

Abstract
Parkinson’s disease (PD) is a motor disorder that involves death of dopaminergic neurons in the substantia nigra pars compacta. Parkin is an autosomal recessive gene that is mutated in early onset PD. We investigated the role of parkin and autophagic clearance in postmortem nigrostriatal tissues from 22 non-familial sporadic PD patients and 15 control samples. Parkin was insoluble with altered cytosolic expression in the nigrostriatum of sporadic PD. Parkin insolubility was associated with lack of degradation of ubiquitinated proteins and accumulation of α-Synuclein and parkin in autophagosomes, suggesting autophagic defects in PD. To test parkin’s role in mediating autophagic clearance, we used lentiviral gene transfer to express human wild type or mutant parkin (T240R) with α-Synuclein in the rat striatum. Lentiviral expression of α-Synuclein led to accumulation of autophagic vacuoles, while co-expression of parkin with α-Synuclein facilitated autophagic clearance. Subcellular fractionation showed accumulation of α-Synuclein and p-Tau in autophagosomes in gene transfer models, similar to the effects observed in PD brains, but parkin expression led to protein deposition into lysosomes. However, parkin loss of function mutation did not affect autophagic clearance. Taken together, these data suggest that functional parkin regulates autophagosome clearance, while decreased parkin solubility may alter normal autophagy in sporadic PD.
Keywords: α-Synuclein, autophagy, striatum, parkin, Parkinson disease
Introduction
Parkinson’s disease (PD) is predominantly sporadic, but some disease-causing mutations suggest a genetic component in disease pathogenesis. Rare mutations in a number of genes are associated with familial forms of PD (Gasser, 2009). Dominantly inherited mutations in leucine-rich repeat kinase 2 (LRRK2) and α-Synuclein cause late onset PD. Genome-wide association studies suggest that naturally occurring sequence variants in α-Synuclein and LRRK2, as well as Tau, constitute an increased risk for late onset sporadic PD (Martin-Villalba et al., 2001, Healy et al., 2004, Cookson and Bandmann, 2010). PD is characterized by death of dopaminergic neurons in the substantia nigra (SN) (Kuhn et al., 2006, Benner et al., 2008, Reynolds et al., 2008) and formation of inclusions known as Lewy bodies (LBs), which primarily contain aggregated α-Synuclein (Spillantini et al., 1997, Wakabayashi et al., 1997, Spillantini et al., 1998a, Spillantini et al., 1998b, Goedert, 1999, Spillantini and Goedert, 2000, Takeda et al., 2000, Goedert, 2001, Trojanowski and Lee, 2003, Lundvig et al., 2005). Mutations in autosomal recessively inherited genes like PARK-2, PTEN-induced kinase-1 (PINK1) and DJ-1, lead to early onset Parkinsonism (Kitada et al., 1998, Lucking et al., 2000, Cookson and Bandmann, 2010). Parkin is an E3 ubiquitin ligase involved in degradation of misfolded proteins (Shimura et al., 2000). Parkin is known to mediate selective autophagy of dysfunctional mitochondria (Narendra et al., 2008, Park et al., 2009, Geisler et al., 2010, Vives-Bauza et al., 2010), while α-Synuclein was suggested to impair autophagy (Winslow et al., 2010, Winslow and Rubinsztein, 2011). Normal autophagy is a multi-step process that involves generation of the phagophores, formation of autophagosomes, which fuse with endosomes to form amphisomes or with lysosomes to form autophagolysosomes (Kovács et al., 1982, Iwata et al., 2005, He and Klionsky, 2009). Changes in autophagy are recognized in neurodegeneration, where accumulation of autophagosomes are characterized by un-degraded autophagic vacuoles in neurons (Kegel et al., 2000, Nixon et al., 2005, Yang et al., 2007, Mizushima et al., 2008, Nixon et al., 2008, Winslow and Rubinsztein, 2008).
To determine the role of parkin and its association with baseline autophagy in sporadic PD, we analyzed human postmortem nigrostriatal tissues via fractionation to determine protein solubility and investigated the effects of parkin on autophagic clearance in lentiviral gene transfer animal models. We sought to determine whether lentiviral expression of α-Synuclein affects autophagy and if parkin activity reverses α-Synuclein effects. We previously generated animal models expressing lentiviral α-Synuclein and found that parkin expression decreases α-Synuclein levels in the absence of ubiquitination (Burns et al., 2009, Khandelwal et al., 2010). Here we tested whether parkin expression regulates α-Synuclein clearance via autophagic degradation in vivo.
Materials and methods
Human postmortem brain tissues
Human postmortem caudate and midbrain regions from 22 PD patients and 15 age matched control subjects were obtained from John’s Hopkins University brain bank. The age, sex, stage of disease and postmortem dissection (PMD) are summarized for each patient in Table 1 and 2. The cause of death is not known. To extract the soluble fraction of proteins, 0.5 g of frozen brain tissues were homogenized in 1x STEN buffer (50 mM Tris (pH 7.6), 150 mM NaCl, 2 mM EDTA, 0.2 % NP-40, 0.2 % BSA, 20 mM PMSF and protease and phosphatase cocktail inhibitor), centrifuged at 10,000 g for 20 min at 4°C, and the supernatants were collected. All samples were then analyzed by ELISA (see below) or Western blot using 30 μg of protein. To extract the insoluble fraction, the pellet was re-suspended in 4M urea solution and centrifuged at 10,000g for 15 min, and the supernatant was collected and 30 μg of protein was analyzed by Western blot. Western blots were quantified by densitometry using Quantity One 4.6.3 software (Bio Rad). Densitometry was obtained as arbitrary numbers measuring band intensity. Data were analyzed as mean±Standard deviation, using Two-tailed t-test (P<0.02) and ANOVA, Neumann Keuls with multiple comparisons (P<0.05) to compare PD and control groups.
Table 1.
Descritpion and clinical diagnosis of human PD patients and control subjects’s tissues analyzed by Western blot and ELISA. PMD: post-mortem dissection.
| BRC # | diagnosis | Age | Sex | Race | PMD | Area |
|---|---|---|---|---|---|---|
| 399 | Control | 79 | F | W | 24 | Caudate |
| 417 | Control | 80 | F | W | 6 | Caudate |
| 487 | Control | 73 | M | W | 22 | Caudate |
| 515 | Control | 62 | M | W | 19 | Caudate |
| 705 | Control | 73 | M | W | 9 | Caudate |
| 1277 | Control | 80 | F | W | 8 | Caudate |
| 2052 | Control | 79 | M | W | 16 | Caudate |
| 1690 | PD | 76 | M | W | 18 | Caudate |
| 1731 | PD | 77 | M | W | 16 | Caudate |
| 2140 | PD with dementia | 84 | F | W | 11 | Caudate |
| 2067 | PD with dementia, cerebrovas. dis (NC) | 76 | M | W | 19 | Caudate |
| 2019 | PD with dementia, cerebrovas. dis | 83 | M | W | 16.5 | Caudate |
| 1989 | PD with dementia, LBD neocortical | 84 | M | W | 5 | Caudate |
| 2074 | PD, cerebrovascular disease | 85 | F | W | 9 | Caudate |
| 1758 | PD, DLB | 81 | M | W | 11 | Caudate |
| 1948 | PD, DLB | 77 | M | W | 5 | Caudate |
| 1796 | PD, Lewy Body CHG Limbic, porencephalic cyst | 81 | M | W | 8.75 | Caudate |
| 1877 | PD, Lewy Body CHG neocortical | 80 | M | W | 19 | Caudate |
| 1955 | PD, Lewy Body CHG neocortical | 84 | M | B | 13 | Caudate |
Table 2.
descritpion and clinical diagnosis of human PD patients and control subjects stained with immonuhistohemistry. PMD: post-mortem dissection, BRAAK: Braal stage, CERAD: Consortium to Establish a Registry for Alzheimer’s Disease.
| BRC # | diagnosis | CERAD | BRAAK | Age | Sex | Race | PMD | area |
|---|---|---|---|---|---|---|---|---|
| 1062 | Control | 58 | M | B | 14 | Hippocampus MB | ||
| 1252 | Control | 70 | M | W | Hippocampus MB | |||
| 1277 | Control | 0 | 80 | F | W | 8 | Caudate, hippocampus, MB | |
| 1352 | Control | 78 | F | 14 | Caudate, hippocampus, MB | |||
| 1615 | Control | 72 | M | W | 20 | Caudate | ||
| 1683 | Control | 1 | 91 | F | W | 8 | Caudate | |
| 1855 | Control | 77 | M | W | Caudate, hippocampus, MB | |||
| 2201 | Control | 0 | 2 | 85 | F | W | 27 | Caudate, hippocampus, MB |
| 2215 | PD with dementia, AD probable | B | 4 | 88 | M | W | 9 | Caudate, MB |
| 2235 | PD, tauopathy non-AD, cerebrovas. dis (NC) | 86 | F | W | 26 | Caudate, MB | ||
| 2243 | PD with dementia | 0 | 0 | 68 | M | W | 50 | Caudate, MB |
| 2253 | PD, contusion | 0 | 1 | 64 | F | W | 15 | Caudate, MB |
| 2267 | PD with dementia, neocortex | 0 | 1 | 75 | M | W | 22 | Caudate, MB |
| 2290 | PD | A | 2 | 82 | M | W | 53 | Caudate, MB |
| 2292 | PD with dementia, AD probable | B | 4 | 77 | M | W | 8 | Caudate, MB |
| 2312 | PD | 0 | 0 | 56 | M | W | 21 | MB |
| 2315 | PD | 0 | 2 | 84 | M | W | 8.5 | Caudate, MB |
| 2352 | PD with dementia, cerebrovas. dis (NC) | 0 | 2 | 83 | F | W | 163 | Caudate, MB |
Immunohistochemistry on slides from human patients was performed on 30 μm thick paraffin embedded brain slices de-paraffinized in Xylenes 2×5 minutes and sequential ethanol concentration, blocked for 1 hour in 10% horse serum and incubated overnight with primary antibodies at 4°C. After 3x 10 minute washes in 1xPBS, the samples were incubated with the secondary antibodies for 1hr at RT, washed 3×10 minutes in 1xPBS. Parkin was immunoprobed (1:200) with mouse anti-parkin (PRK8) antibody that recognizes a.a. 399–465 (Signet Labs, Dedham, MA) or rabbit polyclonal (1:200) anti-parkin (AB5112) antibody that recognizes a.a. 305–323 (Millipore) and counterstained with DAPI. Map 2 was probed (1:300) with mouse monoclonal antibody (Pierce). Glial Fibrillary Acid Protein (GFAP) was probed (1:200) with mouse (GA5) Mouse mAb #3670 (Cell Signaling) or (1:200) rabbit polyclonal (ab4674) antibody (Abcam). Tyrosine Hydroxylase (TH) was probed (1:100) with rabbit polyclonal (AB152) antibody (Millipore) and counterstained with DAB.
Stereotaxic injection
Lentiviral constructs were used to generate the animal models as explained in (Burns et al., 2009, Khandelwal et al., 2010, Herman and Moussa, 2011). Stereotaxic surgery was performed to inject the lentiviral constructs into the striatum of 2-month old male Sprague-Dawley rats. N=8 animals were used in each treatment. A total of 116 animals were used in these studies. All procedures were approved by the Georgetown University Animal Care and Use Committee (GUACUC).
Western blot analysis
To extract the soluble protein fraction, brain tissues were homogenized in 1x STEN buffer, centrifuged at 10,000 × g for 20 min at 4°C, and the supernatants containing the soluble fraction of proteins were collected. To extract the insoluble fraction the pellet was re-suspended in 4M urea or 30% formic acid and adjusted to pH 7 with 1N NaOH and centrifuged at 10,000 × g for 20 min at 4°C, and the supernatant containing the insoluble fraction was collected and analyzed by Western blot. Total parkin was immunoprobed (1:1000) with PRK8 antibody as indicated (Burns et al., 2009) and phospho-parkin was probed (1:1000) with anti-Ser 378 antibodies (Pierce). α-Synuclein was probed with rabbit monoclonal (1:1000) antibody (Santa Cruz). Autophagy antibodies, including beclin-1 (1:1000), autophagy like gene (Atg)-7 (1:1000), Atg12 (1:1000) and LC3-B (1:1000), were used to probe according to autophagy antibody sampler kit 4445 (Cell Signaling, Inc). Histone deacetylase 6 (HDAC6) was probed (1:500) using rabbit polyclonal anti-HDAC6 (Abcam). Rabbit polyclonal anti-SQSTM1/p62 (Cell Signaling Technology) was used (1:500). A rabbit polyclonal (Pierce) anti-LC3 (1:1000) and rabbit polyclonal (Thermo Scientific) anti-actin (1:1000) were used. LAMP-3 was probed (1:500) rabbit polyclonal antibody (Aviva Systems). Rabbit anti-ubiquitin (Santa Cruz Biotechnology) antibody (1:1000) was used. Mitochondrial protein COX-IV was probed (1:1000) with rabbit polyclonal (ab16056) antibody (Abcam) and human poly ADP-ribose polymerase (PARP-1) was probed (1:1500) with monoclonal (MA3-950) antibody (Pierce).
Immunohistochemistry on rat brain tissues was performed on 20 micron-thick 4% paraformaldehyde (PFA) fixed striatal brain sections and compared between treatments. Parkin was probed (1:200) with Rabbit polyclonal antibody (Chemicon). Rabbit polyclonal LC3-B (1:100) was used to probe LC3-B (Cell Signaling, Inc). Thioflavin-S and nuclear DAPI staining were performed according to manufacturer’s instructions (Sigma). Stereological methods- were applied by a blinded investigator using unbiased stereology analysis (Stereologer, Systems Planning and Analysis, Chester, MD) to determine the total positive cell counts in 20 cortical fields on at least 10 brain sections (~400 positive cells per animal) as indicated in (Burns et al., 2009, Khandelwal et al., 2010, Herman and Moussa, 2011).
α-Synuclein, parkin and p-Tau enzyme-linked immunosorbent assay (ELISA)
Specific ELISA (Invitrogen) were performed using 50μl (1μg/μl) of brain lysates detected with 50μl primary antibody (3h) and 100μl anti-rabbit secondary antibody (30 min) at RT. Parkin levels using specific human ELISA (MYBioSource), and p-Tau and α-Synuclein levels were measured using human specific ELISA (Invitrogen) according to manufacturers’ protocols.
Subcellular fractionation to isolate autophagic vacuoles
0.5g of Frozen human or animal brains were homogenized at low speed (Cole-Palmer homogenizer, LabGen 7, 115 Vac) in 1xSTEN buffer and centrifuged at 1,000g for 10 minutes to isolate the supernatant from the pellet. The pellet was re-suspended in 1xSTEN buffer and centrifuged once to increase the recovery of lysosomes. The pooled supernatants were then centrifuged at 100,000 rpm for 1 hour at 4°C to extract the pellet containing autophagic vacuoles (AVs) and lysosomes. The pellet was then re-suspended in 10 ml (0.33 g/ml) 50% Metrizamide and 10 ml in cellulose nitrate tubes. A discontinuous Metrizamide gradient was constructed in layers from bottom to top as follows: 6 ml of pellet suspension, 10 ml of 26%; 5 ml of 24%; 5 ml of 20%; and 5 ml of 10% Metrizamide (Marzella et al., 1982). After centrifugation at 10,000 rpm for 1 hour at 4°C, the fraction floating on the 10% layer (Lysosome) and the fractions banding at the 24%/20% (AV 20) and the 20%/10% (AV10) Metrizamide inter-phases were collected by a syringe and examined.
Transmission Electron Microscopy
Brain tissue were fixed in (1:4, v:v) 4% paraformaldehyde-picric acid solution and 25% glutaraldehyde overnight, and then washed 3x in 0.1M cacodylate buffer and osmicated in 1% osmium tetroxide/1.5% potassium ferrocyanide for 3h, followed by another 3x wash in distilled water. Samples were treated with 1% uranyl acetate in maleate buffer for 1 h, washed 3 x in maleate buffer (pH 5.2), then exposed to a graded cold ethanol series up to 100% and ending with a propylene oxide treatment. Samples are embedded in pure plastic and incubated at 60°C for 1–2 days. Blocks are sectioned on a Leica ultracut microtome at 95 nm, picked up onto 100 nm formvar-coated copper grids, and analyzed using a Philips Technai Spirit transmission EM. All sections were acquired and analyzed by a blind investigator.
Results
Decreased parkin solubility in postmortem striatum of sporadic PD patients
To determine the role of parkin in the brain of sporadic PD patients, we analyzed human postmortem striatal (caudate) tissues from 12 PD patients and 7 age-matched controls as described in Table 1. ELISA measurement of soluble human parkin revealed a significant (P<0.05) decrease (36%) in parkin levels in PD caudate/striatum compared to control (Fig. 1A). Western blot analysis of soluble striatal extracts confirmed the decrease in parkin levels in PD patients compared to control (Fig. 1B and C, 54%). No differences in parkin levels were detected in PD cortex (data not shown). Probing with anti-ubiquitin antibody showed a higher smear of ubiquitinated proteins in PD striatum compared to control (Fig. 1B). However, all samples with PMD greater than 16h showed significantly (P<0.02, two-tailed t-test) higher levels of ubiquitin (48%) in both groups and higher parkin levels (25%) with PMD greater than 13h within the PD group. To further investigate whether the decreased degradation of proteins results in alteration of solubility, we extracted the insoluble proteins in 4M urea (explained in Methods). An increase in the level of parkin was detected in the insoluble fraction (Fig. 1D and E , 82%) in contrast to the soluble extract, which was hardly detected. Parkin phosphorylation at serine 378, which was not detected in the soluble fraction, was observed in the insoluble extract (Fig. 1D and E, 114%). Additionally, more ubiquitinated proteins (Fig. 1D, 3rd blot) were also detected in the insoluble fraction. The variations among the samples are represented to show variation among individual samples, including soluble, insoluble and phospho-parkin (Fig. 1F). Taken together these data suggest decreased parkin solubility and increased phosphorylation in PD.
Figure 1. Parkin is insoluble in post-mortem striatum of human PD patients.
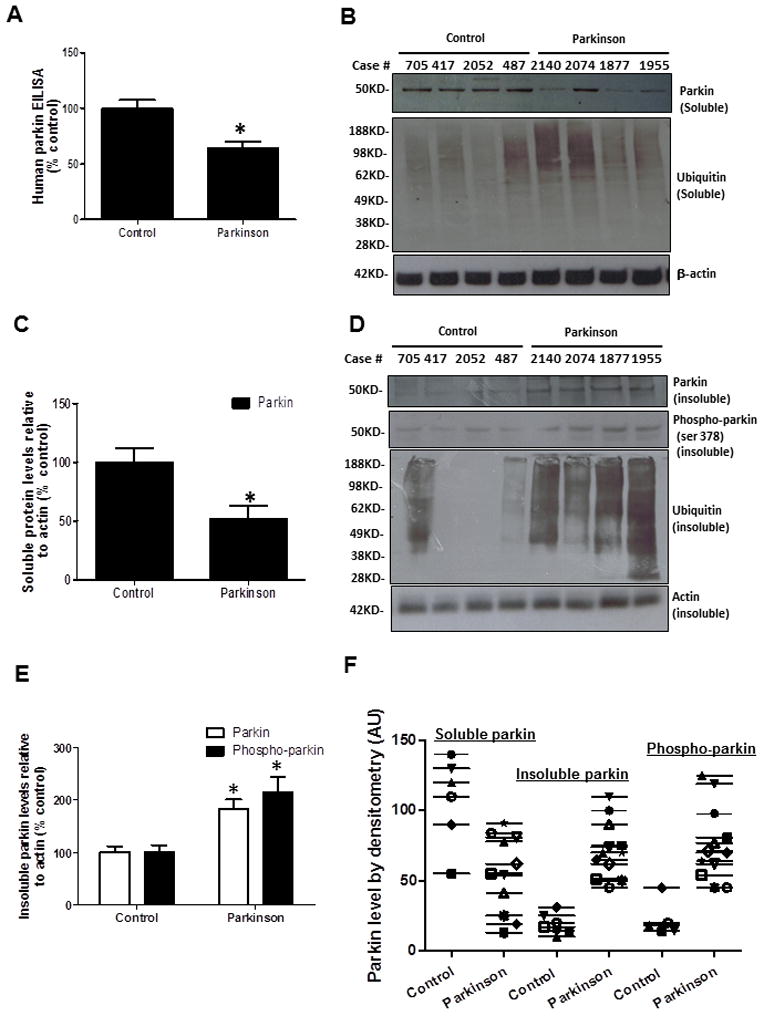
A). Histograms represent ELISA measurement of human parkin in the caudate of PD patients and control subjects. B). WB analysis on 4–12% SDS-NuPAGE gel of soluble human post-mortem striatal lysates in PD patients and control subjects, showing parkin (1st blot) and ubiquitinated proteins (2nd blot) compared to actin loading control. C). Histograms represent quantification of blots. D). WB analysis on 4–12% SDS NuPAGE gel showing the levels of insoluble parkin (1st blot), phospho-parkin (2nd blot), ubiquitinated proteins (3rd blot), and actin (4th blot). E). Histograms represent quantification of blots. Asterisks indicate significantly different. F). Box plot represents individual samples of human PD patients and age-matched controls. Histograms are mean±SD expressed as % to control. ANOVA, Neumann Keuls with multiple comparison, or non-parametirc t-Test. P<0.05. N=12 PD patients and 7 control subjects.
Figure 3. Subcellular fractionation in frozen human PD brain tissues.

A). human anti-parkin (AB5112) staining and counterstaining with nuclear marker DAPI showing cytosolic protein, B). neuronal marker MAP-2 staining and DAPI and C). merged parkin and MAP-2 in serial sections stained with D). TH in the midbrain/SN of a control subject. E). human anti-parkin (AB5112) staining and counterstaining with nuclear marker DAPI showing cytosolic protein, F). neuronal marker MAP-2 staining and DAPI and G). merged parkin and MAP-2 in serial sections stained with H). TH in the midbrain/SN of a PD with Dementia patient. I). WB analysis on 4–12% SDS NuPAGE gel of human striatal lysates showing expression of LC3-I and LC3-II (first panel), LC3-B (second panel) compared to actin loading control (bottom panel) J). Histograms represent densitometry analysis of blots. K). Western blot in subcellular extracts showing LC3-B in AV-10 and AV-20 and LAMP-3 in lysosomal fraction, as well as mitochondrial marker COX-IV and nuclear marker PARP-1. Graphs represent subcellular fractionation and ELISA measurement of L). human α-Synuclein, M). human parkin and N). human p-Tau (AT8). Asterisks indicate significantly different to control. ANOVA, Neumann Keuls with multiple comparison, P<0.05. N=12 PD patients and 7 control subjects.
Altered parkin expression and loss of tyrosine hydroxylase neurons in the nigrostriatum of sporadic PD patients
To determine whether parkin expression is altered in sporadic PD, we analyzed human postmortem midbrain sections from 10 PD patients and 8 control subjects as identified in Table 2. To determine the difference in parkin staining between PD and control brains, we probed serial brain sections collected from each case with human anti-parkin antibody (PRK8) that recognizes a.a. 399–495 and counterstained with either GFAP or DAPI. We used confocal microscopy and observed diffuse parkin cytosolic staining in the caudate (Fig. 2A) and within GFAP-stained astrocytes (yellow stain) of control brain sections (Fig. 2B), and TH staining (Fig. 2C) was also observed in the caudate of a control subject (case 1683). However, intense cytosolic staining in the caudate (Fig. 2D, arrow), and within astrocytes (Fig. 2E), with diminished TH staining (Fig. 2F) were observed in a PD/AD patient (case 2215). To ascertain that parkin or GFAP staining were not due to auto-fluorescence in human slides we incubated slides with and without secondary and or primary antibodies and determined via confocal microscopy the absence of non-specific antibody binding (Data not shown). We further examined parkin expression in midbrain/SN brain regions. Diffuse parkin cytosolic staining (Fig. 2G) and within GFAP-stained astrocytes (Fig. 2H) with TH staining (Fig. 2I) were observed in serial sections of midbrain/SN of control brain (case 1855). Intense cytosolic parkin staining (Fig. 2J, arrow), and within astrocytes (Fig. 2K), with significantly diminished TH staining (Fig. 2L) were observed in a PD patient (case 2315). We then used another combination of antibodies using the AB5112 clone that detects parkin at a.a. 305–323 and GFAP antibodies to verify our results. Intense cytosolic parkin staining (Fig. 2M, arrow), and within astrocytes (Fig. 2N), with significantly diminished TH staining (Fig. 2O) were observed in a PD/dementia patient (case 2243). We then used MAP-2 as a neuronal marker and co-stained with parkin (DAPI counterstain) and TH. Parkin staining (Fig. 3A) was diffuse within the cytosol and was largely localized to MAP-2 labeled neurons (Fig. 3B & C) in the midbrain/SN of a control subject (case 1277). TH staining was also detected in serial brain sections (Fig. 3D). However, more intense and less diffuse parkin staining was detected in the cytosol of DAPI stained cells (Fig. 3E) and parkin staining was localized to MAP-2 stained neurons (Fig. 3F&G), with significantly decreased TH staining (Fig. 3H) in the midbrain/SN of a PD/Dementia patient (case 2267). It is worth it to note that MAP-2 staining was difficult in PD patients as neurons seem to become smaller and less numerous.
Figure 2. Immunostaining of human tissues with human parkin, and GFAP antibodies.
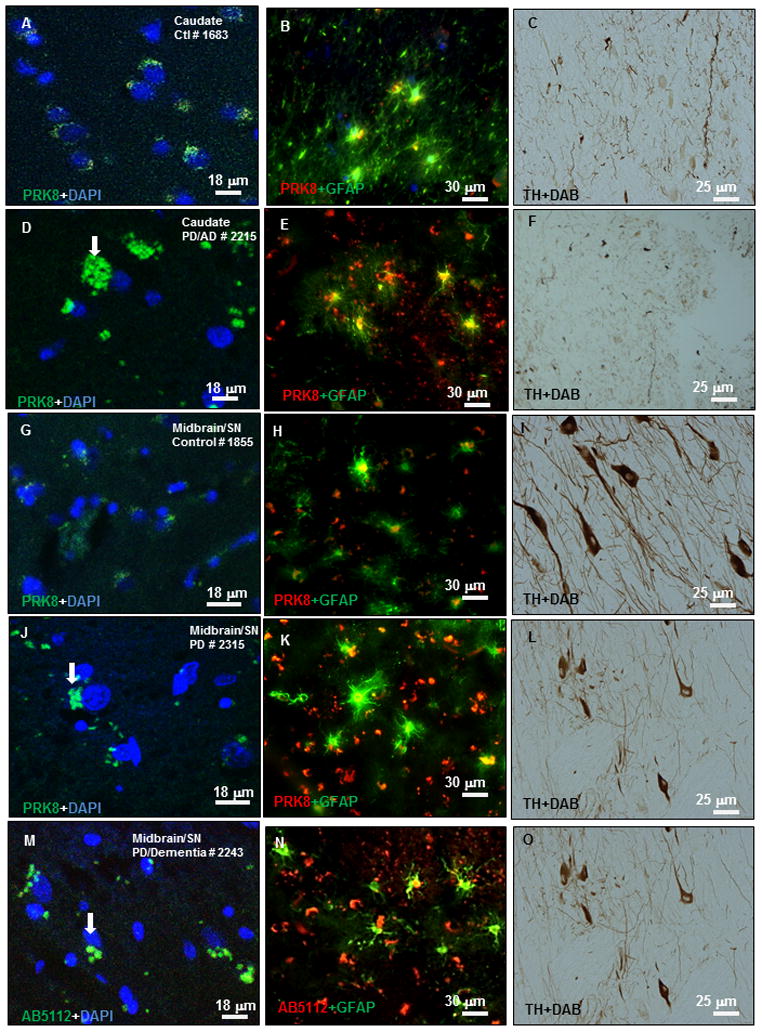
Immunostaining of 20 μm thick paraffin embedded serially sectioned brains with A). human anti-parkin (PRK8) staining and counterstaining with nuclear marker DAPI showing cytosolic protein, B). co-staining with parkin and glial marker GFAP showing parkin expression in astrocytes, C). TH staining in the caudate of a control subject. D). Parkin staining and counterstaining with nuclear marker DAPI showing cytosolic protein, E). co-staining with parkin and glial marker GFAP showing parkin expression in astrocytes, F). TH staining in the caudate of a PD/AD patient. G). Parkin staining and counterstaining with DAPI showing cytosolic protein, H). co-staining with parkin and glial marker GFAP showing parkin expression in astrocytes, I). TH staining in the midbrain/SN of a control subject. J). Parkin staining and counterstaining with DAPI showing cytosolic protein, K). co-staining with parkin and glial marker GFAP showing parkin expression in astrocytes, L). TH staining in the midbrain/SN of a PD patient. M). human anti-parkin (AB5112) staining and counterstaining with nuclear marker DAPI showing cytosolic protein, N). co-staining with parkin and glial marker GFAP showing parkin expression in astrocytes, O). TH staining in the caudate of a control subject.
Alteration of baseline autophagy in post-mortem striatum of PD patients
We previously demonstrated that exogenous parkin expression is associated with autophagic clearance (Herman and Moussa, 2011, Khandelwal et al., 2011b). To determine whether the change in parkin solubility is associated with changes of baseline autophagy, we examined the level of some autophagic markers in human PD striatal extracts. We examined markers of the autophagic cascade, including microtubule-associated light chain protein 3 (LC3). Probing with anti-LC3 antibody suggested an increase in LC3-II levels compared to LC3-I (Fig. 3I&J, 1st blot, 78%, N=12 PD and 7 control), indicating possible conversion and lipidation of LC3. LC3-I is abundant and stable in the brain, the ratio of LC3-II to LC3-I or the amount of LC3-II can be used to monitor the amount of autophagosome (Koike et al., 2005). LC3 is expressed as three isoforms in mammalian cells, LC3-A, LC3-B and LC3-C (He et al., 2003). Because LC3-II itself is degraded by autophagy (Mizushima and Yoshimori, 2007), we measured the amount of LC3 using an antibody specific for the LC3-B isoform. An increase in the level of LC3-B was detected in human striatal extracts from PD patients (N=12) compared to control (N=7) subjects (Fig. 3I&J, 2nd blot, 48%. P<0.05, ANOVA, Neumann Keuls). We then performed subcellular fractionation to isolate autophagic vacuoles and lysosomes and measured the levels of α-Synuclein, parkin and p-Tau using quantitative ELISA. First we determined whether the subcellular fractionation assay successfully extracted autophagosomes from lysosomes in frozen human tissues. Western blot analysis on PD patients brain lysates showed the lysosome-associated membrane glycoprotein 3 (LAMP-3) in the floating fraction containing lysosomes (Fig. 3K, 1st blot), while both the AV-10 and AV-20 fractions contained LC3-B (Fig. 3K, 2nd blot), suggesting that frozen human brains contain autophagic vacuoles and our fractionation did isolate autophagosomes from lysosomes. We also probed for mitochondrial marker cytochrome c oxidase-IV (COX-IV, Fig. 3K, 3rd blot) and nuclear marker Poly ADP-ribose polymerase (PARP-1, Fig. 3K, 4th blot), and detected these markers in all fractions, suggesting that brain samples contained intact organelles. Mitochondrial proteins may not be in autophagosomes, as this gradient subcellular fractionation may also extract mitochondria. A comprehensive assay that clearly shows mitochondria in autophagosomes or lysosomes has to be performed with both IHC co-labeling with LC3-COX-IV (autophagosome) or cathepsin-D-COX-IV (Lysosome) coupled with immuno-EM to determine mitochondrial accumulation in separate autophagic vacuoles. We then used ELISA and measured protein levels in subcellular extracts. The level of α-Synuclein was significantly increased (P<0.05, N=12 PD and 7 control) in AV-10 (31%) and AV-20 (64%) compared to control (Fig. 3L, ANOVA, Neumann Keuls), but no α-Synuclein was detected in the lysosomal fraction. Interestingly, ELISA measurement of parkin levels also showed a significant increase in AV-10 (Fig. 3M, 24%) and AV-20 (Fig. 3M, 23%) and a slight non-significant (9%) increase in the lysosome in PD (N=12) compared to control (N=7) subjects. We also measured the levels of p-Tau as another protein marker that is occasionally associated with PD pathology. Similarly, no p-Tau was detected in the lysosome but the levels of p-Tau were significantly increased in AV-10 (54%) and AV-20 (64%) compared to control (Fig. 3N, N=12 PD and 7 control). Because AV-20 is enriched in autophagosomes (Marzella et al., 1982), these data suggest accumulation of un-degraded proteins in autophagosomes in PD.
Figure 4. Lentiviral expression of α-Synuclein leads to p-Tau and parkin reverses these effects.

A). WB analysis on 4–12% SDS-NuPAGE gel of rat striatal extracts showing levels of parkin (top blot) and α-Synuclein (middle blot) expression and actin levels (lower blot). B). Histograms represent quantification of human α-Synuclein levels by ELISA. C). Histograms represent quantification of human parkin activity. D). ELISA measurement of rat p-Tau. Thioflavin-S staining of 20 μm striatal sections in lentiviral E). parkin, F) α-Synuclein and G). parkin+α-Synuclein injected brains. Human α-Synuclein staining of 20 μm sections cut serially with the thioflavin-S sections in lentiviral K). parkin, L) α-Synuclein and M). parkin+α-Synuclein injected brains. Asterisks indicate significantly different. Histograms are mean±SD expressed as % control. ANOVA, Neumann Keuls with multiple comparison, P<0.05. N=8 animals per treatment for WB and ELISA, 8 for IHC.
Parkin attenuates α-Synuclein-induced protein accumulation in the striatum
Because we observed increased parkin insolubility and decreased soluble parkin levels in association with alteration of autophagy in PD striatum, we sought to over-express parkin and determine whether functional parkin can reverse α-Synuclein effects on autophagic clearance. We generated gene transfer animal models targeting α-Synuclein expression to the striatum of 2-month old rats (Khandelwal et al., 2010). Lentiviral parkin led to significant increases (Fig. 4A, 53% by densitometry, N=8, P<0.05) in parkin levels and lentiviral α-Synuclein led to significant increases (41%) in α-Synuclein levels. Co-expression of parkin with α-Synuclein attenuated the levels of monomeric α-Synuclein (Fig. 4A) and reduced the level of higher molecular weight proteins back to control (LacZ) 4 weeks post injection (Khandelwal et al., 2010). No changes in total parkin levels were observed in brains injected with lentiviral α-Synuclein (Fig. 4A, 1st blot) and no-phosphorylated parkin was detected in rat brains. We then performed independent studies to confirm changes in α-Synuclein levels using quantitative ELISA specific for human α-Synuclein. The levels of human α-Synuclein were significantly increased (Fig. 4B, 54%, N=8) in the striatum of animals injected with lentiviral α-Synuclein compared to LacZ or parkin. Co-injection with lentiviral α-Synuclein and parkin reversed the levels of human α-Synuclein back to control. Lentiviral delivery of parkin into the striatum resulted in a significant increase in parkin when it was expressed alone (Fig. 4C, 44%, N=8) or in the presence of α-Synuclein (53%, N=8).
We previously reported that α-Synuclein expression leads to p-Tau (Khandelwal et al., 2010). Here we used an independent approach to determine changes in rat p-Tau using ELISA. Expression of human α-Synuclein leads to a significant increase (Fig. 4D, 34%, N=8) in p-Tau in the rat striatum, but co-expression of parkin reverses p-Tau back to control. Lentiviral expression of α-Synuclein in the striatum leads to detection of thioflavin-S positive staining (Fig. 4F), compared to lentiviral parkin alone (Fig. 4F). However, co-expression of parkin with α-Synuclein prevents the appearance of thioflavin-S positive staining (Fig. 4G); suggesting that parkin attenuation of α-Synuclein levels may eliminate thioflavin-S positive species in this animal model. To ascertain that thioflavin-S staining is associated with α-Synuclein expression, we stained striatal sections with human α-Synuclein antibody and showed no α-Synuclein staining in sections cut serially with the thioflavin-S sections from lentiviral parkin injected rats (Fig. 4K), compared to an abundant level of α-Synuclein in lentiviral α-Synuclein injected rats, congruent with thioflavin-S staining (Fig. 4L), while parkin co-expression led to disappearance of human α-Synuclein in the rat striatum (Fig. 4M).
Wild type functional parkin, not mutant T240R, mediates clearance of α-Synuclein-induced autophagic vacuoles
We sought to determine whether α-Synuclein expression can change normal autophagy, leading to formation of autophagic vacuoles in vivo. EM images of striatal sections showed no vacuoles in lentiviral LacZ injected animals (Fig. 5A) 4 weeks post injection. Lentiviral expression of α-Synuclein led to cytosolic accumulation of vacuoles (Fig. 5B, asterisks), suggesting that α-Synuclein expression alters autophagy in the rat striatum. Co-expression of parkin with α-Synuclein led to formation of autophagic vacuoles containing debris (Fig. 5C). To ascertain whether parkin function mediates clearance of autophagic vacuoles, we used non-functional T240R parkin, which is a mutant form that loses its E3 ubiquitin ligase activity, leading to ARJPD (Kitada et al., 1998, Lucking et al., 2000, Shimura et al., 2000). Co-expression of mutant T240R parkin with α-Synuclein did not prevent the accumulation of cytosolic vacuoles (Fig. 5D, asterisks), suggesting that parkin mediates autophagic clearance via its E3 ubiquitin ligase function.
Figure 5. Wild type, but not T240R, parkin reverses α-Synuclein-induced accumulation of autophagosomes.

Electron micrographs of striatal sections in rat brains injected with A). Lentiviral LacZ (Lv-LacZ) as control, B). Lentiviral α-Synuclein (Lv-Syn), C). Lentiviral parkin + lentiviral-α-Synuclein (Lv-Syn+Lv-Par), vacuoles contain debris and D). Lentiviral α-Synuclein + lentiviral T240R (Lv-Syn+Lv-T240R), asterisk indicates autophagic vacuoles. N=8. Graphs represent subcellular fractionation and ELISA measurement of E). α-Synuclein and F). p-Tau in gene transfer animal models. ANOVA, Neumann Keuls with multiple comparison, P<0.05. N=5 animals per treatment for subcellular fractionation.
We then measured the levels of human α-Synuclein and p-Tau using quantitative ELISA in subcellular fractions. A significant increase (62%, P<0.05, N=5) in the level of α-Synuclein was detected in AV-10 (Fig. 5E) and AV-20 (19%) compared to LacZ injected animals. However, co-expression of parkin eliminated α-Synuclein from AV-10 and significantly increased its levels in AV-20 (45%) and lysosomes (24%) compared to LacZ (Fig. 5E). Co-expression of α-Synuclein with T240R resulted in significantly elevated (51%) levels of α-Synuclein in AV-10, and unlike wild type parkin, failed to show any deposition in AV-20, which is enriched in autophagosomes (Marzella et al., 1982) or lysosomes. Significantly increased levels (P<0.05, N=5) of p-Tau were detected in AV-10 in animals injected with α-Synuclein (34%) or α-Synuclein+T240R (39%) compared to LacZ. However, wild type parkin expression led to a significant increase of p-Tau in AV-10 (19%) and lysosome (21%) compared to LacZ, α-Synuclein and α-Synuclein+T240R (Fig. 5F). It is worth it to note that no parkin as measured by ELISA was detected in subcellular fractions in these animal models, suggesting that parkin accumulation in autophagic vesicles may take place over a protracted time period in PD.
Functional parkin, not mutant T240R, regulates autophagic clearance in the striatum of α-Synuclein expressing animals
To determine the mechanisms by which parkin can mediate clearance of autophagic vacuoles in the rat striatum, we examined molecular markers of the autophagic pathway. WB analysis showed no difference in beclin-1 levels in animals injected with lentiviral LacZ, parkin or α-Synuclein alone (Fig. 6A). A significant increase in beclin-1 levels (54% by densitometry, N=8, P<0.05) was observed when parkin was co-expressed with α-Synuclein, suggesting that parkin responds to α-Synuclein-induced stress. The levels of Atg7 and Atg12 were also significantly increased by 41% and 33%, respectively, in parkin+α-Synuclein injected animals (Fig. 6A) compared to animals injected with LacZ, parkin or α-Synuclein alone. No changes in LC3-B levels were observed between animals injected with lentiviral LacZ or parkin alone (Fig. 6B) but α-Synuclein expression significantly increased (51%) LC3 levels (Fig. 6B), suggesting increased amount of autophagosomes. Co-expression of parkin and α-Synuclein decreased the levels of LC3-B (29% by densitometry, N=8, P<0.05), suggesting degradation of LC3-B-containing autophagic vacuoles. No changes were also observed in HDAC6 levels (Fig. 6B) between animals injected with LacZ, parkin or α-Synuclein alone, but HDAC6 level was significantly increased (37%) levels (Fig. 6B) when animals were co-injected with parkin and α-Synuclein together, suggesting that parkin expression facilitates fusion between autophagosomes and lysosomes (Iwata et al., 2005). No differences in the levels of molecular markers of autophagy were observed when mutant T240R parkin was injected either alone or with α-Synuclein (data not shown). These data suggest that parkin E3 ubiquitin ligase activity may up-regulate protein levels of the beclin-1-dependent autophagic cascade, facilitating autophagic clearance.
Figure 6. Functional parkin, not mutant T240R, reverses α-Synuclein alteration of normal autophagy.

A). WB analysis on 4–12% SDS NuPAGE gel of rat striatal lysates showing expression of beclin-1 (first panel), Atg7 (second panel) and Atg12 (third panel) compared to actin loading control (bottom panel) in animals injected with Lv-LacZ, Lv-Par, Lv-Syn and Lv-Par+Lv-Syn. B). WB analysis of rat striatal brain lysates showing expression of LC3-B (first panel), and HDAC6 (second panel) compared to actin loading control (bottom panel) in animals injected with Lv-LacZ, Lv-Par, Lv-Syn and Lv-Par+Lv-Syn. Staining of 20 μm thick cortical brain sections injected with C). Lentiviral parkin (Lv-Par), D), Lentiviral α-Synuclein (Lv-Syn) E). Lentiviral parkin + lentiviral α-Synuclein (Lv-Par+Lv-Syn) and F). Lentiviral T240R + lentiviral α-Synuclein (Lv-T240R+Lv-Syn). G). Histograms represent stereological counting of LC3-B positive cells in the striatum. F). Western blot analysis on 4–12% SDS NuPAGE gel with P62 antibody. Asterisks indicate significantly different. Histograms are mean±SD converted to % control. ANOVA, Neumann Keuls with multiple comparison, P<0.05. N=8 animals per treatment for WB and ELISA, 8 for IHC.
We supplemented the EM and WB data with immunohistochemistry to determine the presence of LC3-B. Staining with anti-LC3-B antibody showed no reactivity in the striatum of animals injected with lentiviral parkin (Fig. 6C). Lentiviral expression of α-Synuclein led to an increase in immunoreactivity to LC3-B (Fig. 6D). Stereological counting of LC3-B positive cells revealed a significant increase (Fig. 6G. 43%, P<0.05, N=8) in striata injected with α-Synuclein. Co-injection of lentiviral parkin with α-Synuclein (Fig. 6E) resulted in disappearance of LC3-B from the striatum. To further ascertain that functional E3 ubiquitin ligase parkin mediates autophagic changes, we used LC3-B antibodies in striatal sections co-injected with α-Synuclein and mutant T240R parkin (Fig. 6F) and observed no elimination of LC3-B staining in these animals. Stereological counting of LC3-B stained cells in the striatum co-injected with α-Synuclein and T240R showed a significant increase (37%) in LC3-B reactivity compared to LacZ (Fig. 6F&G). To further determine whether wild type parkin leads to clearance of ubiquitinated proteins via autophagy we stained with anti-P62 antibody. The levels of P62 were significantly (P<0.05, N=8) increased when α-Synuclein (41% by densitometry relative to actin) was expressed compared to LacZ (Fig. 6F). However, parkin co-expression led to complete disappearance of P62 staining, suggesting autophagic degradation of ubiquitinated proteins.
Discussion
These studies show decreased parkin solubility in the striatum of sporadic PD patients, independent of early onset disease-causing mutations. Parkin decreased solubility may result from increased phosphorylation, leading to possible enzyme inactivity in sporadic PD, suggesting that aging is a condition that decreases parkin solubility, leading to progressive susceptibility of the brain to stress (Wang et al., 2005). Parkin was previously shown to be prone to misfolding with many familial-PD mutations (Cookson et al., 2003, Henn et al., 2005, Hampe et al., 2006) and PD-linked stressors, including neurotoxins (MPP+, rotenone, 6-hydroxydopamine) and dopamine, which alter parkin solubility and result in its intracellular aggregation (Wang et al., 2005). Importantly, parkin functions as part of a number of multiprotein complexes, including PINK1 (Greene et al., 2003, Clark et al., 2006, Park et al., 2006), to mediate mitophagy (Narendra et al., 2008, Narendra et al., 2010), the Skp1-Cullin-Fbox (SCF)-like complex to facilitate proteasomal degradation of Cyclin E (Staropoli et al., 2003), and the chaperone Hsp70, and the U-Box protein CHIP (carboxy terminus of Hsc70 interacting protein) (Imai et al., 2002), so allosteric alterations may lead to changes in parkin activity (Wenzel et al., 2011) with partner proteins. We previously demonstrated that wild type and not mutant loss-of-function parkin increases proteasome activity (Burns et al., 2009, Khandelwal et al., 2010, Rosen et al., 2010), leading to degradation of ubiquitinated proteins, while proteasome inhibition leads to ubiquitinated protein accumulation. Furthermore, the polyglutamine expansion of ataxin-3 was recently shown to reduce parkin levels via autophagy in vivo (Durcan and Fon, 2011, Durcan et al., 2011), suggesting that ataxin-3 is another binding partner for parkin. Taken together, our data are consistent with these findings and suggest that parkin solubility is decreased and parkin aggregates may be removed via autophagy in aging brains. Additionally, decreased parkin solubility is associated with alteration of its activity via increased phosphorylation by several kinase activity, including casein kinase 1, protein kinase A, protein kinase C (Yamamoto et al., 2005), cyclin-dependent kinase 5 (Avraham et al., 2007, Rubio de la Torre et al., 2009), c-Abelson (Abl) (Ko et al., 2010, Imam et al., 2011) and potentially PINK1 (Kim et al., 2008, Sha et al., 2010). Therefore, the increased level of insoluble phosphorylated parkin suggests that parkin activity is reduced in sporadic PD.
The human data showed internal variability between samples. Some samples were collected as early as 2 hours post-mortem and there is lack of comprehensive clinical data on patients, implying diagnostic uncertainty. We do also not know the cause of death in these patients. However, these data show that parkin may be degraded in autophagic vacuoles, perhaps as it becomes insoluble and detection of autophagic vacuoles in human frozen tissues is intriguing. Although we performed the COX-IV and PARP WB as controls for fractionation, it is hard not to notice the level of mitochondrial proteins in autophagosomal fractions. The presence of mitochondrial proteins may not be in autophagosomes, as the gradient subcellular fractionation we performed may also extract mitochondria. A comprehensive assay that clearly shows mitochondria in autophagosomes or lysosomes has to be performed via co-labeling with LC3-COX-IV (autophagosome) or cathepsin D-COX-IV (Lysosome) coupled with immuno-EM to determine mitochondrial accumulation in separate autophagic vacuoles. Dysfunction in autophagy, including chaperon-mediated autophagy (CMA), was observed in PD (Ravikumar et al., 2004, Nixon et al., 2005, Sarkar et al., 2007, Pan et al., 2008) and Dementia with LBs (DLB), where α-Synuclein accumulation may alter autophagy and cause lysosomal dysfunction (Cuervo et al., 2004, Martinez-Vicente et al., 2008, Xilouri et al., 2009). The accumulation of P62 in α-Synuclein expressing gene transfer models, suggests impairment of CMA, leading to accumulation of the P62 sensor of ubiquitinated proteins, however, the decrease in P62 when parkin was co-expressed with α-Synuclein suggests clearance of ubiquitinated proteins. Additionally, α-Synuclein aggregates were previously shown to alter parkin solubility in sporadic PD (Kawahara et al., 2008) and inhibit macro-autophagy in cell models and transgenic animals (Winslow et al., 2010). Here we show that parkin insolubility is associated with autophagic defects and accumulation of both α-Synuclein and p-Tau in the autophagosome, suggesting either increased autophagic induction or decreased protein degradation. The subcellular fractionation we performed in human samples sequentially extracts AV-10 first (10% Metrizamide gradient), which is more likely to contain early forming autophagic vacuoles such as the phagophore and autophagosome together. The AV-20 is collected in a second fraction (20% Metrizamide) to collect the remaining autophagosomes, suggesting that α-Synuclein may accumulate in autophagosome and fails to be deposited in the lysosome (last fraction) where it is potentially degraded by proteolytic enzymes. The differential accumulation between parkin and p-Tau in AV-10 compared to α-Synuclein in AV-20 is worth mentioning, as PD patients accumulate α-Synuclein throughout their life, leading to autophagic defects, but when parkin solubility is altered or Tau is hyper-phosphorylated they may accumulate in autophagic vacuoles that form earlier than the autophagosome-rich AV-20, but later in the disease process. In addition, we recently demonstrated in similar studies using post-mortem AD tissues compared to the same control subjects used in the current studies (Lonskaya et al., 2012) that parkin accumulates with intraneuronal Aβ and displays decreased solubility in the hippocampus and cortex in association with identical autophagic defects, suggesting that aging contributes to parkin insolubility and perhaps inactivation. Therefore, parkin expression may contribute to autophagosome maturation, including fusion between the autophagosome and lysosome to facilitate autophagic clearance.
The animal studies demonstrate that α-Synuclein over-expression alters autophagy, in agreement with the human data. Autophagosome accumulation could be due to lack of maturation, leading to inefficient fusion with lysosomes (Kovács et al., 1982, Iwata et al., 2005, He and Klionsky, 2009). Parkin up-regulation of the protein levels of molecular markers of autophagy in the presence of α-Synuclein, suggests that the E3 ubiquitin ligase function may regulate autophagosome maturation and clearance in response to cellular stress, consistent with our previous observation (Lonskaya et al., 2012). Activation of autophagy improves dopaminergic cell survival in parkin deficient and Tau over-expressing mice (Rodriguez-Navarro et al., 2010). Parkin modulates beclin-1-LC3 mediated autophagy (Chen et al., 2010) and our data show that loss of parkin function (T240R) is associated with lack of autophagosome clearance in α-Synuclein gene transfer models. Further studies are needed to address the effects of endogenous parkin on autophagic protein markers in response to α-Synuclein and other amyloid stress.
Expression of α-Synuclein contributes to p-Tau and formation of amyloidogenic proteins, perhaps clogging the cell. The current data suggest that amyloid accumulation results in Tau modification (Khandelwal et al., 2010, Rebeck et al., 2010) and protein aggregation similar to over-expression of Tau (Khandelwal et al., 2011a) or Aβ1–42 (Lonskaya et al., 2012). Accumulation of autophagosomes in neurodegeneration may be due to reduced autophagic flux (Gonzalez-Polo et al., 2005). Parkin expression prevents formation of amyloidogenic proteins and p-Tau, and leads to an increase of HDAC6 levels. Tau regulates HDAC6 function (Perez et al., 2009), suggesting that HDAC6 is a potential modulator of Tau phosphorylation and accumulation (Ding et al., 2008). Autophagosomes recruit lysosomes via retrograde transport on microtubules, requiring an intact microtubule cytoskeleton and cytoplasmic HDAC6 to mediate the fusion of autophagosome with the lysosome (Iwata et al., 2005). Further studies are necessary to better understand the role of Tau in facilitating transport of autophagic organelles in the cell.
In conclusion, decreased parkin solubility may reflect diminished parkin function, which can potentially lead to alteration of baseline autophagy, including parkin, α-Synuclein and p-Tau clearance. Lentiviral expression of α-Synuclein leads to p-Tau and accumulation of autophagic vacuoles. Our data demonstrate an association between α-Synuclein and autophagic dysfunction in PD, and indicate a beneficial role for parkin in autophagic clearance. Parkin role in autophagic clearance could be exploited as a therapeutic strategy in PD.
Highlights.
Parkin is insoluble and accumulates in the nigrostriatum of sporadic PD brains
Decreased parkin solubility may alter normal autophagy in sporadic PD brains
Functional, not mutant, parkin gene transfer enhances autophagic clearance
Aging may reduce parkin solubility and function to regulate autophagic clearance
Acknowledgments
These studies were supported by NIH grant NIA 30378 and Georgetown University funding to Charbel E-H Moussa. The authors would like to thank Dr. Jim Driver from the University of Montana for his support in the EM studies. The authors would also like to thank Dr. O. Pletnikova from Johns Hopkins University Brain Bank for providing the human post-mortem tissues.
Abbreviations
- p-Tau
Tau hyper-phosphorylation
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ARJPD
autosomal recessive juvenile PD
- LC3
microtubule-associated light chain protein-3
- ELISA
enzyme-linked immunosorbent assay
- HDAC6
histone deacetylase 6
Footnotes
Conflicts of interest: The authors declare no conflict of interest whatsoever in association with this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avraham E, Rott R, Liani E, Szargel R, Engelender S. Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J Biol Chem. 2007;282:12842–12850. doi: 10.1074/jbc.M608243200. [DOI] [PubMed] [Google Scholar]
- Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MP, Zhang L, Rebeck GW, Querfurth HW, Moussa CE. Parkin promotes intracellular Abeta1–42 clearance. Hum Mol Genet. 2009;18:3206–3216. doi: 10.1093/hmg/ddp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, Wang G. Parkin Mono-ubiquitinates Bcl-2 and Regulates Autophagy. J Biol Chem. 2010;285:38214–38223. doi: 10.1074/jbc.M110.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Hum Mol Genet. 2010;19:R21–27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, Lockhart PJ, McLendon C, O’Farrell C, Schlossmacher M, Farrer MJ. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet. 2003;12:2957–2965. doi: 10.1093/hmg/ddg328. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Ding H, Dolan PJ, Johnson GV. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J Neurochem. 2008;106:2119–2130. doi: 10.1111/j.1471-4159.2008.05564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7:233–234. doi: 10.4161/auto.7.2.14224. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, Fantaneanu T, Paulson HL, Fon EA. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum Mol Genet. 2011;20:141–154. doi: 10.1093/hmg/ddq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Philos Trans R Soc Lond B Biol Sci. 1999;354:1101–1118. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. Journal of cell science. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe C, Ardila-Osorio H, Fournier M, Brice A, Corti O. Biochemical analysis of Parkinson’s disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum Mol Genet. 2006;15:2059–2075. doi: 10.1093/hmg/ddl131. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Dang Y, Dai F, Guo Z, Wu J, She X, Pei Y, Chen Y, Ling W, Wu C, Zhao S, Liu JO, Yu L. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. The Journal of biological chemistry. 2003;278:29278–29287. doi: 10.1074/jbc.M303800200. [DOI] [PubMed] [Google Scholar]
- Healy DG, Abou-Sleiman PM, Lees AJ, Casas JP, Quinn N, Bhatia K, Hingorani AD, Wood NW. Tau gene and Parkinson’s disease: a case-control study and meta-analysis. J Neurol Neurosurg Psychiatry. 2004;75:962–965. doi: 10.1136/jnnp.2003.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn IH, Gostner JM, Lackner P, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- Herman AM, Moussa CE. The ubiquitin ligase parkin modulates the execution of autophagy. Autophagy. 2011;7:919–921. doi: 10.4161/auto.7.8.15814. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, Roberts JL, Kahle PJ, Clark RA, Li S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson’s disease. J Neurosci. 2011;31:157–163. doi: 10.1523/JNEUROSCI.1833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, Rockenstein E, Imam SZ, Masliah E. alpha-Synuclein aggregates interfere with Parkin solubility and distribution: role in the pathogenesis of Parkinson disease. J Biol Chem. 2008;283:6979–6987. doi: 10.1074/jbc.M710418200. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Feng LR, Maguire-Zeiss K, Rebeck G, Lashuel HA, Moussa CE. Parkinson-related parkin reduces alpha-Synuclein phosphorylation in a gene transfer model. Mol Neurodegener. 2010;5:47. doi: 10.1186/1750-1326-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE. Wild type and P301L mutant Tau promote neuro-inflammation and alpha-Synuclein accumulation in lentiviral gene delivery models. Mol Cell Neurosci. 2011a doi: 10.1016/j.mcn.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011b;20:2091–2102. doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc Natl Acad Sci U S A. 2010;107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, von Figura K, Mizushima N, Saftig P, Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) The American journal of pathology. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács AL, Reith A, Seglen PO. Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Experimental cell research. 1982;137:191–201. doi: 10.1016/0014-4827(82)90020-9. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann N Y Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- Lonskaya I, Shekoyan AR, Hebron ML, Desforges N, Algarzae NK, Moussa CE. Diminished Parkin Solubility and Co-Localization with Intraneuronal Amyloid-beta are Associated with Autophagic Defects in Alzheimer’s Disease. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-121141. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Lundvig D, Lindersson E, Jensen PH. Pathogenic effects of alpha-synuclein aggregation. Brain Res Mol Brain Res. 2005;134:3–17. doi: 10.1016/j.molbrainres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzella L, Ahlberg J, Glaumann H. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. J Cell Biol. 1982;93:144–154. doi: 10.1083/jcb.93.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4:590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Park J, Kim Y, Chung J. Mitochondrial dysfunction and Parkinson’s disease genes: insights from Drosophila. Dis Model Mech. 2009;2:336–340. doi: 10.1242/dmm.003178. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Perez M, Santa-Maria I, Gomez de Barreda E, Zhu X, Cuadros R, Cabrero JR, Sanchez-Madrid F, Dawson HN, Vitek MP, Perry G, Smith MA, Avila J. Tau--an inhibitor of deacetylase HDAC6 function. J Neurochem. 2009;109:1756–1766. doi: 10.1111/j.1471-4159.2009.06102.x. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Hoe HS, Moussa CE. Beta-amyloid1–42 gene transfer model exhibits intraneuronal amyloid, gliosis, tau phosphorylation, and neuronal loss. J Biol Chem. 2010;285:7440–7446. doi: 10.1074/jbc.M109.083915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Kadiu I, Garg SK, Glanzer JG, Nordgren T, Ciborowski P, Banerjee R, Gendelman HE. Nitrated alpha-synuclein and microglial neuroregulatory activities. J Neuroimmune Pharmacol. 2008;3:59–74. doi: 10.1007/s11481-008-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, Cuervo AM, Garcia de Yebenes J, Mena MA. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Rosen KM, Moussa CE, Lee HK, Kumar P, Kitada T, Qin G, Fu Q, Querfurth HW. Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J Neurosci Res. 2010;88:167–178. doi: 10.1002/jnr.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio de la Torre E, Luzon-Toro B, Forte-Lago I, Minguez-Castellanos A, Ferrer I, Hilfiker S. Combined kinase inhibition modulates parkin inactivation. Hum Mol Genet. 2009;18:809–823. doi: 10.1093/hmg/ddn407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet. 2010;19:352–363. doi: 10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998a;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998b;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Masliah E. C-terminal alpha-synuclein immunoreactivity in structures other than Lewy bodies in neurodegenerative disorders. Acta Neuropathol. 2000;99:296–304. doi: 10.1007/pl00007441. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Parkinson’s disease and related alpha-synucleinopathies are brain amyloidoses. Ann N Y Acad Sci. 2003;991:107–110. doi: 10.1111/j.1749-6632.2003.tb07468.x. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett. 1997;239:45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Rubinsztein DC. Autophagy in neurodegeneration and development. Biochim Biophys Acta. 2008;1782:723–729. doi: 10.1016/j.bbadis.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Rubinsztein DC. The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy. 2011;7:429–431. doi: 10.4161/auto.7.4.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Friedlein A, Imai Y, Takahashi R, Kahle PJ, Haass C. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem. 2005;280:3390–3399. doi: 10.1074/jbc.M407724200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–2988. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]