulatory activity. Bioconjug. Chem. 2010;21:39–45. doi: 10.1021/bc900425s. [DOI] [PubMed] [Google Scholar]
- 45.Kandimalla ER, Bhagat L, Wang D, Yu D, Zhu FG, Tang J, Wang H, Huang P, Zhang R, Agrawal S. Divergent synthetic nucleotide motif recognition pattern: design and development of potent immunomodulatory oligodeoxyribonucleotide agents with distinct cytokine induction profiles. Nucleic Acids Res. 2003;31:2393–2400. doi: 10.1093/nar/gkg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Q, Yu D, Agrawal S. Site of chemical modifications in CpG containing phosphorothioate oligodeoxynucleotide modulates its immunostimulatory activity. Bioorg. Med. Chem. Lett. 1999;9:3453–3458. doi: 10.1016/s0960-894x(99)00635-6. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Q, Yu D, Agrawal S. Immunostimulatory activity of CpG containing phosphorothioate oligodeoxynucleotide is modulated by modification of a single deoxynucleoside. Bioorg. Med. Chem. Lett. 2000;10:1051–1054. doi: 10.1016/s0960-894x(00)00157-8. [DOI] [PubMed] [Google Scholar]
- 48.Agrawal S, Kandimalla ER. Antisense and/or immunostimulatory oligonucleotide therapeutics. Curr. Cancer Drug Targets. 2001;1:197–209. doi: 10.2174/1568009013334160. [DOI] [PubMed] [Google Scholar]
- 49.Yu D, Kandimalla ER, Zhao Q, Cong Y, Agrawal S. Immunostimulatory activity of CpG oligonucleotides containing non-ionic methylphosphonate linkages. Bioorg. Med. Chem. 2001;9:2803–2808. doi: 10.1016/s0968-0896(01)00142-0. [DOI] [PubMed] [Google Scholar]
- 50.Wang D, Bhagat L, Yu D, Zhu FG, Tang JX, Kandimalla ER, Agrawal S. Oligodeoxyribonucleotide-based antagonists for Toll-like receptors 7 and 9. J. Med. Chem. 2009;52:551–558. doi: 10.1021/jm8014316. [DOI] [PubMed] [Google Scholar]
- 51.Agrawal S, Jiang Z, Zhao Q, Shaw D, Cai Q, Roskey A, Channavajjala L, Saxinger C, Zhang R. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: in vitro and in vivo studies. Proc. Natl. Acad. Sci. USA. 1997;94:2620–2625. doi: 10.1073/pnas.94.6.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lan T, Dai M, Wang D, Zhu FG, Kandimalla ER, Agrawal S. Toll-like receptor 7 selective synthetic oligoribonucleotide agonists: synthesis and structure-activity relationship studies. J. Med. Chem. 2009;52:6871–6879. doi: 10.1021/jm901145s. [DOI] [PubMed] [Google Scholar]
- 53.Lan T, Kandimalla ER, Yu D, Bhagat L, Li Y, Wang D, Zhu F, Tang JX, Putta MR, Cong Y, et al. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl. Acad. Sci. USA. 2007;104:13750–13755. doi: 10.1073/pnas.0706059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laws PM, Young HS. Current and emerging systemic treatment strategies for psoriasis. Drugs. 2012;72:1867–1880. doi: 10.2165/11634980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 55.Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus-focus on targeting interferon-alpha. Clin. Immunol. 2012;143:210–221. doi: 10.1016/j.clim.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stohl W, Hilbert DM. The discovery and development of belimumab: The anti-BLyS-lupus connection. Nat. Biotech. 2012;30:69–77. doi: 10.1038/nbt.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sullivan T, Atiee G, Bhagat L, Jiang W, Kandimalla E, Murphy J, Precopio M, Putta M, Arbeit R. 6th Annual Meeting of the Oligonucleotide Therapeutic Society. 2010. Safety and pharmacodynamics of IMO-3100, a novel Toll-like receptor antagonist for autoimmune and inflammatory diseases, in a rising single-dose phase 1 clinical trial; pp. 20–23. October, Dana Point, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.