

Abstract
Fluorinated Thomsen-Friedenreich (T) antigens were synthesized efficient from chemically produced fluorinated monosaccharides using a highly efficient one-pot two-enzyme chemoenzymatic approach containing a galactokinase and a D-galactosyl-β1–3-N-acetyl-D-hexosamine phosphorylase. These fluorinated T-antigens were further sialylated to form fluorinated ST-antigens using a one-pot two-enzyme system containing a CMP-sialic acid synthetase and an α2–3-sialyltransferase.
Introduction
Overexpression of tumor-associated carbohydrate antigens (TACAs) on cell surface is a common phenomenon for cancer progression.1,2 In the last two decades, there is a great effort on the synthesis and development of TACA-based vaccines for anti-cancer therapy.3–6 Despite some promises shown in clinical trials, cancer vaccines based on natural TACA structures were not able to induce sufficient T cell-mediated immunity (IgG antibody).2,3 This is many natural TACAs are also presented on normal human tissues at a lower level and are considered as “self” by the human immune system.7,8 In addition, natural TACA-based cancer vaccines may be degraded by glycosidases or transglycosidases in vivo and loss the essential carbohydrate recognition elements. This will dramatically affect the specificity of the antibodies elicited by the vaccines. To overcome these obstacles, chemically modified TACA analogs, including fluoro-sugars,9 C-glycosides,10 and S-glycosides,11 have been developed and tested for vaccine design.
Thomsen-Friedenreich antigen (TF or T-antigen, Galβ1–3GalNAcαSer/Thr) is one of the most common TACAs. T-antigen-presenting mucins are overexpressed on about 90% of human carcinoma cells, including those of breast cancer, prostate cancer, ovarian cancer, and lung carcinomas.12,13 Recently, T-MUC1 glycopeptide analogs containing one or two fluorine substituents on the sugar were synthesized.14–17 They have been conjugated to tetanus toxoid carrier protein and the conjugate vaccines elicited strong and specific immune responses in mice.18–19 These “foreign” fluorinated TACA-based vaccines not only provided enhanced immunogenicity and metabolic stability but also improved bioavailability. Despite the broad application of fluorine substitution in medicinal chemistry for generating several blockbuster drugs in the market (such as Lipitor™ and Prozac™, etc.),20,21 only a few fluorinated T-antigens and derivatives have been synthesized so far.14–17,22–24 Chemical synthesis of fluorinated oligosaccharides including T-antigens and other Galβ1–3GalNAc-containing O-glycans is challenging.14,23,25 It involves multiple protection and deprotection processes and has to deal with the low reactivity of fluorinated acceptors or donors during glycosylation. It usually requires harsh conditions and leads to low yields. Therefore, a more practical high efficient synthetic approach is needed to provide sufficient amount of fluorinated T-antigens for further vaccine development.
Recently, we developed a highly efficient one-pot two-enzyme system containing a novel Bifidobacterium infantis D-galactosyl-β1–3-N-acetyl-D-hexosamine phosphorylase (BiGalHexNAcP) and a recombinant galactokinase (EcGalK) cloned from E. coli K-12 for the synthesis of diverse β1–3-linked galactosides (Scheme 1).26 Compared to galactosyltransferase-catalyzed reaction, the BiGalHexNAcP-catalyzed synthesis uses a galactose-1-phosphate (Gal-1-P) as the donor substrate and does not require multiple enzymes for in situ generation or regeneration of expensive UDP-galactose (the sugar nucleotide for galactosyltransferases). In addition, both recombinant BiGalHexNAcP and EcGalK have high expression levels in E. coli expression system. Furthermore, BiGalHexNAcP has been shown to tolerate some C6-modified GlcNAc as acceptor substrates.26 Therefore, the two-enzyme system offers an efficient and simplified approach for large-scale synthesis of β1–3-linked galactosides and analogs. Here we show that the one-pot two-enzyme system can be used for efficient synthesis of fluorinated T-antigens and their sialylated derivatives.
Scheme 1.
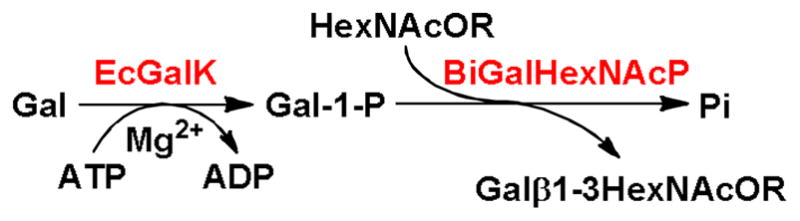
One-pot two-enzyme synthesis of β1–3-linked galactosides. EcGalK, E. coli K-12 galactokinase; BiGalHexNAcP, Bifidobacterium infantis D-galactosyl-1–3-N-acetyl-D-hexosamine phosphorylase.
Results and discussion
Two-step one-pot two-enzyme synthesis of mono- and di-fluorinated T-antigens
The feasibility of applying the BiGalHexNAcP-based one-pot two-enzyme approach in synthesizing fluorinated T-antigens was initially tested at pH 6.0 using 3-azidopropyl 2-acetamido-2,6-dideoxy-6-fluoro-α-galactoside (GalNAc6FαProN3, 4)16 as the acceptor and galactose (Gal) as the donor precursor. Non-fluorinated N-acetyl-galactosamine (GalNAc, 2) and 3-azidopropyl 2-acetamido-2-deoxy-α-galactoside (GalNAcαProN3, 3)27 were used as acceptors for positive controls. Compared to reactions using 2 and 3 as the BiGalHexNAcP acceptors which resulted in good yields (81% and 86%, respectively, for producing galactosides 6 and 7), the yield of reaction using the fluorinated acceptor GalNAc6FαProN3 (4) under the same conditions was quite low (< 10%). Detailed examination found that a slow generation of Gal-1-P in the EcGalK-catalyzed reaction at pH 6.0 and different pH preferences of the two enzymes (pH > 7.0 for EcGalK and 5.0–6.5 for BiGalHexNAcP)26 were the main reasons for the low yield formation of C6-fluorinated T-antigen 8 (Galβ1–3GalNAc6FαProN3). These were confirmed by high yield (>80%) formation of 8 by BiGalHexNAcP-catalyzed reaction at pH 6.0 (optimal pH for BiGalHexNAcP) using Gal-1-P as the donor. To comprise the different pH preferences of EcGalK and BiGalHexNAcP, a two-step process was used for the one-pot two-enzyme synthesis of fluorinated T-antigens. The Gal-1-P and derivatives were generated in situ from Gal and derivatives at pH 7.5 by EcGalK-catalyzed reaction containing ATP, MgCl2, and the acceptor. Once the EcGalK-catalyzed reactions were completed as determined by thin-layer chromatography (TLC) analysis, the pH of the reaction mixtures were adjusted to 6.0 and BiGalHexNAcP was added for the production of disaccharide products.
As shown in Table 1, C6-fluorinated T-antigen 8 was successfully synthesized from GalNAc6FαProN3 (4) in an excellent yield (88%) using the two-step one-pot two-enzyme system without the isolation of the Gal-1-P intermediate generated in situ. Compared to a previously reported chemical synthesis of protected C6-fluorinated T-antigen from Gal and GalNAcαSer which required at least 8 steps with a less than 30% total yield,14 the current chemoenzymatic synthesis of a similar C6-fluorinated T-antigen 8 from Gal and GalNAcαProN3 only required 4 steps and resulted in a total yield of higher than 60%.
Table 1.
Two-step one-pot two-enzyme synthesis of fluorinated T-antigens and derivatives using EcGalK and BiGalHexNAcP.
| Donors | Acceptors | Products | Yields (%) |
|---|---|---|---|
| Gal 1 |
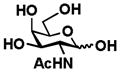 2 GalNAc |
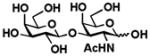 6 Galβ1–3GalNAc |
81 |
| Gal 1 |
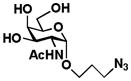 3 GalNAcαProN3 |
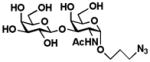 7 Galβ1–3GalNAcαProN3 |
86 |
| Gal 1 |
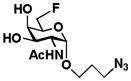 4 GalNAc6FαProN3 |
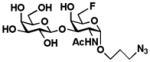 8 Galβ1–3GalNAc6FαProN3 |
88 |
| Gal6F 5 |
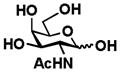 2 GalNAc |
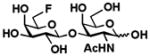 9 Gal6Fβ1–3GalNAc |
80 |
| Gal6F 5 |
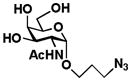 3 GalNAcαProN3 |
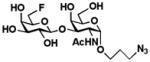 10 Gal6Fβ1–3GalNAcαProN3 |
87 |
| Gal6F 5 |
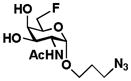 4 GalNAc6FαProN3 |
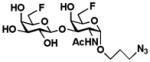 11 Gal6Fβ1–3GalNAc6FαProN3 |
82 |
The two-step one-pot two-enzyme system was further tested for the synthesis of C6′-fluorinated T-antigens 9 (Gal6Fβ1–3GalNAc), 10 (Gal6Fβ1–3GalNAcαProN3), and 11 (Gal6Fβ1–3GalNAc6FαProN3) using C6-fluorinated galactose (Gal6F, 5) as the donor precursor for BiGalHexNAcP. To our delight, the C6-fluorine substitution on Gal was tolerated well by both EcGalK (for the formation of Gal6F-1-P, the donor substrate for BiGalHexNAcP) and BiGalHexNAcP. As shown in Table 1, C6′-fluorinated T-antigens 9–11 were synthesized in very good yields (80–87%). Compared to a previously reported chemical synthesis of C6′, C6-difluorinated T-antigen which required at least 9 steps from Gal and GalNAcαSer with a less than 25% total yield even with an optimization of the glycosylation reaction using microwave radiation (100W, 80°C, 4 h),14,16 the two-step one-pot two-enzyme system synthesis of a similar di-fluorinated T-antigen 11 resulted in a total yield of greater than 50% in 7 steps including chemical synthesis of Gal6F (5) and GalNAc6FαProN3 (4) from non-fluorinated Gal and GalNAcαProN3 using similar processes.14,16
One-pot two-enzyme α2–3-sialylation of fluorinated T-antigens for the formation of fluorinated ST-antigens
The access to fluorinated T-antigens allowed us to explore the acceptor substrate specificity of an α2–3-sialyltransferase for the synthesis of fluorinated sialyl-T (ST) antigens. A one-pot two-enzyme sialylation system containing a Neisseria meningitidis CMP-sialic acid synthetase (NmCSS) and a Pasteurella multocida sialyltransferase 1 (PmST1)27,34 was used for this purpose. In this system, the N-acetylneuraminic acid (Neu5Ac) was activated to form CMP-Neu5Ac by an NmCSS-catalyzed reaction and transferred to the C3 hydroxyl group of the terminal galactose residue in the acceptors by the α2–3-sialyltransferase activity of PmST1 to produce target ST-antigens (Scheme 2). As shown in Table 2, fluorine modification at C6 and/or C6′ of T-antigens did not alter their acceptance as excellent acceptor substrates by PmST1. Sialylated T-antigens with (14–17) or without (12 and 13) fluorine modification were synthesized in excellent yields (92–99%) using the one-pot two-enzyme sialylation system. The structures and the purities of all six sialylated T-antigens (12–17) were confirmed by 1H and 13C NMR spectra and high performance liquid chromatography (HPLC) chromatograms (see ESI† for details).
Scheme 2.
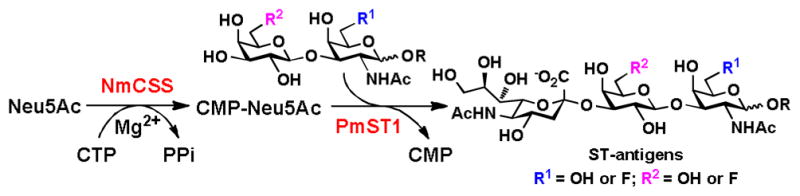
One-pot two-enzyme α2–3-sialylation of fluorinated T antigens for the synthesis of fluorinated ST antigens. NmCSS, Neisseria meningitidis CMP-sialic acid synthetase; PmST1, Pasteurella multocida sialyltransferase 1.
Table 2.
One-pot two-enzyme synthesis of fluorinated ST-antigens and derivatives using NmCSS and PmST1.
| Acceptors | Products | Yields (%) |
|---|---|---|
| 6 |
12 Neu5Acα2–3Galβ1–3GalNAc |
97 |
| 7 |
13 Neu5Acα2–3Galβ1–3GalNAcαProN3 |
97 |
| 8 |
14 Neu5Acα2–3Galβ1–3GalNAc6FαProN3 |
96 |
| 9 |
15 Neu5Acα2–3Gal6Fβ1–3GalNAc |
92 |
| 10 |
16 Neu5Acα2–3Gal6Fβ1–3GalNAcαProN3 |
97 |
| 11 |
17 Neu5Acα2–3Gal6Fβ1–3GalNAc6FαProN3 |
99 |
Despite tremendous advances in the development of chemical glycosylation methods in the last several decades, chemical sialylation remains one of the most challenging glycosylation reactions.28–30 Due to the additional synthetic challenge of fluorinated oligosaccharides, only a few fluorinated sialosides have been synthesized.31–33 The chemoenzymatic method developed here presents an efficient approach to access these challenging and biologically useful carbohydrate antigens.
Chemoenzymatic synthesis of Gal2F-1-P as a potential substrate for enzymatic synthesis of 2-deoxy-2-fluoro-galactosides
Galactosyloligosaccharides or UDP-Gal containing 2-deoxy-2-fluoro-galactose (Gal2F, 18) are general inhibitors and mechanistic probes for galactosidases and galactosyltransferases.35 It has been shown that EcGalK can utilize Gal2F to generate Gal2F-1-P (19) which was used by galactose-1-phosphate uridyltransferase for synthesizing UDP-Gal2F.35 However, none of downstream galactoside processing enzymes (galactosyltransferases, galactosidases, etc.) tested were able to transfer Gal2F for the formation of 2-deoxy-2-fluoro-galactosides. To test whether Gal2F-1-P can be used as a donor substrate for BiGalHexNAcP, Gal2F was synthesized from D-galactose in 5 steps using published procedures36 and tested as a substrate for EcGalK for production of Gal2F-1-P. As shown in Scheme 3, the Gal2F (18) was well tolerated by EcGalK to generate Gal2F-1-P (19) in 93% yield. However, small scale assays indicated that Gal2F-1-P (19) was not a donor substrate by BiGalHexNAcP for transferring Gal2F to either GalNAc or GlcNAc.
Scheme 3.
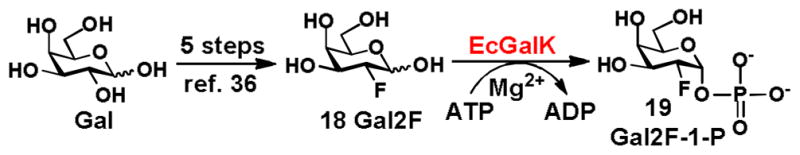
Chemoenzymatic synthesis of Gal2F-1-P.
Conclusions
In summary, a highly efficient two-step one-pot two-enzyme protocol for the preparation of fluorinated T-antigens was developed by adding two enzymes in sequential to accommodate their distinct pH preferences. The substrate promiscuity of EcGalK, BiGalHexNAcP, and PmST1 allow high-yield chemoenzymatic synthesis of fluorinated T- and ST-antigens. In addition, the high expression levels in E. coli expression systems of these enzymes and NmCSS permit their application in large-scale synthesis. Further substrate specificity studies and synthesis of carbohydrate antigens containing fluorine substations at different positions of monosaccharides are underway.
Experimental
General methods
All chemicals were obtained from commercial suppliers and used without further purification unless noted. Anhydrous solvents were dried over 4 Å molecular sieves before being used to carry out organic reactions under nitrogen environment. Thin-layer chromatography (TLC) was performed on silica gel plates 60 GF254 (Yantai, Zhifu) using anisaldehyde sugar stain or 5% sulfuric acid in ethanol for detection of carbohydrates. Silica gel 200–300 mesh (Qingdao, Haiyang) was used for flash column chromatography. Bio-gel P-2 Fine resins (Bio-Rad, Hercules, CA) were used for gel filtration chromatography. 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra were recorded on a Bruker AVANCE-600 spectrometer and 19F NMR (282 MHz) spectra were recorded on a Bruker-AV300 spectrometer. High resolution electrospray ionization (ESI) mass spectra were obtained at National Glycoengineering Research Center and Drug Testing and Analysis Center in Shandong University.
GalNAcαProN3 (3)
Compound 3 was synthesized as reported previously.27 1H NMR (600 MHz, D2O) δ 4.76 (d, J = 3.8 Hz, 1H), 4.01 (dd, J = 7.8, 3.9 Hz, 1H), 3.86–3.76 (m, 3H), 3.69–3.58 (m, 3H), 3.42–3.27 (m, 3H), 1.90 (s, 3H), 1.75 (m, 2H).
GalNAc6FαProN3 (4)
Compound 4 was synthesized from 3 using an approach similar to that reported.14 A solution of compound 3 (1.42 g, 4.69 mmol) and CSA (0.15 g, 0.66 mmol) in 2,2-dimethoxypropane (55 mL) was heated and kept reflux at 110 °C for 70 min. Et3N (0.8 mL) was added and the reaction mixture was concentrated. The syrup residue was re-dissolved in a mixture of MeOH and H2O (20:2 by volume), heated, and kept reflux for 3 h. The solvent was removed by rotovap and the residue was purified by silica gel flash chromatography (PE:EtOAc = 2:1, 2:3) to afford 3,4-actonide protected galactoside intermediate (1.58 g, 98%) as a syrup.
The 3,4-acetonide protected galactoside (0.69 g, 2.00 mmol) was dissolved in dry dichloroethane (5 mL). 2,4,6-Collidine (0.84 g, 6.93 mmol) and N,N-diethylaminosulfurtrifluoride (DAST, 0.51 g, 3.16 mmol) were added. The reaction mixture was irradiated in a CEM Discover™ microwave reactor for 1 h (80 °C, 100 W). The reaction mixture was quenched by adding MeOH (1 mL), concentrated by rotovap, and purified by silica gel flash chromatography (PE:EtOAc = 3:2) to afford the desired protected fluorinated galactoside.
The protected fluorinated galactoside (0.74 g, 2.12 mmol) obtained was dissolved in 80% aq. AcOH (31.25 mL) and stirred at 80 °C for 4 h. The reaction mixture was concentrated under diminished pressure and co-evaporation with toluene (3 × 15 mL). The residue was purified by silica gel flash chromatography (PE:EtOAc = 9:1) to afford the desired product GalNAc6FαProN3 4 (0.58 g, 90%) as a white solid. 1H NMR (600 MHz, D2O) δ 4.96 (d, J = 3.7 Hz, 1H), 4.74 (dt, J = 3.6, 10.2 Hz, 1H), 4.65 (ddd, J = 4.2, 10.8, 30.0 Hz, 1H), 4.26 (ddd, J = 16.8, 7.2, 3.6, 1H), 4.21 (dd, J = 11.4, 3.6 Hz, 1H), 4.08 (d, J = 3.0 Hz, 1H), 3.98 (dd, J = 11.4, 3.6 Hz, 1H), 3.82 (dt, J = 10.2, 6.0 Hz, 1H), 3.57 (dt, J = 10.3, 6.0 Hz, 1H), 3.48 (ddt, J = 18.9, 12.5, 6.2 Hz, 2H), 2.07 (s, 3H), 1.92 (m, 2H). 13C NMR (151 MHz, D2O) δ 174.57, 97.16, 83.51 (d, J = 165.19 Hz), 69.37 (d, J = 19.93 Hz), 68.13 (d, J = 7.85 Hz), 67.31, 65.14, 49.82, 48.11, 27.90, 21.92. 19F NMR (282 MHz, D2O) δ −229.95.
6-Deoxy-6-fluoro-galactose (Gal6F, 5)
Compound 5 was synthesized according to a reported approach.16 1H NMR (600 MHz, D2O) δ 5.25 (d, J = 3.8 Hz, 1H), 4.69–4.49 (m, 8H), 4.30 (ddd, J = 16.5, 7.4, 3.6 Hz, 1H), 4.00 (s, 1H), 3.95 (ddd, J = 14.4, 6.9, 3.3 Hz, 4H), 3.81 (ddd, J = 35.9, 10.3, 3.6 Hz, 2H), 3.63 (dd, J = 10.0, 3.5 Hz, 2H), 3.47 (dd, J = 9.9, 8.0 Hz, 1H). 13C NMR (151 MHz, D2O) δ 96.32, 92.25, 83.39 (d, J = 165.19 Hz), 83.03 (d, J = 165.65 Hz), 82.48, 73.32 (d, J = 20.08 Hz), 72.42, 71.58, 68.92 (d, J = 12.53 Hz), 68.83, 68.74, 68.73, 68.26 (d, J = 7.55 Hz), 68.07. 19F NMR (282 MHz, D2O) δ −229.62, −229.76.
General procedures for two-step one-pot two-enzyme synthesis of fluorinated T-antigens
A hexosamine acceptor (2, 3 or 4, 50–100 mg), galactose (1, 1.5 equiv.) or Gal6F (5, 1.5 equiv.), and ATP (1.5 equiv.) were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 7.5) and MgCl2 (20 mM). After the addition of EcGalK (3.0–4.0 mg), water was added to bring the total volume of the reaction mixture to 10 mL. The reaction mixture was incubated at 37 °C for 15 h with agitation at 140 rpm in an isotherm incubator. When TLC (CH3CN:H2O:HOAc = 2:1:0.2) indicated the completion of EcGalK-catalyzed reaction, the pH of the reaction mixture was brought to pH 6.0 by adding HCl (1.2 M). BiGalHexNAcP (2.0–3.0 mg) was then added and the reaction mixture was incubated at 37 °C for 18–30 h with agitation at 140 rpm. The reaction was stopped by adding the same volume of ice-cold EtOH and the mixture was incubated at 4 °C for 30 min, centrifuged (12,000 rpm) at 4 °C for 21 min, and the supernatant was concentrated and purified by silica gel flash chromatography (CH3CN:H2O = 20:1, 10:1) and Bio-gel P2 gel filtration chromatography.
Galβ1–3GalNAc (6)
97 mg, 81%. NMR data were consistent with those reported previously.26 1H NMR (600 MHz, D2O) δ 5.21 (s, 0.6H), 4.69 (d, J = 8.3 Hz, 0.4H), 4.48 (d, J = 7.2 Hz, 0.6H), 4.44 (d, J = 7.2 Hz, 0.4H), 4.29 (d, J = 10.8 Hz, 0.6H), 4.25 (s, 0.6H), 4.18 (s, 0.4H), 4.13 (s, 0.6H), 4.03 (d, J = 10.8 Hz, 0.6H), 4.00–s3.55 (m, 8H), 3.52 (t, J = 7.8 Hz, 1H), 2.02 (s, 3H).
Galβ1–3GalNAcαProN3 (7)
132 mg, 86%. NMR data were consistent with those reported previously.26 1H NMR (600 MHz, D2O) δ 4.84 (s, 1H), 4.41 (d, J = 7.8 Hz, 1H), 4.28 (d, J = 10.8 Hz, 1H), 4.20 (s, 1H), 3.99 (d, J = 11.4 Hz, 1H), 3.86 (s, 1H), 3.77–3.68 (m, 4H), 3.61–3.57 (m, 2H), 3.50–3.41 (m, 4H), 1.98 (s, 3H), 1.86 (m, 2H).
Galβ1–3GalNAc6FαProN3 (8)
135 mg, 88%. 1H NMR (600 MHz, D2O) δ 4.83 (s, 1H), 4.65–445 (m, 2H), 4.36 (d, J = 7.0 Hz, 1H), 4.26 (d, J = 10.9 Hz, 1H), 4.21 (s, 1H), 4.17 (d, J = 16.4 Hz, 1H), 3.96 (d, J = 10.9 Hz, 1H), 3.81 (s, 1H), 3.71–3.63 (m, 3H), 3.56–3.52 (m, 2H), 3.46–3.36 (m, 4H), 1.93 (s, 3H), 1.81 (m, 2H). 13C NMR (151 MHz, D2O) δ 174.52, 104.71, 97.30, 83.48 (d, J = 165.34 Hz), 76.75, 74.95, 72.43, 70.53, 69.10 (d, J = 19.93 Hz), 68.54, 68.38 (d, J = 8.30 Hz), 65.05, 60.95, 48.47, 48.08, 27.89, 21.93. HRMS (ESI) m/z calcd for C17H30FN4O10 (M + H+) 469.1946, found 469.1939. 19F NMR (282 MHz, D2O) δ −229.40.
Gal6Fβ1–3GalNAc (9)
98 mg, 80%. 1H NMR (600 MHz, D2O) δ 5.22 (s, 0.5H), 4.60–4.50 (m, 2H), 4.43 (d, J = 7.2 Hz, 0.5H), 4.38 (d, J = 5.7 Hz, 0.5H), 4.20 (d, J = 11.4 Hz, 0.5H), 4.13 (s, 0.5H), 4.05 (d, J = 15.4 Hz, 1H), 3.93 (d, J = 11.0 Hz, 0.5H), 3.87–3.80 (m, 2H), 3.76 (d, J = 10.7 Hz, 0.5H), 3.72–3.52 (m, 3H), 3.48–3.40 (m, 1H), 1.94 (s, 3H). 13C NMR (151 MHz, D2O) δ 174.87, 174.59, 104.62, 104.47, 95.06, 91.10, 82.86 (d, J = 165.50 Hz), 82.816 (d, J = 165.65 Hz) 80.02, 77.05, 74.80, 72.94. (d, J = 20.69 Hz), 72.23, 72.19, 70.38, 70.32, 70.13, 68.70, 68.10 (d, J = 6.95 Hz), 68.03, 61.15, 60.93, 52.33, 48.86, 22.15, 21.92. HRMS (ESI) m/z calcd for C14H28FN2O10 (M + NH4+) 403.1728, found 403.1725. 19F NMR (282 MHz, D2O) δ −229.59, −229.74, −229.88, −229.95.
Gal6Fβ1–3GalNAcαProN3 (10)
134 mg, 87%. 1H NMR (600 MHz, D2O) δ 4.85 (s, 1H), 4.66–4.49 (m, 2H), 4.45 (d, J = 7.6 Hz, 1H), 4.29 (d, J = 11.0 Hz, 1H), 4.17 (s, 1H), 3.98–3.87 (m, 4H), 3.73–3.68 (m, 2H), 3.60 (d, J = 9.9 Hz, 1H), 3.50–3.41 (m, 3H), 1.98 (s, 3H), 1.86 (m, 2H). 13C NMR (151 MHz, D2O) δ 174.47, 104.49, 97.11, 82.86 (d, J = 165.65 Hz), 77.18, 72.94 (d, J = 20.54 Hz), 72.16, 70.56, 70.33, 68.69, 68.04 (d, J = 7.10 Hz), 64.84, 61.16, 48.56, 48.09, 27.86, 21.88. HRMS (ESI) m/z calcd for C17H33FN5O10 (M + NH4+) 486.2211, found 486.2201. 19F NMR (282 MHz, D2O) δ −229.86.
Gal6Fβ1–3GalNAc6FαProN3 (11)
126 mg, 82%.1H NMR (600 MHz, D2O) δ 4.83 (s, 1H), 4.64–4.43 (m, 4H), 4.40 (d, J = 7.0 Hz, 1H), 4.26 (d, J = 10.9 Hz, 1H), 4.18 (s, 1H), 3.94 (d, J = 10.9 Hz, 1H), 3.83 (d, J = 13.9 Hz, 1H), 3.70 (d, J = 4.8 Hz, 1H), 3.63 (s, 1H), 3.54 (d, J = 9.7 Hz, 1H), 3.49–3.27 (m, 3H), 1.93 (s, 3H), 1.81 (m, 2H). 13C NMR (151 MHz, D2O) δ 174.53, 104.56, 97.29, 83.50 (d, J = 165.19 Hz), 82.98 (d, J = 165.19 Hz), 76.87, 73.02 (d, J = 20.38 Hz), 72.22, 70.38, 69.10 (d, J = 19.78 Hz), 68.36 (d, J = 7.8 Hz), 68.12 (d, J = 7.10), 65.12, 48.44, 48.10, 27.88, 21.94. HRMS (ESI) m/z calcd for C17H29F2N4O9 (M + H+) 471.1903, found 471.1899. 19F NMR (282 MHz, D2O) δ −229.36, −229.81.
General procedures for one-pot two-enzyme synthesis of fluorinated ST-antigen
A disaccharide acceptor (6, 7, 8, 9, 10 or 11, 50–100 mg), Neu5Ac (1.5 equiv.), and CTP (1.5 equiv.) were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 8.5) and MgCl2 (20 mM). NmCSS (0.5–0.8 mg) and PmST1 (0.2–0.3 mg) were added, and water was added to bring the total volume of the reaction mixture to 10 mL. The reaction mixture was incubated at 37 °C for 30 min with agitation at 140 rpm in an isotherm incubator. The product formation was monitored by TLC (CH3CN:H2O:HOAc = 2:1:0.2 for compounds 12 and 15; EA:CH3OH:H2O: HOAc = 4:2:1:0.1 for compounds 13, 14, 16 and 17), and stained with p-anisaldehyde sugar stain. The reaction was stopped by adding 10 mL of ice-cold EtOH followed by incubation at 4 °C for 30 min. The mixture was centrifuged (12,000 rpm) at 4 °C for 21 min. The supernatant was concentrated and purified by silica gel flash chromatography (EtOAc:MeOH:H2O = 6:2:1) and Bio-gel P2 gel filtration chromatography.
Neu5Acα2–3Galβ1–3GalNAc (12)
87 mg, 97%. NMR data were consistent with those reported previously.27 1H NMR (600 MHz, D2O) δ 5.18 (s, 0.6H), 4.64 (d, J = 7.8 Hz, 0.4H), 4.52 (d, J = 6.7 Hz, 0.6H), 4.46 (d, J = 6.7 Hz, 0.4H), 4.24–4.20 (m, 1H), 4.13–3.79 (m, 7H), 3.72–3.50 (m, 8H), 2.70 (d, J = 11.4 Hz, 1H), 2.00 (s, 3H), 1.98 (s, 3H) 1.76 (t, J = 11.9 Hz, 1H).
Neu5Acα2–3Galβ1–3GalNAcαProN3 (13)
111 mg, 97%. NMR data were consistent with those reported previously.27 1H NMR (600 MHz, D2O) δ 4.86 (s, 1H), 4.49 (d, J = 7.4 Hz, 1H), 4.27 (d, J = 10.8 Hz, 1H), 4.20 (s, 1H), 4.04 (d, J = 9.6 Hz, 1H), 3.99 (d, J = 10.7 Hz, 1H), 3.94 (s, 1H), 3.89 (s, 1H), 3.85–3.56 (m, 12H), 3.54 (d, J = 8.8 Hz, 1H), 3.44–3.41 (m, 5H), 2.70 (d, J = 12.4 Hz, 1H), 1.98 (s, 6H), 1.85 (m, 2H), 1.77 (t, J = 12.2 Hz, 1H). 13C NMR (151 MHz, D2O) δ 174.86, 174.48, 173.24, 104.37, 99.32, 97.09, 77.35, 75.51, 74.63, 72.77, 71.95, 71.53, 68.97, 68.49, 68.09, 67.95, 67.33, 64.79, 62.49, 61.12, 60.84, 51.52, 48.57, 48.06, 39.38, 27.88, 21.94. HRMS (ESI) m/z calcd for C28H48N5O19 (M+H+) 758.2943, found 758.2957.
Neu5Acα2–3Galβ1–3GalNAc6FαProN3 (14)
112 mg, 96%. 1H NMR (600 MHz, D2O) δ 4.89 (s, 1H), 4.74–4.53 (m, 3H), 4.49 (d, J = 7.4 Hz, 1H), 4.29 (d, J = 11.4 Hz, 1H), 4.26 (s, 1H), 4.22 (d, J = 14.1 Hz, 1H), 4.03-4.01 (m, 2H), 3.88 (s, 1H), 3.85–3.77 (m, 2H), 3.75 (d, J = 5.9 Hz, 1H), 3.72–3.56 (m, 4H), 3.54 (d, J = 8.9 Hz, 1H), 3.51–3.41 (m, 4H), 2.70 (d, J = 12.3 Hz, 1H), 1.98 (s, 6H), 1.85 (m, 2H), 1.75 (t, J = 11.9 Hz, 1H). 13C NMR (151 MHz, D2O) δ 174.88, 174.52, 173.59, 104.43, 99.48, 97.23, 83.46 (d, J = 165.50 Hz), 76.94, 75.51, 74.67, 72.73, 71.80 (d, J = 44.85 Hz), 69.07 (d, J = 20.23 Hz), 68.96, 68.21, 67.94, 67.30, 64.97, 62.43, 62.35, 61.34, 60.87, 51.53, 48.41, 48.00, 39.50, 27.85, 21.93. HRMS (ESI) m/z calcd for C28H47FN5O18 (M+H+) 760.2980, found 760.2916. 19F NMR (282 MHz, D2O) δ −229.34.
Neu5Acα2–3Gal6Fβ1–3GalNAc (15)
83 mg, 92%. 1H NMR (600 MHz, D2O) δ 5.19 (s, 0.5H), 4.64 (d, J = 7.8 Hz, 0.5H), 4.60 (s, 0.5H), 4.56 (d, J = 8.4 Hz, 1H), 4.53 (s, 0.5H), 4.50 (d, J = 7.8 Hz, 1H), 4.24 (d, J = 10.8 Hz, 0.5H), 4.19 (s, 0.5H), 4.11–4.02 (m, 1.5H), 4.00 (d, J = 10.8 Hz, 0.5H), 3.94 (s, 1H), 3.90–3.77 (m, 3.5H), 3.74–3.46 (m, 8H), 2.71 (d, J = 12.1 Hz, 1H), 2.01 (s, 3H), 1.98 (s, 3H), 1.74 (t, J = 11.9 Hz, 1H). 13C NMR (151 MHz, D2O) δ 174.91, 174.88, 174.88, 174.61, 173.83, 173.79, 104.32, 104.19, 99.58, 99.58, 95.09, 91.05, 91.05, 82.99 (d, J = 166.70 Hz), 82.93 (d, J = 166.55 Hz), 82.38, 80.36, 80.19, 77.21, 77.21, 75.31, 75.05 (d, J = 66.74 Hz), 72.83 (d, J = 19.02 Hz), 72.76, 72.71, 72.71, 71.95, 71.95, 71.73, 71.73, 70.13, 70.13, 68.83, 68.74, 68.53, 68.53, 68.30, 68.30, 67.94, 67.94, 67.88, 66.98, 66.93, 62.36, 62.36, 61.35, 61.35, 61.16, 61.16, 60.95, 59.18, 52.20, 51.54, 48.86, 39.56, 21.96, 21.93. HRMS (ESI) m/z calcd for C25H42FN2O18 (M+H+) 677.2417, found 677.2412. 19F NMR (282 MHz, D2O) δ −229.19, −229.35, −229.54, −229.64.
Neu5Acα2–3Galβ1–3GalNAc6FαProN3 (16)
111 mg, 97%. 1H NMR (600 MHz, D2O) δ 5.40 (s, 1H), 4.86 (s, 1H), 4.75 (s, 1H), 4.62–4.49 (m, 2H), 4.27 (d, J = 10.7 Hz, 1H), 4.18 (s, 1H), 4.06 (d, J = 9.5 Hz, 1H), 3.99 (d, J = 10.8 Hz, 1H), 3.94 (s, 2H), 3.87 (d, J = 12.2 Hz, 1H), 3.81 (d, J = 10.2 Hz, 2H), 3.76 (d, J = 6.0 Hz, 1H), 3.74–3.56 (m, 5H), 3.54 (d, J = 9.0 Hz, 1H), 3.53–3.46 (m, 2H), 3.45–3.36 (m, 2H), 2.70 (d, J = 12.1 Hz, 1H), 1.98 (s, 6H), 1.86 (m, 2H), 1.75 (t, J = 12.1 Hz, 1H). 13C NMR (151 MHz, D2O) δ 174.86, 174.48, 173.56, 104.17, 99.51, 97.07, 82.95 (d, J = 166.40 Hz), 77.40, 75.25, 72.72, 71.79 (d, J = 43.79 Hz), 70.55, 68.82, 68.50, 68.21, 67.92, 66.94, 64.83, 62.41, 62.34, 61.25 (d, J = 23.71 Hz), 51.52, 48.54, 48.07, 39.45, 27.85, 21.93. HRMS (ESI) m/z calcd for C28H47FN5O18 (M+H+) 760.2900, found 760.2904. 19F NMR (282 MHz, D2O) δ −229.58.
Neu5Acα2–3Gal6Fβ1–3GalNAc6FαProN3 (17)
113 mg, 99%. 1H NMR (600 MHz, D2O) δ 4.90 (s, 1H), 4.68–4.49 (m, 3H), 4.30 (d, J = 10.8 Hz, 1H), 4.21–4.17 (m, 2H), 4.05 (d, J = 9.6 Hz, 1H), 4.01 (d, J = 10.9 Hz, 1H), 3.94 (s, 1H), 3.87 (d, J = 11.5 Hz, 1H), 3.80 (d, J = 10.2 Hz, 2H), 3.75 (d, J = 5.4 Hz, 1H), 3.69–3.51 (m, 5H), 3.45–3.36 (m, 2H), 3.56–3.48 (m, 2H), 3.47–3.34 (m, 2H), 2.70 (d, J = 12.3 Hz, 1H), 1.98 (s, 6H), 1.86 (m, 2H), 1.75 (t, J = 12.0 Hz, 1H). 13C NMR (151 MHz, D2O) δ 174.89, 174.53, 173.64, 104.25, 99.56, 97.23, 83.48 (d, J = 165.65 Hz), 83.00 (d, J = 165.95 Hz) 77.04, 75.25, 72.83, (d, J = 20.54 Hz) 72.74, 71.69, 69.08 (d, J = 19.63 Hz), 68.82, 68.24, 68.18 (d, J = 8.15 Hz), 67.94, 66.98 (d, J = 7.70 Hz), 65.04, 62.43, 61.35, 51.54, 48.38, 48.02, 39.48, 27.83, 21.93. HRMS (ESI) m/z calcd for C28H46F2N5O17 (M+H+) 762.2857, found 762.2857. 19F NMR (282 MHz, D2O) δ −229.29, −229.50.
Chemoenzymatic synthesis of Gal2F-1-P (19)
Gal2F (18) was synthesized similar to that reported.36 Gal2F (18, 100 mg, 1.0 equiv.) and ATP (1.0 equiv.) were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 7.5) and MgCl2 (20 mM). After the addition of EcGalK (3.0–4.0 mg), water was added to bring the total volume of the reaction mixture to 10 mL. The reaction mixture was incubated at 37 °C for 15 h with agitation at 140 rpm in an isotherm incubator. When TLC (CH3CN:H2O:HOAc = 2:1:0.2) indicated that the reaction was completed, the reaction was stopped by adding 10 mL of ice-cold EtOH followed by incubation at 4 °C for 30 min. The mixture was centrifuged (12,000 rpm) at 4 °C for 21 min. The supernatant was concentrated and purified by silica gel flash chromatography (CH3CN:H2O = 10:1) and Bio-gel P2 gel filtration chromatography to provide Gal2F-1-P as a white solid in 93% yield. NMR data were consisted with those reported previously.35 1H NMR (600 MHz, D2O) δ 5.63 (dd, J = 12.0, 6.0 Hz, 1H), 4.61 (dddd, J = 54.0, 12.0, 3.6, 1.8 Hz, 1H), 4.14-4.10 (m, 2H), 4.02 (t, J = 3.6 Hz, 1H), 3.72-3.66 (m, 2H). 19F NMR (282 MHz, D2O) δ −207.75.
Supplementary Material
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 20902087, 21172135 to H.C.), the “973” grant from the Ministry of Science and Technology of China (2012CB822102 to H.C.), National Natural Science Foundation of Shandong Province (ZR2010BM018 to H.C.), NIH grant R01HD065122 and NSF grant CHE-1012511 (to X.C.)
Footnotes
Electronic Supplementary Information (ESI) available: 1H, 13C NMR spectrum. See DOI: 10.1039/b000000x/
Contributor Information
Xi Chen, Email: chen@chem.ucdavis.edu.
Fengshan Wang, Email: fswang@sdu.edu.cn.
Hongzhi Cao, Email: hzcao@sdu.edu.cn.
Notes and references
- 1.Dube DH, Bertozzi CR. Nat Rev Drug Discovry. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 3.Guo ZW, Wang QL. Curr Opin Chem Biol. 2009;13:608–617. doi: 10.1016/j.cbpa.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hevey R, Ling CC. Future Med Chem. 2012;4:545–584. doi: 10.4155/fmc.11.193. [DOI] [PubMed] [Google Scholar]
- 5.Morelli L, Poletti L, Lay L. Eur J Org Chem. 2011:5723–5777. [Google Scholar]
- 6.Yin ZJ, Huang XF. J Carbohydr Chem. 2012;31:143–186. doi: 10.1080/07328303.2012.659364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurianz K, Ziegler S, Garcia-Schuler H, Kraus S, Bohana-Kashtan O, Fishelson Z, Kirschfink M. Mol Immun. 1999;36:929–939. doi: 10.1016/s0161-5890(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 8.Speiser DE, Miranda R, Zakarian A, Bachmann MF, McKallFaienza K, Odermatt B, Hanahan D, Zinkernagel RM, Ohashi PS. J Exp Med. 1997;186:645–653. doi: 10.1084/jem.186.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Y, Guo AL, Guo DS. Curr Org Chem. 2010;14:977–999. [Google Scholar]
- 10.Yuan XJ, Linhardt RJ. Curr Top Med Chem. 2005;5:1393–1430. doi: 10.2174/156802605774643033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pachamuthu K, Schmidt RR. Chem Rev. 2006;106:160–187. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]
- 12.Lin WM, Karsten U, Goletz S, Cheng RC, Cao Y. Int J Exp Pathol. 2011;92:97–105. doi: 10.1111/j.1365-2613.2010.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldus SE, Engelmann K, Hanisch FG. Critical Rev Clin Lab Sci. 2004;41:189–231. doi: 10.1080/10408360490452040. [DOI] [PubMed] [Google Scholar]
- 14.Johannes M, Oberbillig T, Hoffmann-Roder A. Org Biomol Chem. 2011;9:5541–5546. doi: 10.1039/c1ob05373f. [DOI] [PubMed] [Google Scholar]
- 15.Wagner S, Mersch C, Hoffmann-Roder A. Chem Eur J. 2010;16:7319–7330. doi: 10.1002/chem.200903294. [DOI] [PubMed] [Google Scholar]
- 16.Mersch C, Wagner S, Hoffmann-Roder A. Synlett. 2009:2167–2171. [Google Scholar]
- 17.Hoffmann-Roder A, Johannesy M. Chem Comm. 2011;47:9903–9905. doi: 10.1039/c1cc13184b. [DOI] [PubMed] [Google Scholar]
- 18.Oberbillig T, Mersch C, Wagner S, Hoffmann-Roder A. Chem Comm. 2012;48:1487–1489. doi: 10.1039/c1cc15139h. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann-Roder A, Kaiser A, Wagner S, Gaidzik N, Kowalczyk D, Westerlind U, Gerlitzki B, Schmitt E, Kunz H. Angew Chem Int Ed. 2010;49:8498–8503. doi: 10.1002/anie.201003810. [DOI] [PubMed] [Google Scholar]
- 20.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 21.O’Hagan D. J Fluor Chem. 2010;131:1071–1081. [Google Scholar]
- 22.Xue J, Kumar V, Khaja SD, Chandrasekaran EV, Locke RD, Matta KL. Tetrahedron. 2009;65:8325–8335. [Google Scholar]
- 23.Xia J, Xue J, Locke RD, Chandrasekaran EV, Srikrishnan T, Matta KL. J Org Chem. 2006;71:3696–3706. doi: 10.1021/jo052626j. [DOI] [PubMed] [Google Scholar]
- 24.Xia J, Alderfer JL, Piskorz CF, Locke RD, Matta KL. Synlett. 2003:1291–1294. [Google Scholar]
- 25.Bucher C, Gilmour R. Synlett. 2011:1043–1046. [Google Scholar]
- 26.Yu H, Thon V, Lau K, Cai L, Chen Y, Mu SM, Li YH, Wang PG, Chen X. Chem Comm. 2010;46:7507–7509. doi: 10.1039/c0cc02850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau K, Yu H, Thon V, Khedri Z, Leon ME, Tran BK, Chen X. Org Biomol Chem. 2011;9:2784–2789. doi: 10.1039/c0ob01269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halcomb RL, Chappell MD. J Carbohydr Chem. 2002;21:723–768. [Google Scholar]
- 29.Boons GJ, Demchenko AV. Chem Rev. 2000;100:4539–4566. doi: 10.1021/cr990313g. [DOI] [PubMed] [Google Scholar]
- 30.Hanashima S. Trends Glycosci Glycotechnol. 2011;23:111–121. doi: 10.4052/tigg.23.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chokhawala HA, Cao HZ, Yu H, Chen X. J Am Chem Soc. 2007;129:10630–10631. doi: 10.1021/ja072687u. [DOI] [PubMed] [Google Scholar]
- 32.Miyazaki T, Sakakibara T, Sato H, Kajihara Y. J Am Chem Soc. 1999;121:1411–1412. [Google Scholar]
- 33.Stahl W, Sprengard U, Kretzschmar G, Kunz H. Angew Chem Int Ed. 1994;33:2096–2098. [Google Scholar]
- 34.Yu H, Chokhawala H, Karpel R, Wu BY, Zhang JB, Zhang YX, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 35.Burkart MD, Vincent SP, Duffels A, Murray BW, Ley SV, Wong CH. Bioorg Med Chem. 2000;8:1937–1946. doi: 10.1016/s0968-0896(00)00139-5. [DOI] [PubMed] [Google Scholar]
- 36.Burkart MD, Zhang ZY, Hung SC, Wong CH. J Am Chem Soc. 1997;119:11743–11746. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.