

Abstract
Fluids of charged particles act as the supporting medium for chemical reactions and physical, dynamical, and biological processes. The local structure in an electrolytic background is deformed by micro- and nanoscopic polarizable objects. Vice versa, the forces between the objects are regulated by the cohesive properties of the background. We study here the range and strength of these forces and the microscopic origin from which they emerge. We find the forces to be sensitively dependent on the material properties of the charged fluid and the immersed solutes. The induced interactions can be varied over decades, offering high tunability and aided by accurate theory, control in experiments and applications. To distinguish correlational effects from simple ionic screening, we describe electrolyte-induced forces between neutral objects. The interplay of thermal motion, short-range repulsions, and electrostatic forces is responsible for a soft structure in the fluid. This structure changes near polarizable interfaces and causes diverse attractions between confining walls that seem well-exploited by microbiological systems. For parameters that correspond to monovalent electrolytes in biologically and technologically relevant aqueous environments, we find induced forces between nanoscopic areas of the order of piconewtons over a few nanometers.
Keywords: molecular electronics, surface structure, statistical thermodynamics, multicomponent plasmas
The region where different phases meet at an interface hosts the regulatory processes that determine the face of the Earth from a global scale down to the microscopic-length scales. These processes include the chemical reactions in the atmosphere and the Earth’s crust at the water–air and water–solid interfaces (1) as well as the highly specific processes in biology that are coordinated by membranes and complex macromolecular agents (2). Interfaces can localize reactive components, mediate interactions, and allow components in different phases to meet in a selective way, offering tunable reaction rates over a vast range of magnitudes. The catalytic qualities of interfaces are broadly exploited in industrial processing that involves an ample variety of applications, such as metal extraction, water purification, phase transfer catalysis, and reprocessing of nuclear waste. In addition to the chemical aspects, interfaces also have distinct physical properties, which find their applications in transistors and supercapacitors as well as emulsion stabilization.
In this paper, we highlight the physical properties of the liquid–solid interface, with a strong focus on fluids of charged particles that are confined by two dielectric boundaries. In the theoretical analysis, the charged particles are characterized by the charge q± and the maximum coupling strength set by an effective hard-sphere radius a± and the Coulomb potential at contact. The solvent is considered implicitly by a constant permittivity ɛs relative to the vacuum permittivity. By adopting these simplifications that constitute the so-called Primitive Model, we can connect to a tradition of research and expand on familiar knowledge. Moreover, the Primitive Model retains a reasonable transparency and still yields a complexity in phase behavior that matches the diversity of incompletely understood and sometimes counterintuitive experimental observations of like-charge attractions (3–8), collapse and reexpansion of polyelectrolyte chains and gels as the salt concentration increases (9–12), and unexpectedly long-ranged interactions (13–15) and charge inversion (16, 17). We omit a detailed discussion of solvent-specific effects such as hydration, effect of dissolved gases, or contaminants that are common in real solutions (18) in favor of the elucidation of the effects by electrostatic correlations. The subtle interplay between short-range repulsive and long-range attractive forces at a moderate thermal energy is responsible for average (effective) forces between the constituents that can strongly vary in range, magnitude, sign, and shape (from oscillatory to monotonically decaying), especially in multicomponent systems. An accurate thermodynamic average should be taken to recognize these correlational effects, which we do by solving the anisotropic hyper-netted chain (AHNC) (19, 20), comparing with the well-known results from mean-field theories as a valuable reference frame. To isolate correlational effects from the effects that can be approximated by mean-field theories and their corrections, we first discuss charged fluids near interfaces that are neutral and show typical cohesive effects in symmetric electrolytes that cause depletion attractions between parallel walls or accumulation repulsions, depending on the polarizability of the walls and the solvent. Subsequently, we show how charged interfaces reduce the cohesion of the confined ion cloud and induce charge ordering, even in solutions where the bulk can be described as weakly coupled. By calculating the anisotropic pair-correlation functions, we outline the deformation of the local structure in the fluid near the boundaries. The typical soft structure of the fluid is modified by the boundaries and causes effective interactions that cannot be found by mean-field or strong-coupling theories but can be measurably strong, and sufficient to lead to unexpected forces in experiments. We found a remarkably complex behavior for monovalent electrolytes in aqueous solutions at ∼0.1 M concentrations that corresponds to the intracellular environment, where the short-range repulsions and long-range Coulomb forces between the particles induce mean forces near the boundaries that are comparable in magnitude and range but oppositely directed.
The soft structure of charged fluids and its relation to the effective forces between nano- and mesoscopic constituents are poorly understood, despite a long history of research driven by its ubiquitous manifestation in scientific experiments and industrial applications. The limitations are caused by theoretical and computational challenges. Recently, however, methods have become available thanks to a combination of classic liquid-state theory and modern computational power that yields accurate results on practical timescales. We develop here a description of the soft structure between polarizable walls derived from a single theoretical framework, with minimal assumptions, matching the reliability of a Monte Carlo simulation (21–23), and agreeing with experimental observations (24). This description provides a practical understanding of electrostatic correlations in charged fluids and predictability in experiments.
Polarization Depletion
Before embarking on the statistical thermodynamics of a confined charged fluid, we first calculate the electric field generated by two static charges between two parallel polarizable boundaries by means of the method of images. To facilitate the solution of the Maxwell equations, one can replace the boundary conditions by alternative ones that have an identical solution in the region of interest. A flat dielectric discontinuity next to a set of charges can be replaced by a set of so-called image charges with renormalized charge q′ = A12q, depending on the difference in the dielectric constant between medium 1 and 2,
to yield the same boundary conditions and solution for the electric field in the volume around the charges bounded by the discontinuity. An interesting variation is that of two parallel walls, where one would need an infinite set of image charges, because each boundary reflects the images of the other normalized by the factors A12 and A13 with every reflection, with ɛ1 as the relative permittivity of the confined region and ɛ2 and ɛ3 as the relative dielectric constants beyond the boundaries. Fig. 1 shows an example of two oppositely charged spheres between two regions of low permittivity and an overview of the parameterization. The particle’s colors symbolize the sign of their charge. The colors of the images form a pattern depending on the type of the boundary.
Fig. 1.

Visualization of the method of images applied to a system of two charges that are confined between two parallel dielectric boundaries. Two conducting boundaries or boundaries with stronger dielectric media invert the charge with every reflection (SI Text), whereas two boundaries with weaker dielectric media only reduce the charge of the particle (situation above). The total electrostatic potential is obtained by summing the interactions between all of the images, which can be done conveniently in Fourier space. The choice of parameters is shown in Right. Cylindrical coordinates are used, and because of the translational symmetry parallel to the boundaries, there is no angular dependency.
The boundaries with two weaker dielectric media act as (attenuating) mirrors, where the images have the same type of charge as the origin, whereas conducting boundaries reverse the charge with every reflection, which results in an alternating pattern of positive and negative images. Two different boundaries, one with a negative and one with a positive dielectric jump, would give rise to a pattern where two consecutive images are followed by two consecutive images of the opposite charge (shown in SI Text). The total interaction between the spheres involves an infinite summation of image contributions that can be solved conveniently in Fourier space, which is described in SI Text. The self-interaction of the charges (i.e., the interaction of the sphere with its own images) has a lower value between conducting boundaries than between two weak dielectric media, which has significant consequences for the thermodynamic behavior of confined charged spheres. The self-interaction us = use + uso contains, in both cases, a positive term contributed by the even number of reflections:
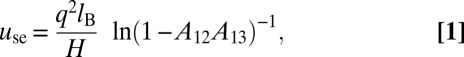 |
with lB = e2/4πɛ1ɛ0kBT being the Bjerrum length, ɛ0 being the vacuum permittivity, kBT being the thermal energy (the choice of the dimension is for later convenience), and H being the separation between the boundaries. The second contribution, by the odd number of reflections, depends on the position z between the boundaries (located at z = 0 and z = H) and the type of the boundary:
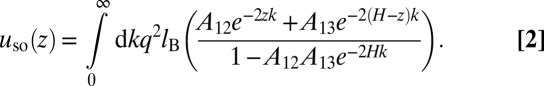 |
For metallic boundaries, the contribution is negative, but for boundaries with weaker dielectrics, it is positive.
In summary, the self-energy of charged particles depends sensitively on the type of the boundaries and the distance between them. One could conclude from the above analysis that charged particles in a fluid would dissolve better between boundaries with a higher permittivity than between boundaries with a lower permittivity. The adsorption behavior may be more nuanced, however, because of electrostatic screening and other thermal effects. An additional statistical framework is needed to grasp those effects.
Thermal Forces and Soft Structure
The interaction between two boundaries and a confined fluid of charged hard spheres depends not only on the polarizability of the boundaries but also on the thermodynamic properties of the fluid, such as the temperature, density of particles, charge, and effective size. At low temperatures, one can ignore the thermal aspects of the fluid and minimize the energy of the ensemble that includes the interparticle interactions and the interactions with the boundaries, treating the ensemble as a solid. At high temperatures, one can estimate the mean forces over a finite timescale, beyond which the system is ergodic, by making use of so-called high-temperature expansions or mean-field approaches that basically overestimate the entropy (take an incomplete thermal average), treating the ensemble as if it were an (almost) ideal gas. Examples of systems can be given where a combination of both types of approaches yields useful insights, for instance in the case of a weakly coupled ionic cloud near a highly charged wall (25–27). Many examples remain, however, where these approaches, patched together or not, do not suffice to distinguish a large variety of thermal effects that is relevant in the liquid-like state of an electrolyte, which is, by far, the most common state in living matter, the oceans, and numerous solutions used by industry. These effects may support functionality in biological processes and provide to nature the potential to tune and invert forces between solutes simply by regulating the background electrolyte. More concretely, even in the Primitive Model, where the complexity is minimized by a small set of parameters, the phase behavior and mean (induced) forces are incompletely understood. Simulation methods have the potential to calculate accurate thermal averages but deal with other challenges, such as an unmanageable number of particles that one has to consider or extreme timescales that the ensemble requires to equilibrate, a characteristic of ionic solutions often reported in experiments. Here, we use a theoretical method based on liquid-state theory that has been developed by Kjellander and Marčelja (19, 20) to focus on the relevant systems that are characterized by a liquid-like or soft structure, where extreme-temperature approaches underestimate the phenomenology and simulation methods are cumbersome. We do ignore the structure of the background medium, assuming a homogeneous constant permittivity (only with a jump at the boundaries) and adopting effective sizes for the ions to account for hydration. One could categorize hydration, the cage of solvent molecules around solutes, as a hard structure that is not easily deformed compared with the soft structure that is considered in this manuscript, although we acknowledge that here, also, nature may have examples where solvent–ion interactions contribute to subtle thermal forces. Just as mean-field theories contributed to the understanding of screened Coulomb interactions (double-layer forces) (28), we hope to expand the understanding by highlighting additional effects predicted by liquid-state theory that are because of the soft structure (electrostatic and hard-core correlations) in confined electrolytes. We find that the local structure, although soft, may lend strong induced interactions between the confining boundaries, even in monovalent aqueous electrolyte solutions and especially at concentrations that correspond to the intracellular environment.
Liquid-State Theory and the AHNC
We use the so-called AHNC approximation for the Ornstein–Zernike equation (29) to calculate the local structure in the confined electrolyte and relate the information to the induced mean forces between the confining boundaries and the thermodynamic properties of the entire system to shed more light on thermal forces between nanoparticles, membranes, and salt-specific effects in colloids. The method is carefully and completely described in the original works by Kjellander and Marčelja (19, 20), and it is shortly outlined below. Instead of introducing an approximation for the direct evaluation of the partition function, we turn to an exact relation between two pair-correlation functions that are defined as the second functional derivative of the partition function with respect to the mean local density ρ(x) [i.e., the direct correlation function c(x, x′)] and with respect to the local fugacity z(x) [that is, the total correlation function h(x, x′) (30)], known as the Ornstein–Zernike equation:
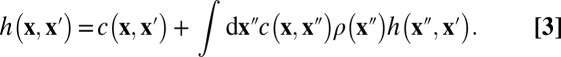 |
A second exact equation can be found by comparing the diagrammatic expansions of c and h in terms of Mayer cluster diagrams (31):
with u as the pair potential between the particles and d as the so-called bridge function (inspired by the topology of its cluster diagrams). Ignoring the bridge function d corresponds to the so-called HNC approximation. The integral equation (Eq. 3) can be solved iteratively with the HNC closure if the mean density is constant in space (i.e., in the absence of external fields), which is done most conveniently with the aid of Fourier transformations. For systems with long-range interactions, such as Coulomb forces, the bridge function seems negligible compared with the other terms over a large-parameter range, such that the HNC yields very accurate pair-correlation functions that are often indistinguishable from the results of Monte Carlo simulations (32, 33). In the presence of external fields or symmetry-breaking boundaries, a third relation is required for the mean local density to close the set of equations:
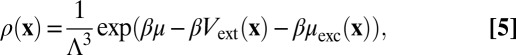 |
where Λ is the de Broglie wavelength, and μexc is the local excess chemical potential, which can be formulated in terms of the above pair-correlation functions within the HNC (34). A direct evaluation by discretizing space with N grid points per dimension would involve  (N9) calculation steps (the correlation functions require six dimensions, and the convolution in Eq. 3 integrates over three dimensions), which easily exceeds any practical timescale, regardless of the current computational possibilities. The dimension of the correlation functions can be reduced, however, if symmetries are present. In the case of two planar, parallel boundaries, the correlation functions can be written as a function of a coordinate r parallel to the boundaries and two coordinates z and z′ along an axis perpendicular to the boundaries thanks to translational symmetry in the directions parallel to the boundaries. If the z axis is discretized, one can simplify the analysis without additional approximations by a mathematical mapping (19, 20), which is illustrated in SI Text. One can map an inhomogeneous 3D system with n components to a 2D system with Mn components that is homogeneous, with M being the number of grid points used for the z axis, by labeling the particles in each layer as a distinct species with a distinct pair-potential uij(r) between the particles with coordinates zi and zj. The Ornstein–Zernike equation (Eq. 3) reduces to a matrix equation, which can be solved with an appropriate closure relation together with the Boltzmann distribution (Eq. 5), to yield information about the local structure as well as the thermodynamic properties on a timescale of minutes on a single central processing unit (if the grid points are chosen cautiously and judiciously). We fix the effective ion radius to a± = 0.3 nm for both species, consider monovalent ions in an aqueous background with a relative permittivity of ɛ1 = 80.1, and focus on the biologically interesting density regime between 1 mM and 1 M, where relevant effects caused by the soft structure appear that are not captured by mean-field theories, high-temperature expansions, and zero-temperature approaches. We consider the environment around a given ion in the form of the mean charge density:
(N9) calculation steps (the correlation functions require six dimensions, and the convolution in Eq. 3 integrates over three dimensions), which easily exceeds any practical timescale, regardless of the current computational possibilities. The dimension of the correlation functions can be reduced, however, if symmetries are present. In the case of two planar, parallel boundaries, the correlation functions can be written as a function of a coordinate r parallel to the boundaries and two coordinates z and z′ along an axis perpendicular to the boundaries thanks to translational symmetry in the directions parallel to the boundaries. If the z axis is discretized, one can simplify the analysis without additional approximations by a mathematical mapping (19, 20), which is illustrated in SI Text. One can map an inhomogeneous 3D system with n components to a 2D system with Mn components that is homogeneous, with M being the number of grid points used for the z axis, by labeling the particles in each layer as a distinct species with a distinct pair-potential uij(r) between the particles with coordinates zi and zj. The Ornstein–Zernike equation (Eq. 3) reduces to a matrix equation, which can be solved with an appropriate closure relation together with the Boltzmann distribution (Eq. 5), to yield information about the local structure as well as the thermodynamic properties on a timescale of minutes on a single central processing unit (if the grid points are chosen cautiously and judiciously). We fix the effective ion radius to a± = 0.3 nm for both species, consider monovalent ions in an aqueous background with a relative permittivity of ɛ1 = 80.1, and focus on the biologically interesting density regime between 1 mM and 1 M, where relevant effects caused by the soft structure appear that are not captured by mean-field theories, high-temperature expansions, and zero-temperature approaches. We consider the environment around a given ion in the form of the mean charge density:
with x as the position of the central ion, α as the sign of its charge, ρ± as the local mean density of ± ions, q = q+ = −q− as the valency of the ions, and g = h + 1 as the pair-distribution function.
Deformation of the Screening Layer—Neutral Boundaries
In the most simple example of two neutral, nonpolarized boundaries, the results of the AHNC reveal two obvious effects that are absent in mean-field theories (Fig. 2). The density of monovalent ions displays a clear depletion near the walls, despite the absence of any external fields in the region between the walls, whereas a pure mean-field theory predicts a constant density. In high-temperature expansions or saddle point approximations (26, 35, 36), this effect appears only in the higher-order corrections and is purely of a correlational origin. Fig. 2 illustrates the mean forces acting on an ion that originate from Coulomb correlations, hard-core correlations, and polarization charge induced by a boundary.
Fig. 2.

The normalized density of monovalent ions between two neutral, nonpolarized boundaries is shown in Left for several values of the reservoir concentration ρs (black, red, and blue at ρs = 0.03, 0.1, and 0.3 M, respectively). The green dashed line corresponds to the density of monovalent ions between two dielectric discontinuities, with ɛ2 = 70, ɛ3 = 93, and ρs = 0.1 M. At low densities, depletion near the boundaries is visible over a range of the order of the Debye length because of the anisotropic screening cloud that pulls ions away from the boundary. The subtle increase in density near contact at ρs = 0.1 M is a signature of hard-core correlations that tend to push ions against the walls, which are caused by anisotropic collisions. At densities above ∼ 0.3 M, the accumulation completely overcomes the depletion, and the Gibbs adsorption becomes positive. The green dashed curve shows additional effects caused by polarization; the right boundary attracts ions because of its polarization charge of opposite sign, whereas the left boundary repels the ions because of its polarization charge of similar sign. The local charge density around an anion at the right boundary is shown in Right, corresponding to the system with the two dielectric discontinuities and ρs = 0.1 M (green dashed lines) with an illustration of the mean forces that act on the ion. The screening cloud around an ion becomes anisotropic near the boundary because of geometric constraints and generates a mean force away from the boundary, which becomes relevant at distances shorter than the typical Debye length. The anisotropic collisions (short-range repulsions) cause a mean force to the wall within a distance of a few particle radii. The induced polarization charge generates a force directed away or to the wall, depending on the dielectric contrast, and is typically of slightly shorter range than the force induced by the anisotropic screening cloud. The typical range of the correlation functions in the direction parallel to the boundaries increases near the boundaries and is more affected by the geometry rather than the dielectric properties of the wall.
The mean force caused by Coulomb correlations is induced by the anisotropic screening cloud around an ion near the wall. Each ion should be neutralized by a screening layer of opposite charge with the same magnitude to satisfy global charge neutrality. In a homogeneous reservoir, the cloud should be spherical on average, and its point of central charge should coincide exactly with the center of the ion. Near a boundary, the cloud has to adapt because of the geometric constraints or energetic considerations if the boundary introduces an external field, such that the point of central charge may shift, which introduces a dipole moment perpendicular to the boundary. The polarization charge of a dielectric boundary adds an extra force, with direction and magnitude that depend on the dielectric contrast (green dashed line in Fig. 2) and affect the deformation of the screening cloud. The hard-core correlations induce a mean force in a similar fashion. Any boundary that breaks the symmetry in the geometric or energetic landscape changes the local environment around the nearby ions and may induce anisotropic collisions. This correlational force is generally directed to the boundary or in any case, in the opposite direction of the density gradient, has a typical short range of the ion size, and becomes relevant at higher densities where the volume fraction of particles has a value above ∼0.01. The total correlation functions have a longer range in the direction parallel to the boundary [visible in the profile of Q(r, z) in Fig. 2 and more drastic in Fig. 3] than the perpendicular direction, and it can be shown that the asymptotic decay of the correlation functions in the parallel direction obeys a power law (37) instead of an exponential law. The reasons for the extension are more subtle, although the trend can be understood from the balance between entropy and electrostatic energy, which gives rise to an isotropic exponential decay in the reservoir but a direction-dependent decay in a geometric confinement. The boundary-induced anisotropy of the screening cloud introduces mean forces perpendicular to the boundary (38), as discussed above, that can be attenuated by shifting the countercharge to the z coordinate of the central charge. However, by shifting the countercharge to the given z coordinate while keeping the r coordinate fixed, one would increase the local density, which is entropically unfavorable. Therefore, the optimal balance is found by shifting the countercharge to the z coordinate of the central particle and spreading the ion cloud in the r direction. The range of the depletion (caused by electrostatic correlations) is dependent on the concentration and becomes smaller at higher densities. Beyond a reservoir density of 0.1 M (corresponding to a packing fraction of ∼0.015) an accumulation near the boundary becomes visible over a range of the ion diameter (because of anisotropic collisions). In the regime roughly between 0.1 and 0.5 M, a transition takes place, where the number of adsorbed particles in excess over the reservoir (Gibbs adsorption) changes from negative to positive.
Fig. 3.
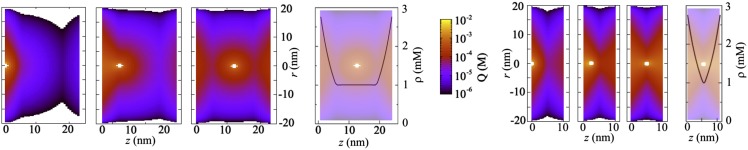
The anisotropic screening cloud around a monovalent anion between two adsorbing boundaries at three different locations, from left to right, in the adsorption layer, near the adsorption layer, and between the walls is shown. The colors indicate the mean total charge around an anion located at the center of the white area. The mean density of ions displays an increase near the boundaries caused by the short-range attraction of the boundary. Although the adsorption is relatively weak (driven by a potential difference of −1 kBT), it is clearly visible that the adsorbed layers contain a large fraction of the screening ions and that the ions within the adsorbed layer correlate visibly with the ions in the other layer, especially at smaller separations (2 Debye lengths for the four left figures and 0.8 Debye length for the four right figures). The figures visualize a type of fluctuation-attraction.
Deformation of the Screening Layer—Interacting Boundaries
The consideration of a system of neutral, noninteracting boundaries helps to reveal and identify several effects that are caused by the local structure in the ion cloud (i.e., ion correlations), such as a long-range depletion originating from the electrostatic interactions and a short-range accumulation stemming from short-range repulsions. Real systems, however, are more complicated, because any soft or hard boundary interacts in general because of polarization, dispersion forces, or surface groups that ionize in solution, which influences the soft structure and therefore, the manifestation of the mean forces that are caused by correlations as well. The previous analysis remains helpful to understand the additional effects caused by the external fields that are generated by the boundary.
As a simple extension, we consider two boundaries that attract anions and cations equally over a short range (39). Fig. 3 shows the local net charge around an anion between the two adsorbing boundaries, separated by 2 (Fig. 3, four left panels) and 0.8 Debye lengths (Fig. 3, four right panels), at different locations. The reservoir concentration is 1 mM and increases near the boundaries up to 3 mM because of the attraction potential of the boundary that decreases linearly by 1 kBT over about 5 nm and is zero at larger distances. Despite the weak attraction, the effect on the soft structure is considerable. The majority of screening ions is located near the walls, regardless of the position of the anion, and most strongly if the anion is close to the boundary. The qualitative features of the typical distribution can be explained from the balance between entropy and electrostatic energy. The screening cloud around the anion can be formed by attracting countercharge from either the dilute bulk phase or the denser adsorption layer, which is entropically more favorable but perhaps energetically less favorable depending on the distance of the anion to the layer. The fraction of countercharge in the adsorption layers depends on the position of the anion, although it is clearly visible in Fig. 3 that an anion in the left layer strongly correlates with the charge in the other layer if the distance is less than 1 Debye length. Fig. 3 shows the presence of fluctuation interactions between the two boundaries. Any fluctuation in the charge in the left adsorption layer induces countercharge in the right layer on average and hence, a mean attractive force between the boundaries. For the given parameters, the effect is negligible, which can be quantified analytically by a model that incorporates Gaussian fluctuations around the mean-field solution (40). Nevertheless, the force can be amplified exponentially by increasing the attraction potential of the boundaries and the surface area of the boundaries to values where higher-order corrections contribute. The AHNC method does not yield explicit analytical expressions for the induced force but does include higher-order fluctuations, and it offers the opportunity to study these fluctuation forces quantitatively.
Phase boundaries generally exert electrostatic forces on nearby electrolytes by means of ionizable surface groups or polarization. Fig. 4 illustrates the deformation of the screening cloud around a negative ion near a positively charged boundary with a surface charge density of σ = +0.01 e/nm2 (1.66 pM/cm2).
Fig. 4.
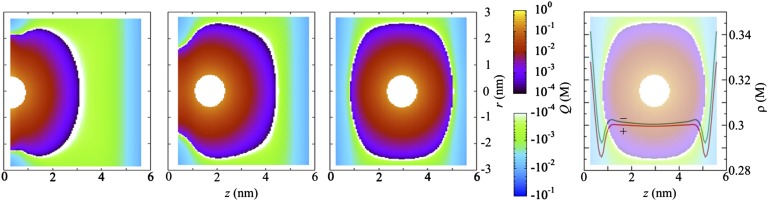
The anisotropic screening cloud around a monovalent anion between two positively charged boundaries of charge density σ = +0.01 e/nm2 in contact with a salt reservoir with concentration ρs = 0.3 M. The mean charge density around the anion changes sign because of the overall excess of negative ions. Near the boundaries, where the local concentrations of both ions and total negative charge are elevated, the screening cloud comprised of cations becomes compressed. The mean concentration around a cation has similar qualitative features as shown in Fig. 3 because of the excess of anions at the boundaries.
The screening cloud behaves differently near the boundaries compared with the system of Fig. 3. The overall mean density of positive ions is lower than the mean density of negative ions, as shown in Fig. 4, Right, to compensate for the charge of the boundaries, but the profiles also display the correlation effects that were found near neutral boundaries (Fig. 2) and deviate significantly from Poisson–Boltzmann results. The negative ion attracts positive counterions and is, on average, surrounded by a positively charged island in a negative background. The largest mean density of positive ions is found in the close vicinity of the boundary but also the largest density of negative ions and the highest value of the electric field, such that the island of positive charge prefers the center, where the mean electric field is weakest and not because of a relative abundance of positive charges (which was the reason for the deformation of the screening cloud near adsorbing boundaries). The mean screening cloud around a positive ion, however, does show similar qualitative features as Fig. 3; in the negative background, a positive ion induces a local maximum at the distance of closest contact and near the boundaries.
Effective Forces Induced by Soft Structure
We have been focusing on the influence of a variety of boundaries on the soft structure in a Coulomb fluid. The analysis of the local structure does not only yield information about the mean forces between the ions and between the ions and the boundaries but also, the pressure between the two boundaries. The deformation of the ionic environment changes the free energy per ion and therefore, the free energy per volume between the boundaries, depending on the separation, as well. Even if the energy change is only a fraction of a kBT per ion, the total change in the free energy can well exceed the thermal energy if the surface area and ion density are large enough. Fig. 5 shows the osmotic pressure as a function of the separation H for a variety of boundaries subtracted by the value at infinite separation, such that it has an asymptote at zero. At small distances, the pressure is negative for all of the curves and becomes stronger on approach, except if the boundaries are adsorbing (exert a short-range attraction). The osmotic pressure is obtained by the contact theorem (41)
 |
with Vi as the external potential (in addition to the hard-core repulsion) caused by the boundary that is experienced by an ion of type i.
Fig. 5.

The pressure as a function of the separation between the boundaries H for neutral boundaries with ρs = 1 mM, 0.1 M, and 0.3 M (black, blue, and green lines, respectively) and the weakly adsorbing boundaries considered in Fig. 3 (red). A subtle negative pressure between the plates is caused by the depletion between the boundaries, which becomes stronger with increasing density over a shorter range. For the adsorbing boundaries, however, the pressure becomes positive at short range, when the adsorption layers overlap. The fluctuation attractions between the boundaries are significantly enhanced because of the adsorbed layer, despite the fact that the adsorption potential in the example is relatively weak. The image charge effects strongly enhance the depletion attraction (low-ɛ boundaries) or the accumulation repulsion (high-ɛ boundaries).
At high ion densities, the pressure is found to decrease sharply at small separations. The attraction can be understood from the electrostatic cohesion in the fluid. The depletion from the boundaries becomes stronger if the boundaries are brought closer together, causing a decrease in the osmotic pressure. The hard-core correlations induce an opposite effect that contributes significantly beyond ∼0.1 M (a packing fraction of ∼0.015). The mean electrostatic force away from the boundary (Fig. 2) decreases if the boundaries approach closer than a few Debye lengths, because the screening cloud becomes less anisotropic. However, the mean force to the boundary caused by the anisotropic collisions remains more or less constant until a separation smaller than a few ion radii, leading to an accumulation near the boundary. The increasing self-energy of the ions (caused by inefficient screening and confinement) is found to become the dominant term, causing a strong depletion and a negative osmotic pressure. The net effect of all contributions is an increased accumulation near the boundary but an overall depletion of ions, causing a negative osmotic pressure. These effects can be tuned slightly by varying the ion radius and more strongly by the polarization of the boundaries (weak dielectric boundaries favoring the depletion attraction and strong dielectric boundaries favoring the accumulation repulsion). Surface charge adds another tuning parameter, with additional possibilities. For a surface charge that is not too high and a moderate reservoir density, it should be possible to create a minimum in the osmotic pressure at a finite distance based on numerical estimates from the above results, but more quantitative study is being performed to outline the parameter regime.
Discussion
At low concentrations (below 1 mM), the mean forces caused by correlations are small for monovalent electrolytes in water and in practice, completely overwhelmed by the forces caused by surface charge and surface polarization that can be approximated by mean-field theories (14). At higher densities (above 0.1 M), the effects remain subtle but contribute to large deviations from mean-field results that could be measurable (42) and add significant forces between confining boundaries that can be either attractive or repulsive depending sensitively on the density of electrolytes, their effective size, and the type of boundaries. The biologically relevant concentration regime between 0.1 ≲ ρs ≲ 1 M is of extraordinary interest for these correlational forces, because it is in this regime that the short-range repulsions and long-range interactions between the ions induce mean forces of comparable magnitude but in opposite direction and over (a slightly) different range; therefore, one can change the induced forces from attractive to repulsive by a small increase in salt concentration or replacement of one ion type by another or induce a local minimum in the interaction potential as potentially observed in the experiments by Kaz et al. (43). We expect that these forces play a role in the (de)stabilization of dense charged colloids (44), emulsion droplets in brackish and saline water, sterically stabilized nanoparticles in a saline environment or with locally increased salt concentrations (45), dense gels and polymers, and solutions with a low permittivity or multivalent ions. In the last example, the correlations are generally much stronger than the ones considered here, leading to clustering, condensation, and a type of local structure that is more rigid (46, 47). Knowledge of the microscopic structure could elucidate the origins for the precipitation and redissolution of polyelectrolytes in monovalent (12) and multivalent salt solutions (5, 11), the interactions between nucleic acids and charged surfaces (7), and the structure and pattern formation in restricted geometries (48, 49), and it could sustain and extrapolate the results of more time-consuming simulation methods (50). The AHNC method provides accurate numerical results within convenient timescales and offers insight in the anisotropic local structure in primitive model electrolytes and the related mean forces. The primitive model displays a remarkable complexity, despite the absence of an explicit solvent, of ion polarization, ion anisotropy, hydration, and other factors that nuance the behavior of real electrolyte solutions. The primitive model does reveal the fingerprints of Coulomb correlations and hard-core interactions, which may play a foremost role in biological matter and provide an invaluable reference framework to distill the contributions of other solvent-specific effects (51). Furthermore, the AHNC offers invaluable reference for the development of (one-particle) density functional theories [DFTs (52, 53)] that generally rely on assumptions for the chemical potential that are based on knowledge of homogeneous systems. Outstandingly successful for hard-sphere systems (54), these DFTs have difficulty describing primitive model electrolytes accurately and consistently when they display soft structure. One has to (silently) assume local charge neutrality for the local or weighted density, because the excess chemical potential is not defined for a homogeneous system that is not globally charge neutral. The AHNC is free of these inconsistencies and additional assumptions, and it also yields the anisotropic pair-correlation functions. The only advantages of such DFTs that are formulated in terms of the one-particle density are the simplicity of the equations and the numerical efficiency, which could be restrictive factors in more complex geometries or for the analysis of dynamic processes, such as dynamic DFTs (55, 56), or the non-equilibrium self-consistent generalized Langevin equation theory (57, 58), which also describes dynamic arrest. For that purpose, one would require an accurate guess of the anisotropic two-point correlation functions, either ab initio or obtained by other effective liquid-state theories (59). The correlation forces considered here are comparable with Casimir (60) or critical Casimir forces (61, 62), with the difference that the boundaries do not constrain the fluctuations in the electromagnetic field or critical solvent mixture, respectively, but the ionic fluid itself. Therefore, it is not surprising that the typical range corresponds to the correlation length in the ionic reservoir, but the magnitude of the force and the dependency on the separation are intricately related to the system parameters and may allow for local minima and nonmonotonic behavior thanks to the complex interplay of the mean forces (22, 23).
Conclusion
We study the soft structure in primitive model electrolytes calculated by an accurate and rigorous liquid-state theory known as the AHNC. This approach permits us to distinguish and quantify the thermal forces in fluids of charged particles, between the charged particles themselves, and between dielectric boundaries that confine the fluid. The intention is motivated by the ubiquitous role that electrolytes play in natural and industrial processes, such as the atmospheric chemistry, colloid stability, protein functionality, selective transport in living matter, drug delivery, water purification, phase transfer catalysis, gel and polymer physics, and many more examples, where the forces at the micro- and nanoscopic length scales are governed by the present electrolytes. We describe several distinct ion-induced forces in aqueous solutions of monovalent ions at salt concentrations ranging roughly between 0.01 and 1 M (a most relevant regime for the examples mentioned above) that are expected to compete for dominance with the well-known screened Coulomb forces. We introduce the concept of soft structure to connect the induced forces to the local microscopic structure in the ionic fluid and visualize the deformation of the local environment around the ions near several types of boundaries that are neutral, adsorbing, charged, polarizable, or a combination of these properties. For monovalent ions, we find important attractive forces above ∼0.1 M because of electrostatic depletion from the boundaries caused by the anisotropic screening of the individual ions, strongly enhanced or counteracted by polarization of the boundary. The considered forces cannot be found by well-known (practical) mean-field theories and are relevant for neutral solutes or solutes close to an isoelectric point as well. We aim to provide an intuitively appealing overview to aid and enhance predictability and control in experiments. The results from the AHNC may provide guidance for the development of simple scaling laws, potentially efficient and practical DFTs, and coarse-grained simulation methods to overcome the disparate length, time, and energy scales that characterize nanomaterials and biological matter.
Supplementary Material
Acknowledgments
R. Kjellander is acknowledged for offering helpful information. David L. Spjut (Carmel, CA) is acknowledged for the artwork of Fig. 1 and Fig. S2. The authors acknowledge funding by the Office of the Director of Defense Research and Engineering (DDR&E) and the Air Force Office of Scientific Research (AFOSR) under Award FA9550-10-1-0167.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302406110/-/DCSupplemental.
References
- 1.Jungwirth P, Tobias D. Ions at the air/water interface. J Phys Chem B. 2002;106(25):6361–6373. [Google Scholar]
- 2.Olvera de la Cruz M. Electrostatic control of self-organization: The role of charge gradients in heterogeneous media. Soft Matter. 2008;4(9):1735–1739. [Google Scholar]
- 3.Swartzen-Allen S, Matijevic E. Colloid and surface properties of clay suspensions. iii. stability of montmorillonite and kaolinite. J Colloid Interface Sci. 1976;56(1):159–167. [Google Scholar]
- 4.Kékicheff P, Marcelja S, Senden T, Shubin V. Charge reversal seen in electrical double layer interaction of surfaces immersed in 2: 1 calcium electrolyte. J Chem Phys. 1993;99(8):6098–6113. [Google Scholar]
- 5.Olvera de la Cruz M, et al. Precipitation of highly charged polyelectrolyte solutions in the presence of multivalent salts. J Chem Phys. 1995;103(13):5781–5791. [Google Scholar]
- 6.Raspaud E, Olvera de la Cruz M, Sikorav JL, Livolant F. Precipitation of DNA by polyamines: A polyelectrolyte behavior. Biophys J. 1998;74(1):381–393. doi: 10.1016/S0006-3495(98)77795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng H, Zhang K, Libera JA, Olvera de la Cruz M, Bedzyk MJ. Polynucleotide adsorption to negatively charged surfaces in divalent salt solutions. Biophys J. 2006;90(4):1164–1174. doi: 10.1529/biophysj.105.070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelini TE, Liang H, Wriggers W, Wong GC. Like-charge attraction between polyelectrolytes induced by counterion charge density waves. Proc Natl Acad Sci USA. 2003;100(15):8634–8637. doi: 10.1073/pnas.1533355100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Mozuelos P, Olvera de la Cruz M. Ion condensation in salt-free polyelectrolyte dilute solutions. J Chem Phys. 1995;103(8):3145–3157. [Google Scholar]
- 10.Solis F, Olvera de la Cruz M. Collapse of flexible polyelectrolytes in multivalent salt solutions. J Chem Phys. 2000;112(4):2030–2035. [Google Scholar]
- 11.Solis F, Olvera de la Cruz M. Flexible linear polyelectrolytes in multivalent salt solutions: Solubility conditions. Eur Phys J Soft Matter. 2001;4(2):143–152. [Google Scholar]
- 12.Jha P, Zwanikken J, Olvera de la Cruz M. Understanding swollen–collapsed and re-entrant transitions in polyelectrolyte nanogels by a modified donnan theory. Soft Matter. 2012;8(37):9519–9522. [Google Scholar]
- 13.Perkin S, Kampf N, Klein J. Long-range attraction between charge-mosaic surfaces across water. Phys Rev Lett. 2006;96(3):038301. doi: 10.1103/PhysRevLett.96.038301. [DOI] [PubMed] [Google Scholar]
- 14.Baumgartl J, Arauz-Lara J, Bechinger C. Like-charge attraction in confinement: Myth or truth? Soft Matter. 2006;2(8):631–635. doi: 10.1039/b603052a. [DOI] [PubMed] [Google Scholar]
- 15.DeRouchey J, Parsegian VA, Rau DC. Cation charge dependence of the forces driving DNA assembly. Biophys J. 2010;99(8):2608–2615. doi: 10.1016/j.bpj.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wernersson E, Kjellander R, Lyklema J. Charge inversion and ion- ion correlation effects at the mercury/aqueous mgso4 interface: Toward the solution of a long-standing issue. J Phys Chem C Nanomater Interface. 2010;114(4):1849–1866. [Google Scholar]
- 17.Guerrero-García G, González-Tovar E, Olvera de la Cruz M. Effects of the ionic size-asymmetry around a charged nanoparticle: Unequal charge neutralization and electrostatic screening. Soft Matter. 2010;6(9):2056–2065. [Google Scholar]
- 18.Ninham B. On progress in forces since the dlvo theory. Adv Colloid Interface Sci. 1999;83(1):1–17. [Google Scholar]
- 19.Kjellander R, Marcělja S. Correlation and image charge effects in electric double layers. Chem Phys Lett. 1984;112(1):49–53. [Google Scholar]
- 20.Kjellander R, Marčelja S. Inhomogeneous coulomb fluids with image interactions between planar surfaces. I. J Chem Phys. 1985;82(4):2122–2135. [Google Scholar]
- 21.Kjellander R, Sarman S. A study of anisotropic pair distribution theories for lennard-jones fluids in narrow slits. Mol Phys. 1990;70(2):215–237. [Google Scholar]
- 22.Greberg H, Kjellander R, Akesson T. Ion-ion correlations in electric double layers from Monte Carlo simulations and integral equation calculations part 2. Case of added salt. Mol Phys. 1997;92(1):35–48. [Google Scholar]
- 23.Greberg H, Kjellander R. Charge inversion in electric double layers and effects of different sizes for counterions and coions. J Chem Phys. 1998;108(7):2940–2953. [Google Scholar]
- 24.Nygård K, et al. Anisotropic pair correlations and structure factors of confined hard-sphere fluids: An experimental and theoretical study. Phys Rev Lett. 2012;108(3):037802. doi: 10.1103/PhysRevLett.108.037802. [DOI] [PubMed] [Google Scholar]
- 25.Rouzina I, Bloomfield V. Macroion attraction due to electrostatic correlation between screening counterions. 1. Mobile surface-adsorbed ions and diffuse ion cloud. J Phys Chem. 1996;100(23):9977–9989. [Google Scholar]
- 26.Netz R, Orland H. Beyond Poisson-Boltzmann: Fluctuation effects and correlation functions. Eur Phys J Soft Matter. 2000;1(2):203–214. [Google Scholar]
- 27.Netz R, Orland H. Variational charge renormalization in charged systems. Eur Phys J Soft Matter. 2003;11(3):301–311. doi: 10.1140/epje/i2002-10159-0. [DOI] [PubMed] [Google Scholar]
- 28.Kornyshev AA. Double-layer in ionic liquids: Paradigm change? J Phys Chem B. 2007;111(20):5545–5557. doi: 10.1021/jp067857o. [DOI] [PubMed] [Google Scholar]
- 29.Ornstein L, Zernike F. Accidental deviations of density and opalescence at the critical point of a single substance. Proc Akad Sci (Amsterdam) 1914;17:793. [Google Scholar]
- 30.Hansen J, McDonald I. Theory of Simple Liquids. London: Academic; 2006. [Google Scholar]
- 31.Mayer J. The theory of ionic solutions. J Chem Phys. 1950;18(11):1426–1436. [Google Scholar]
- 32.Springer J, Pokrant M, Stevens F. Integral equation solutions for the classical electron gas. J Chem Phys. 1973;58(11):4863–4867. [Google Scholar]
- 33.Ng K. Hypernetted chain solutions for the classical one-component plasma up to γ = 7000. J Chem Phys. 1974;61(7):2680–2689. [Google Scholar]
- 34.Hansen J, Torrie G, Vieillefosse P. Statistical mechanics of dense ionized matter. vii. equation of state and phase separation of ionic mixtures in a uniform background. Phys Rev A. 1977;16(5):2153–2168. [Google Scholar]
- 35.Podgornik R, Žekš B. Inhomogeneous coulomb fluid. A functional integral approach. J Chem Soc, Faraday Trans II. 1988;84(6):611–631. [Google Scholar]
- 36.Attard P. 1996. Electrolytes and the electric double layer. Advances in Chemical Physics, eds Prigogine I, Rice SA (John Wiley & Sons, Inc.), Vol 92, pp 1–160.
- 37.Carnie S, Chan D. Correlations in inhomogeneous coulomb systems. Mol Phys. 1984;51(4):1047–1070. [Google Scholar]
- 38.Zwanikken J, van Roij R. Charged colloidal particles and small mobile ions near the oil-water interface: Destruction of colloidal double layer and ionic charge separation. Phys Rev Lett. 2007;99(17):178301. doi: 10.1103/PhysRevLett.99.178301. [DOI] [PubMed] [Google Scholar]
- 39.Schwierz N, Netz RR. Effective interaction between two ion-adsorbing plates: Hofmeister series and salting-in/salting-out phase diagrams from a global mean-field analysis. Langmuir. 2012;28(8):3881–3886. doi: 10.1021/la204060k. [DOI] [PubMed] [Google Scholar]
- 40.Gopinathan A, Zhou T, Coppersmith S, Kadanoff L, Grier D. Weak long-ranged casimir attraction in colloidal crystals. EPL. 2007;57(3):451–457. [Google Scholar]
- 41.Henderson D, Blum L, Lebowitz J. An exact formula for the contact value of the density profile of a system of charged hard spheres near a charged wall. J Electroanal Chem Interfacial Electrochem. 1979;102(3):315–319. [Google Scholar]
- 42.Laanait N, et al. Tuning ion correlations at an electrified soft interface. Proc Natl Acad Sci USA. 2012;109(50):20326–20331. doi: 10.1073/pnas.1214204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaz D, McGorty R, Mani M, Brenner M, Manoharan V. Physical ageing of the contact line on colloidal particles at liquid interfaces. Nat Mater. 2012;11(2):138–142. doi: 10.1038/nmat3190. [DOI] [PubMed] [Google Scholar]
- 44.Von Grünberg H, Van Roij R, Klein G. Gas-liquid phase coexistence in colloidal suspensions? EPL. 2007;55(4):580. [Google Scholar]
- 45.Zwanikken J, Guo P, Mirkin C, Olvera de la Cruz M. Local ionic environment around polyvalent nucleic acid-functionalized nanoparticles. J Phys Chem C Nanomater Interfaces. 2011;115(33):16368–16373. [Google Scholar]
- 46.Valeriani C, Camp P, Zwanikken J, van Roij R, Dijkstra M. Ion association in low-polarity solvents: Comparisons between theory, simulation, and experiment. Soft Matter. 2010;6(12):2793–2800. [Google Scholar]
- 47.Valeriani C, Camp PJ, Zwanikken JW, van Roij R, Dijkstra M. Computer simulations of the restricted primitive model at very low temperature and density. J Phys Condens Matter. 2010;22(10):104122. doi: 10.1088/0953-8984/22/10/104122. [DOI] [PubMed] [Google Scholar]
- 48.Kohlstedt KL, Solis FJ, Vernizzi G, de la Cruz MO. Spontaneous chirality via long-range electrostatic forces. Phys Rev Lett. 2007;99(3):030602. doi: 10.1103/PhysRevLett.99.030602. [DOI] [PubMed] [Google Scholar]
- 49.Solis F, Vernizzi G, Olvera de la Cruz M. Electrostatic-driven pattern formation in fibers, nanotubes and pores. Soft Matter. 2011;7(4):1456–1466. [Google Scholar]
- 50.Jadhao V, Solis F, Olvera de la Cruz M. Electrostatic potential between surfaces bearing ionizable groups in ionic equilibrium with physiologic saline solution. Phys Rev Lett. 2012;109(3):223905. [Google Scholar]
- 51.Espinosa-Marzal RM, Drobek T, Balmer T, Heuberger MP. Hydrated-ion ordering in electrical double layers. Phys Chem Chem Phys. 2012;14(17):6085–6093. doi: 10.1039/c2cp40255f. [DOI] [PubMed] [Google Scholar]
- 52.Evans R. The nature of the liquid-vapour interface and other topics in the statistical mechanics of non-uniform, classical fluids. Advances in Physics. 1979;28(2):143–200. [Google Scholar]
- 53.Zwanikken JW. Moderately coupled charged fluids near dielectric interfaces and in confinement. In: Dean DS, Dobnikar J, Naji A, Podgornik R, editors. New Challenges in Electrostatics of Soft and Disordered Matter. Singapore: Pan Stanford; 2013. [Google Scholar]
- 54.Rosenfeld Y, Schmidt M, Löwen H, Tarazona P. Fundamental-measure free-energy density functional for hard spheres: Dimensional crossover and freezing. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics. 1997;55(4):4245–4263. [Google Scholar]
- 55.Marconi U, Tarazona P. Dynamic density functional theory of fluids. J Chem Phys. 1999;110(16):8032–8044. [Google Scholar]
- 56.Royall CP, Dzubiella J, Schmidt M, van Blaaderen A. Nonequilibrium sedimentation of colloids on the particle scale. Phys Rev Lett. 2007;98(18):188304. doi: 10.1103/PhysRevLett.98.188304. [DOI] [PubMed] [Google Scholar]
- 57.Ramírez-González P, Medina-Noyola M. General nonequilibrium theory of colloid dynamics. Phys Rev E Stat Nonlin Soft Matter Phys. 2010;82(6 Pt 1):061503. doi: 10.1103/PhysRevE.82.061503. [DOI] [PubMed] [Google Scholar]
- 58.Ramírez-González P, Medina-Noyola M. Aging of a homogeneously quenched colloidal glass-forming liquid. Phys Rev E Stat Nonlin Soft Matter Phys. 2010;82(6 Pt 1):061504. doi: 10.1103/PhysRevE.82.061504. [DOI] [PubMed] [Google Scholar]
- 59.Zwanikken JW, Jha PK, Olvera de la Cruz M. A practical integral equation for the structure and thermodynamics of hard sphere Coulomb fluids. J Chem Phys. 2011;135(6):064106. doi: 10.1063/1.3624809. [DOI] [PubMed] [Google Scholar]
- 60.Casimir H. On the attraction between two perfectly conducting plates. Proc K Ned Akad Wet C. 1948;51:793. [Google Scholar]
- 61.Hertlein C, Helden L, Gambassi A, Dietrich S, Bechinger C. Direct measurement of critical Casimir forces. Nature. 2008;451(7175):172–175. doi: 10.1038/nature06443. [DOI] [PubMed] [Google Scholar]
- 62.Bier M, Gambassi A, Oettel M, Dietrich S. Electrostatic interactions in critical solvents. EPL. 2011;95(6):60001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.