

Abstract
Complex biological systems often display a randomness paralleled in processes studied in fundamental physics. This simple stochasticity emerges owing to the complexity of the system and underlies a fundamental aspect of biology called phenotypic stochasticity. Ongoing stochastic fluctuations in phenotype at the single-unit level can contribute to two emergent population phenotypes. Phenotypic stochasticity not only generates heterogeneity within a cell population, but also allows reversible transitions back and forth between multiple states. This phenotypic interconversion tends to restore a population to a previous composition after that population has been depleted of specific members. We call this tendency homeostatic heterogeneity. These concepts of dynamic heterogeneity can be applied to populations composed of molecules, cells, individuals, etc.
Here we discuss the concept that phenotypic stochasticity both underlies the generation of heterogeneity within a cell population and can be used to control population composition, contributing, in particular, to both the ongoing emergence of drug resistance and an opportunity for depleting drug-resistant cells. Using notions of both “large” and “small” numbers of biomolecular components, we rationalize our use of Markov processes to model the generation and eradication of drug-resistant cells. Using these insights, we have developed a graphical tool, called a metronomogram, that we propose will allow us to optimize dosing frequencies and total course durations for clinical benefit.
Keywords: Homeostatic heterogeneity, evolution
1. Introduction
In this report, we will describe the conceptual development of a tool for utilizing dynamic heterogeneity in cancer therapy. In section 2, we argue that the large numbers of interactions between biological components in a population can generate apparent stochasticity in the copy numbers of molecules in a single cell. These fluctuations can manifest as interconversion of individual cells among different phenotypic states, resulting in homeostatic heterogeneity at the cell-population level. In section 3, we provide experimental examples that illustrate the generation of phenotypic heterogeneity and a tendency to restore homeostatic heterogeneity. In sections 4 and 5, we demonstrate how these insights illustrate that dynamic heterogeneity is both a clinical challenge and an opportunity. We develop a tool (metronomogram) to identify variables to consider when optimizing dosing schedules on a patient-individualized basis. For this analysis, we demonstrate that it is important to compare the kinetics of the generation of homeostatic heterogeneity with the kinetics of population expansion. In an accompanying paper we generalize these principles and provide examples of how they scale in time, population number, and spatial extent.
2. Theory
Non-genetic phenotypic variation has now been observed in a multitude of systems, including in the swimming motion of bacteria [1], in the life-cycle of the λ-phage [2], as well as in mammalian cells, where cell-cell variation in protein levels equaling 10%–60% of mean values has been reported in clonal populations [3]. What are the origins of such variety? As reviewed by Raj et al. [4], an entire area of research has developed in the last two decades based on the fundamental assumptions (a) that the numbers of biological molecules, including intracellular mRNA and protein, fluctuate, (b) that these copy-number fluctuations result from stochastic noise in the timings of biochemical reactions, and (c) that these fluctuations manifest as transitions between different biological phenotypes at the single-cell level. A large body of literature has established the importance of these hypotheses in specific biological systems. The purpose of this section is to describe physical sciences rationales for this perspective that are often implied, but not always explicitly developed.
2.1. Fundamental Origins of Stochasticity
It is commonly said that biological networks and cells are stochastic, i.e. they seem to play games of chance. This idea of stochasticity derives from the physicist’s concept that molecules follow a “disorderly heat motion” in environments at finite temperature [5], [6], [7]. While this concept is correct, the complete mechanistic basis of this explanation is not always fully appreciated. We will offer a perspective of how “disorderly” timings of chemical reactions can arise in deterministic systems and generate apparent stochasticity (Sections 2.1.1 and 2.1.2). These mechanisms are distinguished from and layered upon the non-determinism fundamental to the quantum mechanical description of molecular fragments, atoms, and electrons (Section 2.1.3). It is also commonly said that stochastic fluctuations are significant in systems with “small” numbers (not that small numbers cause stochastic fluctuations) [5], [8], [1], [9]. In Section 2.1.4, we will clarify this probabilistic relationship by providing an example and then explaining that dramatic molecular fluctuations can occur even when the copy numbers of some biomolecular components at first appear “large.”
2.1.1. Aspects of stochasticity can be produced by deterministic systems with periodic dynamics
We provide a cartoon example of how the approximate appearance of chance protein translation can arise, at least temporarily, in a highly predictable, deterministic system containing periodic dynamics. Figure 1(a) is a highly deterministic simplification of a cell. DNA “breathes,” periodically moving between accessible (orange) and closed (blue) states. A copy of RNA polymerase (green) moves back and forth through the cell. It crosses the dashed lined representing the spatial position of the gene of interest (green dots). If the gene of interest is accessible, mRNA is transcribed and then becomes available for translation a time interval later when it finally arrives in the cytosol. mRNA also degrades after a precise duration in the cytosol, so the windows of opportunity for translation are sharply defined (green rectangles). A lone ribosome (red) also moves throughout the cell, occasionally passing through the location (dashed line, red dots) where translation can take place. If mRNA is available for translation in the cytosol when the ribosome is nearby (green box and red dot overlap in time), a protein is translated. Consider the case where the time periods for the histone “breaths,” the movement of the RNA polymerase, and movement of the ribosome differ. Initially, the time intervals between consecutive instances of protein translation can appear random. However, single-molecule tracking of the DNA, RNA polymerase, and ribosome in this cartoon would quickly reveal that each molecule undergoes periodic dynamics. Apparent short-time disorder can arise from incommensurate periodic dynamics.
Figure 1.

Fundamental origins of stochasticity and the contribution of “small” numbers to the magnitude of noise. (a) A system of components oscillating individually and deterministically between conditions periodically can sample a variety of waiting times between events. (b) and (c) Small changes in the initial position of the ribosome explosively accumulate in its ensuing trajectory as molecular constituents within a cell ricochet off each other, quickly leading to qualitatively distinct outcomes, i.e. protein production in (b) and the absence of protein production in (c). (d), (e), and (f) The effect of small numbers. The proportion of outcomes that deviate from average behavior decreases as the number of independently fluctuating parts increases. (g), (h), (i), and (j) Central limit theorem and “ ” rule.
2.1.2. Deterministic chaos
In a second example we present a slightly more complicated toy deterministic system familiar to physicists and engineers (Figure 1(b)). Here the timings of chemical reactions appear stochastic for the same reasons that timings of collisions between particular billiards on a pool table can be difficult to predict. Slight differences in initial conditions can explosively accumulate, allowing very similar initial conditions to quickly generate a variety of qualitatively distinct outcomes [10], [11]. For example, a ribosome (red) collides with a protein (blue), which then collides with another protein before deflecting a segment of mRNA toward the right. The ribosome and mRNA then interact, leading to protein translation. In Figure 1(c), we have displaced the initial position of the ribosome slightly to the right. Its initial trajectory is nearly unchanged; it still manages to collide with the same protein at roughly the same place, which deflects it toward the right at roughly the same angle. However, the error in the initial conditions visibly accumulates elsewhere as the two blue proteins graze each other, allowing both molecules to move out of the way of the approaching mRNA, which thus fails to encounter the ribosome. Imagine submerging this reaction in a bath of water molecules. Initially, the cells in Figure 1(b) and Figure 1(c) are nearly identical, but soon one cell has produced a copy of a protein, while the other has not. The time until the next productive chemical reaction is difficult to predict; it can vary, and, in this way, appear stochastic.
When the components within a cell collide and re-shuffle themselves, that cell can “forget” how long it has already persisted in a state with molecules present at their current levels. If a cell cannot remember how long it has resided in a particular state, probability rates for chemical reactions must be the same whether the cell has waited in that state a short or a long time (i.e. memory-free chance). Modeling memory-free chance is particularly simple since only the immediate state of the system needs to be accounted for; the system’s history can be neglected. A familiar example of “memory-free” chance is seen in the probabilities for drawing specific cards: they do not depend on how many times a deck has already been shuffled.
It is important to understand that features of stochasticity can arise in deterministic biological systems by virtue of the large numbers of interactions between parts [12]. Physical scientists often describe noise and randomness as “intrinsic” to biochemical reactions [8], [13], [14], [15], [16]. It is important to avoid misunderstanding these descriptions as claims that the appearance of noise can arise in biological systems only because the constituent molecules are found at the microscopic scale. Instead, “intrinsic” noise should be understood as noise arising from interacting collections of biological components even in the absence of experimental error.
2.1.3. Quantum mechanical non-determinism
The basis for memory-free chance in biological systems we have described, until now, is different from the way that randomness and lack of memory appear at the quantum mechanical level. Quantum mechanics is necessary for accurate calculation of the dynamics of light particles (i.e. atoms, protons, electrons) [17]. In contrast to classical Newtonian mechanics, quantum mechanics does not predict for each point in time a unique outcome for each possible measurement. Instead, the state of the system is fundamentally expressed as a distribution of probability amplitudes for obtaining a variety of different outcomes [18]. For example, such non-determinism can characterize the spin-state (“up” vs. “down”) of an electron or the position of the nitrogen atom in an ammonia molecule. As has been noted in stochastic systems modeling, “Quantum indeterminacy unavoidably enters; e.g., in a unimolecular reaction we can never know exactly when a molecule will transform itself into a different isomeric form” [19]. The timings of chemical reactions are non-deterministic. Furthermore, the probability per unit time for a system to make a transition between quantum states, in many cases, is constant. When calculating the lifetime of an excited atomic state using “Fermi’s Golden Rule,” one determines that the “probability of [an] atom decaying” by emitting a photon in a “time interval dt does not increase the longer the atom survives” [18]. The memory-free aspect of chance in these cases is sometimes rationalized by saying that fundamental particles lack the detailed internal structure to keep track of “age.” They do not contain the gears and springs necessary to function as clocks. In contrast, biological systems may generate memory-free chance because they are filled with colliding and re-shuffling components. Memory-free chance can arise, not only as simple randomness from simple particles, but also as simple randomness from complicated interacting systems. Stochasticity arises in biology in both of these ways. Complicated reshuffling of biological components generates apparent randomness in the times at which molecules encounter each other, and, once molecules are arranged to allow for reactions, quantum indeterminacy characterizes the time intervals that elapse before chemical reactions finally occur, if they occur at all.
2.1.4. Importance of low copy numbers
While multitudes of interacting parts might generate noise in the first place, low copy numbers of some components can allow the magnitude of noise to be significant, leading a system to be noisy. To develop our argument, it would be helpful to describe how the presence of some molecular species in small numbers could influence the magnitude of fluctuations within a population.
If a population consists of members individually displaying random fluctuations, the magnitude of random fluctuations of the population overall, relative to average values, increases as the number of independently fluctuating members decreases. Figure 1(d) shows the two outcomes possible when tossing a fair coin. Half of the tosses will land on heads, and half not on heads (on tails). Figure 1(e) shows the four outcomes that can result from tossing a fair coin twice in succession. Both tosses land on heads in a quarter of the outcomes. Both tosses land not on heads in another quarter of the outcomes. One toss lands on heads and another not on heads in half of the outcomes because this mixture can result either with the first toss not on heads followed by heads or vice versa. Figure 1(f) shows the possible outcomes of tossing a fair coin three times in succession. In this case, we expect, on average, to obtain 1.5 heads. Examples of outcomes within 33% of this result (1 or 2 heads) are highlighted with orange, while examples further away from average (0 or 3 heads) have blue backgrounds. Three-quarters of the outcomes are “near average,” in this sense. When tossing a coin only twice, only half of all possible outcomes are near average, and when tossing a coin once, neither outcome is near average. Whether tossing a coin once or many times, it is always possible to obtain an instance where the deviation between the actual number of heads realized and the number of heads expected is comparable in magnitude to the expected value itself. However, far-from-average fluctuations become relatively more likely as the number of coin tosses decreases [20], [21].
Another way to develop this intuition is to stack the configurations from Figure 1(d), (e), and (f) into the histograms in Figure 1(g), (h), and (i). An additional histogram for N = 25 tosses is shown on the same horizontal scale (Figure 1(j)). As the number of tosses, N, increases, the histograms become increasingly similar to Gaussian distributions. The averages (red circles) and standard deviations (double-headed arrows) of these mound-shaped distributions increase with different power-law scalings with the number of tosses, N. Whereas the average increases linearly in N, the standard deviation increases only as [22], so the ratio of the standard deviation to the mean, or coefficient of variation (CV), decreases as . According to the central limit theorem, the approach toward Gaussian distributions and the power-law scalings for the standard deviation, the average, and the CV that we have just described appear when the random variable of interest is a sum of independently fluctuating “samples,” with no small number of these samples dominating either the fluctuations or the average of the sum. As the number of tosses, or more generally, fluctuating components, N, increases, the relative width of the probability distribution, and the probability of landing in the blue “tails,” decreases. Stochastic fluctuations that result in far from average behavior are expected to be more prevalent in biological systems where some components are present in small numbers.
Physicists often say that stochastic fluctuations are significant in systems with small numbers of parts, i.e. cells with low copy numbers of molecules [5], [8], [1], [15], [14], [9], [23]. These statements are not necessarily claims that random chance is a result of small numbers. Instead, these statements mean that small numbers can contribute to the magnitude of noise in a system already containing noisy components. In this sense, Spudich and Koshland noted that “there may well be certain molecules which are present in such low amounts that they are subject to Poissonian variation. . . . for example, there are only 10–20 molecules of lac repressor per [E. coli bacterium]” [1]. According to the -rule, this corresponds to a CV ~ 20%–30%. It is also important to remember that high copy numbers do not guarantee freedom from noise. Spudich and Koshland explained that for some molecules present in high numbers, “their ultimate number may have been determined by a small number of generating molecules (for example, there are only 6–14 trp mRNA molecules per bacterial cell, and each mRNA molecule is translated an average of 20 times).” In a cascade of reactions leading from species A to B and then to C, a slow reaction from A to B can maintain a low level of B with a relatively broad distribution, which is inherited by the distribution of C. Rapidly producing large numbers of copies of C from B can faithfully propagate, rather than suppress, noise already present in the levels of B. Examples of this principle include translational bursts in the expression of the cI gene in λ-phage [24], transcriptional bursts in mammalian cells [25], and protein number noise in yeast, where proteins are found from “50 to one million copies per cell,” but where 75% of “genes have steady-state transcript levels of one or fewer copies per cell” [26]. Additionally, even small amounts of stochastic noise can trigger dramatic fluctuations in the intracellular molecular atmosphere. In excitable systems, small stochastic excursions from a deterministic stable state can initiate exploration of comparatively large loops in molecular state space. Excitability has been used to describe fluctuations in the pluripotency protein Nanog [27], the competence switch in B. subtilis [28], and circadian clocks [29].
In this section, we explained (i) how the multitude of dynamically interacting parts of intracellular molecular networks could generate stochasticity and (ii) how the presence of some molecular species in small numbers could contribute to the magnitude of fluctuations. Owing in part to subtle ways that “low” copy numbers can be present in a system, the magnitude of fluctuations in the levels of chemical species can be large even for species present in copy numbers much greater than unity. Taken together, these features underlie a physical expectation that biological systems display noise at the single-cell level, and that noise can be comparable in magnitude to average values. Thus, biological systems can generate noise and be noisy.
2.2. Phenotypic interconversion
As mentioned above, fluctuations can occur in units at many levels (molecules, cells, people). The consequences of these fluctuations can result in the generation of variation in observable traits called phenotypes. Phenotypes can be expressed on molecular, morphological, behavioral, or social levels. For example, the expression of the molecule melanin in abundant levels in an individual creates a phenotype of dark skin. Building on our previous examples, we will describe how phenotypes can acquire variability through stochastic fluctuations. This dynamic process is ongoing.
Owing to stochasticity in the timing of synthesis and degradation, a cell can momentarily accumulate a surplus, or experience a trough, in mRNA or protein copy number which generates fluctuating protein levels (Figure 2(a)). The blue and orange subpopulations in the distribution in Figure 2(b) refer to cells with relatively high and low protein levels, respectively. Differences in protein levels can correspond to differences in drug-sensitivity. In support of this association, we note that relatively high protein levels have been suggested [30], [3], [1] and demonstrated [31], [32] to correspond to drug-resistance. The relationship between a cell’s resistance to the drug methotrexate (MTX) and the level of the enzyme dihydrofolate reductase (DHFR) in the cell is a specific example. MTX binds to DHFR, preventing the enzyme from participating in essential steps in DNA synthesis. However, cells can proliferate in the presence of MTX if they contain enough copies of the enzyme so that a sufficient number DHFR proteins continues to contribute to DNA synthesis even after some copies of the enzyme are bound by drug. A higher concentration of MTX requires a higher level of DHFR for cell survival. There is no single threshold level of DHFR that universally separates relative drug-resistance from drug-sensitivity. The vertical line separating the orange and blue portions of the distribution in Figure 2(b) moves to the right when the concentration of MTX is increased and moves to the left when the concentration of MTX is decreased. The timescale characterizing interconversion between relatively drug-sensitive and drug-resistant states depends on the position of this interface. Survival in high concentrations of MTX is made possible by large increases in DHFR protein levels owing to gene amplification, which can reverse on timescales of months or years. Lower concentrations of MTX may, instead, position the interface between DHFR protein levels connected by comparatively rapid proteomic fluctuations. As the level of DHFR fluctuates and momentarily increases, a cell can convert from a relatively drug-sensitive state to a relatively drug-resistant state. Thus, dynamic stochastic fluctuations in protein levels can correspond to stochastic transitions from phenotypes of drug-sensitivity to drug-resistance. Note that these insights also predict that drug-resistant cells can rapidly become drug-sensitive cells. In this view, drug-resistance may be modulated in a continuous fashion by the level of a protein, for example with increasing drug-resistance corresponding to increasing protein level. Drug-resistance can also be simultaneously modulated by the levels of multiple proteins [33]. To simplify our description, we “coarse-grain” the model illustrated in Figure 2(c) by considering the case in which drug-sensitivity depends on one protein, and in which the protein explores two possible levels, as opposed to the continuum of levels in Figure 2(a). The relatively high-protein (blue) and low-protein (orange) conditions thus identify relatively drug-resistant and drug-sensitive states.
Figure 2.

Stochastic fluctuations in mRNA and protein level manifest as phenotypic interconversion at the single-cell level, which generates heterogeneity and a tendency to restore homeostatic heterogeneity at the population level. (a) and (b) Local interconversion exploring a graded phenotypic distribution. (c) Rate coefficients for a minimal Markov model with simplified drug-sensitive and drug-resistant compartments. Drug-sensitive cells replicate with rate coefficient rS, are cleared with rate coefficient mS, and convert to the drug-resistant phenotypic state with rate coefficient cS. Analogous rate coefficients describe the dynamics of the drug-resistant cells. (d) Microscopic description of the minimal model in (c) and equations (1) and (2): Individual cells adopt future phenotypes according to the outcomes of spins of wheels of fortune. The configurations of the roulettes depend on the cells’ immediate phenotypes, rather than their historical states. (e) Gradual approach toward steady-state mixture of drug-sensitive and drug-resistant cells from an initial population of 20 drug-sensitive cells (rS = rR = mS = mR = 0 and cS = cR = 1). (f) Gradual approach toward steady-state heterogeneous population composition from an initial population of 15 drug-resistant cells. (g) Approach toward both steady-state population composition and net expansion rate starting from a purely drug-resistant population (rS − mS = 1, rR = mR = 0, and cS = cR = 1).
Drug-resistance is just one of many examples of phenotypic interconversion found throughout biology. Phenotypic interconversion, often called phenotypic switching, is an active area in stem cell and developmental biology. For example, in an experimental study of the murine hematopoietic system, Chang et al. reported that “EML” progenitor cells jump back and forth between alternative molecular states: one state more rapidly differentiates into erythroid cells while the other state more rapidly differentiates toward the myeloid lineage [34]. In a computational and experimental study by Kalmar et al., fluctuations in the level of the core pluripotency circuit protein Nanog are associated with transient opportunities for individual cells to differentiate [27], [35]. Phenotypic switching may also underlie chronic parasitic infection. The trypanosome, which is the causative agent of sleeping sickness in Africa, can change its coating of variant surface glycoproteins in a process called antigenic variation, which allows it to evade immune clearance [36], [37]. Additional studies of phenotypic switches in bacteria, yeast, and mammalian systems have been recently reviewed [4], [38], [39].
2.3. Stochastic phenotypic interconversion at the individual level generates dynamic heterogeneity at the population level
Over time, stochasticity can create different phenotypes within an individual unit. This is manifested in populations as heterogeneity. We now show how this “dynamic heterogeneity” arises using a mathematical description of interconversion between phenotypes at the individual-cell level. As we will show, this is a novel (emergent) phenotype belonging to the whole population, which cannot be specifically ascribed to any of its individual units.
2.3.1. Markov model
In this subsection we describe a Markov model, and in the next subsection we use it to explain dynamic heterogeneity. Markov processes are transitions between states described by memory-free chance. Such “states” can refer to atomic energy states, ecological habitats, or populations of predators and prey. Here, we use a Markov model to describe the generation and eradication of heterogeneity (for example: drug-sensitive and -resistant cells). This kind of model is traditionally presented using a flow schematic as in Figure 2(c) and the corresponding set of differential equations in (1) and (2). During time intervals when drug is not applied, a cell from the drug-sensitive population, S, replicates with rate coefficient rS, dies (mortality) with rate coefficient mS, and converts to the drug-resistant state with rate coefficient cS. A cell from the drug-resistant population has analogous rate coefficients rR, mR, and cR, leading to the dynamics for the drug-sensitive population, S, and the drug-resistant population, R
| (1) |
| (2) |
To more clearly illustrate how (1) and (2) represent a memory-free process, we provide the equivalent schematic in Figure 2(d). At time t, a population of drug-sensitive cells, here S = 10, stands ready to take turns, each launching a ball onto a roulette. The ball might eventually fall into slots corresponding to replication, mortality, phenotypic conversion, or maintenance of the status quo. The outcomes of the spins of this wheel of fortune are adopted as the fates of the cells at time t + Δt. The proportion of slots, and thus probability, corresponding to replication is rSΔt, the proportion of slots corresponding to death is mSΔt, the proportion corresponding to phenotypic conversion to the drug-resistant state is cSΔt, and the proportion corresponding to maintaining the status quo is, by necessity, the remaining probability 1 − (mS + rS + cS)Δt. The initial population of drug-resistant cells (R = 10 shown) at time t adopts fates specified using a similar roulette. The sizes of the sectors corresponding to replication, mortality, conversion, and maintaining status quo in the two roulettes can differ because the drug-resistant cells have their own rate coefficients mR, rR, and cR. The cells present at time t + Δt adopt subsequent states by reusing the roulettes already illustrated.
The reuse of the same roulettes regardless of the history of the cells’ previous states is the expression of the concept of memory-free chance described previously. This is the graphical representation of the use of constant coefficients in (1) and (2). When modelers use constant rate coefficients, they are not necessarily neglecting long-term changes in biology that alter the kinetics of stochastic fluctuations of molecules inside cells. Instead, they are supposing that a cell can perform many phenotypic state transitions before the rate coefficients need to be appreciably changed. The idea of constant coefficients and memory-free chance are implicitly meant to be temporary.
2.3.2. Generation of homeostatic heterogeneity at the population level
Now that we’ve explained the model, we use it to understand how phenotypic interconversion at the individual cell level can generate a tendency to restore phenotypic heterogeneity at the population level (i.e. homeostatic heterogeneity). In the mathematical field of stochastic matrices, it is known that models with the structure of (1) and (2) have the property that they tend to re-establish heterogeneous steady-state population distributions [40]; they tend to restore demographic homeostasis. While this idea is often understood using eigenvector-eigenvalue analysis, we can also conceptualize this process using the microscopic picture offered by Figure 2(d).
For clarity, we temporarily set the probabilities for proliferation and death to zero. Suppose we start with a pure population of drug-sensitive cells (S > 0, R = 0, as indicated in Figure 2(e)). Upon spinning the roulette, some of the population will remain drug-sensitive, but if cS > 0, other members of the drug-sensitive population will convert to the drug-resistant state. These cells that have newly joined the drug-resistant population can now land on the slots on the roulette corresponding to phenotypic conversion back to the drug-sensitive state. Initially these events of back-conversion are rare owing to the initial scarcity of drug-resistant cells. However, the drug-resistant population accumulates more immigrants from the drug-sensitive population. This increases the number of drug-resistant cells that are available to undergo phenotypic conversion. At the same time, the drainage of the drug-sensitive population reduces the number of drug-sensitive cells that are available for phenotypic conversion in the opposite direction. The number of drug-resistant cells converting to drug-sensitive cells per unit time gradually increases while the number of drug-sensitive cells converting to drug-resistant cells per unit time gradually decreases, and the two rates approach equality. In the limit of long times, the flow of drug-resistant cells to the drug-sensitive population and the flow of cells in the reverse direction cancel. As a consequence, there is no additional net accumulation of drug-resistant cells. This limiting situation is an example of detailed balance, i.e. equilibrium that occurs when opposing flows cancel [41]. Coming from the other direction (Figure 2(f)), a pure drug-resistant population repopulates a heterogeneous mixture in the same qualitative way. Thus, (1) and (2) gradually regenerate a steady-state mixture of drug-sensitive and drug-resistant cells in populations that have been depleted of drug-sensitive cells, or alternatively, depleted of drug-resistant cells.
The same intuition applies when considering proliferation and death, which we initially neglected. The example in Figure 2(g) is modified from the previous two illustrations by assigning drug-sensitive cells a finite rate of expansion (proliferation less death = rS − mS = 1). As previously described in Figure 2(f), a population initially composed entirely of drug-resistant cells relaxes toward a heterogeneous mixture of drug-sensitive and drug-resistant cells. In this example, however, the plot of the drug-sensitive population then crosses and surpasses the plot of the drug-resistant population because the drug-sensitive population is expanding while the drug-resistant cells have a net replication rate of zero. The drug-resistant population eventually increases by accumulating cells that have converted from the expanding drug-sensitive subpopulation. When a steady state ratio of drug-sensitive to drug-resistant cells is achieved, the number of drug-sensitive cells converting to drug-resistant cells is larger than the number of conversions in the opposite direction in the same time period. Some of the proliferation of the drug-sensitive population is “borrowed” to become “effective” proliferation for the drug-resistant population. The two subpopulations then expand at the same rate.
We have just described how a population purified so as to have a “lopsided” distribution (i.e. over-representation of cells with a low protein level or, alternatively, a high protein level) tends to restore its original distribution over time. Not only does interconversion generate phenotypic variation from a single phenotype, interconversion can also underlie this restoration of “homeostatic” heterogeneity.
3. Experimental examples
In this section we present experimental examples of the generation of phenotypic heterogeneity and the tendency to restore the homeostatic heterogeneity we have described in the previous section.
3.1. Generation of phenotypic heterogeneity
We provide an example of the generation of phenotypic heterogeneity from a phenotypically homogenized population using flow cytometry. In Figure 3, we monitored the levels of DHFR in MDA-MB-231 cells using a fluoresceinated methotrexate assay described previously [42]. The parental population in Figure 3(a) was sorted for a subpopulation representing the middle 6.3% of the parent distribution (vertical dashed lines). Figure 3(b) shows the population immediately following enrichment, with a much narrower full-width-at-half-maximum (FWHM) value. The distributions at 30h, 51h, 77h, and 95h following the sort in Figure 3(c), (d), (e), and (f) display broader FWHMs, as quantitated in (g). A population initially purified for a specific range of levels of DHFR explores a variety of DHFR levels over time. The purified population in Figure 3(b) explores phenotypes to the left and to the right. Phenotypic conversion occurs in both directions.
Figure 3.

Experimental examples of the generation of heterogeneity from a purified phenotype and the restoration of homeostatic heterogeneity. Levels of DHFR were measured in single cells from a mammary carcinoma cell line (MDA-MB-231) using flow cytometry. (a)–(g) Generation of heterogeneity from a purified population. (a) Parental cell population with full-width-half-maximum (FWHM) indicated by dots on the logarithmic histogram. (b) The FWHM is narrower after sorting to enrich for the marked fluorescence values (dashed lines). (c), (d), (e), and (f) Histograms with increasing FWHMs at 30h, 51h, 77h, and 95h following sort. (g) Quantification of mean fluorescence (m) and FWHM (w). Broadening FWHM is consistent with generation of phenotypes absent in purified population. (h)–(n) Histograms obtained from the same parental cell population, purified for the most intensely stained members, display a return toward parental mean fluorescence values within a day after sort. The purified population tends to restore the shape and position of the original distribution even though it is initially skewed toward the right. (n) Mean fluorescence values.
3.2. Generation of homeostatic heterogeneity
In a second example, we illustrate the tendency to restore homeostatic heterogeneity. A population purified in a lopsided fashion will restore, not only the breadth, but also the average position of the parent distribution. The same parental population in Figure 3(h) was sorted for a subpopulation representing the 5.3% most intensely stained portion of the parent distribution. The distribution immediately following enrichment has a narrower FWHM and an average fluorescence twice the original value (Figure 3(i)). Over the following 30h, 51h, 77h, and 95h, the distribution broadens and returns to an average fluorescence intensity closer to its parental value (Figure 3 (j), (k), (l), and (m)).
3.3. Previous experimental reports
In addition to the experimental examples we have just provided using single-cell measurements of DHFR levels, the generation of heterogeneity and homeostatic heterogeneity have been reported in a variety of biological systems. In 2006, Sigal et al. observed that single-cell levels of 20 proteins in a human lung cancer cell line wandered up and down, even during time intervals between cell division events [43]. The authors showed that initially well-ordered “rankings” of cellular protein levels became progressively mixed. A subpopulation of cells with relatively high protein levels gradually spreads out across protein-abundance space to populate relatively low-protein-level states. In the study by Chang et al. mentioned above, progenitor cells with low, moderate, or high expression of the surface marker Sca-1 all asymptotically repopulated the broader steady-state phenotypic distribution from which they were drawn [34]. Additional examples in both mammalian and prokaryotic cell populations have been described [44], [45], [1], [46].
4. Clinical consequences of the generation of heterogeneity in drug sensitivity
In light of the prevalence of the generation of homeostatic heterogeneity, investigators have proposed that the process may have clinical consequences. As we now explain, the phenomenon is considered a source of resistance to therapy. We propose that homeostatic heterogeneity also provides a clinical opportunity.
4.1. The tendency to restore heterogeneity in drug-resistance can be a clinical challenge
Dynamic heterogeneity is a source of therapeutic recalcitrance. Of particular relevance to cancer therapy, consider the stochastic variation in DHFR protein levels and the ensuing dynamic heterogeneity. Cells containing relatively greater numbers of DHFR proteins are relatively more resistant to the therapeutic agent MTX. Niepel et al. commented that “natural fluctuations in the proteome and resulting dispersion in drug responsiveness of cells are likely to be important but poorly appreciated factors” contributing to incomplete inhibition of tumor expansion, known as “fractional kill” [3]. “Enduring outlier cells” in a phenotypic distribution could provide a non-genetic source of short-term drug-resistance, which could then serve as targets for more permanent mutations [30], [47], [48]. More recently, Settleman et al. have reported on epigenetic, reversible resistance of cancer cell lines to chemotherapeutic drugs [47], which resembles reversible resistance of bacterial “persisters” to antibiotic drug reported earlier by Balaban et al. [46], [49], [50]. By establishing an equilibrium mixture of cells with cell-cell variation in drug-sensitivity, phenotypic interconversion contributes to the maintenance of a relatively drug-resistant subpopulation prepared to survive the next event of drug kill.
4.2. Homeostatic heterogeneity provides an opportunity to kill newly generated drug-sensitive cells
While we anticipate, alongside these authors, that phenotypic interconversion has the potential to contribute to therapeutic resistance, we also view phenotypic interconversion as a clinical opportunity for cancer treatment. The expansion of a surviving cell population following drug exposure is not merely an expansion in cell number, but also (as we discussed in Section 2.3) a regeneration of a heterogeneous population containing a drug-sensitive subpopulation. As so beautifully explained by computational modeling of antibiotic treatment of bacteria [49] and chemotherapeutic treatment of mammalian cells [51], some populations of phenotypically interconverting cells are amenable to therapeutic ablation if the timing of drug doses appropriately matches the kinetics of the generation of drug-sensitivity.
As we have just described, phenotypic interconversion can be viewed both as a potential source of therapeutic recalcitrance and as a source of therapeutic effectiveness (drug-sensitivity). Both of these perspectives represent the importance of phenotypic interconversion for clinical outcome. We use these insights to develop a tool to understand personalized drug therapy.
5. Developing a tool for choosing dosing frequencies: A metronomogram
Current cancer therapeutic strategies are not always maximally effective. Thus, our goal is to understand the biological basis of drug resistance and how these insights may improve therapy. Traditionally it is thought that drug-resistance results from genetic mutations and that reducing the population of proliferative tumor cells reduces the size of the target that can acquire resistance [52]. In some cases, a higher dose of drug will kill drug-sensitive cells faster than the same drug at a lower concentration [45], [53]. Thus, it makes sense that “oncologists are usually trained to expect maximal benefit at the maximal dose” [54], [55]. The maximal dose administered is known as the maximum-tolerated dose (MTD). Doses exceeding the MTD would cause morbidity and mortality, also known as dose-limiting toxicity (DLT). The current strategy of maximum-tolerated dose means killing the cancer just before killing the patient, more is better, or “no pain no gain” [56]. Many regimens of chemotherapy are, in fact, begun with cycles of maximum-tolerated-dose therapy, a strategy known as debulking or induction [55], [57].
Paradoxically, however, MTD is not always best. This has now been illustrated in a variety of cancers. In 2008, Seidman et al. reported on a clinical trial of women with metastatic breast cancer [58]. Women randomized to one trial arm received the drug paclitaxel at MTD. Doses were separated by rest periods of 3 weeks to allow for patient recovery, e.g. of bone marrow function. Women randomized to an alternative schedule received the same drug paclitaxel, but at a lower dose allowing more frequent administration, once every week, rather than once every three weeks. High-frequency, low-dose therapy has been called “maintenance” or “continuation” therapy and more recently “metronomic” therapy [57], [55], [59]. While the survival rate after several years for the advanced condition in both arms of this study was low, overall survival in the cohort treated metronomically at 6 years was, remarkably, quadruple the overall survival in the cohort treated conventionally.
In another clinical trial, Klingebiel et al. reported on the treatment of children with metastatic rhabdomyosarcoma, a cancer of muscle tissue [60]. Both arms of this trial began with a period of conventional MTD therapy. Children in one study arm were then followed with 2 cycles of very high-dose chemotherapy with autologous stem cell support. Children in the other arm instead received follow-up with metronomic chemotherapy. In this study, the increase in overall survival was dramatic in both absolute and relative terms. Both survival curves plateaued after 3 years, but whereas the high-dose chemotherapy group displayed 15% overall survival, the group treated metronomically displayed 52% overall survival.
These results followed earlier hints of a beneficial role for metronomic scheduling including clinical responses during metronomic paclitaxel treatment in patients already pre-treated with taxanes [61], [62], an advantage in overall survival for children with acute lymphoblastic leukemia (ALL) treated with extended maintenance therapy [63], [64], and an advantage in overall survival for young adults with ALL treated according to metronomic pediatric schedules, rather than adult schedules [65]. For these, and additional applications, high frequency dosing has been reported or postulated to be effective (Supplemental Table 1)
If metronomic therapy has provided clinical benefit and sometimes advantage over conventionally scheduled MTD therapy, why has metronomic therapy not been more broadly adopted as a standard of care? Klement and Kamen have commented that there is “a random and very erratic manner in which doses and frequencies are chosen” [66]. Even though the clinical trials for metastatic breast cancer and metastatic rhabdomyosarcoma described above both demonstrated a survival advantage for metronomic therapy, the advantage in one case was dramatic while in the other minimal. The purpose of designing the metronomogram is to try to identify variables that will maximize effectiveness of metronomic therapy.
5.1. To deplete the therapeutically targeted population, the generation of drug-sensitivity should be faster than net population expansion
Based on the insights we have presented in this paper, we hypothesize that metronomic therapy can be effective when drug-resistant cells convert to a drug-sensitive phenotype. Persistent, frequent application of low-doses of drug then depletes the emerging drug-sensitive cells before they have time to proliferate or become repatriated to the drug-resistant subpopulation. The overall dynamics of the target population are a combined result of drug kill and expansion during periods of drug-free recovery. In order for frequent low doses of therapy to be effective, the rate of restoring full homeostatic heterogeneity in drug-sensitivity must be greater than the rate of population expansion. This requirement relates time-scales for the generation of heterogeneity in drug sensitivity to time-scales for the overall expansion of cell population number. To compare these processes quantitatively, we next introduce a graphical device called a metronomogram that expresses the efficacy of drug administration frequencies applied to a target cell population undergoing phenotypic interconversion.
To relate the cellular processes essential for developing this tool, we provide a toy model (Figure 4(a)). In this timeline, cell populations are cyclically killed. The surviving populations repeatedly regenerate heterogeneous mixtures of drug-sensitive and drug-resistant individuals during time intervals between drug administration. For clarity, we take advantage of the simplifying assumptions, made elsewhere, that drug-resistant cells display no drug response and that drug-sensitive cells die immediately upon exposure to drug [48]. Thus, instantaneous drug-kill occurs periodically at time intervals of duration Δt. The total number of cells at any time is N(t), i.e. the total population of both drug-sensitive and drug-resistant cells immediately following the drug-kill event at time t = 0 is N(0+) while N(Δt−) and N(Δt+), respectively, denote the populations immediately preceding and following drug-kill at time t = Δt.
Figure 4.
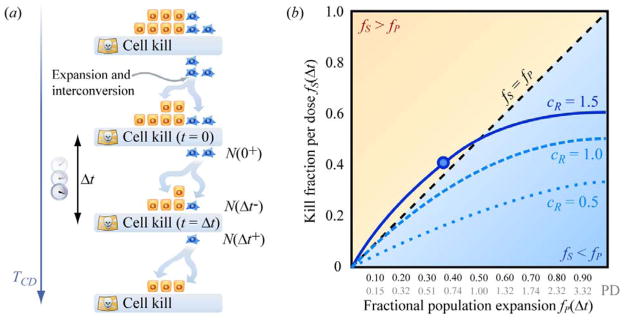
Using a metronomogram to graph the dynamics of phenotypic interconversion and expansion in a heterogeneous population of cells. (a) Dynamics of a mixture of drug-sensitive and drug-resistant subpopulations with rest periods of duration Δt between pulses of cell kill via drug exposure. Therapy continues until all targeted cells have been eradicated. (b) Each curve on a metronomogram expresses the kill fractions fS and population expansion fractions fP explored by varying the interdose rest period Δt for a particular population of cells. The dashed curve labeled cR = 1.0 corresponds to the solution of the model in Figure 2(c) with parameters rR − mR = rS − mS = 1, cR = 1, and cS = 1. The dotted curve labeled cR = 0.5 and the solid curve labeled cR = 1.5 correspond to these alternative values of cR, with the remaining coefficients unchanged.
We seek to reduce the tumor population with each cycle so that the population surviving after drug kill at time t = Δt is smaller than the population surviving the previous event of drug kill at time t = 0.
| (3) |
Adding to both sides the population immediately preceding drug kill at time t = Δt, we determine
| (4) |
that the increase in the population during a kill-free interval of duration Δt (left-hand side of the equation) must be less than the decrease in population associated with drug-kill at the end of the interval (right-hand side of the equation). Defining the kill fraction, or effective drug-sensitized fraction fS(Δt), at time t = Δt as the number of cells killed divided by the number of cells exposed to drug
| (5) |
and defining the population expansion fraction fP(Δt) as the net number of cells added to a population during a drug-free interval divided by the number of cells at the end of the interval
| (6) |
we rewrite (4) concisely as the requirement
| (7) |
that the kill fraction exceed the population expansion fraction. Following a round of drug-kill, the regeneration of the drug-sensitive subpopulation must occur, in this sense, faster than the generation of overall cell population number. If therapy continues until the total cancer cell population N(t) falls below unity, the expected number of drug-resistant cells in the population will also fall below one, and by chance the entire cell population confronting the next dose of drug could be drug-sensitive. Drug-kill then eliminates the last residue of the target cell population. The duration of the total course of therapy is denoted TCD.
5.2. Metronomogram
Equation (7) is a condition for evaluating the efficacy of a particular dosing period Δt. To compare different dosing frequencies, we express this condition graphically. The timeline in Figure 4(a) describes cellular population dynamics for a particular dosing time interval Δt, corresponding to a particular pair of values fP(Δt) and fS(Δt), indicated by a single circle in Figure 4(b). It is important to remember that both the drug-sensitized fraction fS(Δt) and the fraction fP(Δt) by which the population expands in (7) can vary as functions of the drug-free interval Δt. We can repeat the experiment in Figure 4(a) using the same kinds of cells and drug dose per administration, but exploring other values of Δt. This produces additional values of fP(Δt) and fS(Δt) that trace out, for example, the solid curve in Figure 4(b). The example curves in Figure 4(b) illustrate solutions to the Markov model in (1) and (2) with rate coefficients for population expansion and phenotypic interconversion chosen to have similar magnitude (see accompanying paper). We call an fS vs. fP plot a “metronomics nomogram” or “metronomogram.” The dynamics of a population of interconverting drug-sensitive and drug-resistant cells is a combined result of population reduction during periodic drug-kill and population expansion during intervals of drug-free rest. If restoration of homeostatic heterogeneity in drug-sensitivity is faster than population expansion, the population shrinks over time. This corresponds to positions on the metronomogram above the diagonal line fS = fP, which satisfy condition (7). In contrast, the generation of drug-sensitivity is relatively slow in the region below the diagonal. Here, the population expands long term. The diagonal fS = fP distinguishes dosing frequencies that can reduce tumor size (above the diagonal) from dosing frequencies that allow tumor expansion (below the diagonal).
5.3. Choice of drug administration schedule
We now provide an example to illustrate how the metronomogram we have described can be used to choose dosing frequencies (Figure 5(a)). Consider the situation where we measure the cancer cell population at three times in Figure 4(a): right before and right after one of the pulses of drug kill, as well as once immediately preceding the subsequent pulse of drug kill. Then the values of fS and fP can be determined and plotted in Figure 5(a), e.g. circle 1.
Figure 5.
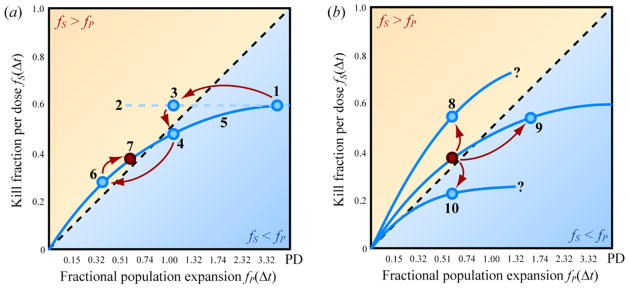
Clinical uses of metronomograms. During therapy, metronomograms could facilitate (a) dosing frequency selection and (b) recommending changes in dosing strategy in response to changes in efficacy of therapy over time.
Because circle 1 turns out to fall below the fS = fP diagonal, another drug administration frequency needs to be found. Extrapolating along horizontal dashed line 2 suggests that a shorter drug-free rest interval Δt might bring us to circle 3, located within the burden-reducing region where fS > fP. As a first approximation we assume that the net replication rate coefficients (proliferation less clearance) of the drug-sensitive and drug-resistant populations are equal. If this approximation is accurate, then an attempt to realize circle 3 will succeed in achieving a data point with the expected fP, corresponding here to ~1.1 population doublings. We might obtain circle 3 itself, or another outcome, such as circle 4. Circle 4 has the expected horizontal position of circle 3, but the actual kill fraction achieved differs, i.e. circle 4 falls vertically below 3. Here circle 4 lies in the region below the fS = fP diagonal, representing dosing frequencies that fail to sustain reduction of tumor burden.
However, such a therapeutic “failure” is actually an opportunity for improving drug administration schedule. The gradual approach toward homeostatic heterogeneity we have described manifests as simple, smooth solutions to the Markov model in (1) and (2), which are plotted in Figure 4(b). This qualitative simplicity makes it easy to use circle 1 and 4 to estimate visually the shape of the blue curve labeled 5 in Figure 5(a). This curve then indicates a range of interdose rest periods that might provide points in the therapeutic region above the fS = fP diagonal. Upon treatment at a fast frequency, we might achieve circle 6 as expected. If toxicity proves intolerable at this schedule, the blue curve we have established can help identify slower dosing frequencies that nevertheless remain in the region fS > fP. For example, the more relaxed schedule represented by circle 7 might be tolerable long-term. Information obtained from “productive failures” would provide data to optimize dosing schedules for each individual.
Proceeding with this schedule, we would continue to monitor tumor burden frequently to look for changes in the rate coefficients in (1) and (2) reflecting long-term changes in tumor biology. Despite maintaining a fixed interval Δt, we might find an increase in kill fraction as in Figure 5(b), circle 8. We might drift toward the right on the previously established blue curve, reaching circle 9. Alternatively, we might fall to another curve containing circle 10. The positions of the curves on the metronomogram suggested by these additional data would help us identify modifications to dosing frequency. If we obtained circle 8, a larger range of dosing frequencies might prove capable of reducing tumor burden with less toxicity. Circle 9 would be consistent with the sharing of molecular mechanisms in phenotypic interconversion and proliferation. In this case, cytostatic agents (which would minimize rR and rS) might move us left, back to the original position above the fS = fP diagonal. We would address circle 10 by investigating ways to hasten the return to homeostatic heterogeneity (maximize cR) in drug-sensitivity in a proliferation-independent, rather than a proliferation-dependent fashion, as discussed in the accompanying paper.
To explain Figure 5(a) and (b), we focused on the simplifying situation where the fitnesses of the drug-sensitive and drug-resistant cells were equal during drug-free rest periods. This allowed the metronomogram to be used literally as a nomogram. However, this need not be the case. We could have obtained a different result for circle 4 in Figure 5(a), horizontally displaced from its illustrated position. If circle 4 had shifted to the right, this would suggest that the fitness of the drug-resistant cells, rR, is greater than the fitness of the drug-sensitive cells, rS. A shift to the left would suggest the opposite. This result would have forced us to consider unequal fitnesses for different phenotypic compartments. Measurements of tumor cell population at various times during a drug-free interval would then be important for constraining the parameters in (1) and (2) necessary for calculating the rest periods Δt capable of reducing tumor burden. Even in these cases, the metronomogram continues to provide a powerful graphical tool for understanding why some ranges of dosing schedules are appropriate for a tumor’s immediate biology, and for proposing revisions to dosing frequency in response to changes in tumor biology during extended therapy.
In the supplemental materials, we discuss how this simplified analysis illustrates a surprising feature. For very high dosing frequencies, the marginal improvement in total course duration becomes minimal. This effect of “diminishing returns” is related to the finite time required for a purified drug-resistant population to generate drug-sensitivity. It may not be worthwhile to increase the dosing frequency arbitrarily at all cost. Even though this simple analysis does not explicitly describe toxicity, it nevertheless contrasts with the idea of “more is better” underlying MTD therapy.
5.4. Calculating total course duration
In addition to choosing appropriate dosing frequencies as we have just described, it is necessary to choose a sufficient total course duration. Therapy concluded prematurely can fail to sustain durable response despite initially successful reduction in tumor burden. The metronomogram can be used to estimate the necessary total course duration as a function of dosing interval Δt. The position of a point (fS(Δt), fP(Δt)) encodes the fold-reduction in tumor size that occurs over a dosing cycle of duration Δt
| (8) |
In (9), TCD is the total course duration that must elapse before a tumor starting with NIM cells at the initiation of metronomic therapy is finally reduced to a size NF. The number of orders of magnitude of fold-reduction in tumor cell population sought during the total course of therapy divided by the number of orders of magnitude of fold-reduction in tumor cell population during each dosing cycle gives a deterministic estimate of the number of dosing cycles required. Multiplied by the duration Δt of each cycle, this ratio provides the total course duration in units of time.
| (9) |
To what size NF do we seek to reduce the tumor burden? By the time we succeed in reducing the tumor cell population to just less than a single cell in the deterministic model, NF < 1, we expect the cell population in the corresponding stochastic dynamics to teeter at the brink of stochastic extinction, if the population has not already become extinct. However, even choices of NF > 1 may provide durable clinical response owing to interactions with the tissue microenvironment. For example, angiogenic and immunological barriers can prevent the expansion of microscopic metastatic colonies for years, e.g. tumor dormancy [67].
In the supplemental information, we discuss how in some cases the total course duration may be of similar order to the length of time the target cell population initially expanded preceding diagnosis and treatment. For some cancers this corresponds to a time-scale of years [68].
In sections 5.3 and 5.4, we sought to identify variables to compare when choosing dosing frequencies and duration. It is important to compare timescales for the generation of heterogeneity with timescales for population expansion in order to evaluate whether the rate of the generation of heterogeneity is faster than the rate of the expansion of the population, as necessary for effective therapy.
Our perspective has been that therapy should be timed to kill drug-sensitized cells before they have time for proliferation and back-conversion to the drug-resistant phenotype. Because phenotypic interconversion is stochastic, drug-sensitive cells can emerge from the drug-resistant population at a variety of times. To kill all of these cells soon after they gain drug-sensitivity, drug doses must be applied frequently. To manage toxicity while sustaining high dosing frequencies over long course durations, we use less than the maximum-tolerated dose. The modeling we have used and its analysis using the metronomogram provide one of the simplest ways we can develop this understanding quantitatively. Understanding these ideas at this basic level is important for being able to recognize the same interplay (between the kinetics of the generation of heterogeneity, the kinetics of population expansion, and therapeutic timing) when it appears in more sophisticated models, e.g. accounting for concentration-dependent drug response and cell-cycle specificity [51], [69] or describing interconversion among continua of drug-sensitive and drug-resistant phenotypes [48].
6. Discussion
In this paper, our goal has been to develop a conceptual tool for understanding and utilizing dynamic heterogeneity in cancer therapy. We argued that the large numbers of interactions between biological components in a population and the small numbers of some molecular species could generate apparent stochasticity at the subcellular level. This, in turn, could manifest as phenotypic interconversion at the single-cell level, resulting in both the generation of heterogeneity and of homeostatic heterogeneity at the population level. In addition to the experimental examples we provided, the processes of the generation of phenotypic heterogeneity and a tendency to restore homeostatic heterogeneity have been reported in other biological studies. Viewing the understanding of dynamic heterogeneity as a clinical opportunity, we developed a tool (metronomogram) to help identify variables to consider when optimizing dosing schedules on a patient-individualized basis. To understand the clinical consequences of stochasticity in the rates of chemical reactions at the cellular level and the dynamic heterogeneity that results at the population level, it is important to compare the kinetics of the generation of homeostatic heterogeneity with the kinetics of population expansion. This way one evaluates whether homeostatic heterogeneity is generated faster than population expansion, as necessary for depletion of the therapeutically targeted population according to a dosing frequency under consideration.
In some clinical applications, the modeling and metronomogram may provide direct insight into a patient’s treatment options. However, the full potential of this conceptual tool can only be appreciated by recognizing the challenges and opportunities it points to in physical, biological, and biomedical engineering sciences. In the remaining discussion, we offer directions for increasing our understanding of the application of the concepts described in this paper to the particular cases of genetic mutations and tumor “stem-cell” biology, and for improving clinical measurement.
6.1. Using dynamic heterogeneity to improve strategies that address genetic mutation
While we have developed our discussion using examples of non-genetic fluctuations in the levels of mRNA and protein, genetic mutations can also produce stochastic transitions in phenotypic states. Genetic point mutations are often found in cancer, and this is the basis for a variety of clinical strategies that have been proposed. These include targeting altered proteins that result from mutations in gene sequences, exploiting fitness costs associated with mutations that confer drug-resistance, controlling tumor burden by targeting drug-sensitive clones even after the appearance of clonal heterogeneity, and targeting the microenvironment or ecology surrounding mutant tumor cells. Because point mutations are potentially permanent, it may be natural to assume that the discussion in this paper is less effective after the acquisition of a mutation. Instead, we propose that it is important to apply the concepts described in this paper in order to most effectively pursue these strategies.
Modern “targeted” therapeutics are designed to bind the proteins resulting from genetic alterations. For example, erlotinib is a tyrosine kinase inhibitor that binds to mutant copies of the protein EGFR. In light of our discussion, it is important to remember that the sensitivity of a cell to erlotinib can be transient even though the sensitizing mutation it harbors is permanent. Even when a drug-sensitizing mutation remains in a cell population, cells can reversibly convert from an erlotinib-sensitive state to an erlotinib-resistant “persister” state [47]. We view redundancy in the pathways through which EGFR stimulates oncogenic phenotypes as an additional possibility [70]. Cells may convert transiently to phenotypic states which rely on other receptors, such as ErbB2, to “stand in” for EGFR. For these reasons, the concepts in this paper may be important for understanding optimal scheduling of targeted therapeutics. Remarkably, most modern therapeutics are currently delivered at daily frequencies. This is consistent with the idea that reduction of tumor burden can be achieved through frequent drug treatment of populations undergoing phenotypic interconversion. Strategies to increase the rate of conversion to the drug-sensitive state or decrease the rate of acquisition of the drug-resistant state may prolong responses to therapy. For example, in the study by Sharma et al., the persister phenotype depended on a temporary chromatin state requiring IGF-1R signaling and activity of the histone deacetylase KDM5A. Inhibiting these molecules reduced the number of persisters surviving treatment by erlotinib and other drugs.
Another consequence of distinguishing between permanent mutations and transient phenotypes is the possibility for continued interconversion between relatively drug-sensitive and drug-resistant states, perhaps with altered kinetics, after the acquisition of “resistance” mutations. Because some mutations increase a cell population’s ability to survive drug at the cost of proliferative capacity (e.g. reduced fitness and a shift to the left), analysis using a metronomogram may still identify dosing frequencies capable of reducing tumor burden. The range of beneficial dosing frequencies for a primary chemotherapeutic may be expanded by using the metronomogram to discover biologic agents that accelerate the generation of drug-sensitivity in proliferation-independent ways (see accompanying paper).
A tumor is not necessarily composed exclusively of cells containing a targetable genetic alteration or, instead, harboring a particular resistance mutation. The genetic variants we have just described may coexist alongside phenotypically and genotypically distinct clonal subpopulations [71], [72]. In some situations, some clones may remain sensitive to drug after other clones have become drug-resistant. In these situations, clinicians may recommend prolonging therapy to slow the expansion of the drug-sensitive clones [73]. Another possibility is that all of the clones in a tumor are sensitive, but to different drugs delivered according to different schedules. We anticipate that assessing tumor burden and adjusting therapeutic administration frequency using a tool like the metronomogram will assist in pursuing both of these strategies, especially when the clonal subpopulations can be measured separately. It is also possible that none of the clones are directly responsive to any drug being applied. In this case early, frequent measurement could indicate the need to consider additional drugs and the opportunity to prevent prolonged exposure to unnecessary side effects. When a targeted cell population does not immediately respond to a therapeutic regimen, it may also prove useful to look for responses in cell types traditionally regarded as being distinct from the frank carcinoma.
One strategy for depleting a robustly resistant population is to kill secondary cell populations upon which the primary target cell population depends for survival. The concepts in this paper can be applied to optimize the scheduling of therapy to deplete these secondary targets. Browder et al. observed more effective control of tumor burden when administering a metronomic schedule of cyclophosphamide as compared with a conventional MTD schedule in the treatment of a preclinical model of Lewis lung carcinoma [74]. The Lewis lung carcinoma cells were already highly resistant to cyclophosphamide before inoculation, indicating that therapeutic efficacy against the carcinoma cells was indirectly mediated through the host. Specifically, Browder et al. proposed that the efficacy of metronomic cyclophosphamide was mediated through death of endothelial cells.
6.2. Tumor “stem-cell” biology
Current research has suggested that tumors contain drug sensitive and drug resistant populations associated with the acquisition of “stem-cell-like” features. It has been suggested that while current therapies are optimized towards removing the bulk of the tumor cells, application of a different class of drugs is needed to remove the tumor “stem-cell” population. If interconversion between the two phenotypes occurs, tumor “stem-cells” will be continuously generated, and therapeutic strategies based on attacking a static cell population alone may not succeed. A more recent rationale has postulated the need to target both compartments [40]. We hypothesize that the properties exhibited by tumor “stem-cells” are a manifestation of the fundamental biological principles and processes described in this manuscript. This would suggest that targeting one phenotype judiciously may suffice to deplete multiple interconverting populations.
6.3. Clinical measurements
In addition to providing additional targets to manipulate therapeutically, identifying multiple cell and tissue types beyond the frank carcinoma may provide more targets for ongoing measurement. In order to re-assess dosing schedules using a tool like the metronomogram (Figure 5(b)), we need to be able to continue to measure the tumor burden and its response to therapy at multiple time points throughout a course of treatment.
For some patients, tumor burden is already assessed long term. The minimal residual burden of leukemic cells can be monitored in a patient’s circulation using PCR methods for years [75], [76]. Unfortunately, the assessment of solid tumor burden continues to be a challenging area of study. Current imaging techniques, including positron-emission tomography (PET) and X-ray CT, have difficulty detecting tumors smaller than about 5 mm in diameter (corresponding to ~108 cells for cell diameters of ~10 microns) [77], [78], [79]. Magnetic resonance imaging (MRI) provides 1 mm x 1 mm x 1 mm spatial resolution in clinical settings, in principle resolving lesions containing ~106 cells. However, distinguishing tumor cells from background tissue remains a challenge, and the development of dynamic contrast-enhanced methods to improve specificity is ongoing [80]. As a consequence, the expansion of many solid tumors cannot be directly imaged using these methods during at least half of the time preceding diagnosis and treatment, as shown in figure 3 from Klein et al. [68]. In the supplemental discussion we show how the initial duration of tumor expansion can be similar to the total course duration subsequently required for therapeutic depletion. This suggests that, in some cases, the solid tumor burden may be technically undetectable for the majority of the total course of therapy.
One strategy for addressing this limitation in detection is to identify other cell populations and biological materials (surrogate markers) that track the tumor burden, but are easier to measure. The detection of circulating tumor cells (CTCs) may provide one such “proxy” for monitoring both early and late stages of treatment. In a recent study, Nagrath et al. isolated CTCs from patient blood using a microchip technique. The CTC populations qualitatively tracked tumor burden for a variety of cancers during treatment measured by CT methods [81]. CTCs have also been detected in blood decades after remission of disease [67], [82]. Because CTCs occur in low abundances (1 out of 109 blood cells), CTC detection likely needs to be combined with measurement of other cell populations and biomarkers for a more precise assessment of overall tumor burden. These may include extracellular matrix, carcinoma-associated fibroblasts, subpopulations of immune cells, or serum biomarkers. Examples of circulating proteins include MUC-1, which is used to monitor metastatic breast cancer [83], and prostate specific antigen (PSA) [84]. Serum biomarkers can also include non-protein materials, such as circulating micro-RNAs [85].
Whichever option we choose for quantifying tumor burden, we need to quantify therapeutic response. We simplified the analysis in this paper by considering the cartoon in which drug-kill occurs instantaneously. In contrast, biological cell death can be delayed by days and rounds of cell division following exposure to a chemotherapeutic agent. For high dosing frequencies, measured changes in cell population on a given day reflect a combination of proliferation and cell death events triggered by various previous doses of drug, making it difficult to tease apart which cells were “killed” by the most recent dose. The diffuse optical spectroscopic imaging (DOSI) method developed by Cerussi et al. may reveal drug-response before the (delayed) manifestation of cell death and clearance. This non-invasive method can be applied daily to assess changes in tissue metabolism by measuring changes in the spectra of infrared light scattered from tumors [86].
These directions will require an interdisciplinary effort, with physicists, chemists, and biologists elucidating mechanisms of stochasticity at various levels, with biologists and clinicians identifying targets for therapy, and with engineers, clinicians, and patients improving techniques for frequent, non-invasive measurement. This integrated effort will someday, hopefully, allow a “quantitative” oncologist to optimally drain a tumor to extinction.
Supplementary Material
Acknowledgments
We thank Dr. Barton Kamen (Robert Wood Johnson Medical Center) for his helpful comments. The research described here was supported by award U54CA143803 from the US National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Cancer Institute or the US National Institutes of Health.
Contributor Information
David Liao, Email: David.Liao@ucsf.edu.
Thea D. Tlsty, Email: Thea.Tlsty@ucsf.edu.
References
- 1.Spudich JL, Koshland DE., Jr Non-genetic individuality: chance in the single cell. Nature. 1976;262:467–471. doi: 10.1038/262467a0. [DOI] [PubMed] [Google Scholar]
- 2.Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage λ-infected Escherichia coli cells. Genetics. 1998;149:1633–1648. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niepel M, Spencer SL, Sorger PK. Non-genetic cell-to-cell variability and the consequences for pharmacology. Curr Opin Chem Biol. 2009;13:556–561. doi: 10.1016/j.cbpa.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raj A, van Oudenaarden A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schrödinger E. Based on lectures at the Dublin Institute for Advances Studies at Trinity College 1943. Dublin: 1944. What is Life? [Google Scholar]
- 6.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- 7.Maheshri N, O’Shea EK. Living with noisy genes: How cells function reliably with inherent variability in gene expression. Annu Rev Biophys Biomol Struct. 2007;36:413–434. doi: 10.1146/annurev.biophys.36.040306.132705. [DOI] [PubMed] [Google Scholar]
- 8.Bartholomay AF. Stochastic models for chemical reactions: I. Theory of the unimolecular reaction process. Bull Math Biophys. 1958;20:175–190. [Google Scholar]
- 9.McAdams HH, Arkin A. It’s a noisy business! Genetic regulation at the nanomolar scale. Trends Genet. 1999;15:65–69. doi: 10.1016/s0168-9525(98)01659-x. [DOI] [PubMed] [Google Scholar]
- 10.Strogatz SH. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering. Boulder, CO: Westview Press; 1994. [Google Scholar]
- 11.Boyce WE, DiPrima RC. Elementary Differential Equations and Boundary Value Problems. 7. New York: John Wiley and Sons; 2001. [Google Scholar]
- 12.Bricmont J. Science of chaos or chaos in science? Physicalia Mag. 1995;17:159–208. [Google Scholar]
- 13.Balázsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: From microbes to mammals. Cell. 2011;144:910–925. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thattai M, van Oudenaarden A. Intrinsic noise in gene regulatory networks. Proc Natl Acad Sci USA. 2001;98:8614–8619. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kepler TB, Elston TC. Stochasticity in transcriptional regulation: Origins, consequences, and mathematical representations. Biophys J. 2001;81:3116–3136. doi: 10.1016/S0006-3495(01)75949-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–3862. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammes-Schiffer S, Tully JC. Proton transfer in solution: Molecular dynamics with quantum transitions. J Chem Phys. 1994;101:4657–4667. [Google Scholar]
- 18.Townsend JS. A Modern Approach to Quantum Mechanics. Sausalito, California: University Science Books; 2000. [Google Scholar]
- 19.Gillespie DT. Stochastic simulation of chemical kinetics. Annu Rev Phys Chem. 2007;58:35–55. doi: 10.1146/annurev.physchem.58.032806.104637. [DOI] [PubMed] [Google Scholar]
- 20.Bernoulli J. Ars Conjectandi. Basel: Thurneysen Brothers; 1713. [Google Scholar]
- 21.Bueche F. Principles of Physics. 4. New York: McGraw-Hill; 1982. pp. 316–319. [Google Scholar]
- 22.Dinov ID, Christou N, Sanchez J. Central limit theorem: New SOCR applet and demonstration activity. J Stat Educ. 2008;16:1–15. doi: 10.1080/10691898.2008.11889560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waks Z, Silver PA. Nuclear origins of cell-to-cell variability. Cold Spring Harb Symp Quant Biol. 2010;75:1–8. doi: 10.1101/sqb.2010.75.027. [DOI] [PubMed] [Google Scholar]
- 24.Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet. 2002;31:69–73. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- 25.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becskei A, Kaufmann BB, van Oudenaarden A. Contributions of low molecule number and chromsomal positioning to stochastic gene expression. Nat Genet. 2005;37:937–944. doi: 10.1038/ng1616. [DOI] [PubMed] [Google Scholar]
- 27.Kalmar T, Lim C, Hayward P, Muñoz-Descalzo, Nichols J, Garcia-Ojalvo J, Arias MAA. Regulated fluctuations in Nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Süel GM, Garcia-Ojalvo J, Liberman LM, Elowitz MB. An excitable gene regulatory circuit induces transient cellular differentiation. Nature. 2006;440:545–550. doi: 10.1038/nature04588. [DOI] [PubMed] [Google Scholar]
- 29.Vilar JMG, Kueh HY, Barkai N, Leibler S. Mechanisms of noise-resistance in genetic oscillators. Proc Natl Acad Sci USA. 2002;99:5988–5992. doi: 10.1073/pnas.092133899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brock A, Chang H, Huang S. Non-genetic heterogeneity -- a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10:336–342. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- 31.Rath H, Tlsty T, Schimke RT. Rapid emergence of methotrexate resistance in cultured mouse cells. Cancer Res. 1984;44:3303–3306. [PubMed] [Google Scholar]
- 32.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y. MET amplification leads to Gefitinib resistance in lung cancer by activating ErbB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 33.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva J, Smith A. Capturing pluripotency. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borst P, Cross GAM. Molecular basis for trypanosome antigenic variation. Cell. 1982;29:291–303. doi: 10.1016/0092-8674(82)90146-5. [DOI] [PubMed] [Google Scholar]
- 37.Cross GAM. Cellular and genetic aspects of antigenic variation in trypanosomes. Annu Rev Immunol. 1990;8:83–110. doi: 10.1146/annurev.iy.08.040190.000503. [DOI] [PubMed] [Google Scholar]
- 38.Raser JM, O’Shea EK. Noise in gene expression: Origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacArthur BD, Ma’ayan A, Lemischka IR. Systems biology of stem cell fate and cellular reprogramming. Nature. 2009;10:672–681. doi: 10.1038/nrm2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Kittel C, Kroemer H. Thermal Physics. 2. Vol. 271. New York: W.H. Freeman and Company; 1980. pp. 407–408. [Google Scholar]
- 42.Johnston RN, Beverley SM, Schimke RT. Rapid spontaneous dihydrofolate reductase gene amplification shown by fluorescence-activated cell sorting. Proc Natl Acad Sci USA. 1983;80:3711–3715. doi: 10.1073/pnas.80.12.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigal A, Milo R, Cohen A, Geva-Zatorsky N, Klein Y, Liron Y, Rosenfeld N, Danon T, Perzov N, Alon U. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- 44.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skipper HE, Schabel FMJ, Mellett LB, Montgomery JA, Wilkoff LJ, Lloyd HH, Brockman RW. Implications of biochemical, cytokinetic, pharmacologic, and toxicologic relationships in the design of optimal therapeutic schedules. Cancer Chemother Rep. 1970;54:431–450. [PubMed] [Google Scholar]
- 46.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 47.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA. A chromatin–mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charlebois DA, Abdennur N, Kaern M. Gene expression noise facilitates adaptation and drug resistance independently of mutation. Phys Rev Lett. 2011;107:218101. doi: 10.1103/PhysRevLett.107.218101. [DOI] [PubMed] [Google Scholar]
- 49.Levin BR. Noninherited resistance to antibiotics. Science. 2004;305:1578–1579. doi: 10.1126/science.1103077. [DOI] [PubMed] [Google Scholar]
- 50.Dhar N, McKinney JD. Microbial phenotypic heterogeneity and antibiotic tolerance. Curr Opin Microbiol. 2007;10:30–38. doi: 10.1016/j.mib.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Webb GF. A cell population model of periodic chemotherapy treatment. In: Eisenfeld J, Levine DS, Witten M, editors. Biomedical Modeling and Simulation. Amsterdam: Elsevier Science Publishers B.V; 1992. pp. 83–92. [Google Scholar]
- 52.Foo J, Michor F. Evolution of resistance to targeted anti-cancer therapies during continuous and pulsed administration strategies. PLoS Comput Biol. 2009;5:e1000557. doi: 10.1371/journal.pcbi.1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hernández-Bronchud M, Molife R. Pharmacology and principles of high dose chemotherapy. In: Lorigan P, Vandenberghe E, editors. High Dose Chemotherapy: Principles and Practice. London: Martin Dunitz; 2002. pp. 15–28. [Google Scholar]
- 54.Cassidy . Drug development and study design. In: Shellens JHM, McLeod HL, Newell DR, editors. Cancer Clinical Pharmacology. Oxford: University Press; 2005. pp. 41–50. [Google Scholar]
- 55.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 56.Kamen BA, Glod J, Cole PD. Metronomic therapy from a pharmacologist’s view. J Pediatr Hematol Oncol. 2006;28:325–327. doi: 10.1097/00043426-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 57.Kerbel RS, Kamen BA. The anti-antiogenic basis of metronomic chemotherapy. Nat Rev Can. 2004;4:423–436. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 58.Seidman AD, et al. Randomized Phase III trial of weekly compared with every-3-weeks paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors. J Clin Oncol. 2008;26:1642–1649. doi: 10.1200/JCO.2007.11.6699. [DOI] [PubMed] [Google Scholar]
- 59.Camitta BM, Kamen BA. Role of methotrexate in the treatment of acute lymphoblastic leukemia. In: Pui CH, editor. Current Clinical Oncology: Treatment of Acute Leukemias: New Directions for Clinical Research. Totowa, New Jersey: Humana Press Ltd; 2003. pp. 357–364. [Google Scholar]
- 60.Klingebiel T, et al. Treatment of children with metastatic soft tissue sarcoma with oral maintenance compared to high dose chemotherapy: Report of the HD CWS-96 trial. Pediatr Blood Cancer. 2008;50:739–745. doi: 10.1002/pbc.21494. [DOI] [PubMed] [Google Scholar]
- 61.Alvarez A, Mickiewicz E, Brosio C, Giglio R, Cinat G, Suarez A. Weekly taxol (T) in patients who had relapsed or remained stable with T in a 21 day schedule. Proc Am Soc Clin Oncol. 1998;17:188a. [Google Scholar]
- 62.Baltali E, Altundag K, Ozisik Y, Guler N, Tekuzman G. Weekly paclitaxel in pretreated metastatic breast cancer: Retrospective analysis of 52 patients. Tohoku J Exp Med. 2004;203:205–210. doi: 10.1620/tjem.203.205. [DOI] [PubMed] [Google Scholar]
- 63.Toyoda Y, Manabe A, Tsuchida M, Hanada R, Ikuta K, Okimoto Y, Ohara A, Ohkawa Y, Mori T. Six months of maintenance chemotherapy after intensified treatment for acute lymphoblastic leukemia of childhood. J Clin Oncol. 2000;18:1508–1516. doi: 10.1200/JCO.2000.18.7.1508. [DOI] [PubMed] [Google Scholar]
- 64.Möricke A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24:265–284. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 65.de Bont JM, van der Holt B, Dekker AW, van der Does-van den Berg A, Sonneveld P, Pieters R. Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia. 2004;18:2032–2035. doi: 10.1038/sj.leu.2403538. [DOI] [PubMed] [Google Scholar]
- 66.Klement GL, Kamen BA. Nontoxic, fiscally responsible, future of oncology: Could it be beginning in the third world? J Pediatr Hematol Oncol. 2011;33:1–3. doi: 10.1097/MPH.0b013e3182024918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Can. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Can. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 69.Panetta JC. A mathematical model of breast and ovarian cancer treated with paclitaxel. Math Biosci. 1997;146:89–113. doi: 10.1016/s0025-5564(97)00077-1. [DOI] [PubMed] [Google Scholar]
- 70.Samaga R, Saez-Rodriguez J, Alexopoulos LG, Sorger PK, Klamt S. The logic of EGFR/ErbB signaling: Theoretical properties and analysis of high-throughput data. PLoS Comput Biol. 2009;5:e1000438. doi: 10.1371/journal.pcbi.1000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol. 2010;4:267–283. doi: 10.1016/j.molonc.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Losi L, Baisse B, Bouzourene H, Benhattar J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis. 2005;26:916–922. doi: 10.1093/carcin/bgi044. [DOI] [PubMed] [Google Scholar]
- 73.Chmielecki J, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Browder T, Butterfield CE, Kräling BM, Shi B, Marshall B, O’Reilly MS, Folkman J. Antiangiogenic scheduling of chemotherapy improves efficacy against expeirmental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- 75.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, Nowak MA. Dynamics of chronic myeloid leukemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 76.Hughes TP, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic meyloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS) Blood. 2010;116:3758–3765. doi: 10.1182/blood-2010-03-273979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lardinois D, Weder W, Hany TF, Kamel EM, Korom S, Seifert B, von Schulthess GK, Steinert HC. Staging of non-small-cell lung cancer with integrated positron-emission tomography and computed tomography. N Engl J Med. 2003;348:2500–2507. doi: 10.1056/NEJMoa022136. [DOI] [PubMed] [Google Scholar]
- 78.Marom EM, Sarvis S, Herndon JE, II, Patz EF. T1 lung cancers: Sensitivity of diagnosis with fluorodeoxyglucose PET. Radiology. 2002;223:453–459. doi: 10.1148/radiol.2232011131. [DOI] [PubMed] [Google Scholar]
- 79.Satti J. The emerging low-dose therapy for advanced cancers. Dose-Response. 2009;7:208–220. doi: 10.2203/dose-response.08-010.Satti. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li KL, Henry RG, Wilmes LJ, Gibbs J, Zhu X, Lu Y, Hylton NM. Kinetic assessment of breast tumors using high spatial resolution Signal Enhancement Ratio (SER) imaging. Magn Reson Med. 2007;58:572–581. doi: 10.1002/mrm.21361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagrath S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meng S, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–8162. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 83.Duffy MJ, Evoy D, McDermott EW. CA 15-3: Uses and limitation as a biomarker for breast cancer. Clin Chim Acta. 2010;411:1869–1874. doi: 10.1016/j.cca.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 84.Gomella LG, Liu XS, Trabulsi EJ, Kelly WK, Myers R, Showalter T, Dicker A, Wender R. Screening for prostate cancer: the current evidence and guidelines controversy. Can J Urol. 2011;18:5875–5883. [Google Scholar]
- 85.Zen K, Zhang CY. Circulating microRNAs: A novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012;32:326–348. doi: 10.1002/med.20215. [DOI] [PubMed] [Google Scholar]
- 86.Cerussi AE, Tanamai VW, Mehta RS, Hsiang D, Butler J, Tromberg BJ. Frequent optical imaging during breast cancer neoadjuvant chemotherapy reveals dynamic tumor physiology in an individual patient. Acad Radiol. 2010;17:1031–1039. doi: 10.1016/j.acra.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.