

Abstract
The flavoprotein cryptochromes (CRYs) act as blue-light receptors in plants and insects, but perform light-independent functions at the core of the mammalian circadian clock. To drive clock oscillations, mammalian CRYs associate with the Period proteins (PERs) and together inhibit the transcription of their own genes. The SCFFbxl3 ubiquitin ligase complex controls this negative feedback loop by promoting CRY ubiquitylation and degradation. Yet, the molecular mechanisms of their interactions and the functional role of flavin adenine dinucleotide (FAD) binding in CRYs remain poorly understood. Here we report crystal structures of mammalian CRY2 in its apo, FAD-bound, and Fbxl3-Skp1-complexed forms. Distinct from other cryptochromes of known structures, mammalian CRY2 binds FAD dynamically with an open cofactor pocket. Strikingly, the F-box protein Fbxl3 captures CRY2 by simultaneously occupying its FAD-binding pocket with a conserved C-terminal tail and burying its PER-binding interface. This novel F-box protein-substrate bipartite interaction is susceptible to disruption by both FAD and PERs, suggesting a new avenue for pharmacological targeting of the complex and a multifaceted regulatory mechanism of CRY ubiquitylation.
INTRODUCTION
Cryptochromes (CRYs) are evolutionarily conserved FAD-binding proteins that share close sequence homology with DNA photolyases and are widespread in both plant and animal kingdoms1,2. Using FAD as a chromophore, plant cryptochromes function as blue light photoreceptors and regulate a broad range of light responses during plant growth and development1,3. In animals, cryptochromes are classified into two types4,5. Type I cryptochromes, exemplified by Drosophila CRY, maintain blue light sensitivity and play a key role in photic entrainment of the insect circadian clock6, whereas Type II cryptochromes are photo-insensitive and evolved as central components of the molecular clock7,8.
As the prototypic Type II cryptochrome, mammalian CRYs act together with PERs, CLOCK, and BMAL1 to constitute a transcription negative feedback loop, which oscillates with a circadian periodicity of ~24 hours9. In this molecular clockwork, CLOCK and BMAL1 heterodimerize and activate the transcription of Cry and Per genes, whose protein products in turn form complexes in the cytoplasm and translocate into the nucleus to suppress their own gene expression by binding and inhibiting CLOCK-BMAL110,11. In mammals, this basic clock circuitry drives the circadian oscillations of both the master clock in the brain and the peripheral clocks throughout the body9,12. Synchronized to light-dark cycles perceived by the retina, the master clock coordinates clocks in the peripheral tissues via a variety of mechanisms, such as feeding behavior, body temperature, and hormonal secretion13–15.
Despite their functional divergence, many plant and animal cryptochromes are functionally linked to protein ubiquitylation and degradation. For example, light-activated Arabidopsis CRY1 mediates seedling photomorphogenesis by inhibiting COP1 ubiquitin ligase16,17, whereas Drosophila CRY modulates the insect circadian rhythm by promoting light-dependent degradation of the clock protein TIM and itself via the JETLAG ubiquitin ligase18,19. In both cases, photo-excitation of FAD is thought to induce conformational changes within the cryptochrome proteins and alter their interactions with the target proteins20,21. Recent biochemical and genetic studies have shown that mammalian CRYs are also functionally connected to the ubiquitin-proteasome system22–24. The SCFFbxl3 ubiquitin ligase complex has been identified to regulate the clock by promoting the degradation of CRYs.
As an integral part of the clock circuitry, SCFFbxl3-catalyzed CRY degradation needs to be tightly regulated. Indeed, the AMP-activated protein kinase (AMPK), a cellular metabolic sensor, has been shown to phosphorylate mouse CRY1 and accelerate its degradation by enhancing CRY1 binding to Fbxl325. Moreover, a recent circadian chemical screen has identified a period-lengthening small molecule that can compete with FAD for CRY binding and stabilize CRYs by inhibiting their ubiquitylation26. These activities of the compound implicate that SCFFbxl3-mediated CRY ubiquitylation might be tunable by the FAD cofactor, whose function in mammalian CRYs remains elusive2. To provide the missing structural framework for understanding the functions of mammalian CRYs and the regulatory mechanisms of CRY-Fbxl3 interaction, here we report the crystal structures of mammalian CRY2 in three different functional forms.
Structure of Apo mCRY2-PHR
Cryptochromes in general contain a conserved photolyase-homology region (PHR) and a unique cryptochrome C-terminal extension (CCE or CCT) (Supplementary Fig. 1a). We first determined the 2.7 Å crystal structure of the proteolytically stable mCRY2-PHR domain (amino acid 1-512) (Supplementary Fig. 2 & Table 1). As expected, mCRY2-PHR adopts a canonical photolyase fold, consisting of an N-terminal α/β domain, a C-terminal α-helical domain, and a connecting linker sequence (Fig. 1a). At the sequence level, vertebrate CRYs are more homologous to metazoan (6-4)-photolyases than plant or Type I animal cryptochromes (Supplementary Table 2). Upon superposition, mCRY2-PHR can be aligned with the most homologous structure of Drosophila (6-4)-photolyase with a root-mean-square deviation (RMSD) of 1.1 Å over 431 equivalent Cα positions (Supplementary Fig. 3). Unlike all known structures of (6-4)-photolyases and cryptochromes27, however, the mCRY2-PHR structure represents the apo form of the flavoprotein (Fig. 1a). We found no electron density of FAD at its expected cofactor-binding pocket, despite the well-defined and conserved FAD-binding cavity28. This feature is in agreement with previous reports showing that only a small fraction of mammalian CRYs contains FAD when isolated from mammalian cells or various heterologous systems2. Similar to Drosophila (6-4)-photolyase, mCRY2-PHR does not co-purify with a second cofactor, which acts as a light-harvesting antenna in some bacterial photolyases20,28–31.
Figure 1. Structure of mCRY2-PHR in apo- and FAD-bound forms.

a, Overall structure of the apo mCRY2-PHR domain. b, mCRY2-PHR-induced quenching of FAD fluorescence. Inset shows FAD fluorescence quenching with mCRY2-PHR concentration titrated (error bars representing s.d.). c, A close-up view of FAD bound to mCRY2-PHR with positive Fo − Fc electron density contoured at 1.5 σ and calculated before the cofactor was built (green mesh). d, mCRY2-PHR and Drosophila (6-4)-photolyase are superimposed with their FAD cofactors shown in sticks and their FAD carbon atoms colored in yellow and green, respectively.
Structure of FAD-bound mCRY2-PHR
Oxidized FAD is naturally fluorescent with an emission maximum at ~525nm. In the presence of excess mCRY2-PHR, FAD fluorescence is significantly quenched, suggesting that mCRY2-PHR still retains specific FAD-binding activity (Fig 1b). By titrating the amount of protein added, we estimated that FAD binds to isolated mCRY2-PHR with an apparent Kd of ~40 μM (Fig. 1b). We next crystallized mCRY2-PHR together with FAD and obtained a 2.2 Å resolution structure of the holoenzyme (Supplementary Table 1). As anticipated, FAD docks into the predicted cofactor pocket of mCRY2-PHR and adopts the characteristic U-shaped conformation as seen in other cryptochrome and photolyase structures (Fig. 1c). Superposition analysis shows that its binding mode is nearly identical to that revealed in the Drosophila (6-4)-photolyase structure (Fig. 1d).
A closer structural comparison, nonetheless, reveals a marked difference at the cofactor-binding site between mCRY2-PHR and other family members. In plant and insect (6-4)-photolyases and cryptochromes, FAD is largely sequestered and buried deep inside the pocket as a prosthetic group20,28–30. In stark contrast, the cofactor is only partially embedded in mCRY2-PHR, with one side of its adenosine diphosphate group fully exposed to the solvent (Fig. 2a, b). This unusual exposure of the cofactor is attributable to the unique features of two adjacent structural elements in mCRY2-PHR, the phosphate(-binding) loop and the protrusion motif. In Drosophila and Arabidopsis (6-4)-photolyases, the adenosine moiety of FAD is mostly hidden under a well-ordered surface loop, which is previously named the phosphate(-binding) loop28,30. This loop harbors a central lysine residue that hydrogen bonds with the FAD adenine N7 atom and is further secured by a nearby sequence known as the protrusion motif (Fig. 2c). In mCRY2-PHR, the protrusion motif is shifted far away from FAD, while the phosphate loop becomes completely disordered in the crystal (Fig. 2c). These local structural variations in mCRY2 result in an overall open conformation of the FAD-binding pocket, which might explain the moderate affinity of FAD.
Figure 2. The open FAD-binding pocket of mCRY2-PHR.
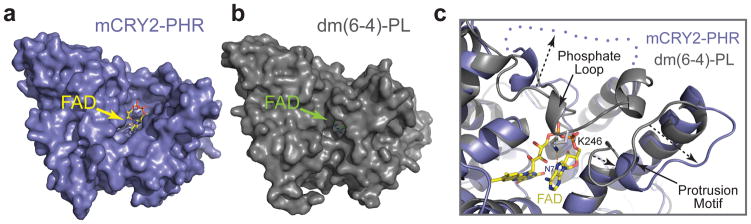
a, b, Surface representations of mCRY2-PHR and Drosophila (6-4)-photolyase with their associated FAD cofactor shown in sticks. c, A close-up view of the open FAD-binding pocket of mCRY2-PHR shown with FAD in sticks and superimposed Drosophila (6-4)-photolyase in ribbon diagrams. A blue dotted line and arrows indicate the disordered mCRY2 phosphate loop and the differences of the phosphate loop and protrusion motif in the two proteins, respectively.
Overall structure of Fbxl3-Skp1-CRY2
The unique CCEs of vertebrate CRYs share a strictly conserved 11 amino acids long segment (CCS for conserved CCE sequence), which is followed by a highly variable C-terminal region (Supplementary Fig. 1b). Using a protein co-expression strategy, we noticed that the nearly full-length form of mCRY2 (amino acid 1-544) with the intact CCS motif can form a stable complex with Fbxl3-Skp1. This prompted us to crystallize and determine a 2.8 Å resolution structure of the heterotrimeric complex (Supplementary Table 1).
The overall architecture of the mCRY2-Fbxl3-Skp1 complex resembles an ice cream cone with the globular mCRY2 sitting on top of the cup-shaped Fbxl3-Skp1 complex (Fig. 3). The Fbxl3 protein harbors a canonical 3-helix F-box motif, which interacts with the SCF adaptor protein Skp1. Its C-terminal leucine-rich-repeat (LRR) domain folds into a curved and sickle-shaped solenoid structure, whose concave surface wraps around the α helical domain and the CCS region of mCRY2 opposite to the α/β domain (Fig. 3, Supplementary Fig. 4). Burying more than 4800 Å2 solvent accessible surface area, the Fbxl3-mCRY2 complex spontaneously assembles in a phosphorylation-independent manner, which was further confirmed by native protein mass spectrometry (Supplementary Fig. 5).
Figure 3. Overall structure of the mCRY2-Fbxl3-Skp1 complex.
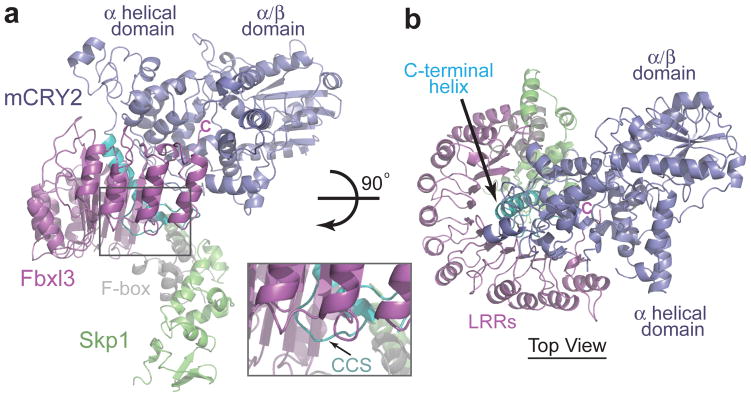
a, Ribbon diagrams of the complex containing mCRY2 (blue), Fbxl3 (magenta), and Skp1 (green). Letter C indicates the carboxyl terminus of Fbxl3. The C-terminal helix and the CCS region in mCRY2 are colored in cyan and the F-box domain of Fbxl3 in grey. The inset figure shows a close-up view of the mCRY2 CCS region. b, Orthogonal view of the mCRY2-Fbxl3-Skp1 complex as shown in Fig. 3a.
The Fbxl3-LRR domain
The Fbxl3-LRR domain contains 12 LRRs, which pack in tandem and give rise to a semicircular arch (Fig. 4a). The solenoid topology of the LRRs dictates that the concave surface of the arch is formed by the parallel β-strands of the repeats and the convex side is decorated by the α helices. We refer to the topside of the arch that is lined with intra-repeat loops as apical ridge (Fig. 4b).
Figure 4. Structure of the Fbxl3 LRR domain.
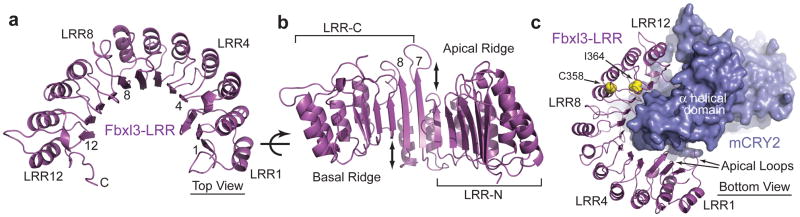
a, Ribbon diagrams of the LRR domain of Fbxl3 along with its complete C-terminal tail. Select LRRs are labeled and numbered at their helices and β-strands. b, An orthogonal view of the LRR domain shown in Fig. 4a. Double-ended arrows indicate the offset between LRR6 and LRR7 and between LRR8 and LRR9. c, A bottom view of the Fbxl3-LRR domain (magenta) bound to mCRY2 (blue). Side chains of the Fbxl3 residues mutated in the overtime and after hours alleles are shown in yellow spheres.
Although the Fbxl3 LRRs fold into a continuous single domain, it can be separated into two obvious halves as suggested by a prominent structural irregularity in LRR7/8. Compared to the N-terminal six LRRs, LRR7 has a much longer β-strand, which causes a significant offset at the apical ridge (Fig. 4b). This feature continues in LRR8, but is lost in LRR9–12. With a similar offset at the basal side between LRR8 and LRR9, the C-terminal four LRRs appear to be up shifted as a whole relative to the N-terminal six repeats. Overall, LRR7 and LRR8 mediate this spatial displacement and hint at a possible gene fusion event when Fbxl3 was evolved in vertebrates. We name the two halves of Fbxl3-LRR as LRR-N (LRR1–6) and LRR-C (LRR7–12).
A quick inspection of the Fbxl3-LRR-mCRY2 interface reveals that the two halves of the Fbxl3-LRR domain play unequal roles in engaging the cryptochrome protein. All six repeats in LRR-C are in close contact with the α helical domain of mCRY2, whereas most parts of LRR-N, except its four apical loops, are separated from mCRY2 by a gap (Fig. 4c). Consistent with a more important role of LRR-C in recruiting mCRY2, the missense mutations in after hours and overtime, C358S and I364T, are mapped to LRR10 and LRR11, respectively (Fig. 4c, Supplementary Fig. 6). Because both residues are located in the hydrophobic core of the LRR fold, their substitution by polar amino acids likely perturbs the local conformation of LRR-C.
The C-terminal tail of Fbxl3
The C-terminal tail of Fbxl3 represents the hallmark of the Fbxl3-mCRY2 interface by sticking out from the flat concave surface of LRR-C and penetrating into mCRY2 (Fig. 5a). The 12 amino acids long C-terminal region of Fbxl3 is mostly invariant in vertebrates and terminates with a tryptophan residue (Supplementary Fig. 6). This sequence caps the LRR solenoid and then takes a sharp turn at Pro422, inserting its extreme C-terminal 5 amino acids into mCRY2’s α helical domain (Supplementary Fig. 7). Strikingly, the last residue of the tail, Trp428, reaches the core of the FAD-binding pocket and physically occupies the cofactor site.
Figure 5. Interaction between the Fbxl3 C-terminal tail and the mCRY2 FAD-binding pocket.

a, Surface representation of the Fbxl3 LRR-C subdomain with its C-terminal tail inserted into the FAD-binding pocket of mCRY2 in blue ribbon diagrams. b, The structure of Drosophila (6-4)-photolyase-DNA complex shown from the same orientation as mCRY2 is displayed in Fig. 5a. FAD is shown in spheres. The damaged DNA substrate is shown in sticks. c, d, A close-up view of the mCRY2 FAD-binding pocket showing key FAD- and Fbxl3-interacting residues. FAD and the Fbxl3 tail are shown in sticks. Yellow dash lines represent hydrogen bonds.
Superposition analysis shows that the Fbxl3 C-terminal tail approaches the FAD-binding pocket from the same angle as the (6-4) DNA lesion does in the Drosophila (6-4)-photolyase-DNA complex structure30 (Fig. 5b). To expand the entrance, Met425 of Fbxl3 pushes aside an important mCRY2 loop, which we name the interface loop. To enter the pocket, the Fbxl3 tail stacks its Pro426 residue against Trp310 of mCRY2 and flips the side chain of mCRY2 His373 with Thr427 (Fig. 5c, d). In the FAD-bound form, these mCRY2 residues directly contact the cofactor. Inside the pocket, the side chain of Fbxl3 Trp428 occupies the central space of the cavity, which corresponds to the narrow gap between the isoalloxazine and adenine rings of FAD. While donating a hydrogen bond to the backbone carbonyl group of Gln307 in mCRY2, the indole ring of Trp428 makes multiple hydrophobic contacts with mCRY2. The carboxyl group of Trp428 forms additional hydrogen bonds with mCRY2 Ser414 and a signature arginine residue, Arg376 (Fig. 5c, d). Overall, this network of interactions anchors the Fbxl3 tail at the very center of the FAD-binding pocket, predicting that FAD and the F-box protein will compete for binding the cryptochrome protein.
Interface between Fbxl3 and mCRY2-PHR
In addition to the C-terminal tail, the Fbxl3-LRR domain also forms extensive interfaces with mCRY2 through its solenoid fold. At the apical ridge of LRR-N, four Fbxl3 loops from LRR1–4 interact with the backside of the α helical domain of mCRY2 opposite to the FAD-binding pocket (Fig. 3b, 4c). This part of the interface is largely mediated by polar interactions (Supplementary Fig. 7).
In contrast to LRR-N, the Fbxl3 LRR-C subdomain binds mCRY2 through a predominantly hydrophobic interface. On its concave wall, LRR-C presents a cluster of more than ten hydrophobic residues strictly conserved in vertebrate CRYs (Fig. 6a, Supplementary Fig. 6). These residues, plus a circle of peripheral polar amino acids, recognize a large complementary surface on mCRY2 adjacent to the cofactor pocket. The interface on the mCRY2 side is constructed by three key structural motifs – the interface loop, the C-terminal helix, and the CCS region (Fig. 6b). With a string of strictly conserved aromatic residues (Supplementary Fig. 8), the mCRY2 interface loop is buttressed by Met425 from the Fbxl3 tail on one side and the mCRY2 C-terminal helix on the other. The mCRY2 CCS region adopts a well-ordered loop structure, which wraps back onto the interface loop and the C-terminal helix. Together, these three motifs contribute a panel of hydrophobic residues grouped at the center of the LRR-C binding interface. By making direct contacts with both Fbxl3 and mCRY2-PHR, the conserved CCS region of mCRY2 likely takes up its structural role at the interface through an “induced fit” mechanism (Supplementary Fig. 9).
Figure 6. Structural and functional analyses of the Fbxl3-mCRY2 interface.
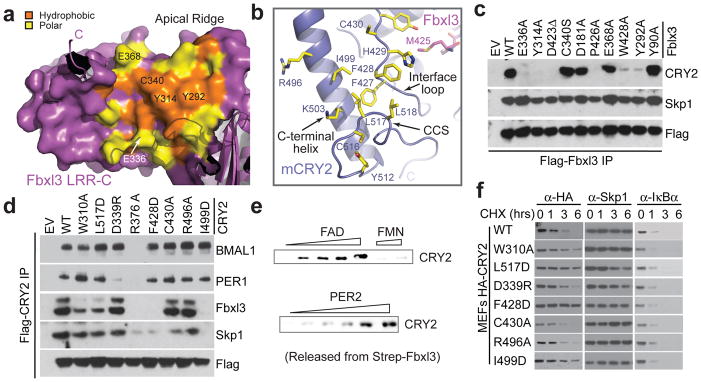
a, Surface and ribbon diagrams of the Fbxl3 LRR-C subdomain showing hydrophobic (orange) and polar (yellow) residues that are involved in mCRY2 binding. Amino acids selected for mutational analyses are labeled. The C-terminal tail and some N-terminal repeats are shown in ribbon diagrams. b, Ribbon diagrams of the mCRY2 structural elements involved in Fbxl3-LRR-C interaction. Select interface residues are shown in sticks. A part of the Fbxl3 tail (magenta) is shown for reference. c, Interactions of retrovirus-expressed Fbxl3 mutants with endogenous mCRY2 in mouse embryonic fibroblasts (MEF) assessed by co-immunoprecipitation and western blot analysis. d, Flag-mCRY2 complexes were immunoprecipitated with an anti-FLAG resin from transfected HEK293T cells and assessed for binding to Fbxl3, PER1, BMAL1 and Skp1. R376A served as a negative control as it is likely detrimental to mCRY2 folding. See Supplementary Discussion for C430A and Fbxl3-C340S mutants. e, Two in vitro competition assays demonstrating the ability of FAD and PER2, but not FMN, to disrupt a preformed Fbxl3-mCRY2 complex. f, MEFs were infected with retroviruses expressing either mCRY2 or mCRY2 mutants and treated with cycloheximide (CHX) for the indicated times. Total extracts were analyzed by immunoblotting.
To map regions that are critical for Fbxl3-mCRY2 interaction, we tested the binding activity of a series of Fbxl3 and mCRY2 mutants. Disruption of interactions at the apical ridge of LRR-N did not affect the complex formation (D181A and Y90A, Fig. 6c; D339R and R496A, Fig. 6d; Supplementary Fig. 7), indicating a supportive role of LRR-N in recruiting mCRY2. By contrast, mCRY2 binding was severely impaired by either single amino acid substitution or truncation of the Fbxl3 tail (P426A, W428A, D423Δ, Fig. 6c). Likewise, alteration of a mCRY2 residue in the cofactor pocket also compromised complex association (W310A, Fig. 5d, 6d). These results pinpoint the Fbxl3 C-terminal tail as a “hotspot” of the interface and suggest a possible antagonistic role of FAD in regulating Fbxl3-mCRY2 interaction. In a purified system, we confirmed that free FAD, but not FMN, was sufficient to disrupt a preformed Fbxl3-mCRY2 complex in a dose dependent manner (Fig. 6e).
Our mutagenesis analysis also established the Fbxl3 LRR-C subdomain as a second critical docking site for mCRY2. Mutations of individual aromatic residues at the hydrophobic concave wall of LRR-C or a nearby CCS-interacting residue effectively abolished mCRY2 binding (Y292A, Y314A, and E336A Fig. 6a, c, Supplementary Fig. 9). The same effect was seen when a single hydrophobic-to-charged mutation was introduced to each of the three LRR-C-interacting structural motifs in mCRY2, i.e., the interface loop (F428D), the C-terminal helix (I499D), and the CCS region (L517D) (Fig. 6b, d). The functional importance of this part of the interface and the Fbxl3 tail-gripping pocket was further underscored by the stabilization of the mCRY2 mutants with “hotspot” residues altered at these sites (Fig. 6f, Supplementary Fig. 9).
The C-terminal helix of mCRY2 and its two basic residues, Arg501 and Lys503, have been documented as essential for PER binding32,33. In complex with Fbxl3, the entire mCRY2 C-terminal helix is masked by the LRR domain of the F-box protein (Fig. 3b, Supplementary Fig. 9). By serendipity, we found that mutation of D339, which is in close proximity to Arg501, also showed defective interaction with PER1 (Fig. 6d, Supplementary Fig. 7). These lines of evidence strongly suggest that PERs and Fbxl3 share an overlapping mCRY2-binding interface, which should prevent them from binding mCRY2 simultaneously. Indeed, PER2-bound mCRY2 is completely devoid of Fbxl3 (Supplementary Fig. 10). Importantly, just like FAD, a purified C-terminal fragment of PER2, which corresponds to a previously mapped minimal CRY-binding region34, was able to dislodge mCRY2 from Fbxl3 (Fig. 6f). We further confirmed that the abundance of PER2 had a direct effect on the stability of both CRY1 and CRY234,35 (Supplementary Fig. 10), likely by competing with the SCFFbxl3 E3 ligase.
Discussion
The structure of the mammalian cryptochrome reveals an unexpected open FAD-binding pocket, which was likely evolved in vertebrates to enable dynamic FAD binding. This unique property is built on the plasticity of the phosphate loop guarding the entrance of the pocket. Intriguingly, this loop is highly conserved among vertebrates (Supplementary Fig. 8), raising the possibility that its alteration by phosphorylation25,36 or CRY-binding proteins might regulate the affinity of FAD (Supplementary Fig. 11).
Importantly, our structural results have unraveled the functional requirement for the dynamic FAD-binding pocket in CRYs and a novel mechanism of substrate-recognition by the SCF ubiquitin ligase. In contrast to the canonical degron-based substrate-engagement scheme37,38, Fbxl3 takes advantage of the deep yet accessible cofactor pocket of CRYs to capture the substrate (Supplementary Fig. 12). This interaction mode establishes FAD and its analogs as potential pharmacological agents for controlling clock oscillation by competing with SCFFbxl3. Such an effect might be achieved by the CRY-stabilizing compounds identified in the recent circadian chemical screen26.
The unexpected interface between mCRY2 and Fbxl3 suggests that their interactions can be regulated by multiple mechanisms. As a common metabolic redox cofactor in the cell, FAD could directly control the stability of CRYs by competing with the ubiquitin ligase. Although oxidized FAD binds mCRY2 with a modest affinity (see Supplementary Discussion), its interaction with the CRY proteins can be influenced by its redox states and multiple aforementioned factors. The mCRY2-Fbxl3 structure also supports a role of PERs in stabilizing CRYs by shielding them from SCFFbxl3. This function is reminiscent of the stabilization of PERs by CRYs against SCFβTrCP-mediated degradation11,34. Such interplay between the two obligate functional partners could serve as a mechanism for synchronizing their stability to ensure robust clock oscillation and phase shift. In fact, the CRY1-destabilizing effect of S71 phosphorylation by AMPK might be partially explained by its reported activity in blocking PER2 binding25.
METHODS SUMMARY
The full Methods provides detailed information about all experimental procedures, including description of (1) protein purification; (2) protein crystallization, data collection, and structure determination; (3) FAD fluorescence assay; (4) in vitro competition assay; and (5) mutagenesis, cell culture, binding, and stability analyses.
METHODS
Recombinant protein purification
The mouse CRY2 (amino acids 1-544) was expressed as a GST fusion protein in Hi5 suspension insect cells and isolated by glutathione affinity chromatography using buffer containing 50 mM Tris-HCl, pH=7.5, 300 mM NaCl, 10% Glycerol, 10 mM DTT and 0.5 mM FAD. The protein was further purified by cation exchange and gel filtration chromatography after on-column cleavage by tobacco etch virus (TEV) protease. During purification, the polypeptide yielded a series of C-terminal degradation products, which were not affected by the presence or absence of FAD and dithionite. Two shorter versions of the protein, mCRY2 (1-527) and mCRY2 (1-512), were subsequently purified. Confirmed by native mass spectrometry analysis, mCRY2 (1-512), but not mCRY2 (1-527), was stable (Supplementary Fig. 1, 2). The mouse CRY2 (1-544) protein was co-expressed with full-length human Fbxl3 and Skp1 (97% identical to mouse Fbxl3 and 3 amino acids different in the F-box-LRR region, Supplementary Fig. 6) in Hi5 suspension insect cells. Fbxl3 was fused with an N-terminal glutathione S-transferase (GST) tag, whereas the other two proteins were tag free. The mCRY2-Fbxl3-Skp1 complex was isolated from the soluble cell lysate by glutathione affinity chromatography. After on-column tag cleavage by TEV, the complex was further purified by anion exchange and gel filtration chromatography and then concentrated by ultrafiltration to 10 mg ml−1 in a buffer of 20 mM Tris-HCl, pH=8.0, 200 mM NaCl, 5 mM DTT. Mouse PER2 (1095-1235) was overexpressed as a GST-fusion protein in E. coli and purified by glutathione affinity, anion exchange, and gel filtration chromatography.
Crystallization, data collection and structure determination
The crystals of the mCRY2-Fbxl3-Skp1 complex were grown at 4 °C by the hanging-drop vapor diffusion method with 1.5 μl protein complex sample mixed with an equal volume of reservoir solution containing 0.2 M ammonium citrate, 13–14% PEG3350 and 7% acetonitrile. Diffraction quality crystals obtained were subjected to a post-crystallization dehydration procedure by gradually increasing the concentration of PEG3350 to 30% and then directly frozen in liquid nitrogen. This procedure consistently improved the resolution of the crystals beyond ~4 Å. The mCRY2-Fbxl3-Skp1 derivative crystals were prepared by soaking the native crystals in the buffer containing 15% PEG3350, and 7% acetonitrile supplemented with 0.2 mM KAu(CN)2 for twelve hours, followed by 0.5 mM KAu(CN)2 for twelve hours and then 1 mM KAu(CN)2 for twelve hours. After soaking, the crystals were dehydrated by the same method as native crystals. The crystals of mCRY2 were also grown at 4°C by the hanging -drop vapor diffusion method with 1.5 μl protein mixed with an equal volume of reservoir solution containing 0.1 M MES-imidazole, pH=6.5, 7.2% PEG8K, 20% ethylene glycol (v/v), 0.03 M NaF, NaBr and NaI. The crystals were directly frozen in liquid nitrogen. The mCRY2-FAD crystals were obtained by soaking the mCRY2 crystals with 1 mM FAD in the reservoir buffer with extra 7.5% Glycerol for two hours before freezing in liquid nitrogen. All data sets were collected at the BL8.2.1 and BL8.2.2 beamlines at the Advanced Light Source in Lawrence Berkeley National Laboratory. Reflection data were indexed, integrated and scaled with the HKL2000 package39. The mCRY2-PHR domain structure was determined by molecular replacement using Drosophila (6-4)-photolyase structure (PDB:3CVU) as the search model. Soaking with FAD altered the unit cell and significantly improved the mCRY2-PHR domain crystals. For the apo-structure, TLS refinement was necessary to reduce the R-free value below 0.30. The mCRY2-Fbxl3-Skp1 structure was solved by molecular replacement combined with Single-wavelength Anomalous Dispersion phases (MR-SAD) using the software PHENIX40. The structural models were manually built, refined, and rebuilt with the program COOT41 and PHENIX40.
FAD fluorescence assay
FAD fluorescence and binding studies were done in a PBS buffer, pH=7.4, with 10% glycerol at room temperature in a ½ area 96 well plate with a total volume of 100 μL. Interaction was quantified by measuring the decrease in FAD fluorescence (excitation at 450 nm and emission at 520 nm) upon binding to mCRY2. FAD concentration was held constant at 10 μM in all wells, and mCRY2-PHR was serially diluted ten times from 300 μM to 292 nM. Nonspecific FAD binding (background) was measured in parallel with equal molar concentrations of ovalbumin or lysozyme, which do not bind FAD. All protein concentrations were run in duplicate. The binding data was fit using a standard nonlinear regression curve fitting method with a log(inhibitor) vs. response model in Prism 5, GraphPad Software (La Jolla, Ca).
In vitro competition assay
Strep-tagged full-length Fbxl3, GST-tagged mCRY2 (1-544), and His-tagged Skp1 were co-expressed in monolayer Hi5 insect cells. The complex was purified without elution by Strep-Tactin affinity resin in a buffer containing 100 mM Tris-HCl, pH=8.0, 300 mM NaCl and 20 mM DTT. The Strep-Tactin resin beads with the immobilized complex were subsequently aliquoted in a volume of 100μl with 1 mg/ml total protein and placed in a panel of 7.5mm gravity columns. The beads were incubated with 300μl of oxidized FAD, FMN or purified mPER2 (1095-1235) in a serial dilution (FAD: 0, 5, 50, 500 μM, and 5 mM; FMN: 50 and 500 μM; mPER2: 0, 2.5, 5, 10, 20, and 40 μM) for three hours at 4 °C. Flow through and subsequent 2 column-volumes wash fractions were collected from each column and the GST-mCRY2 protein released from the column was analyzed by SDS-PAGE and detected by western blotting using Anti-GST antibody.
Cell Culture
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% bovine serum (BS). Immortalized mouse embryonic fibroblasts (MEF) were maintained in DMEM containing 10% fetal bovine serum (FBS). HEK293T cells were transfected with PEI (Polysciences) according to manufacture instructions. Cyclohexemide (Sigma) was used at a final concentration of 100μM. For retrovirus production, GP-293 packaging cells (Clontech) were used. Forty-eight hours after transfection, the virus-containing medium was collected and supplemented with 8 μg/ml polybrene (Sigma). Cells were then infected by replacing the cell culture medium with the viral supernatant for six hours. Forty-eight hours after infection, cells were selected with 1 μg/ml puromycin.
Co-immunoprecipitation binding studies
Cells were harvested and subsequently lysed in lysis buffer (LB: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 0.5% NP40, plus protease and phosphatase inhibitors). Immunoprecipitation was performed with anti-FLAG agarose beads (Sigma). Immuno-complexes were washed 5 times with LB and eluted in Laemmli buffer containing 1% SDS. Cell extracts or Flag-immunocomplexes were analyzed by SDS-PAGE followed by immunoblot analysis with the following antibodies: anti-Flag (Sigma), anti-CRY1 and CRY2 (Bethyl), anti-PER1 (Alpha Diagnostic), anti-PER2 (Bethyl), anti-BMAL1 (Bethyl), anti-Skp1 (Invitrogen), anti-Fbxl322.
Plasmids
Wild type and mutant mouse CRY2 cDNAs were subcloned into pcDNA3.1-FLAG. mPER2 was cloned into pMT-Myc. For retrovirus production, cDNAs encoding FLAG or HA tagged, Fbxl3 or CRY2 wild type and mutants, were subcloned into the retroviral vector pBabePuro. Point mutants were generated using the QuikChange Site-directed Mutagenesis kit (Stratagene).
Supplementary Material
Acknowledgments
We thank the beamline staff of the Advanced Light Source at the University of California at Berkeley for help with data collection, and members of the Zheng laboratory for discussion. This work is supported by the Howard Hughes Medical Institute (N. Z. and M. P.), the National Institutes of Health (R01-CA107134 to N.Z., 5T32-HL007151 to L.B., and R01-GM057587, R37-CA-076584, and R21-CA161108 to M.P.), and the University of Washington (S.T.M. and M.F.B.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions. W.X., L.B., M.P., and N.Z. conceived, N.H.S. initiated, and W.X. conducted the protein purification and crystallization experiments. W.X. and N.Z. determined and analyzed the structures. W.X., T.R.H., and N.Z. conceived and W.X. and T.R.H. conducted FAD fluorescence and in vitro competition experiments. L.B., M.P., W.X., and N.Z. conceived and L.B. conducted mutational and binding studies and stability analyses. S.T.M. and M.F.B. conducted native mass spectrometry experiments.
Structural coordinates and structural factors for Fbxl3-CRY2-Skp1, CRY2-FAD, and CRY2 are deposited in the Protein Data Bank under accession numbers 4I6J, 4I6G, 4I6E.
Authors declare no financial interest.
References
- 1.Chaves I, et al. The cryptochromes: blue light photoreceptors in plants and animals. Annu Rev Plant Biol. 2011;62:335–364. doi: 10.1146/annurev-arplant-042110-103759. [DOI] [PubMed] [Google Scholar]
- 2.Oztürk N, et al. Structure and function of animal cryptochromes. Cold Spring Harb Symp Quant Biol. 2007;72:119–131. doi: 10.1101/sqb.2007.72.015. [DOI] [PubMed] [Google Scholar]
- 3.Lin C, Shalitin D. Cryptochrome structure and signal transduction. Annu Rev Plant Biol. 2003;54:469–496. doi: 10.1146/annurev.arplant.54.110901.160901. [DOI] [PubMed] [Google Scholar]
- 4.Yuan Q, Metterville D, Briscoe AD, Reppert SM. Insect cryptochromes: gene duplication and loss define diverse ways to construct insect circadian clocks. Mol Biol Evol. 2007;24:948–955. doi: 10.1093/molbev/msm011. [DOI] [PubMed] [Google Scholar]
- 5.Zhu H, et al. The two CRYs of the butterfly. Curr Biol. 2005;1(5):R953–954. doi: 10.1016/j.cub.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 6.Stanewsky R, et al. The cryb mutation identifies cryptochrome as a circadian photoreceptor in Drosophila. Cell. 1998;95:681–692. doi: 10.1016/s0092-8674(00)81638-4. [DOI] [PubMed] [Google Scholar]
- 7.van der Horst GT, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;39(8):627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 8.Griffin EA, Staknis D, Weitz CJ. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;28(6):768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- 9.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;41(8):935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 10.Shearman LP, et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 11.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 12.Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–549. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 13.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13(1):25–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Yang HQ, Tang RH, Cashmore AR. The signaling mechanism of Arabidopsis CRY1 involves direct interaction with COP1. Plant Cell. 2001;13(25):73–2587. doi: 10.1105/tpc.010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Ma LG, Li JM, Zhao HY, Deng XW. Direct interaction of Arabidopsis cryptochromes with COP1 in light control development. Science. 2001;294(1):54–158. doi: 10.1126/science.1063630. [DOI] [PubMed] [Google Scholar]
- 18.Peschel N, Chen KF, Szabo G, Stanewsky R. Light-dependent interactions between the Drosophila circadian clock factors cryptochrome, jetlag, and timeless. Curr Biol. 2009;19:241–247. doi: 10.1016/j.cub.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 19.Koh K, Zheng X, Sehgal A. JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science. 2006;3(12):1809–1812. doi: 10.1126/science.1124951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoltowski BD, et al. Structure of full-length Drosophila cryptochrome. Nature. 2011;480:396–399. doi: 10.1038/nature10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B, Liu H, Zhong D, Lin C. Searching for a photocycle of the cryptochrome photoreceptors. Curr Opin Plant Biol. 2010;13:578–586. doi: 10.1016/j.pbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busino L, et al. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 23.Godinho SI, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 24.Siepka SM, et al. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamia KA, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirota T, et al. Identification of Small Molecule Activators of Cryptochrome. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller M, Carell T. Structural biology of DNA photolyases and cryptochromes. Curr Opin Struct Biol. 2009;19:277–285. doi: 10.1016/j.sbi.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Hitomi K, et al. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc Natl Acad Sci U S A. 2009;106:6962–6967. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brautigam CA, et al. Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2004;101:12142–12147. doi: 10.1073/pnas.0404851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maul MJ, et al. Crystal structure and mechanism of a DNA (6-4) photolyase. Angew Chem Int Ed Engl. 2008;47:10076–10080. doi: 10.1002/anie.200804268. [DOI] [PubMed] [Google Scholar]
- 31.Park HW, Kim ST, Sancar A, Deisenhofer J. Crystal structure of DNA photolyase from Escherichia coli. Science. 1995;268:1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 32.Ozber N, et al. Identification of two amino acids in the C-terminal domain of mouse CRY2 essential for PER2 interaction. BMC Mol Biol. 2010;11:69. doi: 10.1186/1471-2199-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaves I, et al. Functional evolution of the photolyase/cryptochrome protein family: importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Mol Cell Biol. 2006;26:1743–1753. doi: 10.1128/MCB.26.5.1743-1753.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yagita K, et al. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 2002;21:1301–1314. doi: 10.1093/emboj/21.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen R, et al. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol Cell. 2009;36:417–430. doi: 10.1016/j.molcel.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanada K, Harada Y, Sakai M, Todo T, Fukada Y. Serine phosphorylation of mCRY1 and mCRY2 by mitogen-activated protein kinase. Genes Cells. 2004;9:697–708. doi: 10.1111/j.1356-9597.2004.00758.x. [DOI] [PubMed] [Google Scholar]
- 37.Hao B, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Tan X, et al. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature. 2007;446:640–645. doi: 10.1038/nature05731. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. In: Methods in Enzymology. Carter CW, Sweet RM, editors. 276 . Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 40.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 41.CCP4. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.