

Abstract
Nicotinic acetylcholine receptors (nAChRs), being responsible for mediating key physiological functions, are ubiquitous in the central and peripheral nervous systems. As members of the Cys loop ligand-gated ion channel family, neuronal nA-ChRs are pentameric, composed of various permutations of α (α2 to α10) and β (β2 to β4) subunits forming functional heteromeric or homomeric receptors. Diversity in nAChR subunit composition complicates development of selective ligands for specific subtypes, since the five binding sites reside at the subunit interfaces. The acetylcholine binding protein (AChBP), a soluble extracellular domain homologue secreted by mollusks, serves as a general structural surrogate for the nAChRs. In this work, homomeric AChBPs from Lymnaea and Aplysia snails were used as in situ templates for the generation of novel and potent ligands that selectively bind to these proteins. The cycloaddition reaction between building block azides and alkynes to form stable 1,2,3-triazoles generated the leads. The extent of triazole formation on the AChBP template correlated with the affinity of the triazole product at the nicotinic ligand binding site. Instead of the in situ protein-templated azide-alkyne cycloaddition reaction occurring at a localized, sequestered enzyme active center as previously shown, we demonstrate that the in situ reaction can take place at subunit interfaces of an oligomeric protein and can thus be used as a tool for identification of novel candidate nAChR ligands. The crystal structure of one of the in situ formed triazole–AChBP complexes shows binding poses and molecular determinants of interactions predicted from structures of known agonists and antagonists. Hence, the click chemistry approach with an in situ template of a receptor provides a novel synthetic avenue for generating candidate agonists and antagonists for ligand-gated ion channels.
1. INTRODUCTION
Nicotinic acetylcholine receptors (nAChRs) belong to a superfamily of neurotransmitter ligand-gated ion channels characterized by a pentameric structure of Cys loop containing subunits.1,2 This family of proteins is actuated upon binding of a specific ligand and includes other key neurotransmitter receptors, such as glycine, GABA-A, and 5-HT3 types.3 The nAChRs are currently being considered as therapeutic targets for CNS disorders, such as schizophrenia, nicotine addiction, and Alzheimer’s disease among other cognitive disorders.2,4,5 An extensive variety of synthetic and naturally occurring ligands for nAChRs of variable selectivity are known. Agonists and competitive antagonists bind in the extracellular domain at the interface between subunits, and agonists transmit ligand occupation through conformational changes to open an internally located ion channel effecting a rapid depolarization.1,6
The acetylcholine binding proteins (AChBPs), homologous to the extracellular domain of pentameric ligand-gated ion channels, have been found to express as soluble pentamers in select gastropods and polychaetes.7–11 Homologous to the N-terminal ~210 amino acids in the extracellular receptor domain, AChBP mimics the recognition properties of nAChRs, providing a critical template describing the shape and spatial disposition of residues contributing to the binding site.7,12 The five acetylcholine binding sites of the AChBPs lie at the subunit interfaces and are partially surrounded by a flexible loop C found between sheets β9–10 that extends across the subunit interface.
Structure based drug design and screening of compound libraries have been a laborious task for the nAChRs. The membrane disposition of the nAChR, the diversity of receptor subtypes, and the dynamic nature of the binding site complicate classical drug discovery approaches. By contrast, target-guided in situ synthesis is an attractive and efficient approach to drug discovery because it directly employs the biological target for the assembly of candidate leads from their own building blocks. Independent of the knowledge of the geometry of the protein target, it allows fast screening of potential ligands whose building blocks are “templated” by the target protein. Although this concept has been previously demonstrated using different connecting reactions,13–16 the in situ click chemistry approach is unique with its reliance upon the nearly bioorthogonal 1,3-dipolar cycloaddition between azides and alkynes, a reaction that is fully compatible with functional groups found in normal physiologic environment.
These building blocks readily react with each other when in proximity but remain inert to side chains and amide backbones of proteins. This highly exergonic reaction produces five-membered nitrogen heterocycles, anti- or syn- 1,2,3-triazoles, that are stable to acidic and basic hydrolysis, as well as to severe red-ox conditions.17 A target protein, employed as a template, can generate a high affinity ligand from monovalent building blocks using the in situ click chemistry approach.18 The process begins with selective binding of anchor molecules to specific areas of the protein binding site. Subsequently, if two of the bound anchor molecules are in proximity, they will irreversibly link together within the confines of their binding pockets and thereby allow formation of an energetically more favorable new complex with the protein. The resultant compound can then be detected by liquid chromatography mass spectrometry (LC-MS).
Since this approach employs the biological target to assemble its own ligands from a library of reagents that can be combined in a multitude of different ways,18 rather than requiring the synthesis, purification and screening of each possible library product, the use of in situ click chemistry promises more efficiency than traditional combinatorial chemistry or fragment-based drug design. Moreover, the triazole product may induce or select a conformation of the macromolecule so adapted in conformation to the binding of the ligand.19 The template-generated compounds are then further screened to assess binding affinity and specificity for the target.
In this work, we used AChBPs from Lymnaea stagnalis (Ls), Aplysia californica (Ac), and the Y55W Aplysia mutant (AcY55W) as template surrogates for nAChRs. Due to the dynamic molecular motion of the protein subunit interface, in situ click chemistry facilitates identification of potential ligands for different conformational states of a protein without the need to synthesize large screening libraries. We initially prepared an artificial triazole containing AChBP ligand, via Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) conditions,20,21 and after confirmation of the triazole’s affinity subsequently utilized the compound’s components to validate the AChBPs as in situ click chemistry templates. The alkyne component and subsequent in situ azide component that formed product with higher efficiency and affinity were then used as leads for the refinement of building blocks to form triazole structures. These derivatives should mimic the properties of the natural nAChR neurotransmitter, acetylcholine (ACh), along with naturally occurring and synthetic congeners, thereby providing a starting point for developing selective ligands that address the binding site. Further, we demonstrate that when a mixture of building blocks is screened simultaneously, then multiple ligands may be formed whereby the amount of the triazole product produced by the template is proportional to its affinity for the binding site. Structurally we demonstrate in situ product formation at a subunit interface binding region of a multi-subunit protein, the AChBP. This receptor surrogate serves as a template for the generation of nicotinic receptor ligands.
2. EXPERIMENTAL METHODS
Synthetic Compound Preparations
All azides were synthesized by the displacement of either a halide, mesylate or tosylate with sodium azide in dimethyl formamide. Propargyl phenol ethers were prepared using literature conditions.22 The 1,4-triazole products 2, 14, 15, 18, 19, 27, 28 and 29 were synthesized using CuAAC, whereas 1,5-triazole 18a was synthesized using ruthenium-catalyzed azide-alkyne cycloaddition (RuAAC,23,24 see supporting information for details).
Procedure for the in situ formation of 2 from a binary component mixture
Azide 4 (1 μL, 100 mM in 0.1 M aq. sodium phosphate buffer, pH 7.0 (PBS)) was added to 98 μL of a solution of the protein of interest (~1 mg/mL in PBS) in a microfuge tube, followed immediately by alkyne 3 (1 μL, 50 mM in DMSO) to give final concentrations of 1 mM azide 4 and 0.5 mM alkyne 3. The reaction was briefly vortexed and then incubated at room temperature. In a separate microfuge tube, azide 4 (1 μL, 100 mM in PBS) and alkyne 3 (1 μL, 50 mM in DMSO) were diluted with water (87 μL) before aq. copper sulfate (1 μL, 0.05 M) and aq. sodium ascorbate (10 μL, 0.1 M) were added. The reaction was briefly vortexed and then incubated at room temperature. After 3 days, three samples (25 μL) from each tube were directly injected into the LC/MS instrument to perform LC/MS-SIM analysis: Zorbax, 4.6 mm × 3 cm, SB-C18 (rapid resolution) reverse phase column, preceded by a Phenomenex C18 guard column, flow rate 0.5 mL/min, using a gradient elution as follows: (H2O + 0.05% TFA):(MeCN + 0.05% TFA) 100:0 to 0:100 over 15 min; then 100% MeCN + 0.05% TFA for 5 min with a post run time of 5 min using the starting solvent ratio. Detection was by electrospray ionization and mass spectrometric detection in the positive selected-ion monitoring, tuned to the molecular mass of 2 (M+). The cycloaddition product was identified by comparison of the retention time, determined by analysis of the copper catalyzed reaction, and its molecular weight. Control experiments in the presence of BSA (1 mg/mL) instead of the binding protein, as well as in the presence of Ls and the known receptor inhibitor MLA (1 mM final concentration) were run as described above.
Procedure for the in situ screening of single component libraries
Library 1a: Azides 4, 5, 6, 7 and 8 were dissolved in 0.1 M sodium phosphate buffer pH 7.0 (PBS, 100 mM) and the solutions combined. The solution of azides (1 μL) was added to the protein of interest (~1 mg/mL in PBS, 98 μL) in a microfuge tube, followed immediately by alkyne 3 (1 μL, 50 mM in DMSO). The reaction and controls were performed similarly to the binary component mixture. After 3 days the reactions were analyzed, in triplicate, as described for the binary mixture, with the mass spectroscopic detection in the positive selected-ion monitoring tuned to the 5 expected molecular masses of the products (M+). The cycloaddition products were identified by comparison of the retention time, determined by analysis of the copper catalyzed reaction, and their molecular weights. Azide library 1b was in situ click chemistry screened as described above substituting azides 4, 5, 6, 7 and 8 with azides 9, 10, 11, 12 and 13. The alkyne library was again screened as described above, using azide 9 (100 mM in PBS) and a combined DMSO solution of alkynes 3, 23, 24, 25 and 26 at a final concentration of 0.5 mM.
Procedure for the in situ screening of azide and alkyne libraries
Azides 4, 5, 6, 7, 8, 9, 10, 11, 12 and 13 were dissolved in PBS (100 mM) and 25 μL of each solution taken and combined. Alkynes 3, 23, 24, 25 and 26 were dissolved in DMSO (50 mM) and the solutions combined. The combined solution of azides (10 μL) was added to a solution of Ls (~1 mg/mL in PBS, 980 μL) in a microfuge tube, followed immediately by the combined solution of alkynes (10 μL). The reaction was briefly vortexed and then incubated at room temperature. After 10 days, triplicate analysis of the protein-catalyzed reaction was performed using the chromatography conditions described above. To improve MS detection sensitivity, 10 injections (each 25 μL) were performed; each injection was tuned to detect 5 of the expected 50 molecular weights. A control experiment in the presence of BSA (1 mg/mL) instead of the binding protein was performed. As some of the potential in situ click chemistry products have the same molecular weights, the corresponding triazoles were prepared by reacting each azide with the five alkynes under CuAAC conditions. Therefore, azides 4, 5, 6, 7, 8, 9, 10, 11, 12 and 13 (1 μL, 100 mM in PBS) were charged to 10 separate microfuge tubes and water (87 μL), combined alkyne solution (1 μL) as described above, aq. copper sulfate (1 μL, 0.05 M) and aq. sodium ascorbate (10 μL, 0.1 M) were added. The reactions were briefly vortexed and then incubated at room temperature. After 10 days, analyses of the copper catalyzed reactions were performed as described above. The cycloaddition products of the in situ screen were identified by their molecular weights and by comparison of the retention time of the formed products with the values determined by analysis of the copper catalyzed reactions.
Preparation of AChBPs
The AChBPs from Ls, Ac, and the Ac-Y55W mutant were expressed and purified as previously described.25,26 Briefly, AChBPs were expressed with an amino-terminal FLAG epitope tag and secreted from stably transfected HEK293S cells lacking the N-acetyglucosaminyltransferase I (GnTI−) gene.27 Protein was purified with FLAG-antibody resin and eluted with FLAG peptide (Sigma). Affinity purified protein was further characterized by size exclusion chromatography, to ascertain pentameric association, in a Superose 6 10/300 GL column (GE Healthcare) in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.02% NaN3. Purified AChBP pentamers were then concentrated in a YM50 Centri-con ultrafiltration unit (Millipore) to a final concentration of approximately 5 mg/ml, removing monomeric subunits and trace contaminants.
Radioligand Binding Assays
A scintillation proximity assay was used to determine the apparent Kd value of the compounds as reported previously.26 Briefly, AChBP (0.5–1.0 nM final concentration in binding sites), polyvinyltoluene anti-mouse SPA scintillation beads (0.17mg/ml final concentration, GE Healthcare), monoclonal anti-FLAG M2 antibody from mouse 1:8000 dilution (Sigma), and (±)-[3H]-epibatidine (5–20 nM final concentration, GE Healthcare) were combined with PBS. A quick screen was performed to determine relative binding affinities of compounds by adding compound at a final concentration of 10 μM to the previously mentioned solution. Apparent Kd values were then determined for compounds that reduced normalized counts per minute below 50%. Saturation binding of (±)-[3H]-epibatidine was measured by adding increasing concentrations of (±)-[3H]-epibatidine in a constant volume. Nonspecific binding was determined in parallel by adding a saturating concentration (12.5 μM) of methyllycaconitine (Tocris) to an identical set of wells. Competition assays were conducted in a similar manner except that the concentration of (±)-[3H]-epibatidine was held constant (5–20 nM final concentration) and varying concentrations of competing ligand were added to the wells in a constant volume. The resulting mixtures were allowed to equilibrate at room temperature for a minimum of 1 h and measured on a 1450 MicroBeta TriLux liquid scintillation counter (Wallac). The data obtained were normalized and the Kd calculated from the observed EC5028 value using GraphPad Prism 4.02 (San Diego, CA). A minimum of three independent experiments, performed in duplicates, were used to determine the reported Kd values.
Complex formation and crystallization
The 18/Ac AChBP complex was formed by combining 2 μl of a 10 mmol 18 dissolved in DMSO with 48 μl of purified concentrated protein at a concentration of 5 mg/ml to achieve a stoichiometric excess of ligand to binding sites. The 18/Ac AChBP complex co-crystals were prepared by the vapor diffusion hanging drop method. Concentrated protein complex was mixed in a 1 μl: 1 μl solution consisting of 0.1 M Tris-HCl, pH 8.0, 0.25 M MgCl2, 20% (w/v) PEG 4000, incubated at 22°C and suspended over 500 μl of the solution. Crystals of final size 0.3 × 0.3 × 0.2 mm appeared after a few weeks.
X-ray Diffraction Data Collection
18/Ac AChBP complex co-crystals were transferred to a cryoprotective harvest solution consisting of 0.1 M Tris-HCl, pH 8.0, 0.25 M MgCl2, 12% (w/v) PEG 4000, and 10% (v/v) glycerol and flash cooled directly in liquid nitrogen. A full set of X-ray diffraction data was collected at 100K on ALS beamline 8.2.2 at Lawrence Berkeley National Laboratory. Data were processed by the HKL2000 program.29 Data collection statistics are given in Supplementary Table 7.
Structure Refinement
The 18/Ac AChBP complex structure was solved by the molecular replacement method using the PHASER30 software using an ensemble of AChBP structures (PDB codes 2BYN, 2PGZ, 2BYP, 2BYR, 2BYS, and 3C79) as the search model. The electron density maps were fitted with COOT31 and structure refinement used the program REFMAC5.32 Refinement statistics are listed in Supplementary Table 7. Atomic coordinates and structure factors of the complex have been deposited in the PDB (ID code 4DBM). The structural figures were generated with PyMOL33 and Discovery Studio 3.0 (Accelrys).
3. RESULTS
We hypothesized that substituting a 1,2,3-triazole unit for the ester moiety of ACh would mimic its hydrogen bond acceptor character, (Figure 1), an interaction that has previously been observed in triazole-containing peptidomimetics.34–36 Furthermore, the conservation of the trimethylammonium-containing moiety of ACh would maintain the crucial cation-π interaction with Trp147 and other proximal aromatic amino acid side chains (AcAChBP numbering).25,37–40
Figure 1.
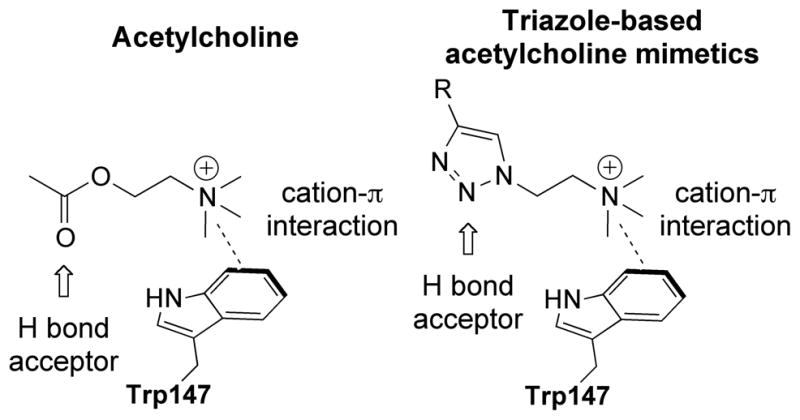
The binding poses of acetylcholine (left) and proposed binding poses of triazole-based acetylcholine mimetic.
Conforming to our hypothesis, azide 1 was reacted, under standard CuAAC reaction conditions, with a range of alkynes and the resultant tertiary amine quaternized with methyl iodide (Scheme 1 – equation 1). Screening of the compounds (data not shown) against Ac, Ls and AcY55W AChBPs identified compound 2 as having a relatively strong association with all three AChR surrogates (Table 1).
Scheme 1.
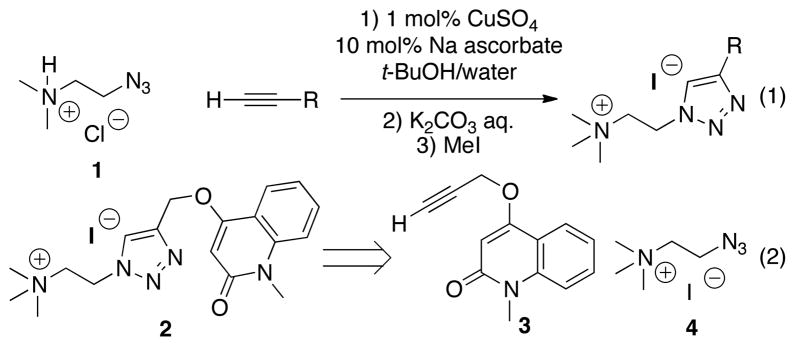
Synthesis of triazole library using standard CuAAC reaction conditions.
Table 1.
Proof of concept: Amount of triazole 2 formed in situ in the presence of AChBP templates compared with the respective affinities of the triazole product.
| AChBP species | MS counts[2] | Kd [nM] | Affinity [μM−1] | Structure[3] | # |
|---|---|---|---|---|---|
| Ls | 100.0 ± 0.3 | 75 ± 26 | 13 |
|
2 |
| Ac | 8.0 ±0.1 | 1900± 800 | 0.53 | ||
| AcY55W | 8.9 ±0.2 | 370± 50 | 2.7 | ||
| Ls + MLA | 2.8 ±0.0 | n/a | n/a | ||
| BSA[1] | 2.8 ±0.1 | n/a | n/a |
BSA control protein replacing the AChBP template.
MS counts were corrected for background via BSA controls and normalized to Ls.
Cation detected by MS.
We first wanted to confirm that the flexible subunit interfaces in the AChBPs were capable of catalyzing the formation of 2 in situ, as most previous examples have relied upon well-defined sites internal to the subunit.41 To validate our hypothesis, the constituent alkyne 3 and azide 4 (Scheme 1 – equation 2) were incubated in the presence of Ls, Ac, and AcY55W AChBPs in PBS at room temperature for 3 days (see Experimental Methods). In addition, two control reactions were performed: incubating the reactants in the presence of bovine serum albumin (BSA), to determine whether non-specific protein catalysis of the triazole-forming cycloaddition was occurring; and in the presence of Ls with a known competing ligand, methyllycaconitine (MLA), to confirm that the protein-templated reaction was occurring at the ACh orthosteric binding site.
Triplicate samples of the reaction mixtures were analyzed by liquid chromatography-mass spectrometry (LC-MS) using selected-ion monitoring (detection window set to the molecular weight corresponding to the most abundant peak) was used in all cases and compared to the retention time of the previously synthesized sample of 2. Analysis of these data showed that Ls efficiently templated the formation of ligand 2 under the reaction conditions, while both Ac and AcY55W AChBP also produced the product, but much less efficiently (Table 1).
Comparison of the in situ click chemistry reactions performed in the presence of AChBPs to the BSA control confirmed that triazole formation was selectively accelerated in the presence of the receptor surrogates. The control reaction showed that MLA, a nicotinic antagonist, returned product formation to background levels, thus demonstrating that the flexible, subunit-interface ACh-binding site was indeed serving as the template for the cycloaddition reaction. It is noted that the amount of product formed in the presence of the template correlates with the affinities (1/Kd, μM−1) determined for the tested AChBPs. This suggests that product selectivity toward a specific target over closely related receptor subtypes could be inferred from the bioorthogonal triazole formation, without the need to acquire individual binding data. Unfortunately the 1,4- (anti-) and 1,5- (syn-) 1,2,3-triazole isomers proved inseparable by LC-MS, despite considerable method development, and thus subsequent experiments considered both regioisomers together, while comparisons are made to the 1,4-isomers due to ease of synthesis via CuAAC.20,21 The outcome of the in situ triazole templation at a flexible-binding site expands the templation potential of in situ click chemistry to receptor relevant targets and to intersubunit binding sites.
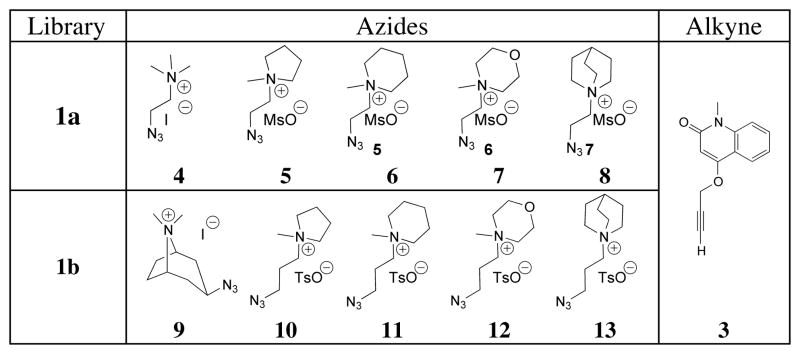
Triazole 2, the in situ formed ligand with the dissociation constant in the nanomolar range for Ls, was next used as a lead for the discovery of analogs with improved affinity and selectivity for the closely related AChBPs. Selective target-catalyzed synthesis of a new ligand from a library of building blocks simultaneously present in a single reaction mixture would be a significant advantage in drug discovery, enabling rapid screening of many building block pools or combinations and reducing the amount of protein required for each individual analysis. To this end, azide building blocks containing a variety of quaternary nitrogen centers were synthesized (Table 2). This library was designed to systematically probe the effect of the quaternary amine using ring systems of increasing complexity, 4, 5–8 (Library 1a, Table 2) and 9 (Library 1b, Table 2), while also extending the linker between the azide and the amine for each iterative compound, 10–13 (Library 1b, Table 2).
Table 2.
Azide libraries 1a and 1b: in situ click chemistry screen against alkyne 3.
|
The in situ reactions for library 1a and 1b were allowed to proceed at room temperature for 3 days. LC-MS analysis, performed in triplicate, of the reaction mixtures revealed several interesting phenomena (Tables 3a, b). As demonstrated above, Ls catalyzed the formation of the triazole products more efficiently than Ac and AcY55W AChBPs, while no products were detected in the BSA control reaction. However, in contrast to the binary mixture, there was no pronounced increase in the formation of triazole 2 when library 1a was used. This finding can be explained by the preferential binding of azides 6 and 8 to the template and favoring the formation of products 15 and 14, respectively.42 These compounds were synthesized via CuAAC, and their Kd values determined. Comparison of the amount of product formed in situ with the affinity of these compounds (1/Kd, μM−1) revealed a clear trend (Table 3a) which suggests that the amount of product formed is related to its affinity for the specific AChBP, relative to the other members of the library. A similar correlation was observed when azide library 1b (9–13) was screened against alkyne 3 (Table 3b). Under those conditions, triazole 18 was formed in the greatest amount by Ls and was also shown to have the highest affinity, Kd = 0.96 ± 0.22 nM, consistent with the observations from library 1a. In contrast, pyrrolidinium derivative 19, which had formed in lower amounts had a significantly lower affinity, Kd = 82 ± 14 nM. In contrast, the amount of triazoles 16, 17 and 20–22 were comparatively low and therefore they were not synthesized. The capability to identify only combinations that warrant larger scale synthesis and further investigation demonstrates another advantage of the in situ click chemistry approach.
Table 3a.
Templation and binding data for the triazole derivatives of the in situ click chemistry screen of azide building blocks (library 1a).
| AChBP species | MS counts[1] | Kd [nM] | Affinity [μM−1] | Structure[3] | # |
|---|---|---|---|---|---|
| Ls | 3.8 ± 0.1 | 75 ± 26 | 13 |
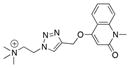
|
2 |
| Ac | 0.0 | 1900 ± 800 | 0.53 | ||
| AcY55W | 0.0 | 370 ± 50 | 2.7 | ||
|
| |||||
| Ls | 100 ± 2.4 | 9 ± 0.4 | 110 |
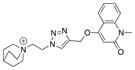
|
14 |
| Ac | 8.0 ± 0.1 | 120 ± 25 | 8.3 | ||
| AcY55W | 15.9 ± 0.1 | 88 ± 4.2 | 11 | ||
|
| |||||
| Ls | 97.3 ± 3.3 | 10 ± 1.7 | 100 |
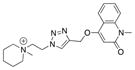
|
15 |
| Ac | 10.1 ± 0.0 | 210 ± 46 | 4.8 | ||
| AcY55W | 12.8 ± 0.2 | 50 ± 4.7 | 20 | ||
|
| |||||
| Ls | 26.7 ± 0.6 | n.d.[2] | n.d. |
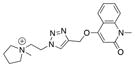
|
16 |
| Ac | 5.6 ± 0.1 | n.d. | n.d. | ||
| AcY55W | 3.9 ±1.7 | n.d. | n.d. | ||
|
| |||||
| Ls | 0.0 | n.d. | n.d. |
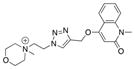
|
17 |
| Ac | 0.0 | n.d. | n.d. | ||
| AcY55W | 0.0 | n.d. | n.d. | ||
MS counts were corrected for background via BSA controls and normalized.
n.d. represents no data as compound was not synthesized to generate affinity values.
Cation detected by MS.
Table 3b.
Templation and binding data for the triazole derivatives of the in situ click chemistry screen of azide building blocks (library 1b).
| AChBP species | MS counts[1] | Kd [nM] | Affinity [μM−1] | Structure[3] | # |
|---|---|---|---|---|---|
| Ls | 100.0 ± 0.5 | 0.96 ± 0.22 | 1040 |
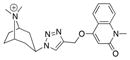
|
18 |
| Ac | 14.3 ± 0.1 | 24 ± 6.8 | 42 | ||
| AcY55W | 13.3 ± 0.3 | 17 ± 4.8 | 59 | ||
|
| |||||
| Ls | 40.0 ± 1.5 | 82 ± 14 | 12 |
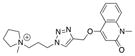
|
19 |
| Ac | 13.3 ± 0.5 | 470 ± 65 | 2.1 | ||
| AcY55W | 12.3 ± 0.3 | 280 ± 21 | 3.6 | ||
|
| |||||
| Ls | 29.2 ± 1.1 | n.d.[2] | n.d. |
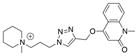
|
20 |
| Ac | 14.1 ± 0.3 | n.d. | n.d. | ||
| AcY55W | 14.8 ± 0.3 | n.d. | n.d. | ||
|
| |||||
| Ls | 0.0 | n.d. | n.d. |
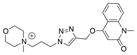
|
21 |
| Ac | 0.0 | n.d. | n.d. | ||
| AcY55W | 0.0 | n.d. | n.d. | ||
|
| |||||
| Ls | 28.4 ± 0.7 | n.d. | n.d. |
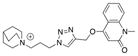
|
22 |
| Ac | 8.0 ± 0.2 | n.d. | n.d. | ||
| AcY55W | 12.1 ± 0.3 | n.d. | n.d. | ||
MS counts were corrected for background via BSA controls and normalized.
n.d. represents no data as compound was not synthesized to generate affinity values.
Cation detected by MS.
Having identified tropane derivative 18 as the compound of highest affinity, a set of alkynes was designed and reacted with azide 9. This library, comprising the previously tested quinolinone derivative 3 and variously substituted aryl propargyl ethers 23–26 (Table 4), was incubated with Ls, Ac and AcY55W AChBPs. LC-MS analysis, performed in triplicate, of the reaction mixtures showed that all tested alkynes underwent AChBP-templated cycloaddition reactions with azide 9. As before, the reactions against the BSA control produced no detectable products. Triazoles 27, 28 and 29 were synthesized using standard CuAAC conditions, and their Kd values determined (Table 5). Again it is worth noting that compounds 27–29 shared both comparable amounts of product formation and similar magnitude of affinities. Only triazole 30 was not synthesized in the presence of Ac or AcY55W AChBPs, and only small amounts were detected in the reaction catalyzed by Ls. The previously discovered triazole 18, originating from alkyne 3 and azide 9, formed in significantly higher quantity and, as expected, exhibited a substantially higher affinity for all of the binding proteins (Table 5).
Table 4.
Alkyne library 2 utilized for in situ screen against azide 9.
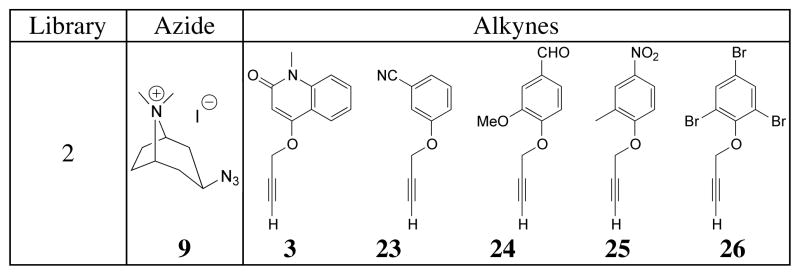
|
Table 5.
Templation and binding data for the triazole derivatives of the in situ screen of alkyne building blocks (library 2).
| AChBP species | MS countsa | Kd [nM] | Affinity [μM−1] | Structurec | # |
|---|---|---|---|---|---|
| Ls | 100.0 ± 2.2 | 0.96 ± 0.22 | 1040 |
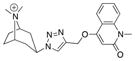
|
18 |
| Ac | 11.4 ± 0.4 | 24 ± 6.8 | 42 | ||
| AcY55W | 10.3 ± 0.3 | 17 ± 4.8 | 59 | ||
|
| |||||
| Ls | 17.0 ± 0.2 | 42 ± 19 | 24 |
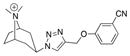
|
27 |
| Ac | 6.7 ± 0.3 | 220 ± 87 | 4.5 | ||
| AcY55W | 6.2 ± 0.1 | 130 ± 35 | 7.7 | ||
|
| |||||
| Ls | 20.3 ± 0.4 | 64 ± 26 | 16 |
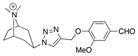
|
28 |
| Ac | 7.7 ± 0.5 | 260 ± 80 | 3.8 | ||
| AcY55W | 11.3 ± 0.1 | 130 ± 44 | 7.7 | ||
|
| |||||
| Ls | 10.6 ± 0.6 | 13 ± 6.5 | 77 |
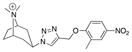
|
29 |
| Ac | 6.1 ± 0.4 | 61 ± 27 | 16 | ||
| AcY55W | 4.2 ± 0.1 | 30 ± 18 | 33 | ||
|
| |||||
| Ls | 4.3 ± 0.1 | n.d.b | n.d. |
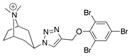
|
30 |
| Ac | 0.0 | n.d. | n.d. | ||
| AcY55W | 0.0 | n.d. | n.d. | ||
MS counts were corrected for background via BSA controls and normalized;
n.d. represents no data as compound was not synthesized to generate affinity values;
cation detected by MS.
Having demonstrated that AChBPs selectively catalyze the formation of high affinity ligands from libraries of alkynes and azides, we next examined whether the template could select the most productive combinations (i.e. those resulting in the most potent ligands) from a pool of various azides and alkynes simultaneously present in the reaction mixture. To this end, azides 4 and 5–13 were reacted with alkynes 3 and 23–26 in a combined mixture using Ls as the template (see Experimental Methods for the details). Ls was chosen because of the higher yields obtained with the previous reactions shown in Tables 3a, 3b, and and 5. To compensate for the reduced concentrations of the individual building blocks, the incubation time was increased to 10 days. The results showed that several triazole products could be detected (Figure 2) in comparison to the BSA control reaction, which exhibited no product formation.
Figure 2.

Templation data for the triazole derivatives of the in situ screen of azides versus alkynes building blocks on Ls AChBP (data have been normalized to the largest peak).
The majority of the products formed in the presence of the Ls template had been previously identified in simpler in situ reaction pools. Triazole 18, the product of the cycloaddition between alkyne 3 and azide 9, was again formed in the greatest amounts, which is consistent with the affinity of this triazole for Ls. Compound 14, with the second highest affinity discovered in this study, formed in the second largest amount. Similar trends were also observed between the affinity of previously examined triazole products and the amount formed, despite the increased complexity of the system. Interestingly, the combination of quinuclidinium azide 8 and alkynes 25 and 26, and the cycloaddition products of piperidinium azide 6 with alkynes 23, 24, and 25 were identified by LC-MS analysis. These products were the result of combinations that had not previously been screened. Although it is of note that these compounds formed in situ, the relatively low amounts formed suggests they have lower affinities and were therefore accorded lower priority.
We have therefore demonstrated that it is possible to screen libraries of azides and alkynes simultaneously and detect the formation of triazole products, while generating valuable information regarding the affinity of the compounds for the target protein. The increased number of building blocks screened in a single reaction further increases the throughput of the in situ click chemistry approach. Moreover, the observed positive correlation between the amount of product formed and its affinity for the target could be useful for subsequent structure-activity studies, whether through traditional medicinal chemistry methods or further iterations of in situ target-templated screening.
Having demonstrated that triazole 18 exhibited the highest affinity of all the compounds detected throughout this study, we determined binding parameters of its 1,5-isomer, 18a, which was prepared using RuAAC as shown in Scheme 2. Although the Kd of 18a for Ls was only slightly higher than of 18, its binding to Ac and AcY55W was significantly weaker, and this and other 1,5-disubstituted triazoles were not examined further in the present study.
Scheme 2.

RuAAC synthesis of the 1,5-triazole isomer 18a.
To define the binding site determinants governing the reaction, we were interested in deciphering the binding pose of triazole 18, the most potent ligand found in this study. Since our conditions of crystallization typically yield higher resolution structures with Ac-AChBP, we concentrated on refining the structure of this complex, yielding a structure with a resolution of 2.3Å. The 3σ omit map of the best represented ligand is shown in Figure 3A.
Figure 3.

Crystallographic analysis of triazole 18 in complex with Ac AChBP. Complementary subunit is represented with green labels, while the primary subunit is denoted with black labels. (A) Surface representation of ligand interpolated charges and F0-FC omit map at 3σ. (B) Triazole 18 in principal subunit binding pocket highlights the interactions of the dimethyl-tropane aza-nitrogen with surrounding aromatic residues Trp147, Tyr93, Tyr188 in the principal subunit viewed radially at the subunit interface. The triazole nitrogen’s (N-3) interaction with a structural water is also shown. Residues shown are within 4Å. (C) Connolly surface representation of quaternary tropane moiety as viewed from the membrane side of the binding site. Stabilization is achieved by an aromatic nest of residues of the principal subunit noted in Panel B and Tyr55 from the complementary subunit. (D) Surface representation of quinolinone, triazole and tropane rings (dark grey sticks, white label) showing the participation of Arg79, Met116, Ile118 and Tyr55 of the complementary subunit in addition to those noted in the principal subunit.
We found the density for the quinolinone to be much weaker than that of the triazole and tropane ring systems in most of the binding sites. The faint density suggests a flexibility of the quinolinone exists which is most likely due to the rotatable methylene carbon bond that links the quinolinone to the triazole. Indeed, the binding mode shows that the quaternary bridged-nitrogen forms a cation-π interaction with the Trp147, Tyr188, and Tyr93 side chains, a previously noted observation for quaternary amines that bind nAChRs.38 Figure 3B and Supplementary Figure 1C show distances from the quaternary tropane nitrogen and the closest atoms in the aromatic side chain of Trp147, Try93 and Tyr188 to be between 4.3–5.2Å, as has been noted to be optimal for cation-π interactions.43 The tight fit of the ligand is achieved by the aromatic side chains, including Y195 from the principal subunit and Y55 from the complementary subunit, which surround the quaternary nitrogen and provide an anchor for the azide building block 9 (Figure 3C). The propargyloxy quinolinone ring system stacks between the vicinal disulfide bridge between Cys190 and Cys191 of the β9–10 linker (Loop-C) and Met116 (Figure 3B and D, Supplementary Figure 1B). The triazole is positioned such that the N-3 hydrogen bonds with a neighboring water molecule, similarly to that reported for the pyridine ring systems (Figure 3B, Supplementary Figure 1A).37,39,44 This water molecule forms a hydrogen bond linkage with other water molecules extending toward the complementary subunit and the vestibule of the nAChR channel. The ether oxygen also forms a hydrogen bonding system with that of Arg79 and Tyr195. The hydroxyl of Tyr195 approaches basally, while the protonated nitrogen of Arg79 reaches down from the apical region. The carbonyl of Val148, binding to the complementary hydrogen of Arg79’s protonated nitrogen, and a structural water associated between Tyr195 and Glu193 completes the hydrogen bonding system (Supplemental Figure 1A).
4. DISCUSSION
In situ click chemistry has previously been employed in the development of high affinity ligands for the active sites of acetylcholinesterase, HIV protease, and carbonic anhydrase.18,41,45,46 These ligands bind within a gorge centro-symmetric to a subunit in the oligomeric AChE, the shallow binding site in the HIV protease, or the single subunit in monomeric carbonic anhydrase. The AChBPs, with their binding sites at the interface of two subunits, provide a distinct template for in situ click chemistry, yet the bioorthogonal reaction forming the heterocyclic triazole can take place in the presumed more flexible and exposed subunit interface.
In this study we also demonstrate that the efficiency of the target-catalyzed synthesis of the ligands is partially determined by the affinity of the reaction product, i.e. the amount of the formed triazole increases with the strength of its binding to the target. This was also the case when we compared the preference of anti- and syn-regioisomer formation on acetyl-cholinesterase with the equilibrium affinities of the respective triazoles.47 This correlation between preference of formation and equilibrium affinity may suggest that the transition state more closely resembles the reaction product than the simple bimolecular associations of the building blocks with the template. With the acetylcholinesterase complexes, we also demonstrated that the higher affinity syn- triazole regioisomer induced a change in conformation enhancing aromatic π-π association at the peripheral site, whereas a change in conformation did not occur with the lower affinity anti-isomer.19 Accordingly, one of the tenets of selectivity for the in situ click chemistry reaction may arise from the precursors driving a conformation preferred by the triazole reaction product rather than accommodating a conformation of the unbound protein. This bodes well for the ability of the technique to select conformations of the ligand-protein complex that differ from the unbound protein.
Further support for the concept of the triazole product adopting a binding pose and selecting a conformation most compatible with the product association comes from examination of the crystal structure of triazole 18. Here we observe a bound conformation and pose that has been predicted from the conformation of other quaternary amine containing ligands that bind to AChBP through cation-quadrupole interactions involving the electron-rich aromatic side chains (e.g. tryptophan). The positioning of the triazole such that it may hydrogen bond within the pocket supports the notion that precursors drive a conformation change needed for the reaction between them. The in situ reaction may be influenced by the binding strength of the component containing the tertiary or quaternary nitrogen (in our case the quaternary tropane), and the ability of the complementary component to form the triazole in the proper orientation allowing for the formation of a highly selective molecule. Comparison of precursor component conformations with those of the final triazole compound will be of interest in the future. However, accomplishing this task may be difficult as the precursor affinities are typically low, so their crystal structures would not be well resolved in the pocket of the AChBP.
5. CONCLUSIONS
We have successfully used a combination of CuAAC and in situ click chemistry to rapidly convert the endogenous ligand of nAChRs, acetylcholine, into a highly potent and selective ligand, 18, for Ls AChBP. This work represents the first example of in situ click chemistry performed at a flexible subunit interface of an oligomeric protein, and thus increases the number of biologically relevant targets that can be investigated using this methodology. In addition, we have demonstrated a positive correlation between the amount of product generated in situ and its affinity for the target binding protein. Therefore, this approach enables rapid refinement of structures, while providing valuable information about binding and selectivity over closely related targets without the need to synthesize and assay individual compounds. Furthermore, we have shown that building block sets can be screened in a single reaction pool with 10 azides and 5 alkynes allowing for the possible generation and detection of the highest affinity compounds from several discrete sets of congeneric building blocks. The variety of possible combinations enhances the efficiency of the technique and reduces the amount of protein required for the experiments. Finally, we have co-crystallized the most active compound that emerged from these studies, 18, with Ac-AChBP and shown that our initial hypothesis about specific interactions was indeed valid. We are currently continuing to expand the utility of the combination of these methodologies to further refine our lead molecules for both affinity and selectivity towards Ls AChBP, and subsequently investigating whether these properties can translate the ligand’s affinity to human nAChRs. Historically, the AChBPs have provided significant insights into the determinants of ligand recognition by nAChRs. In particular, crystal structures of Ls and Ac ligand complexes have elucidated and confirmed residues involved in the ligand binding domain as well as given rise to information regarding the global extracellular domain. It has also been possible to modify the side chains of AChBP so that its subunit interface more closely resembles that of the homomeric α7 nAChR.48 This target-templated ligand identification approach, coupled with a multiarray synthesis of triazoles from azide and alkyne building blocks, offers an efficient means of identifying and then refining candidate ligands specifically engineered towards the nAChRs and other flexible targets.
Supplementary Material
Acknowledgments
Funding Sources
This study was supported by USPHS grants UO1 DA19320-05 and R01 GM18360-37. B.S. acknowledges a fellowship from the Swiss National Science Foundation (SNF). JGY and AN were supported by NIH training grant GM-07752. JGY was also supported from NSF GK-12 0742551. C.K. is supported by NIH grant R01 GM090161-01 and R. A. Welch Foundation Chemistry and Biology Collaborative Grant. V.V.F. acknowledges support by NIH (NIGMS) grant R01 GM087620. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Supporting Information. Synthetic methods and characterization of new compounds and additional data on the crystallographic analysis of the Ac AChBP co-crystallized with triazole 18. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Karlin A. Nat Rev Neurosc. 2002;3:102. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP, Taly A. Trends Mol Med. 2008;14:93. doi: 10.1016/j.molmed.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Thompson AJ, Lester HA, Lummis SC. Q Rev Biophys. 2010;43:449. doi: 10.1017/S0033583510000168. [DOI] [PubMed] [Google Scholar]
- 4.Changeux JP, Edelstein SJ. Nicotinic Acetylcholine Receptors: From Molecular Biology to Cognition. Odile Jacob/Johns Hopkins University Press; New York: 2005. [Google Scholar]
- 5.Changeux JP. Nat Rev Neurosc. 2010;11:389. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 6.Vernallis AB, Conroy WG, Berg DK. Neuron. 1993;10:451. doi: 10.1016/0896-6273(93)90333-m. [DOI] [PubMed] [Google Scholar]
- 7.Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP. Nature. 2001;411:261. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- 8.Hansen SB, Talley TT, Radic Z, Taylor P. J Biol Chem. 2004;279:24197. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celie PH, Klaassen RV, van Rossum-Fikkert SE, van Elk R, van Nierop P, Smit AB, Sixma TK. J Biol Chem. 2005;280:26457. doi: 10.1074/jbc.M414476200. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Wang H, Cui Y, Zhang G, Zheng G, Liu S, Xie L, Zhang R. Mar Biotech. 2009;11:596. doi: 10.1007/s10126-008-9176-8. [DOI] [PubMed] [Google Scholar]
- 11.McCormack T, Petrovich RM, Mercier KA, DeRose EF, Cuneo MJ, Williams J, Johnson KL, Lamb PW, London RE, Yakel JL. Biochemistry. 2010;49:2279. doi: 10.1021/bi902023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Nature. 2001;411:269. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 13.Furlan RLE, Otto S, Sanders JKM. Proc Natl Acad Sci USA. 2002;99:4801. doi: 10.1073/pnas.022643699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rideout D. Cancer Inv. 1994;12:189. doi: 10.3109/07357909409024874. [DOI] [PubMed] [Google Scholar]
- 15.Rideout D, Calogeropoulou T, Jaworski J, McCarthy M. Biopolymers. 1990;29:247. doi: 10.1002/bip.360290129. [DOI] [PubMed] [Google Scholar]
- 16.Eliseev AV. Pharmaceutical News. 2002;9:207. [Google Scholar]
- 17.Tomé AC. In: Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. Storr R, Gilchrist T, editors. Vol. 13. Thieme; New York: 2003. p. 544. [Google Scholar]
- 18.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew Chem Int Ed. 2002;41:1053. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Bourne Y, Kolb HC, Radic Z, Sharpless KB, Taylor P, Marchot P. Proc Natl Acad Sci U S A. 2004;101:1449. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 21.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 22.Majumdar KC, Ghosh M, Jana M. Synthesis. 2002:669. [Google Scholar]
- 23.Zhang L, Chen X, Xue P, Sun HHY, Williams ID, Sharpless KB, Fokin VV, Jia G. J Am Chem Soc. 2005;127:15998. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]
- 24.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J Am Chem Soc. 2008;130:8923. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 25.Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. EMBO J. 2005;24:3635. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talley TT, Yalda S, Ho KY, Tor Y, Soti FS, Kem WR, Taylor P. Biochemistry. 2006;45:8894. doi: 10.1021/bi060534y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Proc Natl Acad Sci U S A. 2002;99:13419. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Methods Enzymol. Vol. 276. Academic Press; 1997. p. 307. [DOI] [PubMed] [Google Scholar]
- 30.Storoni LC, McCoy AJ, Read RJ. Acta Crustallogr, Sect D: Biol Crystallogr. 2004;60:432. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Acta Crustallogr, Sect D: Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Acta Crustallogr, Sect D: Biol Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 34.Horne WS, Stout CD, Ghadiri MR. J Am Chem Soc. 2003;125:9372. doi: 10.1021/ja034358h. [DOI] [PubMed] [Google Scholar]
- 35.Van Maarseveen JH, Horne WS, Ghadiri MR. Org Lett. 2005;7:4503. doi: 10.1021/ol0518028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Angell YL, Burgess K. Chem Soc Rev. 2007;36:1674. doi: 10.1039/b701444a. [DOI] [PubMed] [Google Scholar]
- 37.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Neuron. 2004;41:907. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 38.Xiu X, Puskar NL, Shanata JA, Lester HA, Dougherty DA. Nature. 2009;458:534. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hibbs RE, Sulzenbacher G, Shi J, Talley TT, Conrod S, Kem WR, Taylor P, Marchot P, Bourne Y. EMBO J. 2009;28:3040. doi: 10.1038/emboj.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourne Y, Radic Z, Araoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgo J, Marchot P. Proc Natl Acad Sci U S A. 2010;107:6076. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mamidyala SK, Finn MG. Chem Soc Rev. 2010;39:1252. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 42.While mass spectrometry can not give a quantitative measurement of the amount of product formed, our investigations demonstrate that qualitative relationships can be ascertained (see Supporting Information for details).
- 43.Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, Benz J, Stihle M, Diederich F. Angew Chem Int Ed. 2005;44:4400. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- 44.Talley TT, Harel M, Hibbs RE, Radic Z, Tomizawa M, Casida JE, Taylor P. Proc Natl Acad Sci U S A. 2008;105:7606. doi: 10.1073/pnas.0802197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mocharla VP, Colasson B, Lee LV, Roeper S, Sharpless KB, Wong CH, Kolb HC. Angew Chem Int Ed. 2005;44:116. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 46.Whiting M, Muldoon J, Lin YC, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Angew Chem Int Ed. 2006;45:1435. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 47.Bourne Y, Radic Z, Taylor P, Marchot P. J Am Chem Soc. 2010;132:18292. doi: 10.1021/ja106820e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nemecz A, Taylor PW. J Bio Chem. 2011;286:42555. doi: 10.1074/jbc.M111.286583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.