

Summary
Excessive mitochondrial fission is associated with the pathology of a number of neurodegenerative diseases. Therefore, inhibitors of aberrant mitochondrial fission could provide important research tools in addition to potential leads for drug development. Using a rational approach, we designed a novel and selective peptide inhibitor, P110, of excessive mitochondrial fission. P110 inhibits Drp1 enzyme activity and blocks Drp1/Fis1 interaction in vitro and in cultured neurons, whereas it has no effect on the interaction between Drp1 and other mitochondrial adaptors, as demonstrated by co-immunoprecipitation. Furthermore, using a model of Parkinson's disease (PD) in culture, we demonstrated that P110 is neuroprotective by inhibiting mitochondrial fragmentation and reactive oxygen species (ROS) production and subsequently improving mitochondrial membrane potential and mitochondrial integrity. P110 increased neuronal cell viability by reducing apoptosis and autophagic cell death, and reduced neurite loss of primary dopaminergic neurons in this PD cell culture model. We also found that P110 treatment appears to have minimal effects on mitochondrial fission and cell viability under basal conditions. Finally, P110 required the presence of Drp1 to inhibit mitochondrial fission under oxidative stress conditions. Taken together, our findings suggest that P110, as a selective peptide inhibitor of Drp1, might be useful for the treatment of diseases in which excessive mitochondrial fission and mitochondrial dysfunction occur.
Key words: Dynamin related protein 1, Mitochondrial fission, Neuronal cell death, Peptide inhibitor
Introduction
Impairment of mitochondrial integrity results in reactive oxygen species (ROS) production, and induces protein and lipid oxidation, DNA damage and activation of programmed cell death (PCD) (Mammucari and Rizzuto, 2010). Thus, mitochondrial dysfunction contributes to the pathology of a number of diseases including neurodegenerative (Kim and Sack, 2012), cardiovascular (Palaniyandi et al., 2010) and metabolic (Rolo et al., 2011).
Mitochondria are organized in a highly dynamic tubular network that is continuously reshaped by opposing processes of fusion and fission (Chan, 2006b). This dynamic process controls not only mitochondrial morphology, but also the subcellular location and function of mitochondria. Defects in either fusion or fission limit mitochondrial motility, decrease energy production and increase oxidative stress, thereby promoting cell dysfunction and death (Jahani-Asl et al., 2010; Scott and Youle, 2010). The two opposing processes, fusion and fission, are controlled by evolutionarily conserved large GTPases that belong to the dynamin family of proteins. In mammalian cells, mitochondrial fusion is regulated by mitofusin-1 and -2 (MFN-1/2) and optic atrophy 1 (OPA1), whereas mitochondrial fission is controlled by dynamin-1-related protein, Drp1 (Chan, 2006a; Scott and Youle, 2010) and its mitochondrial adaptors such as Fis1, Mff and MIEF1 (Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011).
Drp1 is primarily found in the cytosol, but it translocates from the cytosol to the mitochondrial surface in response to various cellular stimuli to regulate mitochondrial morphology (Chang and Blackstone, 2010). At the mitochondrial surface, Drp1 is thought to wrap around the mitochondria to induce fission powered by its GTPase activity (Smirnova et al., 2001). Cell culture studies demonstrated that Drp1-induced excessive mitochondrial fission and fragmentation plays an active role in apoptosis (Estaquier and Arnoult, 2007; Frank et al., 2001), autophagic cell death (Barsoum et al., 2006; Twig et al., 2008) and necrosis (Wang et al., 2012). Inhibition of Drp1 by either expression of a Drp1-dominant negative mutant or by RNA interference leads to decreased mitochondrial fragmentation. This reduction in mitochondrial fission impairment results in longer and more interconnected mitochondrial tubules, increased ATP production, and the prevention of cell death (Barsoum et al., 2006; Frank et al., 2001; Yuan et al., 2007).
The association of Drp1 with the mitochondrial outer membrane and its activity in mammalian cells depends on various accessory proteins. Fis1 is an integral mitochondrial outer membrane protein that recruits Drp1 to promote fission (James et al., 2003; Yoon et al., 2003). In yeast, recruitment of Dnm1 (yeast Drp1) from the cytosol and assembly in punctate structures on the mitochondrial surface depends on Fis1 (Fannjiang et al., 2004; Suzuki et al., 2005). hFis1 is the ortholog of the yeast mitochondrial fission factor, Fis1, and was therefore thought to be involved in the mitochondrial recruitment of Drp1; hFis1 overexpression promotes mitochondrial fragmentation and hFis1 depletion produces an interconnected mitochondrial network in HEK293 and COS-7 cells (James et al., 2003; Yoon et al., 2003). However, recent data suggest that other proteins [Mff, MiD51 (MIEF1) and MiD49] substitute for Fis1 in mammals (Otera et al., 2010; Zhao et al., 2011). In addition, Fis1 has been recently shown to regulate mitochondrial fission independently of Drp1 in HeLa cells (Onoue et al., 2012). Thus, the role of Fis1 in Drp1-mediated mitochondrial fission and how Fis1 contributes to mitochondrial function under normal and stressed conditions need to be determined.
Since protein-protein interaction (PPI) between Drp1 and its mitochondrial adaptor appears to be required for mitochondrial fission, an inhibitor of this interaction may have a therapeutic utility. To test this hypothesis, we used a rational design to identify short peptide inhibitors of the PPI between Drp1 and Fis1. A novel selective peptide inhibitor of Drp1 has been identified and its use as an inhibitor of Drp1-mediated mitochondrial dysfunction in a cell culture model of Parkinson's disease (PD) was examined.
Results
Rational design of peptide inhibitor for mitochondrial fission
We have previously demonstrated that two non-related proteins that interact in an inducible manner often have shared short sequences of homology that represent sites of both inter- and intra-molecular interactions (Qvit and Mochly-Rosen, 2010; Ron and Mochly-Rosen, 1995; Souroujon and Mochly-Rosen, 1998). For example, a peptide corresponding to a homologous sequence between protein kinase C (PKC) and its scaffold protein, RACK, serves as a selective inhibitor of the function of PKC, as determined in culture and in in vivo animal models of acute myocardial infarction (Chen et al., 2001a; Dorn et al., 1999; Kheifets et al., 2006), heart failure (Inagaki et al., 2008), pain (Sweitzer et al., 2004), and cancer (Kim et al., 2011). Applying the same approach, we used L-ALIGN sequence alignment software (Huang, 1991) and identified three different regions of homology between Drp1 (Drp1, human, O00429) and Fis1 (Fis1, human, Q9Y3D6) (Fig. 1A; the six regions are marked as regions 108 through 113). The amino acid sequence of these regions, listed in Fig. 1B, are identified by color in the crystal structure of Fis1 (1NZN) and rat dynamin-1 (3ZVR) which is highly similar to Drp1 (Fig. 1C). These six regions are present on the surface of Drp1 and Fis1, thus likely accessible for PPI. Furthermore, using similar principles to the evolutionary trace method of Lichtarge and colleagues (Lichtarge et al., 1996), we found that all the homologous sequences are conserved in a variety of species (Fig. 1D; supplementary material Fig. S1). However, only the sequence in region 110 is identical in mammalians, fish, chicken and yeast, suggesting that this region is most likely critical for the function of Drp1. Another filter to determine whether region 110 in Drp1 represents a unique site for protein-protein interaction is to determine whether it is present in other proteins in the human genome. In addition to Drp1, 16 other proteins have a sequence that is at least 80% similar to the sequence in region 110 (Fig. 1E). However, Fis1 was the only protein in which this sequence was 100% identical in other mammalians (and 50% identical in yeast; Fig. 1E), further supporting the hypothesis that 110 represents an important interaction region in Drp1 for Fis1.
Fig. 1.

Rational design of peptide inhibitors that interfere with the interaction between Drp1 and Fis1. (A) Cartoons of the main domains of Drp1 (human, O00429) and Fis1 (human, Q9Y3D6). Highlighted in the same colors are the three regions of homology between the two proteins, regions 108, 109 and 110 in Drp1 and the corresponding regions 111, 112 and 113 in Fis1. (B) Sequence of homology between Drp1 and Fis1. Amino acids are represented by the one-letter code; asterisks (*) indicate identical amino acids; colons (:) indicate high similarity between amino acids. Peptides P108–113 correspond to these homologous regions. (C) Ribbon presentation of the 3D structure of rat dynamin 1 (3ZVR) and human Fis1 (1NZN). [Because the crystal structure of Drp1 is not available, we provided the structure of dynamin 1, which is 40% identical and 57% similar to human Drp1 (using the European Molecular Biology Open Software Suite, EMBOSS). We highlighted the positions of the homologous regions 108–110 in Drp1 on the dynamin 1 structure, and of regions 111–113 on the Fis1 structure (using PyMol).] (D) Conservation of the homologous sequences between Drp1 and Fis1 in human, rat, mouse and yeast is provided in color code (for full alignment, see supplementary material Fig. S1). Blue, all the amino acids are identical or similar; green, up to 36% difference in amino acid composition; yellow, 37–63% difference in amino acid composition; orange, up to 86% difference in amino acid composition; red, all the amino acids are different. Phylogenetic analysis was constructed on sequences obtained from the National Center for Biotechnology Information (NCBI), using the Molecular Evolutionary Genetics Analysis (MEGA) package version 5.05 (Tamura et al., 2011). Multiple alignments used the Muscle program (http://nar.oxfordjournals.org/content/32/5/1792.full.pdf+html), and calculation of phylogenetic tree is based on neighbor-joining algorithms. Scale bar: 0.1 substitutions/site. (E) Presence of the sequence represented by region 110 (DLLPRGT) in human proteins and conservation of this sequence in orthologs of these proteins in mouse, rat, and yeast is provided in color code. P110-like sequences were found in Drp1, Fis1, TOM22 (mitochondrial import receptor subunit, TOM22), DYN1 (dynamin-1), DYN2 (dynamin-2), DYN3 (dynamin-3), MIA3 (melanoma inhibitory activity protein 3), SCN5A (sodium channel protein type 5, subunit alpha), HIP1 (Huntingtin-interacting protein 1), PCDGK (protocadherin gamma-C3), BI2L2 (brain-specific angiogenesis inhibitor 1-associated protein 2-like protein 2), ZSWIM5 (zinc finger, SWIM-type 5), ADAM 17 (disintegrin and metalloproteinase domain-containing protein 17), AP2B (AP-2 complex subunit beta), ZSWM4 (zinc finger SWIM domain-containing protein 4), CYP2W1 (cytochrome P450, 2W1) and MSLN (mesothelin). The conservation of these sequences is given in the same color code as in D. P110 sequence was 100% identical/similar among these four species only in Drp1 (blue), 100% identical/similar between all the mammalian Fis1 proteins examined and 50% identical/similar (yellow) in yeast. All the other human proteins that had a P110-like sequence shared only 80% or less homology (green), which was less or not conserved in other mammals (green, yellow, orange and red for 80, 50, 25 and 0% homology) and, with the exception of AP2B, was completely missing in yeast (red).
We next synthesized peptides corresponding to region 110, in addition to the five other homologous regions between Fis1 and Drp1 (Fig. 1B) and conjugated them to the cell permeating TAT protein-derived peptide, TAT47–57, as we described (Chen et al., 2001a; Chen et al., 2001b). These peptides are referred to as P110, P108, P109, P111, P112 and P113 and their effect on Drp1/Fis1 functions was tested next.
Selection of peptide inhibitor of mitochondrial fission
Drp1 is a large GTPase and its mitochondrial fission activity is dependent on its GTP hydrolysis (Chang and Blackstone, 2010). Because P109 and P110 are derived from the GTP exchange domain (GED) and the GTPase domain in Drp1, respectively (Fig. 1A), we first determined whether these peptides affect the enzymatic activity of Drp1. P109 and P110 inhibited 40% and 50% of the GTPase activity of recombinant Drp1, respectively (Fig. 2A); the other peptides, including the corresponding homologous peptides derived from Fis1, P112 and P113, respectively, exerted no significant effect. These data are surprising, because they indicate that these two Drp1-derived peptides interact with Drp1, thus suggesting that the sequence corresponding to P109 and P110 are also involved in intra-molecular interactions or in intermolecular interactions between oligomers of Drp1 (Zhu et al., 2004). As expected, a dominant negative mutant of Drp1, Drp1 K38A, which inhibits Drp1's GTPase activity and fission activity (Barsoum et al., 2006), reduced ∼70% of the Drp1 GTPase activity (Fig. 2A). Importantly, peptide P110 had no effects on the GTPase activities of other mitochondrial dynamics-related proteins, such as MFN1 and OPA1 (Fig. 2B), which regulate mitochondrial fusion (Chan, 2006b), or on dynamin-1 (Fig. 2B), a member of the Drp family that mediates endocytosis of the plasma membrane (McClure and Robinson, 1996). These data indicate that peptide P110 is a selective inhibitor of the GTPase activity of Drp1. In contrast, P109 increased the GTPase activity of MFN1 and OPA1 in the same assay (MFN1, 229±10%; OPA1, 177±13%; supplementary material Fig. S2, P<0.05, n = 3, respectively), indicating that P109 is a non-selective regulator of large GTPases.
Fig. 2.

Characterization of the Fis1- and Drp1-derived peptides in vitro. (A) GTPase activity of Drp1 was determined using Drp1 recombinant protein (GST-Drp1, 25 ng) in the presence or absence of peptides P108, P109, P110, P111, P112 and P113 (1 µM each). Peptides P108–P110 were derived from human Drp1 and peptides 111–113 were from Fis1, as indicated in Fig. 1, and conjugated to the cell-permeable peptide TAT47–57. TAT47–57 was used as a control peptide carrier, and Drp1K38A (25 ng) dominant negative was used as a positive control. The data are expressed as means ± s.e. of four independent experiments (*P<0.05; **P<0.01 versus Drp1 recombinant protein alone). (B) GTPase activities of human MFN1, OPA1 and dynamin-1 (25 ng each) were determined in the presence or absence of peptide P110 (1 µM). (C) Mitochondria-enriched fraction (100 µg mitochondrial protein) was isolated from mouse liver and incubated with GST-Drp1 (50 ng) in the presence of the indicated peptides. Upper panel: western blot analysis of GST-Drp1 association with the mitochondrial fraction using anti-Drp1 antibodies. Lower panel: histograms depicting the amounts of GST-Drp1 associated with the mitochondria of mouse liver. The data are expressed as means ± s.e. of three independent experiments; *P<0.05 versus control group. (D) P110 blocked the interaction between Drp1 and Fis1. Peptides P109, P112, P110 and P113 (1 µM, each) or peptide carrier, TAT47–57 (1 µM), were incubated with Drp1 recombinant proteins (100 ng) for 30 minutes prior to the addition of Fis1 recombinant protein (100 ng). The mixed proteins were treated with a chemical cross-linker DSP (1 mM for 30 min). Immunoprecipitates (IP) with anti-Drp1 antibodies were analyzed by immunoblotting (IB) with anti-Fis1. Quantitative data are provided in the histogram. The data are expressed as means ± s.e. of three independent experiments; **P<0.01 versus control group. (E) Human SH-SY5Y neuronal cells were treated with the indicated peptides (1 µM) for 1 hour followed by treatment with MPP+ (2 mM for 1 hour). Following a brief in vivo cross-linking, cells were homogenized. Total cell lysates were then subjected to immunoprecipitation (IP) with anti-Fis1, anti-Mff or anti-MIEF1 antibodies, respectively, and the immunoprecipitates were analyzed by immunoblotting (IB) with anti-Drp1 antibodies. Quantitative data are provided in the histogram. The data are expressed as means ± s.e. of four independent experiments; *P<0.05 versus control group; #P<0.05 versus MPP+-treated group. Input lanes in D and E represent 10% of the sample used for immunoprecipitation.
Our initial assumption was that the peptides we designed represent PPI surfaces between Drp1 and Fis1. If that is the case, they should inhibit the association of Drp1 with the mitochondria. We isolated mouse liver mitochondria and incubated them with recombinant Drp1 (GST-Drp1). Drp1 bound to mitochondrial preparations and only P110 (1 µM) reduced this association by ∼50% (P<0.05, Fig. 2C); peptide carrier TAT47–57 or the other Drp1- or Fis1-derived peptides, including the P110 homolog, P113, had no effect (Fig. 2C). In another set of experiments, we incubated human Drp1 and Fis1 recombinant proteins and immunoprecipitated the complex with anti-Drp1 antibody followed by immunoblotting with anti-Fis1 antibody. Drp1 bound Fis1 in this assay and this interaction was blocked by the addition of P110 but not by other peptides, including P113 (Fig. 2D), the Fis1 homologous peptide to P110 (Fig. 1B). Using cultured human neuroblastoma SH-SY5Y cells, we next determined the effect of peptide P110 (1 µM) on the interaction between Drp1 and Fis1 in the presence or absence of the mitochondrial stressor 1-methyl-4-phenylpyridinium (MPP+) (a specific mitochondrial complex I inhibitor and a neurotoxin causing Parkinsonism). Immunoprecipitation analyses demonstrated a great increase in the Drp1/Fis1 interaction by MPP+ treatment as compared to that in control cells. Treatment with P110 abolished this interaction in MPP+-treated cells (Fig. 2E). Importantly, the interaction of Drp1 with other mitochondrial adaptors, including Mff and MIEF1, did not change after MPP+ treatment and was unaffected by P110 treatment (Fig. 2E). These data further support our hypothesis that P110 selectively inhibits Drp1/Fis1 interaction and not the interaction with other mitochondrial fission proteins. Thus, we selected P110 as a peptide candidate to further determine its activity in cell culture.
P110 inhibited Drp1 translocation to the mitochondria and mitochondrial fission in cultured cells subjected to mitochondrial stressors
Drp1 translocation from cytosol to the mitochondria is a hallmark of mitochondrial fission. We determined whether peptide inhibitor P110 inhibited the translocation of Drp1 to the mitochondria in cultured SH-SY5Y cells treated with MPP+ (Fig. 3), CCCP (carbonyl cyanide m-chloro phenyl hydrazone, a mitochondrial uncoupler, Fig. 3), H2O2 (hydrogen peroxide, an oxidative stress inducer, supplementary material Fig. S3) or rotenone (a mitochondrial complex I inhibitor and a neurotoxin causing Parkinsonism, supplementary material Fig. S3). Treatment with P110 (1 µM) abolished the increase in Drp1 association with the mitochondrial fractions under all stress conditions, suggesting that P110 blocked Drp1 translocation to the mitochondria induced by a number of mitochondrial stressors (Fig. 3A,B). Drp1 likely functions through self-assembly to drive the membrane constriction event that is associated with mitochondrial division (Ingerman et al., 2005; Zhu et al., 2004). We found that P110 treatment inhibited tetramer formation of Drp1 induced by MPP+ in cultured SH-SY5Y cells (Fig. 3C). Moreover, P110 treatment inhibited the increase in GTPase activity of Drp1 following MPP+ treatment, but did not affect the GTPase activity under basal conditions (Fig. 3D). However, P110 treatment did not affect the total levels of Drp1 or other proteins associated with mitochondrial fusion and fission in any of the treatment groups (supplementary material Fig. S3B). These findings confirmed the selectivity of P110 for activated Drp1. Finally, confocal imaging analysis demonstrated that Drp1 is localized to mitochondria, after the SH-SY5Y neuronal cells were exposed to MPP+ or CCCP (Fig. 3E) and P110 treatment abolished induced co-localization (Fig. 3E). In contrast, P113, the Fis1-derived homolog of P110, had no effects on blocking the association of Drp1 with the mitochondria under the same conditions (Fig. 3E). Taken together, these data indicate that P110 is a selective inhibitor of Drp1 translocation to the mitochondria in cultured cells subjected to mitochondrial stressors.
Fig. 3.

Peptide P110 specifically inhibited Drp1 interaction with the mitochondria in cultured SH-SY5Y neuronal cells exposed to mitochondrial stressors. Cultured human SH-SY5Y neuronal cells were treated with peptide P110 (1 µM) for 30 minutes prior to a 1 hour incubation in the absence or presence of MPP+ (2 mM) or CCCP (10 µM). (A) Western blot analysis of mitochondrial fractions was determined by the indicated antibodies. VDAC was used as a loading control. (B) Quantification of the levels of Drp1 is provided in a histogram. Data are expressed as means ± s.e. of three independent experiments. (C) Total lysates of SH-SY5Y cells from the indicated groups were subjected to western blot analysis using reducing or non-reducing gels, and monomeric (∼75 kDa) and tetrameric (∼200 kDa) Drp1 levels were determined. (D) The GTPase activity of immunoprecipitated Drp1 from cultured cells was expressed as the mean ± s.e. of three independent experiments. (E) Confocal microscopy of stained SH-SY5Y cells with anti-Drp1 (1:500 dilution) and anti-Tom20 (a marker of mitochondria, 1:500 dilution) antibodies following incubation with either MPP+ (2 mM for 1 hour) or CCCP (5 µM for 30 min). Lower panels show enlarged areas of the white boxes in the above panels. Scale bar: 0.5 µm. Pearson's coefficient/cell (Drp1/Tom20 co-localization) was determined using confocal microscopy (Fluoview FV100, Olympus) and is provided as a histogram, as mean ± s.e. of three independent experiments. (#P<0.05 versus control cells; *P<0.05 versus cells treated with MPP+ or CCCP.)
P110 corrected dysregulation of mitochondrial morphology following stress
Disruption of the balanced process between mitochondrial fusion and fission leads to excessive mitochondrial fragmentation, which has been linked to mitochondrial dysfunction and cell death (Qi et al., 2011; Westermann, 2010). We determined the effects of Drp1 peptide inhibitor, P110, on mitochondrial morphology in cultured SH-SY5Y cells in response to mitochondrial stressors. Extensive mitochondrial fragmentation evidenced by small, round or dot-like staining patterns was induced in cultures treated with MPP+ or CCCP and P110 greatly reduced this mitochondrial fragmentation in cells exposed to MPP+ (from 50% to 14% of the cells; P<0.05) and CCCP (from 63% to 23% of the cells; P<0.05; Fig. 4A). The extent of inhibition of mitochondrial fragmentation by P110 treatment was similar to that from the group treated with Drp1 siRNA under the same conditions. Again, P113 has no effect on stress-induced mitochondrial fragmentation (Fig. 4A). Importantly, P110 did not affect mitochondrial network integrity under basal conditions (Fig. 4A). Furthermore, electron microscopy analysis supported our observation that P110 treatment had minimal effect on mitochondrial morphology in normal cells (Fig. 4B). To determine whether the effect of P110 is specific for Drp1, we treated Drp1 wild-type (WT) and Drp1 knockout (KO) mouse embryonic fibroblasts (MEF) with MPP+. We found that P110 treatment consistently reduced mitochondrial fragmentation induced by MPP+ in Drp1 WT MEF cells. However, there was no improvement of mitochondrial morphology by P110 treatment in Drp1 KO MEF under the same conditions (Fig. 4C). Given that MPP+ induced mitochondrial fragmentation in Drp1 KO MEF cells, it remains to be determined whether mitochondrial fragmentation in these cells represents a compensatory effect of other proteins involved in regulation of mitochondrial morphology.
Fig. 4.

P110 treatment reduced mitochondrial fragmentation. (A) Cultured SH-SY5Y cells were treated with P110 (1 µM) or P113 (1 µM) for 30 minutes followed by incubation with MPP+ (2 mM, for 4 hours) or CCCP (5 µM, for 30 minutes). The cells were then stained with anti-Tom20 antibody (green) and Hoechst stain (scale bar: 0.5 µm). Mitochondrial morphology was analyzed using a 63× oil immersion lens. The boxed area in each upper micrograph is enlarged under each micrograph. (B) Mitochondrial morphology was analyzed by electron microscopy of the indicated groups. A total of 100 cells in each experimental group was observed (scale bars: 1 µm). (C) Drp1 wild-type or knockout MEF were treated with MPP+ (2 mM for 8 hours) following the addition of P110 (1 µM). The cells were then stained with anti-Tom20 antibodies (green) and Hoechst stain (scale bar: 0.5 µm). Histogram: the percentage of cells with fragmented mitochondria relative to the total number of cells is presented as the mean ± s.e. of three independent experiments, counted by an observer blinded to the experimental conditions. At least 200 cells per group were counted. Con, control.
Treatment with P110 reduced mitochondrial dysfunction by suppressing the production of mitochondrial ROS
Drp1-dependent mitochondrial fission impairment has been shown to occur during the early stage of mitochondrial dysfunction (Barsoum et al., 2006; Yuan et al., 2007). We therefore determined whether the Drp1 inhibitor, P110, affects mitochondrial dysfunction. Treatment of SH-SY5Y cells with P110, but not its Fis1-derived homolog, P113, abolished MPP+-induced production of mitochondrial superoxide, a major source of mitochondrial ROS (Fig. 5A; supplementary material Fig. S4).
Fig. 5.

Treatment with P110 reduced mitochondrial dysfunction. SH-SY5Y cells were treated with P110 (1 µM) or P113 (1 µM) followed by incubation of MPP+ (2 mM) for 2 hours. (A) Mitochondrial superoxide production was determined using the mitochondrial superoxide indicator MitoSOXTM Red. Nuclei were stained with Hoechst (blue). Upper panel: representative data. Scale bar: 0.5 µm. Lower panel: quantification of the red fluorescence of three independent experiments was provided in a histogram. P110 Ala-scan analogs (1–7) were tested in the same assay at 1 µM each. (B) Mitochondrial membrane potential was determined using TMRM. The fluorescence was quantified using a fluorescence reader, and the data are presented as means ± s.e. of percentage relative to control mitochondria from three independent experiments. #P<0.05 versus control cells; *P<0.05, **P<0.01 versus MPP+- or CCCP-treated cells. (C) Isolated mouse liver mitochondria (50 µg) were incubated with Drp1 recombinant protein (50 ng) in the presence or absence of P110 (1 µM). Production of mitochondrial superoxide was determined over 8 minutes using MitoSOXTM. The data are percentage change relative to the basal levels presented as means ± s.e. of three independent experiments. #P<0.05 versus control mitochondria; *P<0.05 versus mitochondria treated with Drp1 alone. (D) Mitochondrial membrane potentials of isolated mouse liver mitochondria were determined using TMRM, as described in the Materials and Methods section. NAC (2.5 mM) was used as a positive control. Upper panel: protein levels of cytochrome c in the mitochondria were determined by western blot analysis with anti-cytochrome c antibodies in the indicated groups. (E) Total ROS production was determined in SH-SY5Y cells using CM2-HDCFA (1 µM for 30 minutes at 37°C) in the indicated groups. The data (means ± s.e. of three independent experiments) are presented as percentage change relative to the control group. *P<0.05 versus either MPP+- or CCCP-treated group; #P<0.05 versus control group. Con, control.
We were struck by the findings that although P113 is different from P110 by only three conservative substitutions (Glu for Asp, Lys for Arg and Ser for Thr), only P110 exerted biological activity. To further determine the contribution of each of the amino acids of P110 on the bioactivity of the peptide, we used an Ala-scan (Brunel et al., 2006) approach, in which each amino acid in the peptide was substituted individually with an alanine (supplementary material Fig. S5). The Ala-scan analogs of P110 had limited or no effect relative to the effects of P110 on mitochondrial ROS production (Fig. 5A, lower panel), suggesting that each of the 7 amino acids of P110 contributes to the biological effects of the peptide.
We also found that P110 treatment significantly recovered mitochondrial membrane potential (MMP) in the presence of MPP+ or CCCP (Fig. 5B) and improved the assembly of the mitochondrial electron transport chain (ETC) (supplementary material Fig. S6) in cultured SH-SY5Y cells treated with MPP+.
To determine the direct effects of activated Drp1 on the mitochondria, we incubated Drp1 recombinant protein with isolated mouse liver mitochondria, using the approach as in (Johnson-Cadwell et al., 2007) with some modification. Mitochondrial superoxide production followed by the addition of Drp1 recombinant increased by ∼50% (P<0.05, n = 3) relative to basal conditions (Fig. 5C). Importantly, adding either P110 or Drp1 dominant negative mutant (Drp1K38A) abolished Drp1-induced ROS elevation in the isolated mitochondria (Fig. 5C, P<0.05, n = 3). Therefore, addition of activated Drp1 directly caused mitochondrial ROS production. The mechanism by which Drp1 induces mitochondrial ROS production, in vitro, and the role of GTPase activity in this effect is currently under investigation.
Drp1 incubation with mitochondria resulted in a decline in cytochrome c in the mitochondrial fraction, an effect that was inhibited in the presence of P110 (Fig. 5D, upper panel), indicating that P110 inhibited cytochrome c release from the mitochondria under these conditions. Cytochrome c release is one of the signs of mitochondrial membrane potential (MMP) dissipation. We therefore examined the MMP of these isolated mouse liver mitochondria in the presence or absence of P110. Drp1 addition caused a significant reduction of MMP, and treatment with P110 improved MMP, similar to that of the Drp1K38A-treated group or to a group treated with the anti-oxidant, N-acetyl cysteine (NAC; Fig. 5D). Finally, P110 treatment greatly reduced total ROS produced by the mitochondrial stressors, MPP+ or CCCP (Fig. 5E). Taken together, these data demonstrated that inhibition of stressor-induced hyper-activation of Drp1 by P110 reduced mitochondrial damage by suppressing mitochondrial ROS production, and improving mitochondrial membrane potential and mitochondrial integrity.
P110 reduced programmed cell death and improved cell viability by protecting mitochondrial integrity
Impairment of mitochondrial fission is closely linked with increased apoptosis and autophagic cell death in response to various stimuli by increasing mitochondrial depolarization and ROS production (Wikstrom et al., 2009). Moreover, Drp1 hyper-activation on the mitochondria has been recently demonstrated to participate in TNFα-induced necrotic cell death (Wang et al., 2012), suggesting that Drp1-dependent mitochondrial dysfunction may represent a convergent point of several programed cell death (PCD) pathways. Thus, we determined whether Drp1 peptide inhibitor, P110, affected PCD under stress conditions by inhibiting aberrant mitochondrial fission. P110 greatly reduced the accumulation of active Bax on the mitochondria, blocked the release of cytochrome c from the mitochondria and improved decreased Bcl-2 levels on the mitochondria in cultured SH-SY5Y neuronal cells treated with MPP+ (Fig. 6A). Thus, P110 treatment inhibited the early stage of apoptosis. The number of apoptotic SH-SY5Y cells after exposure to MPP+ was also greatly reduced by P110 treatment (Fig. 6B). Autophagy is another consequence of excessive mitochondrial fission (Twig et al., 2008; Wikstrom et al., 2009). Consistent with previous studies (Zhu et al., 2007), we found that MPP+ caused excessive autophagy, as evidenced by the induction of autophagic marker LC3 (microtubule-associated protein 1, light chain 3, also known as ATG8). Treatment with P110 reduced the number of LC3-positive puncti in the cells and LC3 cleavage (LC3 I to LC3 II) (Fig. 6C,D), suggesting an inhibition of excessive autophagy. Finally, we found that reduction in the PCD by treatment with the peptide inhibitor, P110, was associated with improved cell viability in cultured SH-SY5Y cells in response to stress (Fig. 6E). Again, treatment with P113, the Fis1-derived P110 homologous peptide, or with the seven P110 Ala-scan analogs had no significant effect on cell viability under the same conditions (Fig. 6E). Taken together, our data are consistent with the previous study that Drp1 hyper-activation plays an active role in different types of cell death. Importantly, the selective mitochondrial fission inhibitor, peptide P110, rescued cells from these cell death pathways.
Fig. 6.

Treatment with P110 reduced cell death induced by mitochondrial stressors. (A) SH-SY5Y neuronal cells were treated with P110 followed by exposure to MPP+ (2 mM for 1 hour). The active form of Bax (NT-Bax), cytochrome c and Bcl-2 on the mitochondria were determined by western blot analysis with the indicated antibodies. Shown are representative data of three independent experiments. (B) Apoptosis was determined using annexin V staining 8 hours after MPP+ exposure. The number of apoptotic cells was expressed as mean ± s.e. of percentage relative to total number of cells from three independent experiments. (C) Cultured SH-SY5Y cells were stained with anti-LC3 antibodies (green); punctate LC3 staining is an indicator of autophagy. Nuclei were stained with Hoechst dye. Upper panel: representative images. Lower panel: histogram depicting the number of LC3-positive puncta/cell in the indicated groups. The data are presented as the means ± s.e. of three independent experiments, scored by an observer blinded to the experimental conditions. (D) Western blot analysis of LC3 I versus LC3 II was determined with anti-LC3 antibodies. Histogram: the data are expressed as means ± s.e. of three independent experiments. (E) Cell viability was measured by MTT assay. SH-SY5Y cells were treated with MPP+ (2 mM for 24 hours) following treatment with TAT, P110, P113 or P110 Ala-scan analogs (1–7, 1 µM each). #P<0.05 versus control group; *P<0.05, **P<0.01 versus MPP+-treated cells. Con, control.
P110 treatment reduced neurite degeneration of dopaminergic neurons in a Parkinsonism culture model
Aberrant mitochondrial fission has been highlighted in a number of neurodegenerative diseases, such as Parkinsonism, indicating a potential mechanism by which mitochondrial dysfunction contributes to neurodegenerative diseases (Reddy et al., 2011). MPP+ induces selective degeneration of dopaminergic neurons in a chemical experimental model of Parkinsonism. We therefore determined the effects of P110 treatment on the viability of primary dopaminergic neuronal cells in response to MPP+ exposure. Consistent with our findings above, treatment with the Drp1 peptide inhibitor, P110, reduced mitochondrial fragmentation and mitochondrial ROS production in primary dopaminergic neurons exposed to MPP+ (Fig. 7A–D). Importantly, we found that P110 treatment reduced neurite loss of dopaminergic neurons, which were identified by tyrosine hydroxylase (TH), a marker of dopaminergic neurons (Fig. 7B,E). These data suggest that inhibition of Drp1-induced mitochondrial dysfunction by P110 decreased neuronal degeneration in a cell culture model of Parkinsonism. It remains to be determined whether the peptide inhibitor P110 is useful in preventing or slowing down the progression of Parkinsonism, in vivo.
Fig. 7.

Inhibition of Drp1 by P110 reduces loss of neurites in primary dopaminergic neuronal cells exposed to MPP+. Primary rat dopaminergic neurons (cultured for 6 days) were treated without or with P110 (1 µM) followed by treatment with or without MPP+ (1 µM). (A) At 2 hours following MPP+ treatment, the cells were stained with MitoSOXTM (to measure mitochondrial superoxide production) and anti-TH antibodies (a marker of dopaminergic neurons). (B) At 15 hours after MPP+ treatment, cells were stained with anti-TH antibody and anti-Tom20 antibody (a marker of mitochondria). Right-hand panels are enlarged images of boxed areas in each left-hand panel. Scale bar: 0.5 µm. Quantification of mitochondrial superoxide production (C), mitochondrial fragmentation (D) and neurite loss (E) are provided in histograms as means ± s.e. of three independent experiments. Con, control.
Discussion
In this study, we report the design and use of a novel selective peptide inhibitor of excessive mitochondrial fission, P110. P110 selectively inhibited the activation of fission protein, Drp1, and the interaction of Drp1 with Fis1 on mitochondria only under stressed conditions in cultured neuronal cells, leading to protection from mitochondrial dysfunction and excessive fission and from cell death (Fig. 8). The protective effects of P110 are supported by several lines of evidence. a) P110 treatment reduced mitochondrial fragmentation in neuronal cells exposed to several mitochondrial toxins; b) P110 treatment reduced mitochondrial ROS (O2−) production and subsequently improved mitochondrial membrane potential and mitochondrial integrity; c) P110 increased cell viability by reducing apoptosis and autophagic cell death; d) P110 treatment reduced loss of neurite in primary dopaminergic neurons in a Parkinsonism cell culture model by reducing mitochondrial fragmentation and mitochondrial ROS production. Importantly, we also found that P110 treatment had minimal effects on mitochondrial fission and cell viability under non-stressed conditions. Taken together, these data suggest that peptide inhibitor P110 is a specific inhibitor for activated Drp1 under oxidative stress, but not under basal conditions.
Fig. 8.
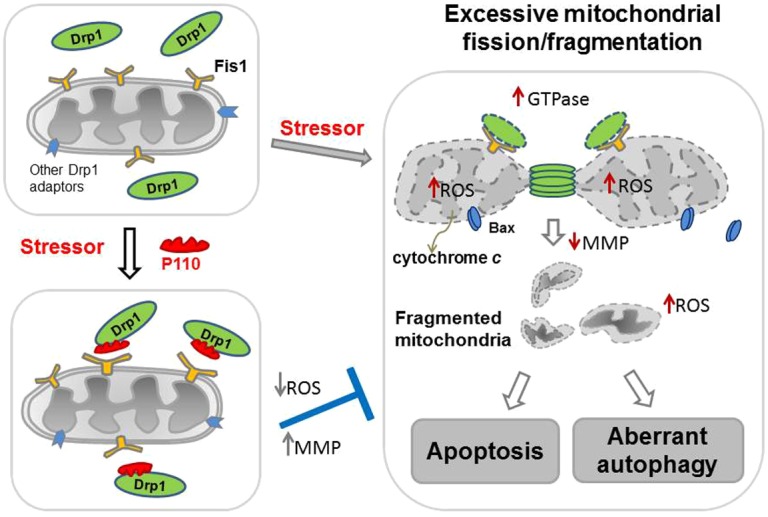
Summary scheme. Under normal conditions (upper left-hand panel), Drp1 (green) is located mostly in the cytosol. Following application of a variety of cell stressors (right-hand panel), Drp1 translocates to the mitochondria. The mitochondria-associated Drp1 has increased GTPase activity. On the mitochondrial surface, Drp1 binds to its mitochondrial adaptors, such as Fis1, a second component of the mitochondrial fission machinery, under stress conditions, and wraps around the mitochondrial tubules to drive mitochondrial division (fission). Under stress conditions, association of Drp1 with the mitochondria causes an increase in mitochondrial ROS production, which subsequently leads to dissipation of mitochondrial membrane potential (MMP). As a consequence, Bax, a pro-apoptotic factor, translocates from the cytosol to the mitochondria and the mitochondrial inter-membrane space protein, cytochrome c, is released to the cytosol, leading to increased apoptosis. Increased mitochondrial ROS production and decreased MMP also lead to excessive mitochondria-related autophagy, another type of cell death. Drp1-derived peptide inhibitor, P110 (red), binds to Drp1 and inhibits Drp1's GTPase activity and association with the mitochondria (lower left-hand panel), probably by blocking the interaction between Drp1 and Fis1 on the mitochondria. P110 treatment thus prevents the Drp1-induced cascade of mitochondria-induced excessive fission and cell death by inhibiting the translocation of Drp1 to the mitochondria.
What is the molecular mechanism by which P110 inhibits Drp1's functions? Although P110 is derived from Drp1 and represents a homologous sequence to Fis1 (Fig. 1), we suggest that it directly binds to Drp1 (Fig. 8), because it inhibits the GTPase activity of Drp1 in the absence of Fis1 (Fig. 2A), whereas the homologous peptide derived from Fis1 (P113) did not. Furthermore, the peptide is derived from the GTPase domain of Drp1 (Fig. 1A), yet it does not affect the GTPase activity of a number of other GTPases including, OPA1, MFN1 and dynamin 1 (Fig. 2B). Finally, P110 inhibits the direct interaction between Drp1 and Fis1 in vitro (Fig. 2D) and in SH-SY5Y neuronal cells in culture (Fig. 2E), suggesting that it either directly competes with Fis1 binding or that it allosterically prevents the exposure of the Fis1-binding site on Drp1. Given that P110 suppressed Drp1 polymerization induced by mitochondrial stress in SH-SY5Y cells (Fig. 3C), and since Drp1 polymers are required for its increased GTPase and fission activity (Zhu et al., 2004), it is possible that P110 inhibition of either or both GTPase and polymerization of Drp1 affect directly or indirectly its interaction with Fis1. These possibilities remain to be examined. In addition, whether MPP+-induced Drp1 tetramer formation through increased cellular oxidative stress remains to be determined.
Our findings that any change in the composition of P110 (conservative substitution of the charged amino acids, represented by P113 and using sequential substitution of each amino acid with alanine) resulted in a loss of all the effects on mitochondrial ROS and cell death induced by mitochondrial toxins is of interest. Given that the sequence in Drp1 may be derived from an α-helix structure (Fig. 1C), this region could provide more than one interaction surface; it can bind directly to Drp1, and inhibit its GTPase activity, in addition to competing for Fis1 interaction with Drp1. Alternatively, inhibition of the GTPase activity by P110 may cause a conformational (allosteric) change in Drp1 that prevents its interaction with Fis1.
Our previous studies showed that short peptides derived from interaction sites between two proteins act as highly specific inhibitors and are effective drugs in basic research and in animal models of human diseases, such as myocardial infarction and hypertension (Inagaki et al., 2003; Palaniyandi et al., 2009; Qi et al., 2008). We proposed that because the peptides are flexible and represent part of the natural binding site, they may be superior and more selective inhibitors of PPI as compared with more rigid small molecules (Qvit and Mochly-Rosen, 2010; Ron and Mochly-Rosen, 1995; Souroujon and Mochly-Rosen, 1998).
While our study characterizing peptide P110 was carried out, Mff and MIEF1 have been identified as additional adaptors of Drp1 on the mitochondria in mammals (Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011). We therefore conducted additional studies and found that P110 had no effect on Mff and MIEF1 protein levels (supplementary material Fig. S3B) and did not block the interaction between Drp1 and Mff or between Drp1 and MIEF1 (Fig. 2E, right-hand panels). Thus, P110 inhibition of oxidative stress induced fission is independent of Mff and MIEF1, at least in SH-SY5Y neuronal cell cultures. Consistent with previous studies (Otera et al., 2010; Zhao et al., 2011), the interaction between Drp1 and Fis1 in control cells was weaker when compared to those on Drp1/Mff and Drp1/MIEF1 (Fig. 2E) and peptide 110 had no effect on these interactions (Fig. 2E). The data are consistent with the finding that Fis1 is not essential for basal mitochondrial fission in mammalian cells, as fission was unaffected in Fis1 deleted HeLa cells (Otera et al., 2010). However, we found that in SH-SY5Y cells treated with MPP+, there was a great increase in the Drp1/Fis1 interaction, which was abolished in the presence of P110. These data suggest a new role for Fis1 in regulating excessive mitochondrial fission induced by mitochondrial oxidative stress. It may also explain our observation that P110 treatment inhibited excessive mitochondrial fission under stressed conditions, but exerted no significant effect on mitochondrial morphology in normal cells.
Developing strategies to limit mitochondrial damage and to ensure cellular integrity by inhibiting mitochondrial fission impairment can identify important new therapeutics. The first inhibitor of mitochondrial fission, Mdivi-1, was identified using a chemical screen in yeast cells harboring a mitochondrial fusion-defective mutant fzo1-1 (human mitofusin 1) (Cassidy-Stone et al., 2008). Mdivi-1 increases growth rate in yeast and inhibits mitochondrial division in yeast and mammals by blocking Dnm1 (yeast Drp1) polymerization (Cassidy-Stone et al., 2008). However, whether Mdivi-1 affects mammalian Drp1 in the same way as in yeast Dnm1 remains to be determined.
Although P110 is a peptide, it is bioavailable. It is a conjugate between the Drp1-dervied sequence in region 110 (cargo) and the cell-permeable peptide carrier TAT47–57, which has been widely used for cargo delivery in cultures and in vivo. Our previous studies showed that TAT-conjugated peptides can quickly enter cells and pass through the blood-brain barrier (Bright et al., 2004; Qi et al., 2008) and have extensive bio-distribution within minutes after single dose treatment in vivo and in cultures (Begley et al., 2004; Miyaji et al., 2011). Thus, P110 may be useful in treatment of diseases associated with excessive mitochondrial fission, such as Parkinsonism.
A causal role for Drp1 in impairment of mitochondrial fission in the pathogenesis of Parkinsonism has been recently reported (Büeler, 2009). The Parkinsonism-inducing neurotoxins, 6-hydroxy dopamine, rotenone, and MPP+, all trigger Drp1 translocation to the mitochondria and mitochondrial fragmentation (fission), thus leading to dopaminergic cell death in neuronal cultures. Parkinsonism-related proteins PINK1, parkin, DJ-1 and alpha-synuclein appear to control mitochondrial function by associating with Drp1 and regulating mitochondrial fusion/fission events. It is clear that neurotoxins causing Parkinsonism and Parkinsonism-associated genes are related to mitochondrial dynamics. Thus, controlling Drp1-mediated mitochondrial fission impairment in Parkinsonism may be of particular importance to inhibit neurodegeneration. Using cell culture models of Parkinsonism, we found that in response to MPP+, P110 reduced dopaminergic neuronal degeneration by inhibiting Drp1-mediated mitochondrial dysfunction. In addition, impaired mitochondrial dynamics and excessive mitochondrial fission have been connected to a number of neurological disorders, including neurodegenerative diseases (Knott et al., 2008), hypertensive encephalopathy (Qi et al., 2008), stroke (Liu et al., 2012) and neurologic pain (Ferrari et al., 2011). Therefore, an inhibitor of Drp1, such as P110, may be a useful treatment for diseases in which impairment of mitochondrial dynamics occurs.
Materials and Methods
Materials
MPP+, CCCP, NAC, protease inhibitor cocktail and phosphatase inhibitor cocktail were purchased from Sigma-Aldrich (USA). Recombinant protein Drp1, Fis1, MFN1 and OPA1 were from Abnova (Walnut, CA, USA). Antibodies for Tom20 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), Drp1 (DLP1) and OPA1 were from BD Biosciences (Rockville, MD, USA). Antibodies for Fis1, Mff and MIEF1 were from Proteintech (Chicago, IL, USA), VDAC and cytochrome c were from MitoScience (Eugene, OR, USA) and pan-actin was from Sigma-Aldrich (USA). Antibodies to Bcl-2 and Bax were from Millipore (Billerica, USA), antibodies to mitofusin 1 and 2 were from Abcam (Cambridge, MA, USA) and antibodies to LC3 were from Cell Signaling Biotechnology (Danvers, MA, USA). Anti-mouse IgG and anti-rabbit IgG, peroxidase-linked species-specific antibodies were from Thermo Scientific (Rockford, IL, USA). The Drp1/Fis1 peptides were synthesized at Dr Daria Mochly-Rosen's lab at Stanford University and conjugated to TAT-carrier peptide (amino acids 47–57).
Peptide synthesis
Peptides were synthesized using microwave chemistry on a Liberty Microwave Peptide Synthesizer (CEM Corporation, Matthews, NC, USA). Peptides were synthesized as one polypeptide with TAT47–57 carrier in the following order: N-terminus–TAT–spacer (Gly-Gly) – cargo (corresponding to regions 108,109, 110, 111, 112 or 113)–C-terminus. The C-termini of the peptides were modified to C(O)-NH2 using Rink Amide AM resin to increase stability (Sabatino and Papini, 2008). Peptides were analyzed by analytical reverse-phase high-pressure liquid chromatography (RP-HPLC) (Shimadzu, MD, USA) and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) and purified by preparative RP-HPLC (Shimadzu, MD, USA). Peptides were 85–95% pure. The detailed procedure is described below.
All commercially available solvents and reagents were used without further purification: dichloromethane (DCM), N-methyl-2-pyrrolidone (NMP), triisopropylsilane (TIS), N,Ndiisopropylethylamine (DIEA), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU), 1-hydroxybenzotriazole (HOBt) and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich Chemicals (MO, USA); dimethylformamide (DMF) (Alfa Aesar, MA, USA); piperidine (Anaspec, CA, USA) rink amide AM resin (substitution 0.49 mmol/g, CBL Biopharma LLC, CO, USA), Fmoc-protected amino acids were obtained from Advanced ChemTech and GL Biochem (KY, USA and Shanghai, China). Side chains of the amino acids used in the synthesis were protected as follows: Boc (Lys/Trp), But (Ser/Thr/Tyr), OBut (Asp/Glu), Pbf (Arg) and Trt (Asn/Cys/Gln/His). Peptides were chemically synthesized using Liberty Microwave Peptide Synthesizer (CEM Corporation, Matthews, NC, USA) with an additional module of Discover (CEM Corporation, Matthews, NC, USA) equipped with a fiberoptic temperature probe for controlling the microwave power delivery following the fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (tBu) strategy in a 30 ml teflon reaction vessel. Deprotection was performed with 20% piperidine in DMF with 0.1 M HOBt. Coupling reactions were performed with HBTU in DMF (0.45 M, 0.9 eq), amino acids in DMF (0.2 M, 1 eq) and DIEA in NMP (2 M, 2 eq). Each deprotection and coupling reaction was performed with microwave energy and nitrogen bubbling. Microwave cycle included two deprotection steps of 30 sec and 180 sec, each. All coupling reactions lasted 300 sec. Peptide cleavage from the resin and deprotection of the amino acids side chain were carried out with TFA/TIS/H2O /phenolsolution (90:2.5:2.5:5 v/v/v/v) for 4 h at room temperature. The resin was washed with TFA. The crude products were precipitated with diethyl ether, collected by centrifugation, dissolved in H2O/CH3CN and lyophilized. Products were analyzed by analytical RP-HPLC (Shimadzu LC-20 equipped with: CBM-20A system controller, SPD-20A detector, CTO-20A column oven, 2× LC-20AD solvent delivery unit, SIL-20AC autosampler, DGU-20A5 degasser from Shimadzu, MD, USA) using an ultro 120 5μm C18Q (4.6 mm ID 150 mm) column (Peeke Scientific, CA, USA) at 1 ml/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). For separation, a linear gradient of 5–95% B in 45 min was applied and the detection was at 215 nm. Products were purified by preparative reverse-phase high-pressure liquid chromatography (RPHPLC; Shimadzu LC-20) equipped with CBM-20A system controller, SPD-20A detector, CTO20A column oven, 2× LC-6AD solvent delivery unit and FRC-10A fraction collector from Shimadzu, MD, USA), using an XBridge Prep OBD C18 5μm (19 mm × 150 mm) column (Waters, MA, USA) at 10 ml/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). For separation, a linear gradient of 5–95% B in 45 min was applied and the detection was at 215 nm. Where indicated, peptides were conjugated to Tat47-57 carrier peptide through a spacer of two Gly amino acids added to each of the peptides (at the C terminus) as part of the synthesis.
Cell culture
Human neuroblastoma SH-5YSY cells were maintained in modified Eagle's medium (50%) and F12 medium (50%) supplemented with 10% (v/v) heat-inactivated fetal calf serum and 1% (v/v) penicillin/streptomycin.
Primary neurons from E18 rat midbrain tissue (BrainBits, Springfield, IL, USA) were seeded at about 50000 cells on cover slides coated with poly-d-lysine/laminin in neurobasal medium supplemented with 2% B27 and 0.5 mM glutamate. At 6 DIV, cells were treated with 1 µM MPP+. All cultured cells were maintained at 37°C in 5% CO2/95% air.
Mouse embryonic fibroblasts (MEF) of Drp1 wild-type and knockout were a generous gift of Dr Hiromi Sesaki from John Hopkins University. The cells were maintained in DMEM supplemented with 10% (v/v) heat-inactivated fetal calf serum and 1% (v/v) penicillin/streptomycin.
GTPase activity assay
Recombinant Drp1, MFN1, OPA1 or dynamin-1 (25 ng) were incubated with the indicated peptides for 30 min. GTPase activity of the proteins was determined using a GTPase assay kit (Novus Biologicals, Littleton, CO) according to manufacturer's instructions.
To determine GTPase activity of Drp1 in cultured cells, a total of 1 mg of whole-cell extract was immunoprecipitated overnight with 20 µg of anti-Drp1 antibodies and 50 µl of Protein A/G–agarose, as described in a previous study (Wang et al., 2012). After three washes with lysis buffer and three washes with GTPase buffer (50 mM Tris [pH 7.5], 2.5 mM MgCl2, and 0.02% 2-mercaptoethanol), the beads were incubated with 0.5 mM GTP at 30°C for 1 hr. The released free phosphate was quantified using the GTPase assay kit, as above.
Isolation of mitochondrial-enriched fraction and lysate preparation
SH-5YSHY cells were washed with cold phosphate-buffered saline (PBS) and incubated on ice in lysis buffer (250 mM sucrose, 20 mM HEPES-NaOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, protease inhibitor cocktail, phosphatase inhibitor cocktail) for 30 minutes. Cells were scraped and then disrupted 10 times by repeated aspiration through a 25 gauge needle, followed by a 30 gauge needle. Mouse liver tissue was minced and ground by pestle in lysis buffer. The homogenates were spun at 800 g for 10 min at 4°C and the resulting supernatants were spun at 10,000 g for 20 minutes at 4°C. The pellets were washed with lysis buffer and spun at 10,000 g again for 20 minutes at 4°C. The final pellets were suspended in lysis buffer containing 1% Triton X-100 and were noted as mitochondrial-rich lysate fractions. The mitochondrial membrane protein VDAC was used as marker and loading control.
Chemical cross-linking and immunoprecipitation
For direct binding between Drp1 and Fis1, recombinant Drp1 (100 ng) and Fis1 (100 ng) were incubated in PBS containing 1 mM dithiobis[succinimidyl propionate] (DSP) for 30 min in the presence or absence of Drp1/Fis1 peptides (1 µM each) or control peptide TAT. After termination of the reaction by 10 mM Tris-HCl, pH 7.5, reaction mixtures were subjected to immunoprecipitation with anti-Drp1 antibody in the presence of 1% Triton X-100. Immunoprecipitates were washed with PBS containing 1% Triton X-100 and analyzed by SDS-PAGE and subsequent immunoblotting with antibodies to Fis1 and Drp1.
Co-immunoprecipitation experiments in cultured SH-SY5Y cells were carried out followed by formaldehyde (FA) in vivo crosslinking according to the method described (Hájek et al., 2007) with some modification. Briefly, SH-SY5Y cells were washed in PBS buffer, and proteins were cross-linked by incubating the cells in PBS buffer containing 1% FA for 20 min at room temperature. The crosslinking reaction was terminated by washing in PBS containing 100 mM glycine. The cells were then washed in PBS and lysed in 0.5 ml PBS buffer containing 1% Triton X-100 and protease inhibitor cocktail. Cell suspensions were sonicated and then vortexed, and centrifuged to remove insoluble debris. The resultant supernatants were incubated with antibodies overnight at 4°Cand the agarose was washed in PBS containing 1% Trion X-100 followed by PBS. The immunoprecipitates were dissolved in SDS-sample buffer and analyzed by SDS-PAGE and immunoblotting.
Association of Drp1 with mitochondria
Mouse liver mitochondria were isolated as described above. GST-Drp1 (50 ng) was incubated with the mitochondrial fraction (100 µg protein) in mitochondrial isolation buffer (250 mM sucrose, 20 mM HEPES, 5 mM KCl, 1.5 mM MgCl2, 5 mM glutamate and 5 mM malate). After 8 minutes of incubation, the reaction solution was centrifuged at 15,000 rpm for 2 min at 4°C. The pellet was then washed with the buffer three times. The final pellet was dissolved in the mitochondrial isolation buffer containing 1% Triton X-100. The levels of Drp1 and cytochrome c were then detected by western blot analysis.
Immunocytochemistry
Cells cultured on coverslips were washed with cold PBS, fixed in 4% formaldehyde and permeabilized with 0.1% Triton X-100. After incubation with 2% normal goat serum (to block non-specific staining), fixed cells were incubated overnight at 4°C with antibodies against Drp1 (1:500), Tom20 (1:500), LC3 (1:1000), or tyrosine hydroxylase (TH; Millipore, 1:200). Cells were washed with PBS and incubated for 60 minutes with FITC-labeled goat anti-rabbit antibody and rhodamine-labeled goat anti-mouse antibody (1:500, Invitrogen, USA) followed by incubation with Hoechst dye (1:10000) for 10 minutes. Coverslips were mounted and slides were imaged by confocal microscopy (Olympus, Fluoview FV100). To determine mitochondrial superoxide production in cultures, cells were incubated with 5 µM MitoSOXTM red mitochondrial superoxide indicator (Invitrogen) for 10 min at 37°C and imaged by microscopy. Quantification was carried out using Image J software.
Electron microscopy
Cultured SH-SY5Y cells were treated with peptide P110 followed by treatment with MPP+ (2 mM). Four hours later, the cells were fixed by 2.5% glutaraldehyde in 0.1 M cacodylate buffer. Cells on the film (2 mm thickness, Electron Microscopy Sciences, Hatfield, PA) were post-fixed in ferrocyanide-reduced osmium tetroxide, and soaked in acidified uranyl acetate for 2 hrs. Dehydration at ascending concentrations of ethanol was followed by passage through propylene oxide and embedding in PolyBed 812 resin (Polysciences). Horizontal thin sections of cells were sequentially stained with acidified uranyl acetate, followed by Sato's triple lead stain and examined in a JEOL 1200EX electron microscope. A total of 100 cells in each experimental group were observed. The observer was blinded to the treatment groups.
Transfection with siRNA for Drp1
Small interfering siRNA duplexes for Drp1 or negative control were obtained from Amcon Biotechnology (CA, USA). Adhered SH-5YSY cells at 50% confluency were transfected for 48 hrs with siRNA of Drp1 or control siRNA using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions.
Mitochondrial function measurements in cultured cells
Cultured SH-SY5Y cells in black 96-well plates were treated without or with Drp1 peptide inhibitor, P110 (1 µM), 30 min prior to treatment with MPP+ (2 mM) or CCCP (5 uM). After 2 hrs of treatment, cells were stained with MitoSOX (5 uM, for 10 min, mitochondrial ROS) or tetramethyl rhodamine methyl ester (TMRM) (0.5 uM, for 20 min, MMP) at 37°C. For total ROS detection, cells were treated with MPP+ or CCCP for 24 hrs. ROS level was measured by CM-H2DCFDA (1 µM for 30 min, Invitrogen). Cells were washed three times with PBS. The detection was performed in a fluorescence microplate reader (Tecan, Infinite M1000) according to manufacturer's instructions. All measurements were normalized to the cell number counted using Hoechst stain.
Determination of mitochondrial function in isolated mouse liver mitochondria
Mitochondria were isolated from mouse liver as described above. Mitochondria (0.5 mg protein/ml) were suspended in the mitochondrial buffer containing 250 mM sucrose, 20 mM HEPES-NaOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 5 mM malate and 5 mM glutamate. A 50 µg portion of mitochondrial protein samples were incubated with the indicated recombinant proteins (50 ng) followed by incubation with MitoSOXTM red (10 µM) in the presence or absence of the Drp1 peptide, P110 (1 µM) at 37°C. At 10 min after equilibration, a kinetic detection of MitoSOXTM was performed using black 96-well plates in a fluorescence microplate reader at 510 nm excitation and 580 nm emission. To measure mitochondrial membrane potential (MMP), 50 µg of mitochondria were incubated with recombinant Drp1 (50 ng) followed by incubation with TMRM (0.5 µM) in the presence or absence of P110 (1 µM) or NAC (2.5 mM). Fluorescence detection was performed using black 96-well plates in a fluorescence microplate reader at 560 nm excitation and 690 nm emission and potential was assessed by quenching of the fluorescent signal.
Measurement of cell viability
Human SH-SY5Y cells were treated with Drp1 peptide inhibitor, P110, P110 Ala-scan analogs or control peptide, TAT, for 30 min followed by incubation without or with MPP+ (2 mM for 24 hours). Cell viability was measured using an in vitro toxicology assay, MTT-based kit (Sigma, USA), according to the manufacturers′ instruction.
Detection of apoptosis using binding of fluorescent annexin V
SH-SY5Y cells were treated without or with MPP+ (2 mM) for 8 hrs in the presence or absence of the peptide inhibitor, P110 (1 µM), at 37°C. Cells were washed twice and stained with annexin V-FITC (Biovision, USA) according to manufacturer's instructions, and imaged using a microscope (Nikon Eclipse E600).
Western blot analysis
Protein concentrations were determined by Bradford assay. A 20 µg portion of proteins was resuspended in Laemmli buffer, loaded on SDS-PAGE and transferred on to nitrocellulose membranes. To determine Drp1 polymers, 50 µg total protein were suspended in Laemmli buffer without 2-mercaptoethanol. Membranes were probed with the indicated antibody followed by visualization by ECL.
Statistical methods
Data are expressed as means ± s.e. Unpaired t-test for differences between two groups, one-factor ANOVA with Fisher's test for differences among more than two groups, and Fisher test for categorical data were used to assess significance (P<0.05).
Supplementary Material
Acknowledgments
We thank Dr Hiromi Sesaki for providing Drp1 wild-type and knockout MEF cells, Dr Hisashi Fujioka for carrying out the EM analysis. A patent on the design and application of mitochondrial fission peptide inhibitors has been filed. The authors claim that there is no conflict interest on this work.
Footnotes
Author contributions
X.Q. designed the experiments, performed and analyzed the study, and wrote the manuscript; N.Q. synthesized the peptides and analyzed peptide sequences and evolution; Y.-C.S. determined the effects of Drp1 peptide on mitochondrial ETC complexes, Drp1 association with mouse liver mitochondria and effects of P110 on mitochondrial function and apoptotic cell death; D.M.-R. supervised the design of Drp1 peptides, helped the design and analysis of the study and revised the manuscript.
Funding
This work was partly supported by the University Hospitals Case Medical Center Spitz Brain Health Fund (to X.Q.), the National Institutes of Health [grant number HL52141 to D.M.-R.]; and the Stanford Institute for Neuro-Innovation and Translational Neurosciences (SINTN) (to D.M.-R.). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.114439/-/DC1
References
- Barsoum M. J., Yuan H., Gerencser A. A., Liot G., Kushnareva Y., Gräber S., Kovacs I., Lee W. D., Waggoner J., Cui J.et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911 10.1038/sj.emboj.7601253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley R., Liron T., Baryza J., Mochly–Rosen D. (2004). Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem. Biophys. Res. Commun. 318, 949–954 10.1016/j.bbrc.2004.04.121 [DOI] [PubMed] [Google Scholar]
- Bright R., Raval A. P., Dembner J. M., Pérez–Pinzón M. A., Steinberg G. K., Yenari M. A., Mochly–Rosen D. (2004). Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. 24, 6880–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel F. M., Zwick M. B., Cardoso R. M., Nelson J. D., Wilson I. A., Burton D. R., Dawson P. E. (2006). Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. J. Virol. 80, 1680–1687 10.1128/JVI.80.4.1680-1687.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H. (2009). Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 218, 235–246 10.1016/j.expneurol.2009.03.006 [DOI] [PubMed] [Google Scholar]
- Cassidy–Stone A., Chipuk J. E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M. J., Shaw J. T., Hinshaw J. E., Green D. R.et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. C. (2006a). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- Chan D. C. (2006b). Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99 10.1146/annurev.cellbio.22.010305.104638 [DOI] [PubMed] [Google Scholar]
- Chang C. R., Blackstone C. (2010). Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 1201, 34–39 10.1111/j.1749-6632.2010.05629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Hahn H., Wu G., Chen C. H., Liron T., Schechtman D., Cavallaro G., Banci L., Guo Y., Bolli R.et al. (2001a). Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc. Natl. Acad. Sci. USA 98, 11114–11119 10.1073/pnas.191369098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Wright L. R., Chen C. H., Oliver S. F., Wender P. A., Mochly–Rosen D. (2001b). Molecular transporters for peptides: delivery of a cardioprotective epsilonPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7). Chem. Biol. 8, 1123–1129 10.1016/S1074-5521(01)00076-X [DOI] [PubMed] [Google Scholar]
- Dorn G. W., 2nd, Souroujon M. C., Liron T., Chen C. H., Gray M. O., Zhou H. Z., Csukai M., Wu G., Lorenz J. N., Mochly–Rosen D. (1999). Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc. Natl. Acad. Sci. USA 96, 12798–12803 10.1073/pnas.96.22.12798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estaquier J., Arnoult D. (2007). Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 14, 1086–1094 10.1038/sj.cdd.4402107 [DOI] [PubMed] [Google Scholar]
- Fannjiang Y., Cheng W. C., Lee S. J., Qi B., Pevsner J., McCaffery J. M., Hill R. B., Basañez G., Hardwick J. M. (2004). Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 18, 2785–2797 10.1101/gad.1247904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari L. F., Chum A., Bogen O., Reichling D. B., Levine J. D. (2011). Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J. Neurosci. 31, 11404–11410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S., Gaume B., Bergmann–Leitner E. S., Leitner W. W., Robert E. G., Catez F., Smith C. L., Youle R. J. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525 10.1016/S1534-5807(01)00055-7 [DOI] [PubMed] [Google Scholar]
- Hájek P., Chomyn A., Attardi G. (2007). Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin-like protein 2. J. Biol. Chem. 282, 5670–5681 10.1074/jbc.M608168200 [DOI] [PubMed] [Google Scholar]
- Huang X., Miller W. (1991). Time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 12, 337–357 10.1016/0196-8858(91)90017-D [DOI] [Google Scholar]
- Inagaki K., Chen L., Ikeno F., Lee F. H., Imahashi K., Bouley D. M., Rezaee M., Yock P. G., Murphy E., Mochly–Rosen D. (2003). Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 108, 2304–2307 10.1161/01.CIR.0000101682.24138.36 [DOI] [PubMed] [Google Scholar]
- Inagaki K., Koyanagi T., Berry N. C., Sun L., Mochly–Rosen D. (2008). Pharmacological inhibition of epsilon-protein kinase C attenuates cardiac fibrosis and dysfunction in hypertension-induced heart failure. Hypertension 51, 1565–1569 10.1161/HYPERTENSIONAHA.107.109637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerman E., Perkins E. M., Marino M., Mears J. A., McCaffery J. M., Hinshaw J. E., Nunnari J. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027 10.1083/jcb.200506078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani–Asl A., Germain M., Slack R. S. (2010). Mitochondria: joining forces to thwart cell death. Biochim. Biophys. Acta 1802, 162–166 10.1016/j.bbadis.2009.09.006 [DOI] [PubMed] [Google Scholar]
- James D. I., Parone P. A., Mattenberger Y., Martinou J. C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 278, 36373–36379 10.1074/jbc.M303758200 [DOI] [PubMed] [Google Scholar]
- Johnson–Cadwell L. I., Jekabsons M. B., Wang A., Polster B. M., Nicholls D. G. (2007). ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 101, 1619–1631 10.1111/j.1471-4159.2007.04516.x [DOI] [PubMed] [Google Scholar]
- Kheifets V., Bright R., Inagaki K., Schechtman D., Mochly–Rosen D. (2006). Protein kinase C delta (deltaPKC)-annexin V interaction: a required step in deltaPKC translocation and function. J. Biol. Chem. 281, 23218–23226 10.1074/jbc.M602075200 [DOI] [PubMed] [Google Scholar]
- Kim K. Y., Sack M. N. (2012). Parkin in the regulation of fat uptake and mitochondrial biology: emerging links in the pathophysiology of Parkinson’s disease. Curr. Opin. Lipidol. 23, 201–205 10.1097/MOL.0b013e328352dc5d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Koyanagi T., Mochly–Rosen D. (2011). PKCδ activation mediates angiogenesis via NADPH oxidase activity in PC-3 prostate cancer cells. Prostate 71, 946–954 10.1002/pros.21310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott A. B., Perkins G., Schwarzenbacher R., Bossy–Wetzel E. (2008). Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518 10.1038/nrn2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O., Bourne H. R., Cohen F. E. (1996). An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257, 342–358 10.1006/jmbi.1996.0167 [DOI] [PubMed] [Google Scholar]
- Liu W., Tian F., Kurata T., Morimoto N., Abe K. (2012). Dynamic changes of mitochondrial fission proteins after transient cerebral ischemia in mice. Brain Res. 1456, 94–99 10.1016/j.brainres.2012.03.038 [DOI] [PubMed] [Google Scholar]
- Mammucari C., Rizzuto R. (2010). Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 131, 536–543 10.1016/j.mad.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure S. J., Robinson P. J. (1996). Dynamin, endocytosis and intracellular signalling (review). Mol. Membr. Biol. 13, 189–215 10.3109/09687689609160598 [DOI] [PubMed] [Google Scholar]
- Miyaji Y., Walter S., Chen L., Kurihara A., Ishizuka T., Saito M., Kawai K., Okazaki O. (2011). Distribution of KAI-9803, a novel δ-protein kinase C inhibitor, after intravenous administration to rats. Drug Metab. Dispos. 39, 1946–1953 10.1124/dmd.111.040725 [DOI] [PubMed] [Google Scholar]
- Onoue K., Jofuku A., Ban–Ishihara R., Ishihara T., Maeda M., Koshiba T., Itoh T., Fukuda M., Otera H., Oka T.et al. (2012). Fis1 acts as mitochondrial recruitment factor for TBC1D15 that involved in regulation of mitochondrial morphology. J. Cell Sci. 126, 176–185 10.1242/jcs.111211 [DOI] [PubMed] [Google Scholar]
- Otera H., Wang C., Cleland M. M., Setoguchi K., Yokota S., Youle R. J., Mihara K. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158 10.1083/jcb.201007152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniyandi S. S., Sun L., Ferreira J. C., Mochly–Rosen D. (2009). Protein kinase C in heart failure: a therapeutic target? Cardiovasc. Res. 82, 229–239 10.1093/cvr/cvp001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniyandi S. S., Qi X., Yogalingam G., Ferreira J. C., Mochly–Rosen D. (2010). Regulation of mitochondrial processes: a target for heart failure. Drug Discov. Today Dis. Mech. 7, e95–e102 10.1016/j.ddmec.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C. S., Osellame L. D., Laine D., Koutsopoulos O. S., Frazier A. E., Ryan M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573 10.1038/embor.2011.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Inagaki K., Sobel R. A., Mochly–Rosen D. (2008). Sustained pharmacological inhibition of deltaPKC protects against hypertensive encephalopathy through prevention of blood-brain barrier breakdown in rats. J. Clin. Invest. 118, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Disatnik M. H., Shen N., Sobel R. A., Mochly–Rosen D. (2011). Aberrant mitochondrial fission in neurons induced by protein kinase Cdelta under oxidative stress conditions in vivo. Mol. Biol. Cell 22, 256–265 10.1091/mbc.E10-06-0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvit N., Mochly–Rosen D. (2010). Highly specific modulators of protein kinase C localization: applications to heart failure. Drug Discov. Today Dis. Mech. 7, e87–e93 10.1016/j.ddmec.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P. H., Reddy T. P., Manczak M., Calkins M. J., Shirendeb U., Mao P. (2011). Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118 10.1016/j.brainresrev.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolo A. P., Gomes A. P., Palmeira C. M. (2011). Regulation of mitochondrial biogenesis in metabolic syndrome. Curr. Drug Targets 12, 872–878 10.2174/138945011795529056 [DOI] [PubMed] [Google Scholar]
- Ron D., Mochly–Rosen D. (1995). An autoregulatory region in protein kinase C: the pseudoanchoring site. Proc. Natl. Acad. Sci. USA 92, 492–496 10.1073/pnas.92.2.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino G., Papini A. M. (2008). Advances in automatic, manual and microwave-assisted solid-phase peptide synthesis. Curr. Opin. Drug Discov. Devel. 11, 762–770 [PubMed] [Google Scholar]
- Scott I., Youle R. J. (2010). Mitochondrial fission and fusion. Essays Biochem. 47, 85–98 10.1042/bse0470085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E., Griparic L., Shurland D. L., van der Bliek A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroujon M. C., Mochly–Rosen D. (1998). Peptide modulators of protein-protein interactions in intracellular signaling. Nat. Biotechnol. 16, 919–924 10.1038/nbt1098-919 [DOI] [PubMed] [Google Scholar]
- Suzuki M., Neutzner A., Tjandra N., Youle R. J. (2005). Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J. Biol. Chem. 280, 21444–21452 10.1074/jbc.M414092200 [DOI] [PubMed] [Google Scholar]
- Sweitzer S. M., Wong S. M., Tjolsen A., Allen C. P., Mochly–Rosen D., Kendig J. J. (2004). Exaggerated nociceptive responses on morphine withdrawal: roles of protein kinase C epsilon and gamma. Pain 110, 281–289 10.1016/j.pain.2004.04.004 [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G.et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Jiang H., Chen S., Du F., Wang X. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 10.1016/j.cell.2011.11.030 [DOI] [PubMed] [Google Scholar]
- Westermann B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- Wikstrom J. D., Twig G., Shirihai O. S. (2009). What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 41, 1914–1927 10.1016/j.biocel.2009.06.006 [DOI] [PubMed] [Google Scholar]
- Yoon Y., Krueger E. W., Oswald B. J., McNiven M. A. (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23, 5409–5420 10.1128/MCB.23.15.5409-5420.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Gerencser A. A., Liot G., Lipton S. A., Ellisman M., Perkins G. A., Bossy–Wetzel E. (2007). Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 14, 462–471 10.1038/sj.cdd.4402046 [DOI] [PubMed] [Google Scholar]
- Zhao J., Liu T., Jin S., Wang X., Qu M., Uhlén P., Tomilin N., Shupliakov O., Lendahl U., Nistér M. (2011). Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 30, 2762–2778 10.1038/emboj.2011.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P. P., Patterson A., Stadler J., Seeburg D. P., Sheng M., Blackstone C. (2004). Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 279, 35967–35974 10.1074/jbc.M404105200 [DOI] [PubMed] [Google Scholar]
- Zhu J. H., Horbinski C., Guo F., Watkins S., Uchiyama Y., Chu C. T. (2007). Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 170, 75–86 10.2353/ajpath.2007.060524 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.