

Abstract
The neuronal loss associated with Alzheimer's disease (AD) affects areas of the brain that are vital to cognition. Although recent studies have shown that new neurons can be generated from progenitor cells in the neocortices of healthy adults, the neurogenic potential of the stem/progenitor cells of AD patients is not known. To answer this question, we compared the properties of glial progenitor cells (GPCs) from the cortices of healthy control (HC) and AD subjects. The GPCs from AD brain samples displayed reduced renewal capability and reduced neurogenesis compared with GPCs from HC brains. To investigate the mechanisms underlying this difference, we compared β-catenin signaling proteins in GPCs from AD versus HC subjects and studied the effect of amyloid β peptide (Aβ, a hallmark of AD pathology) on GPCs. Interestingly, GPCs from AD patients exhibited elevated levels of glycogen synthase kinase 3β (GSK-3β, an enzyme known to phosphorylate β-catenin), accompanied by an increase in phosphorylated β-catenin and a decrease in nonphosphorylated β-catenin compared with HC counterparts. Furthermore. we found that Aβ treatment impaired the ability of GPCs from HC subjects to generate new neurons and caused changes in β-catenin signaling proteins similar to those observed in GPCs from AD patients. Similar results were observed in GPCs isolated from AD transgenic mice. These results suggest that Aβ-induced interruption of β-catenin signaling may contribute to the impairment of neurogenesis in AD progenitor cells.
Introduction
Studies have shown that large pools of glial progenitor cells (GPCs) are located in the human neocortex (Gogate et al., 1994; Pincus et al., 1997; Roy et al., 1999; Chang et al., 2000; Arsenijevic et al., 2001; Palmer et al., 2001; Windrem et al., 2002; Nunes et al., 2003; Goldman and Sim, 2005). These cells are classified by their antigen phenotypes, such as A2B5-positive cells (Kondo and Raff, 2000; Nunes et al., 2003) and NG2-expressing cells (Belachew et al., 2003; Aguirre and Gallo, 2004; Aguirre et al., 2004; Chittajallu et al., 2004; Rivers et al., 2008). NG2-positive (NG2+) cells are the predominant type of GPC in injured brains (Chang et al., 2000; Horky et al., 2006). Although GPCs' ability to generate new neurons in local brain regions remains controversial (Spalding et al., 2005; Bhardwaj et al., 2006), stem/progenitor cells from non-neurogenic regions have been shown to possess self-renewal and neurogenic capabilities after exposure to environmental stressors (Armstrong et al., 1992; Gogate et al., 1994; Pincus et al., 1997; Scolding et al., 1998; Wolswijk, 1998; Roy et al., 1999; Arsenijevic et al., 2001; Palmer et al., 2001; Nunes et al., 2003). Additionally, recent studies have shown that NG2+ GPCs isolated from postnatal rodents are able to form neurospheres and to generate new neurons and glia (Belachew et al., 2003; Aguirre and Gallo, 2004; Aguirre et al., 2004; Chittajallu et al., 2004).
Alzheimer's disease (AD) is a neurodegenerative disease characterized by amyloid plaque deposits and neurofibrillary tangles (Selkoe, 2003; Tanzi and Bertram, 2005). While the progressive neuronal loss that occurs in AD has not been found to induce neurogenesis (Regeur et al., 1994; Bundgaard et al., 2001), significant gliogenesis was observed in the degenerative cortex (Otero et al., 2004). The mechanism that underlies the fate of GPCs remains unclear; however, several signal transduction pathway cascades have been shown to control progenitor cell fate (Anderton et al., 2000). Wnt/β-catenin signaling, for example, has been shown to be involved in progenitor cell lineage decisions (Zhang et al., 1998; He et al., 2004; Hirabayashi et al., 2004) in developmental stages (Wright et al., 1997; Lee et al., 2004; Logan and Nusse, 2004; Otero et al., 2004) as well as in adult brains (Lie et al., 2005). Reduced Wnt/β-catenin signaling has been reported in AD brains (Gavrieli et al., 1992; Zhou et al., 2008), and decreased expression of β-catenin was also found in AD patients carrying presenilin-1-inherited mutations (Anderton et al., 2000; Hirabayashi and Gotoh, 2005; Lie et al., 2005).
In this study, we found that the levels of neurogenesis by GPCs from AD brains were significantly reduced compared with those from HC brains. This reduction in neurogenesis was also accompanied by disrupted β-catenin signaling in AD versus HC GPCs and their progeny. We found that the treatment of HC GPCs with exogenous Aβ induced changes in neurogenesis, β-catenin signaling, and proneural gene expression that were similar to those exhibited by AD GPCs and their progeny. Furthermore. we found that the overexpression of β-catenin partially restored neurogenesis from AD GPCs, while the reduction of β-catenin using β-catenin siRNA decreased neurogenesis from HC GPCs. These results suggest that elevated Aβ levels in AD brains may impair neuron induction from GPCs by interfering with the β-catenin signaling pathway.
Materials and Methods
Collection of human cortical tissue.
Brain tissues were obtained from rapidly autopsied geriatric patients (n = 14 HC, n = 14 AD) enrolled in the Brain Donation Program at Sun Health Research Institute. The average postmortem interval (PMI) was <3 h. The average ages of the HC and AD subjects were 84.3 and 83.7 years, respectively. The tissues were isolated and experimental manipulations were performed with sex-matched HC and AD (supplemental Table 1, available at www.jneurosci.org as supplemental material). To avoid deep formations, the superficial gray matter of the superior temporal cortex was harvested.
Collection of mouse cortical tissue.
Cortical tissues were harvested from wild-type (WT) and AD-like transgenic (APP23) mice of 12 and 24 months of age (n = 20 for each group). The mice were from a C57BL/6 background. The transgenic APP23 mice were provided by Novartis Institute for Biomedical Research; these mice express mutant human βAPP (Swedish double mutation, KM670/671NL) under the control of a brain- and neuron-specific murine Thy-1 promoter element.
Isolation of cortical NG2+ GPCs by immunomagnetic selection.
The cortical tissue was cut into 200 μm slices, digested with 20 U of papain/DNase I (Worthington), and overlaid onto a discontinuous density gradient of Optiprep (Nycomed). After gradient centrifugation for 60 min, cells were incubated with a polyclonal antibody that binds to cell-surface chondroitin sulfate proteoglycan (NG2, Millipore Bioscience Research Reagents) as described previously (Wright et al., 1997; Konishi et al., 2002; Othman et al., 2005). GPCs were further separated by the addition of Dynabeads (Dynal) that had previously been coated with recombinant streptavidin via DNA linker (Dynal) and conjugated to a biotinylated goat anti-rabbit IgG (Jackson ImmunoResearch). The binding cells were selected with a Dynal magnetic particle concentrator and were removed from the beads with DNase I.
Cell culture.
Single NG2-expressing cells from human or mouse brains were cultured in flasks that were precoated with 10 mg/ml poly(2-hydroxyethyl methacrylate) (Sigma, catalog: P3932) to avoid cell adhesion (Zhang et al., 2001). Primary neurospheres were generated by suspending the NG2-expressing cells in Neurobasal A medium containing 5 ng/ml bFGF, 10 ng/ml EGF, and 20 ng/ml PDGF-AA (growth medium) for 14 d. Secondary neurospheres were generated by dissociating the primary neurospheres with papain and resuspending the cells in identical growth medium for another 14 d. Before analyses, cells from neurospheres were dissociated and seeded on chamber slides or plates that were precoated with 100 μg/ml poly-l-lysine hydrobromide (PLL, Sigma, catalog: P9155). For differentiation induction, Neurobasal A was supplemented with 2% B27, 1% N-2 supplement, and 10 ng/ml BDNF (for maintaining cell survival). The medium was deprived of mitogenic growth factors, PDGF-AA, bFGF, and EGF. The neurotrophic factors and culture supplements used in the differentiation medium were obtained from Invitrogen, except for the other supplies mentioned above.
Aβ1–42 treatment.
Before treating the cultures, Aβ1–42 (PP69, EMD Biosciences) was preaggregated at 37°C overnight (Haughey et al., 2002b). Dissociated GPCs were grown in differentiation medium and were exposed to various concentrations of Aβ1–42 (0, 0.1, 1.0, or 5.0 μm) 3 h after plating (Haughey et al., 2002b) for 2 d. The medium was replaced with differentiation medium that lacked Aβ1–42 on the day 3 and at various time points for analyses.
Aβ ELISA.
Human GPCs, their progeny cells, and the incubation medium were collected. Neocortical tissues from APP23 and WT mice (n = 3 per group) were harvested at 12 and 24 months of age. The cells were sonicated, and the brain tissue was homogenized in homogenization buffer. The Aβ ELISA was performed as described previously (He et al., 2007). In brief, protein concentrations were measured by protein assays (Bio-Rad). For total Aβ ELISA, the capture antibody was monoclonal anti-Aβ antibody 4G8 (Millipore Bioscience Research Reagents), and the detection antibody was biotinylated monoclonal antibody anti-Aβ 6E10 (Serotec). Aβ40 and Aβ42 were measured with Aβ40 and Aβ42 ELISA kits (KHB3481 and KHB3544, Biosource). The ELISA system has been extensively tested and no cross-reactivity between Aβ40 and Aβ42 was observed (data not shown). Four technical replicates were performed for each Aβ determination.
RT-PCR.
For RT-PCR, the sequences of primers for Ngn1 were 5′-AGTGACCTATCCGGCTTCCT and 5′-AGCGTCTGATTTTGGTGAG, and the primers for Ngn2 were 5′-GCTGGGTCTGGRACACGTT and 5′-AACACTGCCTCGGAGAAGAG. RT-PCR was performed using the One-Step RT-PCR kit (Invitrogen) according to the manufacturer's instructions.
β-Catenin recombinant retrovirus preparation.
The β-catenin full cDNA clone was purchased from OpenBiosystems. The open reading frame was subcloned to a pLXRN retrovirus vector from Clontech. A FLAG tag peptide sequence was inserted into the C-terminal of β-catenin open reading open reading frame before the stop codon. Retrovirus was produced by cotransfecting a GP2–293 packaging cell line with the pLXRN vector (containing the β-catenin open reading frame) and a VSV-G expression vector via calcium phosphate transfection (Clontech). The recombinant virus was collected and stored at −80°C.
β-Catenin transfection.
Suspended NG2+ cells from 4 AD patient brains were cultured in the growth medium for 14 d. The spheres were dissociated into single cells. Cells were put into the growth medium containing ∼1 × 106 β-catenin recombinant retrovirus particles for 5 h. The medium was changed every 3 d, and the culture was extended for 14 d. The β-catenin-transfected neurosphere was harvested and dissociated into single cells. Eighty percent of the cells were plated in a 6-well plate for Western blot analysis, and 20% of the cells in an 8-well plate for immunochemistry after 4 d culture.
siRNA introduction.
β-Catenin small interference RNA (siRNA) (sc-29210) (Zeng et al., 2007), GSK-3β siRNA (sc-35525) (Zhou et al., 2008), and control nonspecific siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. Passage GPCs from WT or APP23 mouse brains were dissociated and plated on 24-well plates in differentiation medium and were transiently transfected with siRNAs using Lipofectamine 2000 (Invitrogen). The cells were collected for Western blot assay at 3 d after transfection.
Immunofluorescence.
Cells were fixed on 8-well chamber slides (∼1 × 105 cells/ml) with 4% paraformaldehyde in 4°C for 15 min and were sequentially treated with 0.15% Triton X-100 (for cell permeability) for 10 min and 10% goat serum (for blockage of nonspecific binding) for 30 min. The cells were immunostained with specific antibodies at 2, 7, or 14 d in vitro. Primary antibodies were applied as follows: rabbit anti-doublecortin (DCX, Abcam, ab18723, 1:400), mouse anti-PSA-NCAM (Millipore Bioscience Research Reagents, MAB5324, 1:500, IgM), mouse anti-βIII tubulin (Covance, 1:500), rabbit anti-MAP-2 (Millipore Bioscience Research Reagents, MAB3418, 1:400), rabbit anti-GFAP (DAKO, 1:3000), mouse anti-GFAP (Covance, SMI-22R, 1: 10,000), mouse anti-preoligodendroglial antigen O1 (R&D, 1:2000, IgM), rabbit anti-β-catenin (Millipore Bioscience Research Reagents, AB19022, 1: 1000), rabbit anti-β-catenin (Sigma, C2206, 1:2000), and rabbit anti-FLAG (Sigma, F7425, 1:10,000). Fluorescent-labeling Alexa Fluor 488- or 568-conjugated secondary antibodies against rabbit IgG, mouse IgG or IgM were used for detection (Invitrogen, 1:1000).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (Gavrieli et al., 1992).
The cells were fixed with 4% paraformaldehyde at 4°C, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 3% BSA for 30 min. The cells were then incubated with the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reaction mixture from the Boehringer Mannheim kit according to the manufacturer's procedures (Boehringer Mannheim). Negative controls were treated with Label Solution (without terminal transferase) instead of the TUNEL reaction mixture. Positive controls were incubated with DNase I to induce DNA strand breaks and were labeled with the TUNEL reaction mixture. Incubation was performed in a humidified atmosphere for 60 min at 37°C in the dark. Samples were analyzed using a fluorescent microscope. Normal nuclei, which contained only insignificant amounts of DNA 3′-OH ends, did not stain with this technique; however, cells with apoptotic morphology exhibited condensed nuclei labeling.
Nuclear extraction.
Cell nuclei were extracted based on the protocol of Nuclear Extraction Kit by IMAGENEX Corporation (catalog: 10081K). In brief, cells were collected and resuspended in 300 μl of 1× Hypotonic Buffer. The suspension was added to 15 μl of the 10% Detergent Solution. The whole cell lysate was centrifuged at 14,000 rpm for 20 min, thereby isolating the cytoplasmic fraction in the supernatant. The nuclear fraction was obtained by resuspending the pellet in 30 μl of Nuclear Lysis Buffer and centrifuging the suspension, thereby isolating the nuclear fraction in the supernatant.
Western blot.
GPC neurosphere cells or their daughter cells were lysed using cell lysis buffer (10 mm Tris-HCl pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 0.1 m Na3VO4, 0.5% Triton X-100). Twenty-five micrograms of protein were separated on an 8% SDS-PAGE gel and transferred to a PVDF membrane. Proteins were probed with the following polyclonal rabbit antibodies: nestin (Santa Cruz, 1:100), Dlx2 (Millipore Bioscience Research Reagents, Ab5726, 1:5000), Wnt3 (Zymed, catalog: 38–2700, 1:2000), frizzled (Santa Cruz, sc-9169, 1:5000, H-300), Neurogenin 2 (Ngn2, Abcam, ab38550, 1:1000), ASH1 (MASH1, Millipore Bioscience Research Reagents, AB15582, 1:4000), β-catenin (Millipore Bioscience Research Reagents, AB19022, 1:10,000), PARP (Santa Cruz, sc-7150, 1:1000), and β-catenin [phosphoserines 33 and 37] (Zymed, catalog: 486000, 1:2000). Monoclonal mouse antibodies were applied against GSK-3β (Zymed, catalog: 39–9500, 1:5000), NeuN (Millipore Bioscience Research Reagents, MAB377, 1:500), and β-actin (Sigma, 1:20,000). Similar procedures were applied for neocortical samples from mice. Western blot measurements were repeated 3 times independently.
Quantification of immunoreactive structures.
Digitized images were obtained with a DEI-470 digital camera (Optronics) on a Leica fluorescence microscope (Leica). Immunopositive cells were counted in four 40× microscope fields per sample. Blue nuclei (DAPI positive) were counted as the total cell number. Results were analyzed with Image-pro Plus Analysis by an investigator who was blinded to the experimental subjects and treatment.
Statistical analyses.
Results were expressed as mean ± SD. All analyses were performed using a software program (SPSS version 11.5.1; SPSS). Differences between two groups were assessed using Student's t tests. Differences between three or more groups were evaluated by one-way ANOVAs. The level of significance was p ≤ 0.05.
Results
Aβ is increased and neurogenesis is decreased in GPC cultures from AD patients
Neurons in AD brains have been shown to produce increased levels of Aβ, especially Aβ40 and Aβ42, compared with neurons in healthy brains (Selkoe, 2003; Tanzi and Bertram, 2005). To investigate whether Aβ production is also promoted in GPCs in AD brains, Aβ40 and Aβ42 levels in GPCs from AD and HC brains were measured via ELISA. We found a twofold increase in both Aβ40 and Aβ42 levels in AD GPCs compared with their HC counterparts (Fig. 1a). AD GPCs' progeny also exhibited increased Aβ40 and Aβ42 compared with their HC counterparts (Fig. 1a). Because Aβ is generated in a vesicular pathway, it has little effect on the extra-vesicular components of the cell. Therefore, we tested Aβ levels in the medium, and normalized both Aβ40 and Aβ42 to the total protein levels of the corresponding cell lysates. As expected, both Aβ40 and Aβ42 levels increased in the medium of AD GPCs, with a prominent increase of Aβ42 levels in the medium containing AD daughter cells (Fig. 1b).
Figure 1.
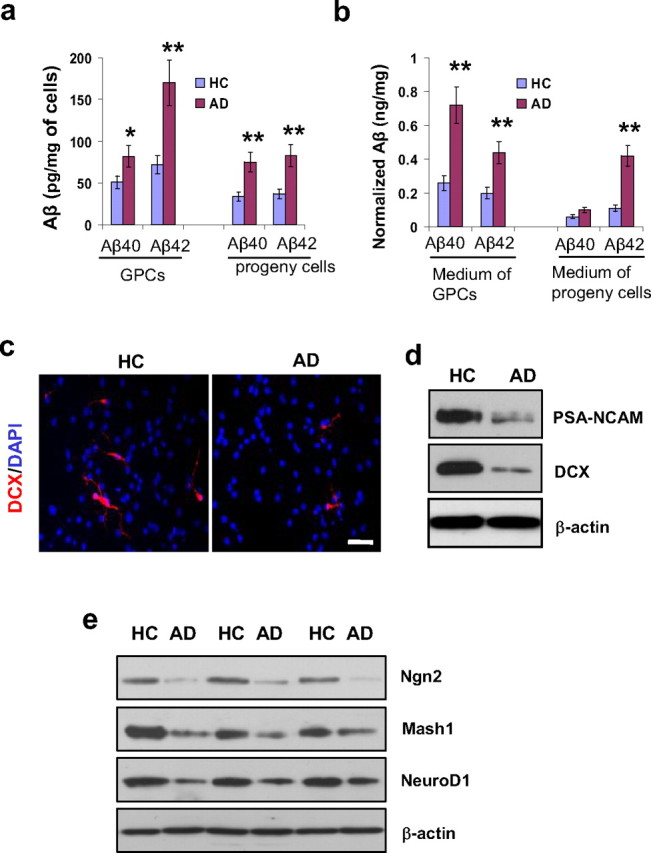
Aβ production, neurogenesis, and proneural gene products in AD versus HC progenitor cells. Second passage GPCs from AD and HC subjects (n = 3 per group) were cultured in differentiation medium for 2 d. a, Aβ ELISA revealed increased levels of Aβ40 and Aβ42 in AD GPCs and in their daughter cells compared with their HC counterparts (n = 3 per group, Student's t test, *p < 0.05, **p < 0.01). b, Increased levels of Aβ40 and Aβ42 were also observed in the medium surrounding AD GPCs and their daughter cells compared with the medium surrounding HC cells (n = 3 per group, Student's t test, **p < 0.01). c, The levels of neurogenesis among the AD and HC GPC progeny were visualized by immunostaining new neurons with an anti-DCX antibody and labeling all cell nuclei with DAPI. Scale bar, 50 μm. d, A representative Western blot showed significantly decreased levels of neuroblast markers PSA-NCAM and DCX in AD progeny cells compared with their HC counterparts. e, Proneural gene expressions of Ngn2, Mash1, and NeuroD1 were also decreased in AD progeny cells compared with their HC counterparts.
To compare the cell fate decisions of AD versus HC GPCs as they relate to neurogenesis, we observed the differentiation of second passage GPC progeny toward neurons after 2 d of being cultured in differentiation medium by immunolabeling the cells with DAPI for nuclei detection and with doublecortin (DCX) for detection of immature neurons (Fig. 1c). We compared the proportions of GPC progeny that developed into neurons in HC versus AD subjects by counting the DAPI-positive and DCX-positive cells in random fields. In the HC progeny, there were 82 DCX-positive cells out of 1526 DAPI-labeled nuclei, and, in the AD progeny cells, there were 26 DCX-positive cells out of 1438 DAPI-labeled nuclei. Statistical analysis revealed that a significantly smaller portion of AD GPC progeny developed into immature neurons compared with HC GPC progeny (1.8 ± 0.4% in AD vs 5.4 ± 1.9% in HC, n = 3 per group, Student's t test, p < 0.05). To confirm the immunostaining results, we compared the levels of DCX and polysialic acid-neural cell adhesion molecule (PSA-NCAM), another marker of immature neurons, in AD versus HC GPC progeny after 2 d in differentiation medium (Fig. 1d). The results indicate a decline in the production of new neurons from AD GPCs compared with HC GPCs.
To determine whether the decline in neurogenesis from AD GPCs is due to changes in proneural gene expression, we measured the expression levels of neurogenin 2 (Ngn2), mammalian achaete-scute homolog 1 (Mash1), and neurogenic differentiation factor 1 (NeuroD1) by Western blot. AD GPC progeny exhibited reduced expression of Ngn2, Mash1, and NeuroD1 compared with HC progeny after 2 d in differentiation medium (Fig. 1e), suggesting that the decline of neurogenic gene expression may be associated with the reduction of differentiation toward neurons in AD versus HC GPC progeny.
Aβ promotes apoptosis in newly generated neurons
To ensure that the observed decline in visible new neurons in AD GPC progeny is not due to insufficient time for differentiation, we extended our observation time point to the seventh day of culture. We detected various differentiated cell types by triple immunostaining for βIII tubulin (a neuron-selective protein), GFAP (an astrocyte protein), and O1 (an oligodendrocyte marker) (Fig. 2a). Fewer AD GPC progeny differentiated into neurons than did HC progeny. To compare GPC differentiation in a more robust manner, the GPC progeny were costained with DAPI and one cell type-specific marker, and the number of type-specific cells out of the total number of cells in four randomly selected 40× microscope fields was determined. One hundred sixteen out of 1367 HC progeny cells differentiated into neurons (indicated by βIII tubulin labeling), whereas only 8 out of 1496 AD progeny cells differentiated into neurons. Statistical analysis indicated a significant decline in the percentage of neurons in AD versus HC GPC progeny (0.5 ± 0.3% vs 8.5 ± 1.3%, respectively, n = 3 per group, Student's t test, p < 0.01). In the same manner, we analyzed the proportions of astrocytes (indicated by GFAP+ labeling) and oligodendrocytes (indicated by O1+ labeling) in HC and AD GPC progeny. For HC progeny, 94.6 ± 5.2% differentiated into astrocytes and 2.4 ± 1.1% differentiated into oligodendrocytes, and, for AD progeny, 94.7 ± 6.8% differentiated into astrocytes and 5.1 ± 1.6% differentiated into oligodendrocytes.
Figure 2.
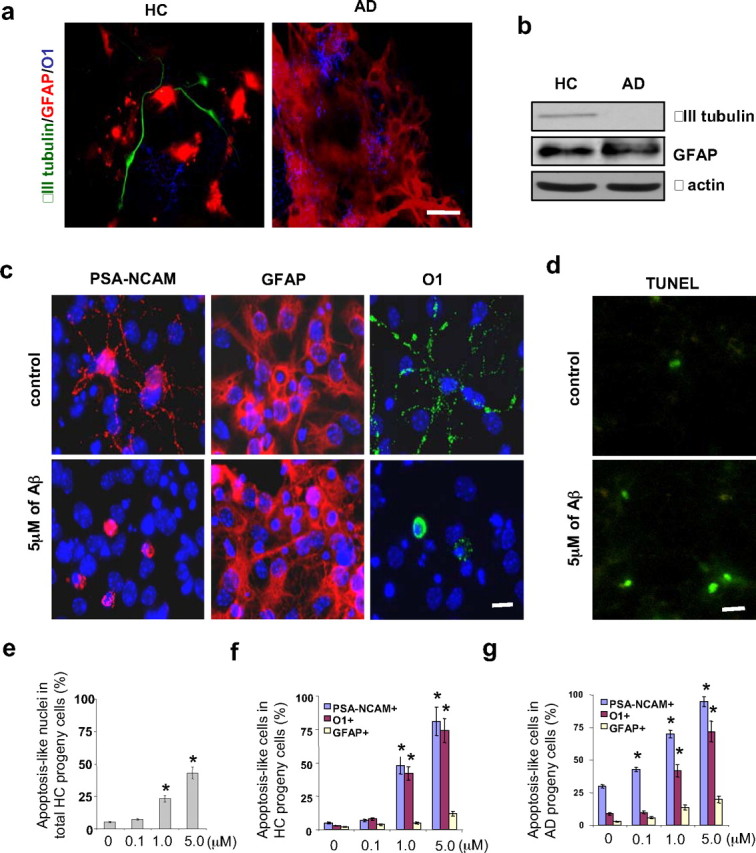
Cell fate decisions of AD and HC GPCs and the apoptotic effect of Aβ treatment on newly generated cells. Second passage GPCs from AD and HC subjects (n = 3 per group) were cultured in either Aβ-treated or nontreated differentiation medium for 2 d followed by another 5 d in Aβ-free differentiation medium. a, The cell fate decisions of nontreated AD and HC GPC progeny were visualized by immunostaining the astrocytes (red) with an anti-GFAP antibody, the oligodendrocytes (blue) with an anti-O1 antibody, and the mature neurons (green) with an anti-βIII tubulin antibody. There were fewer neurons (green) among the AD GPC progeny than among the HC progeny. b, A representative Western blot demonstrated reduced βIII tubulin levels in the AD progeny compared with the HC progeny (both nontreated), while no significant differences in GFAP expression were observed. c, Differentiated progeny from HC GPCs treated with 5 μm Aβ1–42 were visualized by immunostaining for PSA-NCAM (a neuroblast marker), GFAP (an astrocyte marker), O1 (an oligodendrocyte marker), and DAPI (a cell nucleus marker). d, A representative micrograph with TUNEL staining showed an increased number of apoptotic cells in progeny from Aβ-treated versus nontreated HC GPCs. e, Quantification of immunoreactive structures revealed a significant increase in the number of apoptotic nuclei among the progeny of HC GPCs treated with ≥1 μm Aβ compared with their nontreated counterparts (n = 3 for each group, ANOVA, *p < 0.01). f, Quantification also revealed an increase in the number of apoptosis-like PSA-NCAM+ and O1+ cells among the progeny of HC GPCs exposed to ≥1 μm Aβ (n = 3 per group, ANOVA, *p < 0.01). g, Nearly all of the PSA-NCAM+ cells and most of the O1+ cells were apoptosis-like in the AD GPC progeny treated with 5 μm Aβ (n = 3 per group, ANOVA, *p < 0.01). Experiments were repeated three times per condition. Scale bars: a, 25 μm; c, d, 10 μm.
To confirm the immunostaining results, the β-III tubulin and GFAP levels in AD and HC GPC progeny after 7 d in differentiation medium were analyzed by Western blot (Fig. 2b). The βIII tubulin levels were significantly lower than the GFAP levels in both the HC and AD progeny, confirming that the GPCs are more likely to differentiate into astrocytes than neurons. Additionally, the βIII tubulin levels in the AD progeny were reduced compared with the HC progeny, confirming that mature neuron formation by GPCs is impaired in AD.
To determine whether Aβ-induced, premature neuronal death contributes to the lack of mature neurons in AD GPC progeny cells, we observed cell death in HC and AD GPC progeny after exposure to various doses of preaggregated Aβ1–42 (0, 0.1, 1, 5 μm) for 2 d followed by 5 d in Aβ-free differentiation medium. The cells with condensed or fragmented nuclei were considered apoptotic using DAPI staining. The neuronal-restricted progenitors were verified with an antibody against PSA-NCAM, astrocytes with an antibody against GFAP, and preoligodendroglial cells with an antibody against surface antigen O1 (Fig. 2c). Apoptotic cells were also visualized using TUNEL staining, which revealed that HC progeny that were treated with 5 μm Aβ1–42 exhibited more apoptotic cells than did nontreated HC progeny (Fig. 2d). The proportion of differentiated GPC progeny that were apoptotic was determined by costaining with DAPI and a cell type-specific marker (either PSA-NCAM for neuronal progenitors, GFAP for astrocytes, or O1 for oligodendrocytes) and determining the proportion of differentiated cells (indicated by a specific marker) whose nuclei (indicated by DAPI) were condensed or fragmented out of the total number of differentiated cells of that cell type. When the different cell types (neuronal progenitors, oligodendrocytes, and astrocytes) in the HC progeny were grouped together, treatment with ≥1 μm Aβ increased the overall proportion of apoptotic cells (Fig. 2e). For both HC and AD progeny cells, treatment with ≥1 μm Aβ significantly and dramatically increased the proportion of neuronal progenitors and oligodendrocytes that were apoptotic, so that, with 5 μm Aβ treatment, ∼90% of neuronal progenitors and ∼70% of oligodendrocytes were apoptotic (Fig. 2f,g). Aβ treatment did not significantly alter the percentage of astrocytes that were apoptotic (Fig. 2f,g), suggesting that neuronal- and oligodendroglial-restricted progenitors were more susceptible to Aβ toxicity than were newly generated astrocytes. These data demonstrate that Aβ may induce apoptosis in newly generated neurons in a dose-dependent manner.
β-Catenin decreases in AD glial progenitor cells and their daughter cells
To determine whether the Wnt/β-catenin signaling pathway contributes to the observed differences in cell fate decision of AD and HC GPCs and their progeny, we used Western blot analysis to determine the levels of various molecules involved in the pathway. We found no significant differences in the expression levels of Wnt3 or frizzled (a Wnt3 receptor) between AD and HC GPCs or between their progeny cells (Fig. 3a). However, nonphosphorylated β-catenin, which is the downstream effector of Wnt3 signaling, was significantly decreased in AD GPCs and their progeny cells compared with the HC counterparts, whereas phosphorylated β-catenin and GSK-3β (an enzyme that phosphorylates β-catenin) were increased (Fig. 3a). We also examined the expression and subcellular localization of β-catenin in the HC and AD GPC progeny by immunohistochemistry and found that the expression of β-catenin in the nuclei of AD progeny was decreased compared with the HC progeny (Fig. 3b).
Figure 3.
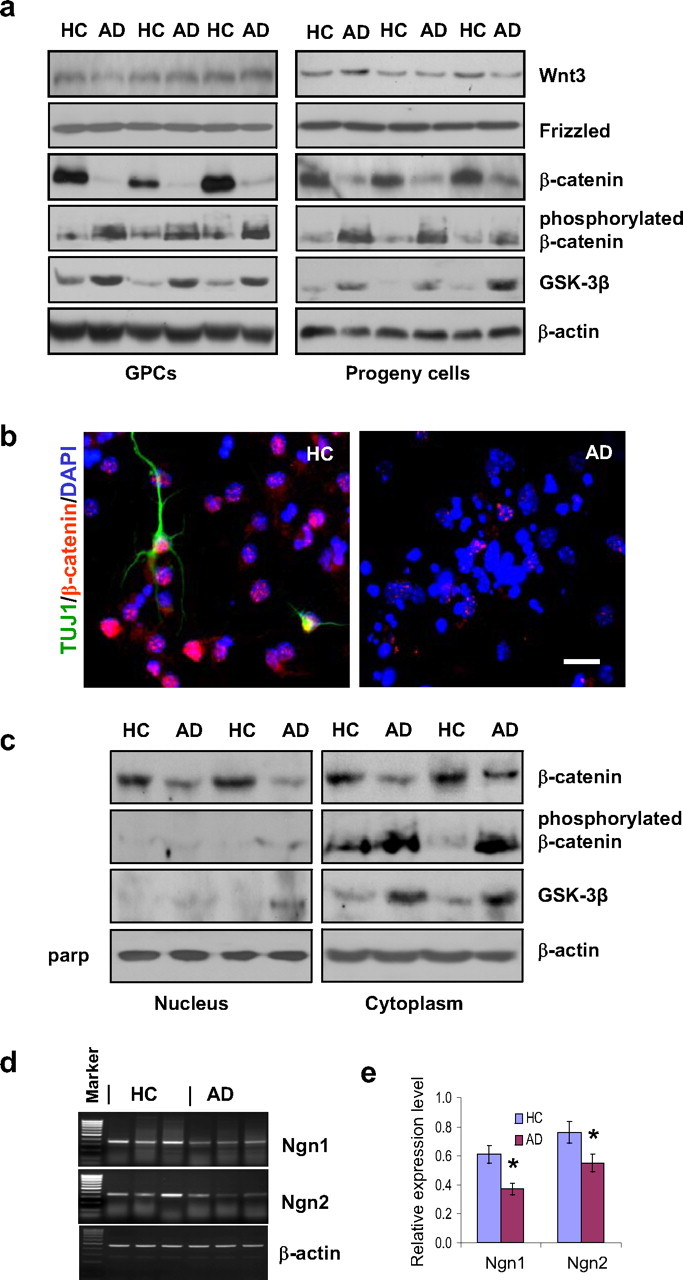
β-Catenin signaling and proneural gene transcription in AD versus HC GPCs and their progeny. a, Western blot showed a reduction of nonphosphorylated β-catenin levels and an increase in both phosphorylated β-catenin and GSK-3β levels in AD GPCs and their progeny compared with their HC counterparts, without significant differences in either Wnt3 or frizzled expression. b, Immunostaining for βIII-tubulin (mature neurons), β-catenin, and DAPI (all nuclei) showed a decrease in β-catenin expression in AD differentiating cells compared with HC cells. c, Western blot showed a decrease in nonphosphorylated β-catenin expression in the nuclear fractions of AD GPC progeny compared with HC progeny with no detectable GSK-3β or phosphorylated β-catenin expression in AD or HC progeny. The nonphosphorylated β-catenin levels also decreased in the cytoplasmic fraction of AD progeny compared with HC progeny; however, this change was accompanied with increased levels of GSK-3β and phosphorylated β-catenin. d, RT-PCR revealed that mRNA levels of Ngn1 and Ngn2 were reduced in AD GPC progeny compared with HC progeny. e, Spot densitometric analysis by FluorChem 8900 software was used to quantify the RT-PCR relative expression levels of Ngn1 and Ngn2 mRNA in the AD and HC GPC progeny. The expression levels were normalized to the respective β-actin levels (Student's t test, *p < 0.05, n = 3 per group).
The subcellular expression levels of phosphorylated and nonphosphorylated β-catenin as well as the levels of GSK-3β in HC and AD GPC progeny were examined further by Western blot. For both HC and AD progeny, phosphorylated β-catenin and GSK-3β were found mostly in the cytoplasm, whereas nonphosphorylated β-catenin was found in both the nucleus and the cytoplasm. The levels of nonphosphorylated β-catenin throughout the AD progeny cells were significantly reduced compared with HC progeny. This was accompanied by increased levels of phosphorylated β-catenin and GSK-3β in the cytoplasm of the AD progeny (Fig. 3c).
To determine the whether the observed reduction of β-catenin levels in AD GPCs and their daughter cells is accompanied by decreased transcription of proneuronal genes, we performed RT-PCR to examine the mRNA levels of the proneural genes Ngn1 and Ngn2 in AD versus HC GPC daughter cells. The results showed that both Ngn1 and Ngn2 mRNA levels were decreased in AD daughter cells, suggesting that decreased β-catenin may be related to decreased proneural gene expression and less differentiation toward neurons in AD GPCs and their progeny (Fig. 3d,e).
β-Catenin transfection restores the neuronal induction of AD GPCs
To determine whether β-catenin levels affect neurogenesis in AD GPCs, we exposed the AD GPCs to retroviruses containing the β-catenin open reading frame for 5 h and extended the culture for 14 d to see whether the overexpression of β-catenin would increase neuronal production from AD neurospheres. The transfected neurospheres were dissociated into single cells and cultured in the differentiating medium for 4 d. The controls were nontransfected GPCs from HC and AD brains. To verify the successful transfection of the AD GPC progeny, we used immunostaining to visualize cells that were successfully transfected (labeled with an anti-FLAG-tag antibody) out of the total number of cells (labeled with DAPI) (Fig. 4b). We found that 39.32 ± 13.23% (n = 3) of the AD GPC progeny that were exposed to the retrovirus were successfully transfected (FLAG+), while none of the control group AD GPC progeny were FLAG+. Western blot revealed that the expression level of β-catenin in the transfected AD GPC progeny was greater than in the nontransfected AD progeny but lower than the β-catenin level in the nontransfected HC progeny (Fig. 4a).
Figure 4.
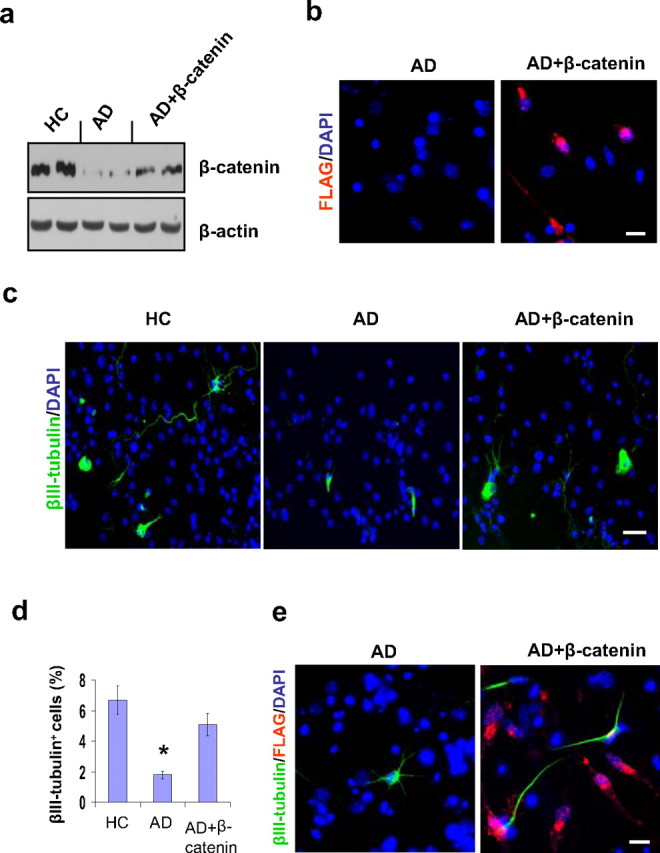
Effect of β-catenin transfection on neurogenesis in AD GPC progeny. Single passage NG2+ cells from AD brains were virally transfected with either β-catenin cDNA or a control vector for 5 h, and the culture was extended to 14 d. The transfected AD, control AD, and control HC neurospheres were dissociated into single cells and were cultured in differentiation medium for another 4 d. a, Western blot revealed that the expression level of β-catenin in the transfected AD GPC progeny was significantly greater than in the control AD progeny but less than in the control HC progeny. b, The success of the β-catenin transfection was confirmed by immunostaining the transfected and control AD GPC progeny with an anti-FLAG-tag antibody. Approximately 40% of the DAPI-labeled cells in the transfected AD GPC progeny were FLAG+, while no control AD progeny were FLAG+. c, Among the control HC, control AD, and β-catenin-transfected AD GPC progeny, the neurons were visualized by βIII tubulin staining (green), and the nuclei were counterstained with DAPI (blue) (n = 2 per group). d, Immunoreactive quantification among the control HC, control AD, and transfected AD progeny revealed that the percentage of mature neurons among the β-catenin-transfected AD GPC progeny was significantly restored (n = 3 per group, ANOVA, *p < 0.05). e, The β-catenin-transfected AD GPCs produced more new neurons (βIII-tubulin+) than did the control AD GPCs. Among the transfected AD GPC progeny, ∼70% of the new neurons developed from virally infected AD GPCs (indicated by the colocalization of βIII-tubulin and FLAG-tag immunostaining). Scale bars: c, 50 μm; b, e, 30 μm.
To determine whether the partially restored β-catenin levels in the transfected AD progeny can restore neuron production, we used immunostaining to visualize the number of mature neurons (labeled with an anti-βIII tubulin antibody) out of the total number of cells (labeled with DAPI) in the transfected AD versus the nontransfected AD and HC GPC progeny (Fig. 4c). The percentage of mature neurons in transfected AD GPC progeny was significantly greater than in the nontransfected AD GPC progeny and did not differ significantly from the percentage in the nontransfected HC progeny (Fig. 4d), suggesting that β-catenin transfection in AD GPCs may increase the levels of neurogenesis. To test the notion that β-catenin transfection increases the likelihood of AD GPC differentiation into mature neurons, we used immunostaining to label the cells that were transfected with viral vector (FLAG+), the mature neurons (βIII-tubulin+), and all the cell nuclei (DAPI+) in the transfected and nontransfected AD GPC progeny (Fig. 4e). A greater percentage of neurons occurred among the β-catenin-transfected progeny compared with the nontransfected progeny, with 72.65 ± 11.81% (n = 3) of the neurons among the transfected group arising from successfully transfected cells (FLAG+, β-III tubulin+ cells rather than FLAG−, β-III tubulin+ cells). Together, these data suggest that the reduced β-catenin levels exhibited by AD GPCs compared with HC GPCs can be restored and that this restoration of β-catenin has the ability to rescue the levels of neurogenesis in AD GPCs.
β-Catenin and GSK-3β siRNA transfection alters the expression of a proneural gene
To examine the relationship between the β-catenin signaling pathway and potential downstream proneural target genes, we suppressed the expression of β-catenin in GPCs from 12-month-old WT mice via the introduction of β-catenin siRNA. The Western blot showed that siRNA treatment caused a significant reduction in β-catenin levels but no significant changes in GSK-3β levels (Fig. 5a). We tested the expression of Ngn 2, which is coded for by a proneuronal gene, via Western blot and found a significant decrease in Ngn 2 expression in the β-catenin siRNA treated cells compared with the control cells (Fig. 5b), suggesting that reduced β-catenin may lead to decreased neurogenesis. Our earlier data revealed that both the GSK-3β levels and the relative levels of phosphorylated versus nonphosphorylated β-catenin were altered in GPCs from AD versus HC human subjects (Fig. 3a–c), suggesting that GSK-3β may be involved in the regulation of β-catenin. To confirm the role of GSK-3β in the regulation of β-catenin, we transfected GSK-3β siRNA into GPCs from 12-month-old APP23 mice. The results of the Western blot demonstrated that treatment with GSK-3β siRNA decreased the GSK-3β expression levels and increased the β-catenin levels in the GPCs from APP23 mice (Fig. 5c). We also found that the introduction of GSK-3β siRNA significantly increased the expression of Ngn2 (Fig. 5d).
Figure 5.
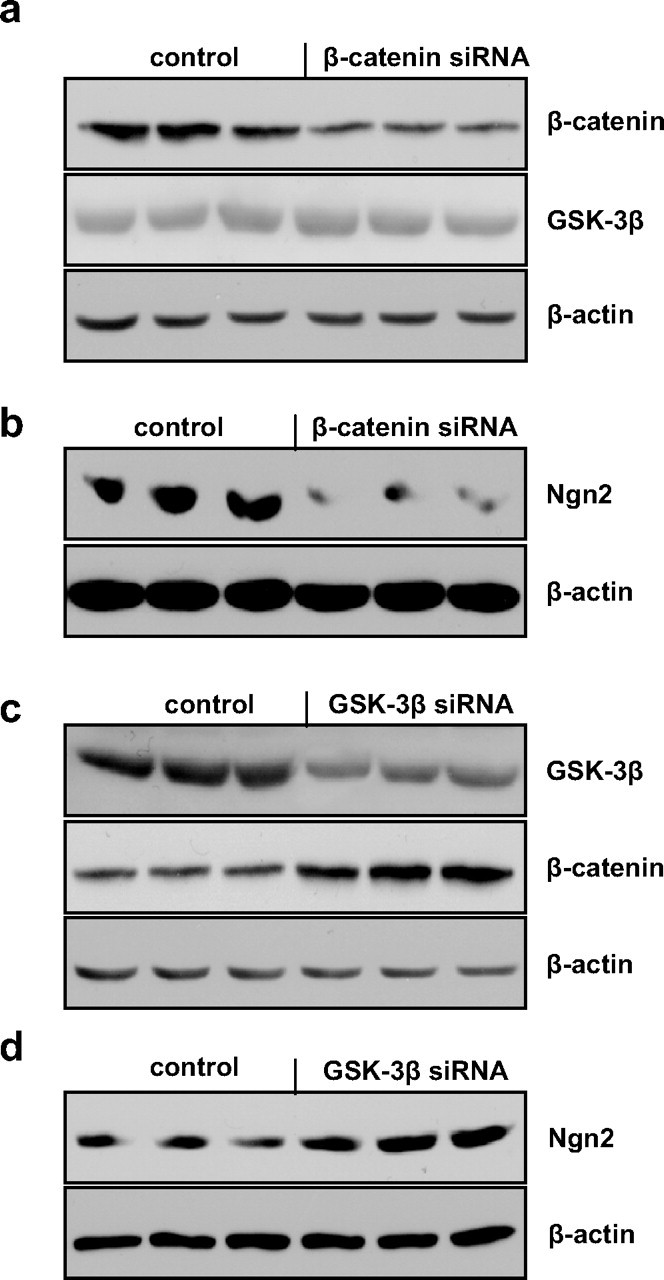
Effects of β-catenin and GSK-3β siRNA on proneuronal gene expression. GPCs from 12-month-old WT or APP23 mouse brains were cultured in differentiation medium and then siRNAs were introduced. Western blot revealed that the transfection of WT GPCs with β-catenin siRNA decreased the expression of β-catenin without significantly changing the expression of GSK-3β (a) and also decreased the expression of Ngn2 (b) compared with control WT GPCs. Alternately, the transfection of APP23 GPCs with GSK-3β siRNA reduced GSK-3β expression and promoted the expression of β-catenin (c), while also enhancing Ngn2 protein expression (d) compared with control APP23 GPCs.
Neuron induction and β-catenin signaling are reduced in GPCs from aged APP23 mice
To confirm the effect of chronic Aβ accumulation in vivo on β-catenin signaling and cell fate decisions of GPCs toward neurons, we used APP23 transgenic mice, which overproduce Aβ and exhibit AD-like pathology but lack tau-related pathology (Garrido et al., 2002; De Ferrari et al., 2003; Salins et al., 2007), as a model. NG2+ GPCs from the neocortices of 12- and 24-month-old APP23 and wild-type (WT) mice were isolated and cultured in the same manner as were the GPCs from the human subjects. We compared the cell growth and self-renewal rates of the human and mouse GPCs and found them to be similar (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
We performed Aβ ELISA to compare the amounts of Aβ in the neocortices of the WT and APP23 mice and found that the levels of Aβ40, Aβ42, and total Aβ in the 12- and 14-month-old WT mice were nondetectable compared with the levels in the age-matched APP23 mice (Fig. 6a–c). The amount of total Aβ in the neocortices of the APP23 mice was 121 ± 32 ng/mg protein (n = 3) at 12 months of age, and, at 24 months of age, the total Aβ level increased more than fourfold to 693 ± 28 ng/mg protein (Fig. 6a). Similar trends were also found for Aβ40 and Aβ42 levels (Fig. 6b,c).
Figure 6.
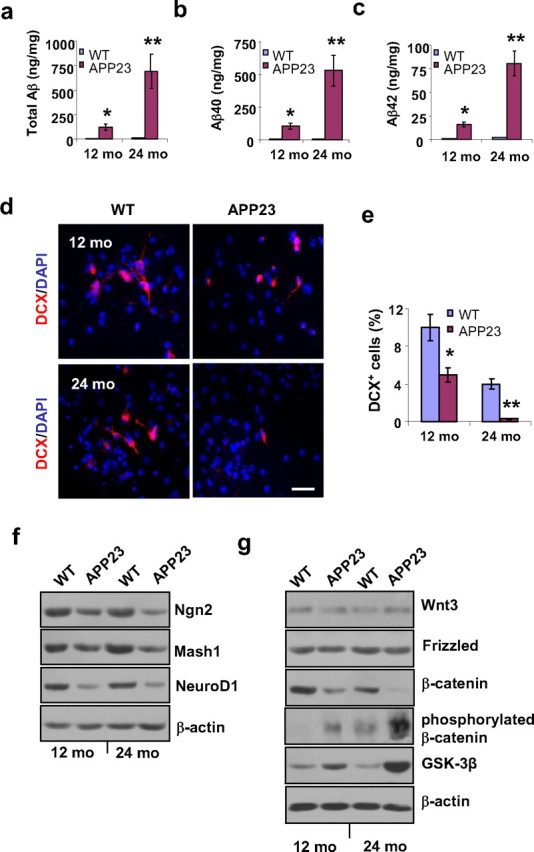
Aβ production, neurogenesis, proneural gene products, and β-catenin signaling in GPC progeny from APP23 versus WT mice at 12 and 24 months of age. a–c, Aβ ELISAs of neocortical samples from 12- and 24-month-old APP23 and WT mice revealed that total Aβ levels (a), Aβ40 levels (b), and Aβ42 levels (c) were significantly higher in the APP23 mice than in the WT mice, and the Aβ levels increased with age in the APP23 mice (Student's t test, *p < 0.05, **p < 0.01, n = 3 per group). d, After 2 d in differentiation medium, immunostaining showed a reduced number of newborn neurons (DCX+) among the progeny of GPCs (labeled with DAPI) from APP23 mice at 12 and 24 months of age compared with age-matched WT mice. e, Quantification of the immunoreactive structures revealed a significant reduction in the percentage of newborn neurons among the APP23 progeny compared with age-matched WT progeny (Student's t test, *p < 0.05, **p < 0.01, n = 3 per group). f, After 7 d in differentiation medium, the expression levels of the proneural gene products Ngn 2, Mash1, and NeuroD1 were reduced in the GPC progeny from 12- and 24-month-old APP23 mice compared with those from age-matched WT mice (n = 3 per group). g, After 7 d in differentiation medium, nonphosphorylated β-catenin levels decreased and both GSK-3β and phosphorylated β-catenin levels increased in the GPC progeny of 12- and 24-month-old APP23 mice compared with age-matched WT mice; however, no significant changes in the levels of Wnt3 or frizzled were observed (n = 3 per group). Scale bar, 50 μm.
To compare the levels of differentiation toward neurons in second passage WT versus APP23 GPCs after 2 d in differentiation medium, we immunostained the cells with DCX (to label the neuroblasts) and DAPI (to label the nuclei of all the cells) (Fig. 6d). We found that the percentages of neuroblasts among the GPC progeny were significantly reduced in the GPCs from 12- and 24-month-old APP23 mice compared with those from age-matched WT mice (Fig. 6e).
To investigate whether the decreased production of new neurons in APP23 mice is associated with the decreased expression of proteins coded for by neurogenesis-associated genes, we measured the levels of Ngn2, Mash1, and NeuroD1 (coded for by neurogenesis-associated genes) by Western blot. The levels of Ngn2, Mash1, and NeuroD1 were significantly reduced in the progeny of GPCs from 12- and 24-month-old APP23 mice compared with those from age-matched WT mice (Fig. 6f).
We also examined the expression levels of Wnt3/β-catenin signal molecules by Western blot, and found that neither Wnt3 nor frizzled expression levels were affected by age or by genotype. However, nonphosphorylated β-catenin levels were significantly decreased while both phosphorylated β-catenin and GSK-3β levels were significantly increased in the progeny cells from APP23 GPCs compared with their WT counterparts (Fig. 6g), which is consistent with our results in human samples.
Aβ impairs neuronal induction by reducing β-catenin levels
To determine whether Aβ impairs the ability of NG2+ GPCs to generate new neurons in humans, HC GPC neurospheres were treated with different concentrations of Aβ1–42 peptide for 2 d. Then, the Aβ was removed, and the cultures were tested after either 7 or 14 d in differentiation medium. To compare the levels of neuronal induction following the various Aβ treatments, HC GPC progeny were immunolabeled to detect the number of mature neurons (βIII tubulin+) out of the total number of cells (DAPI+) (Fig. 7a). Exposure to ≥1 μm Aβ1–42 was found to decrease the percentage of new neurons among the progeny in a dose-dependent manner (Fig. 7b).
Figure 7.
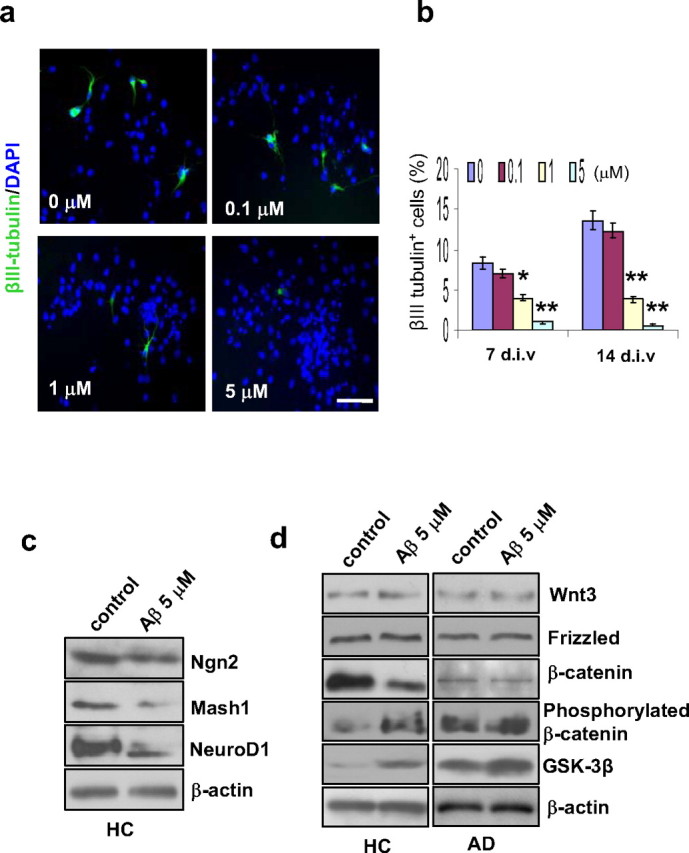
Effect of Aβ treatment on neurogenesis and β-catenin signaling. HC GPC neurospheres were treated with different concentrations of Aβ1–42 peptide for 2 d. Then, the Aβ was removed, and the cultures were tested after either 7 or 14 d in differentiation medium. a, After 7 d of differentiation, immunostaining revealed a dose-related reduction in the percentage of neurons (βIII tubulin+) among the Aβ-treated HC GPC progeny (labeled with DAPI). Scale bar, 50 μm. b, Quantification of immunoreactive structures after 7 and 14 d of differentiation showed that the percentage of new neurons among HC GPC progeny decreased significantly with ≥1.0 μm Aβ1–42 exposure compared with controls (0 μm Aβ1–42). c, A representative Western blot of HC GPC progeny treated with 5 μm Aβ1–42 revealed reduced levels of proneural gene products Ngn2, Mash1, and NeuroD1 compared with control HC progeny. d, HC and AD GPCs were treated with 5 μm Aβ1–42 for 2 d, and the culture was extended to 7 d in Aβ-free differentiation medium. Western blot showed a reduction in nonphosphorylated β-catenin and increases in both GSK-3β and phosphorylated β-catenin in Aβ-treated HC GPC progeny compared with control progeny. In AD GPC progeny, the blot did not show any significant change in the levels of nonphosphorylated or phosphorylated β-catenin between the Aβ-treated and control progeny; however, the Aβ-treated AD GPC progeny did exhibit a significant increase in GSK-3β expression compared with control AD progeny. No significant differences in either Wnt3 or frizzled levels were observed between Aβ-treated and control progeny from HC or AD GPCs (n = 3 per group).
To determine whether the Aβ treatment-induced impairment of neurogenesis by HC GPCs is partially due to changes in expression of proneural genes, HC progeny cells were exposed to 5 μm Aβ1–42 for 2 d, and the culture was kept for 14 d after the removal of Aβ. The results of the Western blot demonstrated reduced expressions of the neurogenic genes Ngn2, Mash1, and NeuroD1 in the Aβ-treated cells compared with the control cells (Fig. 7c), suggesting that transient treatment with Aβ1–42 has a long lasting effect on the cell fate decisions of GPCs.
To determine whether the mechanism by which Aβ alters proneural gene expression and impairs neuron induction in HC GPCs involves the same signal pathway that is disrupted in AD GPCs, we used Western blot analysis to detect the levels of the Wnt3/β-catenin signaling molecules in the progeny of Aβ-treated versus nontreated HC and AD GPCs. Similar to the results we found in the AD versus HC GPC progeny, the levels of Wnt3 and frizzled were not significantly altered in the Aβ-treated HC progeny compared with the nontreated HC progeny (Fig. 7d). However, the level of nonphosphorylated β-catenin was significantly decreased while the levels of phosphorylated β-catenin and GSK-3β were significantly increased in the Aβ-treated versus the nontreated HC progeny(Fig. 7d). Meanwhile, Aβ treatment further promoted GSK-3β expression in AD GPC progeny cells, whose nontreated levels of GSK-3β are already elevated compared with HC cells. However, significant changes in the expression of either phosphorylated or nonphosphorylated β-catenin in the Aβ-treated versus the nontreated AD progeny did not occur (Fig. 7d).
Discussion
Recently, the statement “neurons do not regenerate during adulthood” has been challenged, and increasing evidence shows that mature brains are capable of regenerating neurons (Sugaya, 2005). Certain “neurogenic” regions of the dentate gyrus and the olfactory bulbs display enhanced adult neurogenesis in response to neuronal damage (Gould and Tanapat, 1997; Parent, 2003; Koketsu et al., 2006; Gould, 2007). Recent studies have also demonstrated that increased neurogenesis occurs in the hippocampus and ependyma of patients with chronic neurodegenerative disorders such as AD (Jin et al., 2004) and Huntington's disease (Curtis et al., 2003).
Under normal conditions, little or no neurogenesis occurs in the “non-neurogenic” regions of the brain, such as the neocortex, after birth (Spalding et al., 2005; Bhardwaj et al., 2006) even though many glial progenitor cells are present in these regions (Gogate et al., 1994; Pincus et al., 1997; Roy et al., 1999; Chang et al., 2000; Arsenijevic et al., 2001; Palmer et al., 2001; Windrem et al., 2002; Nunes et al., 2003; Goldman and Sim, 2005). Recent investigations have found new neuron production in situ after targeted ablation of neurons in the rodent neocortex (Magavi et al., 2000; Chen et al., 2004), as well as ischemic lesion (Jiang et al., 2001), indicating that multipotential recovery (or reprogramming) of the progenitor cells in the parenchyma is possible after lesion stimulation. However, the neocortices of AD patients, which undergo progressive neuron loss due to damage by toxic Aβ deposits (Probst et al., 1991; Selkoe, 1999) have not been found to exhibit damage-induced neurogenesis (Regeur et al., 1994; Bundgaard et al., 2001) even though significant gliogenesis was observed (Bondolfi et al., 2002). Related to these discoveries, Sugaya found that only glial differentiation took place following the transplantation of stem cells into the brains of Alzheimer's transgenic mice (Sugaya et al., 2006), even though neurogenesis occurred following the xenograft of human progenitor cells into healthy rat brains (Nunes et al., 2003).
To investigate the fate of the progenitor cells derived from AD versus HC subjects, we isolated NG2+ progenitor cells from the human neocortex and found that, even at advanced ages (>80 years old), the progenitor cells still have the competence to renew themselves and to generate new neurons. However, in the progenitor cells derived from AD patients' brains, the self-renewal capability was diminished compared with the rate of renewal in progenitor cells from the healthy brain samples. The amount of neurogenesis was also significantly reduced in progenitor cells from AD patients compared with those from HC subjects.
We also found that the treatment of HC progenitor cells with Aβ caused a significant dose-related reduction in the amount of neurogenesis. Together, the results of our in vitro experiments suggest that elevated levels of Aβ in the AD progenitor cells and in the Aβ-treated HC progenitor cells inhibit neuronal induction by interfering with the β-catenin signaling pathway and thereby decreasing the expression of proneural genes.
Previous studies have reported that Aβ promotes neuronal apoptosis in AD by (1) causing ectopic cell cycle reentry (Busser et al., 1998; Wu et al., 2000; Yang et al., 2001; Herrup and Arendt, 2002; Yang et al., 2003; Herrup et al., 2004; Cicero and Herrup, 2005; Yang et al., 2006; Yang and Herrup, 2007; Zhang et al., 2008) and (2) activating GSK-3β (Pei et al., 1999; Moon et al., 2004), thereby leading to the degradation of β-catenin and the inactivation of Wnt signaling (Salins et al., 2007). The activation of Wnt signaling has been shown to reverse β-amyloid fibril-induced neurodegeneration and behavioral impairments (De Ferrari et al., 2003; Alvarez et al., 2004). Similarly, a study found that the PKC-mediated inhibition of GSK-3β activity protected rat hippocampal neurons from Aβ toxicity by leading to the accumulation of cytoplasmic β-catenin and the transcriptional activation of Wnt target genes via the formation of the β-catenin/T-cell factor/lymphoid enhancer factor-1 (TCF/LEF-1) complex (Garrido et al., 2002).
The formation of the β-catenin/TCF/LEF-1 complex is key to the transcriptional regulation of target genes by the Wnt/β-catenin signaling pathway. LEF-1 and β-catenin form a ternary complex with DNA that displays an altered DNA bend (Behrens et al., 1996). Additionally, bipartite transcription factor β-catenin/TCF is a key effector of the canonical Wnt/β-catenin signaling pathway (He et al., 1998). In the absence of Wnt/β-catenin signaling, TCF forms a complex with transcriptional corepressors, including Groucho (Cavallo et al., 1998) and C-terminal-binding protein 1 (Valenta et al., 2003), and downregulates the transcription of the Wnt target genes (Chen et al., 1999). In the presence of Wnt/β-catenin signaling, however, β-catenin converts TCF into a transcriptional activator for the same panel of genes that are repressed by TCF in the absence of β-catenin (Nusse, 1999; Brantjes et al., 2001). A dominant-negative form of TCF inhibits the ability of constitutively active β-catenin in the stimulation of TCF-dependent transcription (Kolligs et al., 1999).
Wnt/β-catenin signaling pathway has been known to promote self-renewal in a variety of tissue stem cells (Dravid et al., 2005), including neural stem cells (Chenn and Walsh, 2002; Zechner et al., 2003) and hematopoietic stem cells (Reya et al., 2003; Willert et al., 2003). In addition to affecting the self-renewal of stem cells, activation of the Wnt/β-catenin pathway has been shown to promote the differentiation of neural precursor cells, while inhibition of the pathway has been shown to prevent the differentiation of the precursor cells (Lie et al., 2005; Teo et al., 2005). In this study, we found that even a transient toxic dose of Aβ can cause permanent damage to GPCs by increasing the levels of GSK-3β, which in turn cause β-catenin levels to decrease leading to the downregulation proneural gene transcription and an impairment of neuron induction. These results are consistent with the results of previous studies and suggest that the toxic effects of Aβ on neurons and progenitor cells involve similar changes to the Wnt/β-catenin signaling pathway (Haughey et al., 2002a,b; Millet et al., 2005; Eucher et al., 2007). If this mechanism is correct, then Aβ-induced neuronal damage could be treated with reagents like PKC agonists or Lovastatin, a reagent that may affect cholesterol synthesis and reduce Aβ production (Garrido et al., 2002; Salins et al., 2007). Whether these reagents could be used to restore the levels of neuron induction from AD GPCs is an area for future research.
In conclusion, the Aβ toxicity may diminish the multipotential capability of GPCs and other neural precursor cells by disrupting the β-catenin signaling such that GSK-3β levels increase, causing the phosphorylation and degradation β-catenin, which leads to reduced proneural gene expression (Fig. 8). Therefore, even though GPCs still exist inside the brains of AD patients, the GPCs are unable to generate adequate numbers of new neurons to compensate for the neuronal loss caused by Aβ aggregation. The development of therapeutic approaches to inhibit GSK-3β and/or elevate β-catenin in GPCs or other neural progenitor cells may allow us to mitigate the toxic effects of Aβ and to promote neurogenesis in AD patients (Stambolic et al., 1996; De Ferrari and Inestrosa, 2000; Inestrosa et al., 2002; Castelo-Branco et al., 2004).
Figure 8.
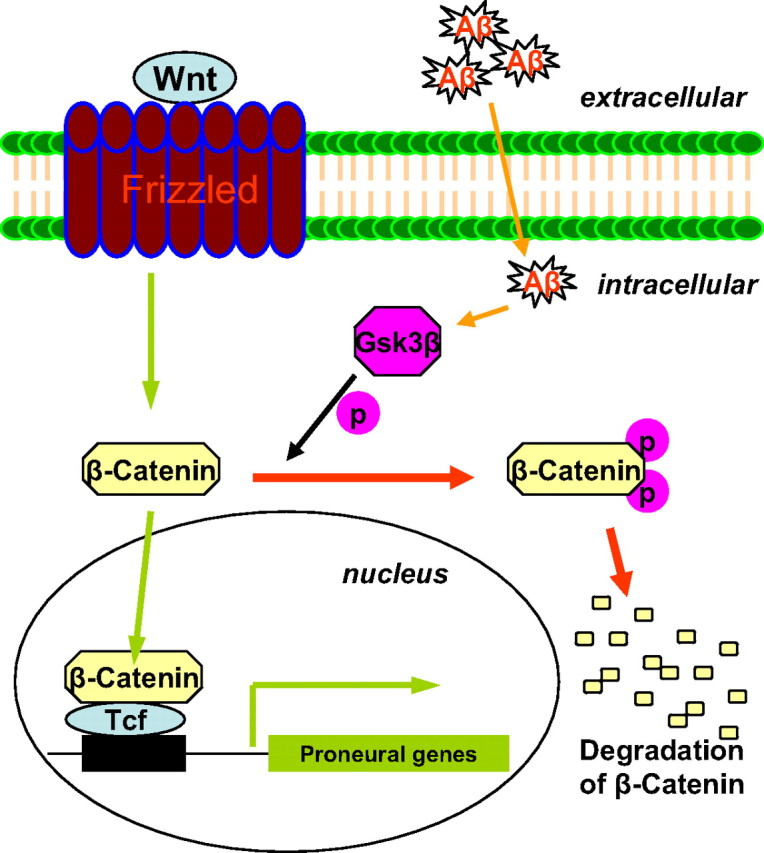
Aβ-induced interruption of β-catenin signaling and its effect on proneural gene expression. The schematic presentation shows that Aβ elevates GSK-3β, which in turn promotes the phosphorylation and degradation of β-catenin. Reduced β-catenin signaling downregulates the expression of proneural genes.
Footnotes
This work was supported by the American Health Assistance Foundation, the Alzheimer's Association Zenith Award, the Arizona Alzheimer Consortium, and the National Institute on Aging (NIAAG025888). We sincerely thank Dr. Fred H. Gage from the Laboratory of Genetics at the Salk Institute for Biological Studies for his critical reading and valuable comments on our manuscript and enthusiastic support. We thank Adrienne Bowers, Kristina Rogers, Gina Ciavarella, Nezahet Mutlu, and Zhenyu Zhong for their editorial assistance and for providing technical assistance in this study.
References
- Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–10541. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AA, Chittajallu R, Belachew S, Gallo V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–589. doi: 10.1083/jcb.200311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res. 2004;297:186–196. doi: 10.1016/j.yexcr.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Anderton BH, Dayanandan R, Killick R, Lovestone S. Does dysregulation of the Notch and wingless/Wnt pathways underlie the pathogenesis of Alzheimer's disease? Mol Med Today. 2000;6:54–59. doi: 10.1016/s1357-4310(99)01640-8. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12:1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic Y, Villemure JG, Brunet JF, Bloch JJ, Déglon N, Kostic C, Zurn A, Aebischer P. Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp Neurol. 2001;170:48–62. doi: 10.1006/exnr.2001.7691. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–186. doi: 10.1083/jcb.200210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk-Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, Frisén J. Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci U S A. 2006;103:12564–12568. doi: 10.1073/pnas.0605177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantjes H, Roose J, van De Wetering M, Clevers H. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res. 2001;29:1410–1419. doi: 10.1093/nar/29.7.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundgaard MJ, Regeur L, Gundersen HJ, Pakkenberg B. Size of neocortical neurons in control subjects and in Alzheimer's disease. J Anat. 2001;198:481–489. doi: 10.1046/j.1469-7580.2001.19840481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco G, Rawal N, Arenas E. GSK-3beta inhibition/beta-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J Cell Sci. 2004;117:5731–5737. doi: 10.1242/jcs.01505. [DOI] [PubMed] [Google Scholar]
- Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Fernandez J, Mische S, Courey AJ. A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev. 1999;13:2218–2230. doi: 10.1101/gad.13.17.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Magavi SS, Macklis JD. Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A. 2004;101:16357–16362. doi: 10.1073/pnas.0406795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol. 2004;561:109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero S, Herrup K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci. 2005;25:9658–9668. doi: 10.1523/JNEUROSCI.1773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzheimer's disease. Brain Res Brain Res Rev. 2000;33:1–12. doi: 10.1016/s0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Chacón MA, Barría MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- Dravid G, Ye Z, Hammond H, Chen G, Pyle A, Donovan P, Yu X, Cheng L. Defining the role of Wnt/beta-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells. 2005;23:1489–1501. doi: 10.1634/stemcells.2005-0034. [DOI] [PubMed] [Google Scholar]
- Eucher JN, Uemura E, Sakaguchi DS, Greenlee MH. Amyloid-beta peptide affects viability but not differentiation of embryonic and adult rat hippocampal progenitor cells. Exp Neurol. 2007;203:486–492. doi: 10.1016/j.expneurol.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002;16:1982–1984. doi: 10.1096/fj.02-0327fje. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogate N, Verma L, Zhou JM, Milward E, Rusten R, O'Connor M, Kufta C, Kim J, Hudson L, Dubois-Dalcq M. Plasticity in the adult human oligodendrocyte lineage. J Neurosci. 1994;14:4571–4587. doi: 10.1523/JNEUROSCI.14-08-04571.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Sim F. Neural progenitor cells of the adult brain. Novartis Found Symp. 2005;265:66–80. discussion 82–97. [PubMed] [Google Scholar]
- Gould E. How widespread is adult neurogenesis in mammals? Nat Rev Neurosci. 2007;8:481–488. doi: 10.1038/nrn2147. [DOI] [PubMed] [Google Scholar]
- Gould E, Tanapat P. Lesion-induced proliferation of neuronal progenitors in the dentate gyrus of the adult rat. Neuroscience. 1997;80:427–436. doi: 10.1016/s0306-4522(97)00127-9. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med. 2002a;1:125–135. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002b;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, Mishina Y, Li L. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- Herrup K, Arendt T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer's disease. J Alzheimers Dis. 2002;4:243–247. doi: 10.3233/jad-2002-4315. [DOI] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci Res. 2005;51:331–336. doi: 10.1016/j.neures.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development. 2004;131:2791–2801. doi: 10.1242/dev.01165. [DOI] [PubMed] [Google Scholar]
- Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J Comp Neurol. 2006;498:525–538. doi: 10.1002/cne.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa N, De Ferrari GV, Garrido JL, Alvarez A, Olivares GH, Barría MI, Bronfman M, Chacón MA. Wnt signaling involvement in beta-amyloid-dependent neurodegeneration. Neurochem Int. 2002;41:341–344. doi: 10.1016/s0197-0186(02)00056-6. [DOI] [PubMed] [Google Scholar]
- Jiang W, Gu W, Brännström T, Rosqvist R, Wester P. Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke. 2001;32:1201–1207. doi: 10.1161/01.str.32.5.1201. [DOI] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koketsu D, Furuichi Y, Maeda M, Matsuoka N, Miyamoto Y, Hisatsune T. Increased number of new neurons in the olfactory bulb and hippocampus of adult non-human primates after focal ischemia. Exp Neurol. 2006;199:92–102. doi: 10.1016/j.expneurol.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Kolligs FT, Hu G, Dang CV, Fearon ER. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19:5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science. 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lindholm K, Yang LB, Li R, Shen Y. Isolation of living neurons from human elderly brains using the immunomagnetic sorting DNA-linker system. Am J Pathol. 2002;161:1567–1576. doi: 10.1016/S0002-9440(10)64435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Kléber M, Hari L, Brault V, Suter U, Taketo MM, Kemler R, Sommer L. Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science. 2004;303:1020–1023. doi: 10.1126/science.1091611. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- Millet P, Lages CS, Haïk S, Nowak E, Allemand I, Granotier C, Boussin FD. Amyloid-beta peptide triggers Fas-independent apoptosis and differentiation of neural progenitor cells. Neurobiol Dis. 2005;19:57–65. doi: 10.1016/j.nbd.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- Nusse R. WNT targets. Repression and activation. Trends Genet. 1999;15:1–3. doi: 10.1016/s0168-9525(98)01634-5. [DOI] [PubMed] [Google Scholar]
- Otero JJ, Fu W, Kan L, Cuadra AE, Kessler JA. Beta-catenin signaling is required for neural differentiation of embryonic stem cells. Development. 2004;131:3545–3557. doi: 10.1242/dev.01218. [DOI] [PubMed] [Google Scholar]
- Othman M, Klueber K, Lu C, Winstead W, Roisen F. Immunomagnetic separation of adult human olfactory neural progenitors. Biotech Histochem. 2005;80:177–188. doi: 10.1080/10520290500469769. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH. Cell culture. Progenitor cells from human brain after death. Nature. 2001;411:42–43. doi: 10.1038/35075141. [DOI] [PubMed] [Google Scholar]
- Parent JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9:261–272. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Pincus DW, Harrison-Restelli C, Barry J, Goodman RR, Fraser RA, Nedergaard M, Goldman SA. In vitro neurogenesis by adult human epileptic temporal neocortex. Clin Neurosurg. 1997;44:17–25. [PubMed] [Google Scholar]
- Probst A, Langui D, Ulrich J. Alzheimer's disease: a description of the structural lesions. Brain Pathol. 1991;1:229–239. doi: 10.1111/j.1750-3639.1991.tb00666.x. [DOI] [PubMed] [Google Scholar]
- Regeur L, Jensen GB, Pakkenberg H, Evans SM, Pakkenberg B. No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer's type. Neurobiol Aging. 1994;15:347–352. doi: 10.1016/0197-4580(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy NS, Wang S, Harrison-Restelli C, Benraiss A, Fraser RA, Gravel M, Braun PE, Goldman SA. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci. 1999;19:9986–9995. doi: 10.1523/JNEUROSCI.19-22-09986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salins P, Shawesh S, He Y, Dibrov A, Kashour T, Arthur G, Amara F. Lovastatin protects human neurons against Abeta-induced toxicity and causes activation of beta-catenin-TCF/LEF signaling. Neurosci Lett. 2007;412:211–216. doi: 10.1016/j.neulet.2006.07.045. [DOI] [PubMed] [Google Scholar]
- Scolding N, Franklin R, Stevens S, Heldin CH, Compston A, Newcombe J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain. 1998;121:2221–2228. doi: 10.1093/brain/121.12.2221. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Aging, amyloid, and Alzheimer's disease: a perspective in honor of Carl Cotman. Neurochem Res. 2003;28:1705–1713. doi: 10.1023/a:1026065122854. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Sugaya K. Possible use of autologous stem cell therapies for Alzheimer's disease. Curr Alzheimer Res. 2005;2:367–376. doi: 10.2174/1567205054367919. [DOI] [PubMed] [Google Scholar]
- Sugaya K, Alvarez A, Marutle A, Kwak YD, Choumkina E. Stem cell strategies for Alzheimer's disease therapy. Panminerva Med. 2006;48:87–96. [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Teo JL, Ma H, Nguyen C, Lam C, Kahn M. Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc Natl Acad Sci U S A. 2005;102:12171–12176. doi: 10.1073/pnas.0504600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Lukas J, Korinek V. HMG box transcription factor TCF-4's interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res. 2003;31:2369–2380. doi: 10.1093/nar/gkg346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Windrem MS, Roy NS, Wang J, Nunes M, Benraiss A, Goodman R, McKhann GM, 2nd, Goldman SA. Progenitor cells derived from the adult human subcortical white matter disperse and differentiate as oligodendrocytes within demyelinated lesions of the rat brain. J Neurosci Res. 2002;69:966–975. doi: 10.1002/jnr.10397. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AP, Fitzgerald JJ, Colello RJ. Rapid purification of glial cells using immunomagnetic separation. J Neurosci Methods. 1997;74:37–44. doi: 10.1016/s0165-0270(97)22400-3. [DOI] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21:797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Herrup K. Cell division in the CNS: protective response or lethal event in post-mitotic neurons? Biochim Biophys Acta. 2007;1772:457–466. doi: 10.1016/j.bbadis.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer's disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner D, Fujita Y, Hülsken J, Müller T, Walther I, Taketo MM, Crenshaw EB, 3rd, Birchmeier W, Birchmeier C. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–418. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]
- Zeng G, Apte U, Cieply B, Singh S, Monga SP. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9:951–959. doi: 10.1593/neo.07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci U S A. 2008;105:8772–8777. doi: 10.1073/pnas.0711355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- Zhou F, Zhang L, Wang A, Song B, Gong K, Zhang L, Hu M, Zhang X, Zhao N, Gong Y. The association of GSK3 beta with E2F1 facilitates nerve growth factor-induced neural cell differentiation. J Biol Chem. 2008;283:14506–14515. doi: 10.1074/jbc.M706136200. [DOI] [PubMed] [Google Scholar]