

Abstract
Background: In support of the Integrated Risk Information System (IRIS), the U.S. Environmental Protection Agency (EPA) completed a toxicological review of trichloroethylene (TCE) in September 2011, which was the result of an effort spanning > 20 years.
Objectives: We summarized the key findings and scientific issues regarding the human health effects of TCE in the U.S. EPA’s toxicological review.
Methods: In this assessment we synthesized and characterized thousands of epidemiologic, experimental animal, and mechanistic studies, and addressed several key scientific issues through modeling of TCE toxicokinetics, meta-analyses of epidemiologic studies, and analyses of mechanistic data.
Discussion: Toxicokinetic modeling aided in characterizing the toxicological role of the complex metabolism and multiple metabolites of TCE. Meta-analyses of the epidemiologic data strongly supported the conclusions that TCE causes kidney cancer in humans and that TCE may also cause liver cancer and non-Hodgkin lymphoma. Mechanistic analyses support a key role for mutagenicity in TCE-induced kidney carcinogenicity. Recent evidence from studies in both humans and experimental animals point to the involvement of TCE exposure in autoimmune disease and hypersensitivity. Recent avian and in vitro mechanistic studies provided biological plausibility that TCE plays a role in developmental cardiac toxicity, the subject of substantial debate due to mixed results from epidemiologic and rodent studies.
Conclusions: TCE is carcinogenic to humans by all routes of exposure and poses a potential human health hazard for noncancer toxicity to the central nervous system, kidney, liver, immune system, male reproductive system, and the developing embryo/fetus.
Keywords: assessment, cancer/tumors, cardiovascular, epidemiology, immunologic response, Integrated Risk Information System (IRIS), meta-analysis, mode of action, physiologically based pharmacokinetic (PBPK) modeling, trichloroethylene
Trichloroethylene (TCE) is a chlorinated solvent once widely used as a metal degreaser, chemical intermediate and extractant, and component of some consumer products. Total releases to the environment reported to the U.S. Environmental Protection Agency (EPA) Toxics Release Inventory have declined from > 57 million pounds in 1988 to about 2.4 million pounds in 2010 (U.S. EPA 2012b). Because it has a relatively short half-life, TCE is not commonly detected in biomonitoring surveys, and the percentage of subjects with detectable levels (> 0.1 ng/mL) has declined from about 10% to 1% between samples collected in 1988–1994 and those collected in 2003–2004 (Centers for Disease Control and Prevention 2009; Wu and Schaum 2000]. From a regulatory and environmental-cleanup perspective, TCE has been identified in soil or groundwater at > 700 of approximately 1,300 Superfund hazardous waste sites listed by the U.S. EPA (2011c). Additionally, the U.S. EPA has identified TCE as one of the volatile organic compounds to be regulated as a group in drinking water (U.S. EPA 2010, 2011a) and as one of the priority existing chemicals under review for regulatory action under the Toxic Substances Control Act (U.S. EPA 2012a). Indeed, because of TCE’s continued presence in the environment, most people are likely to have some exposure to the compound through contaminated drinking water, ambient outdoor or indoor air, or, less commonly, contaminated foods.
The U.S. EPA’s Integrated Risk Information System (IRIS) program released an updated human health risk assessment of TCE in September 2011 (U.S. EPA 2011d). This assessment was developed over a period of > 20 years and underwent many stages of both internal and external peer review. Key inputs were recommendations for additional analysis and research from a National Research Council (NRC) panel report reviewing the key scientific issues pertaining to TCE hazard and dose–response assessment (NRC 2006). This report, together with a series of issue papers developed by U.S. EPA scientists (Caldwell and Keshava 2006; Chiu et al. 2006a, 2006b; Keshava and Caldwell 2006; Scott and Chiu 2006), provided the foundation for developing an objective, scientifically rigorous human health risk assessment for TCE. The U.S. EPA’s final assessment also incorporated input from two independent peer reviews by the U.S. EPA’s Science Advisory Board (U.S. EPA SAB 2002, 2011), other federal agencies (U.S. EPA 2009b, 2011b), and the public (U.S. EPA 2009a).
Here we describe key findings and scientific issues addressed in the U.S. EPA’s toxicological review of TCE (U.S. EPA 2011d), covering the following topics: a) the role of metabolism in TCE toxicity, which was informed by the development and use of an updated physiologically based pharmacokinetic (PBPK) model; b) the carcinogenicity of TCE, including the development of meta-analyses of epidemiologic studies for informing causal inferences, as recommended by the NRC (2006), and analyses of laboratory animal mechanistic and toxicokinetic data contributing to the evaluation of biological plausibility of the epidemiologic data; and c) noncancer toxicity related to two end points—immunotoxicity and developmental cardiac toxicity—for which substantial new data have become available. Findings and issues related to other important topics not discussed here (e.g., susceptibility, mixtures/coexposures, and dose–response assessment) have been described previously (e.g., Caldwell JC et al. 2008; NRC 2006; U.S. EPA 2011d).
Role of Metabolism in TCE Toxicity
A broad and complex range of relevant information for assessing human health effects of TCE is available. Previous reviews have found TCE to adversely affect the central nervous system (Bale et al. 2011), liver (Bull 2000), kidney (Lash et al. 2000b), immune system (Cooper et al. 2009), and reproductive systems and developing embryo/fetus (NRC 2006). As shown in Figure 1, TCE is metabolized in humans and experimental animal species by both oxidation and glutathione (GSH)-conjugation metabolic pathways, with subsequent production of numerous toxicologically active compounds (Chiu et al. 2006b; Lash et al. 2000a). These include the oxidative metabolites chloral hydrate, trichloroacetic acid (TCA), and dichloroacetic acid, and the GSH conjugation metabolites dichlorovinyl glutathione and dichlorovinyl cysteine. This complex assortment of metabolic compounds is generated from and transported across multiple tissues, making evaluation of mechanistic data especially challenging (Caldwell JC et al. 2008). Liver effects of TCE are thought to result from oxidative metabolites (Buben and O’Flaherty 1985; Bull 2000), whereas effects on kidney are generally associated with metabolites resulting from GSH conjugation (Lash et al. 2000b). The identity of TCE metabolites involved in the induction of other health effects of TCE is less clear, although similarities have been observed between TCE and its oxidative metabolites in the respiratory tract (e.g., Odum et al. 1992) and developmental toxicity (e.g., Johnson et al. 1998a).
Figure 1.
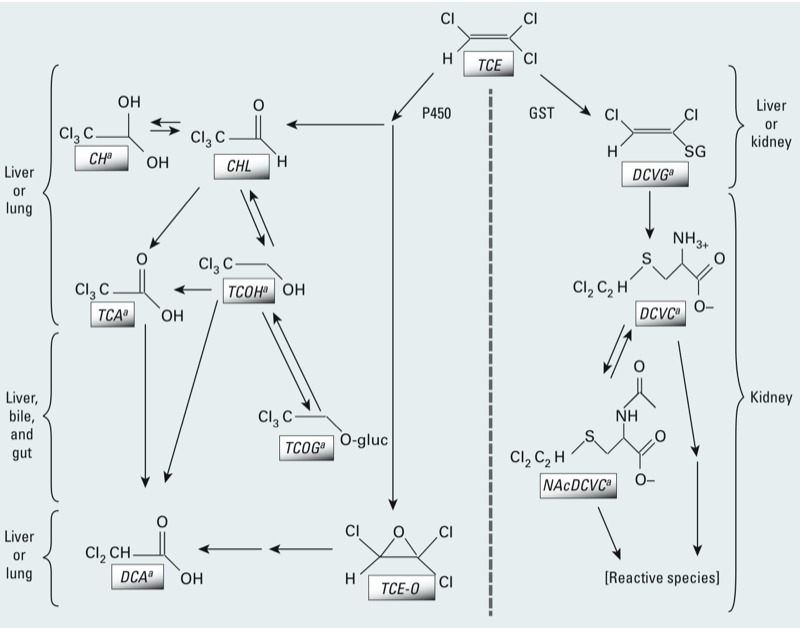
Simplified TCE metabolism scheme. Metabolism of TCE occurs through two main irreversible pathways: oxidation via the microsomal mixed-function oxidase system (i.e., cytochrome P450s; left) and conjugation with GSH by glutathione S-transferases (GSTs; right). Oxidation occurs predominantly in the liver, and to a lesser extent in the lung; the first metabolic products are TCE-oxide (TCE‑O), chloral (CHL), and chloral hydrate (CH), with the latter two quickly transformed to trichloroethanol (TCOH; a reversible reaction) and trichloroacetic acid (TCA). TCOH is glucuronidated to form TCOH-glucuronide (TCOG), which undergoes enterohepatic recirculation (excretion in bile with regeneration and reabsorption of TCOH from the gut). TCA and TCOG are excreted in urine. Further metabolism of TCA and TCOH has not been well characterized but may include dichloroacetic acid (DCA) (Lash et al. 2000a). TCE-O may also form DCA, among other species (Cai and Guengerich 1999). TCE conjugation with GSH in the liver or kidney form dichlorovinyl glutathione (DCVG), which is further processed in the kidney, forming the cysteine conjugate S-dichlorovinyl-L-cysteine (DCVC). DCVC may be bioactivated by beta-lyase or flavin-containing monooxygenases to reactive species (Anders et al. 1988; Krause et al. 2003; Lash et al. 2003), or (reversibly) undergo N-acetylation to the mercapturate N-acetyl dichlorovinyl cysteine (NAcDCVC), which is then excreted in urine or sulfoxidated by CYP3A to reactive species (Bernauer et al. 1996; Birner et al. 1993; Werner et al. 1995a, 1995b).
Tools such as PBPK models can be very useful for integrating complex toxicokinetic information on absorption, distribution, metabolism, and excretion of TCE and its metabolites. Many PBPK models for TCE have been developed to predict the relationship between external measures of exposure and internal dose measures (Bois 2000a, 2000b; Clewell et al. 2000; Fisher 2000; Hack et al. 2006). Chiu et al. (2009) and Evans et al. (2009) updated and “harmonized” these efforts into a new model for use in the IRIS assessment.
For example, Evans et al. (2009) and Chiu (2011) illustrated the importance of internal dose in investigating mechanisms of TCE toxicity, addressing the key question of whether the TCE metabolite TCA can account for mouse hepatomegaly caused by TCE. They used the TCE PBPK model to compare the hepatomegaly response after TCE administration with the response after direct administration of its metabolite TCA, using the common internal dose measure of TCA liver concentration. If TCA were the only contributor to TCE-induced hepatomegaly, this comparison would show equal changes in liver weight for equal TCA liver concentrations, regardless of whether TCA was the result of TCE metabolism or the result of direct TCA administration. However, as reported by Evans et al. (2009) and Chiu (2011), TCA appears to account for no more than half of the hepatomegaly that resulted from TCE exposure, implying that effects related to TCE exposure beyond those accounted for by TCA are also operative in TCE-induced hepatomegaly.
Carcinogenicity
Evaluation of cancer epidemiology for kidney cancer, liver cancer, and non-Hodgkin lymphoma (NHL). The U.S. EPA conducted a systematic review of 76 human epidemiologic studies on TCE and cancer (Scott and Jinot 2011; U.S. EPA 2011d). Each study was evaluated with respect to explicitly identified characteristics of epidemiologic design and analysis to examine whether chance, bias, or confounding could be alternative explanations for the study’s results. A more in-depth analysis (including meta-analysis) of the epidemiologic studies was conducted for kidney cancer, liver cancer, and NHL. These end points were of a priori interest based on the results of a preliminary review of the epidemiologic data and the findings from rodent bioassays of TCE exposure.
Meta-analysis approach and results. Meta-analyses can be used to combine underpowered studies, to evaluate effects across the set of studies, and to examine consistency (or heterogeneity) of results. The NRC (2006) identified a number of weaknesses in previous meta-analyses of TCE carcinogenicity, such as subjective assessment of quality and lack of sensitivity analyses. Thus, the U.S. EPA conducted new meta-analyses to support evaluation of the epidemiologic data on TCE (Scott and Jinot 2011; U.S. EPA 2011d). As recommended by the NRC (2006), the U.S. EPA (2011d) a) established objective study inclusion criteria; b) fit the data to both fixed-effect and random-effects models; c) evaluated statistical heterogeneity across the studies; d) performed sensitivity analyses examining the influence of individual studies and of different measures of relative risk (RR) from studies presenting alternative estimates (e.g., incidence or mortality); and e) conducted tests for potential publication bias (which may occur if positive studies are more likely to be published). Figure 2 presents the meta-analysis summary effect estimates (RRm) from the random-effects models for any TCE exposure (Figure 2A) and for the highest TCE exposure groups (Figure 2B).
Figure 2.
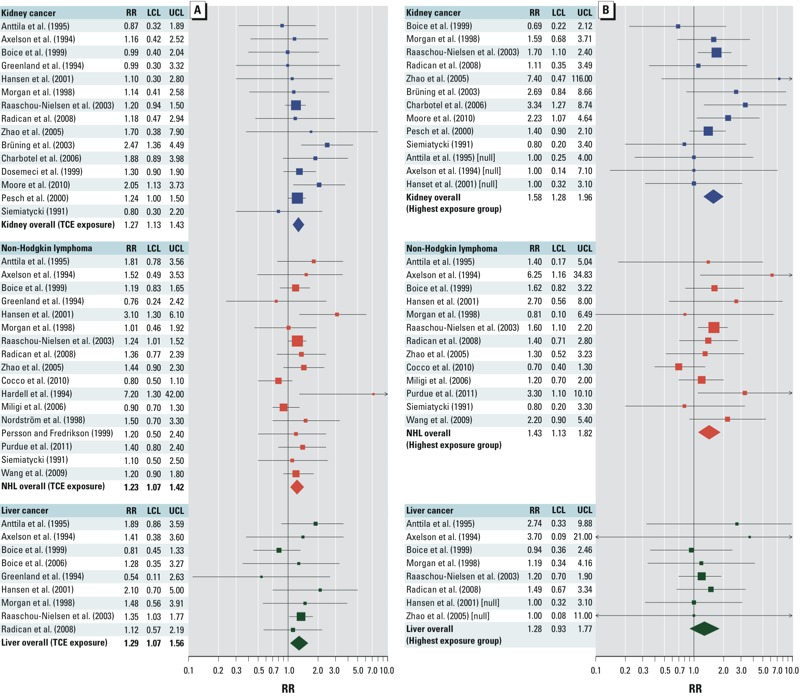
Forest plots from random-effects models of overall (i.e., “ever” or “any”) TCE exposure (A) and highest TCE exposure groups (B), adapted from Scott and Jinot (2011). Individual study RR (squares) and RRm (diamonds) values are plotted with 95% CIs (LCL, lower confidence limit; UCL, upper confidence limit) for each cancer type. Symbol sizes reflect relative weight of the studies.
Issues in the interpretation of cancer epidemiologic evidence. Two additional key issues regarding the U.S. EPA’s interpretation (U.S. EPA 2011d) of the cancer epidemiologic evidence for kidney cancer, NHL, and liver cancer have been raised in peer review and public comments: the modest magnitude of the RRm estimates for the three cancer types, and the role of meta-analysis within a causality determination.
The RRm estimates from the U.S. EPA (2011d) meta-analyses for the three cancer types were modest {e.g., with overall exposure (Figure 2A): 1.27 [95% confidence interval (CI): 1.13, 1.43] for kidney cancer; 1.23 (95% CI: 1.07, 1.42) for NHL, and 1.29 (95% CI: 1.07, 1.56) for liver cancer (Scott and Jinot 2011)}, raising the possibility that the observed associations could be the result of confounding. However, a detailed examination by the U.S. EPA of potential confounding from lifestyle factors or other occupational exposures concluded that confounding was not supported as an alternative explanation for the observed excesses (U.S. EPA 2011d).
For example, although smoking can potentially confound kidney cancer results, several kidney cancer case–control studies included in the meta-analysis (U.S. EPA 2011d) reported associations with TCE exposure even after controlling for smoking in statistical analyses. In addition, if the cohort studies had been confounded by smoking, increased lung cancer risk would be expected. However, increases in lung cancer risk in individual studies were either absent or insufficient to account for the observed excess kidney cancer risk. Overall, after combining studies, RRm estimates for lung cancer were 0.96 (95% CI: 0.76, 1.21) for overall TCE exposure and 0.96 (95% CI: 0.72, 1.27) for the highest exposure groups (Scott and Jinot 2011; U.S. EPA 2011d).
Another key issue is the role of meta-analysis in the overall evaluation of causality. Meta-analysis can provide an objective, quantitative method to increase statistical power and precision because the resultant summary effect estimate is based on multiple studies. Strengths of the meta-analyses (U.S. EPA 2011d) include study identification based on a systematic and transparent review, evaluations of potential publication bias, examinations of the sensitivity of the overall effect to different inputs, and investigations of possible factors responsible for any statistical heterogeneity observed across studies. However, the U.S. EPA’s characterization of the epidemiologic evidence (U.S. EPA 2011d) considered multiple aspects of the data as a whole and did not rely solely on the meta-analysis findings.
Synthesis of epidemiologic evidence. Table 1 summarizes the epidemiologic evidence according to the key concepts proposed by Hill (1965). For TCE and kidney cancer, there was convincing evidence of a causal association in humans. Particularly compelling was the consistency of increased RR estimates for kidney cancer across the 15 independent epidemiologic studies of different designs and populations from different countries that met the criteria for inclusion in the meta-analysis (Figure 2). The U.S. EPA (2011d) observed increased RRm estimates for kidney cancer that were robust, not being sensitive to different study or RR inputs. The U.S. EPA (2011d) also found no evidence of heterogeneity among studies or publication bias. The observations of a greater RRm estimate with the highest exposure groups (Figure 2B) and of statistically significant trends between TCE exposure and kidney cancer in two high-quality epidemiologic studies (Charbotel et al. 2006; Moore et al. 2010) support an exposure–response gradient. Finally, potential confounding from smoking or other occupational exposures was unlikely to explain the association of TCE exposure with kidney cancer.
Table 1.
Primary components for a causality determination based on the epidemiologic database for TCE.
| Consideration | Summary of weight of evidence |
|---|---|
| Consistency of observed association | Strong evidence of consistency for kidney cancer (consistently elevated RRs). Meta‑analysis yielded robust, statistically significant summary RR, with no evidence of heterogeneity or potential publication bias. |
| Moderate evidence of consistency for NHL (consistently elevated RRs); RR estimates more variable compared with kidney cancer. Meta-analysis yielded robust, statistically significant summary RR, with some heterogeneity (not statistically significant) and some evidence for potential publication bias. | |
| Limited evidence of consistency for liver cancer (fewer studies overall, more variable results). Meta-analysis showed no evidence of heterogeneity or potential publication bias, but the statistical significance of the summary estimate depends on the large study by Raaschou-Nielsen et al. (2003). | |
| Strength of observed association | Strength of association is modest. Other known or suspected risk factors (smoking, body mass index, hypertension, or coexposure to other occupational agents such as cutting or petroleum oils) cannot fully explain the observed elevations in kidney cancer RRs. The alternative explanation of smoking was ruled out by the finding of no increased risk of lung cancer. Indirect examination of some specific risk factors for liver cancer or NHL did not suggest confounding as an alternative explanation. |
| Specificity | Limited evidence suggesting that particular von Hippel-Lindau mutations in kidney tumors may be caused by TCE (Brauch et al. 1999, 2004; Brüning et al. 1997; Nickerson et al. 2008; Schraml et al. 1999); additional research addressing this issue is warranted. |
| Biological gradient (exposure–response relationship) | Only a few epidemiologic studies examined exposure–response relationships. Studies with well-designed exposure assessments reported a statistically significant trend of increasing risk of kidney cancer (Charbotel et al. 2006; Moore et al. 2010; Zhao et al. 2005) or NHL (Purdue et al. 2011) with increasing TCE exposure. Further support was provided by the meta-analyses; higher summary RR estimates for kidney cancer and NHL were observed for the highest exposure groups than for overall TCE exposure, taking possible reporting bias into account. Liver cancer studies generally had few cases, limiting the ability to assess exposure–response relationships. The meta-analysis for liver cancer did not provide support for a biological gradient (lower summary RR estimate for highest exposure groups than for overall TCE exposure, taking possible reporting bias into account). |
| Biological plausibility and coherence | TCE metabolism results in reactive, genotoxic, and/or toxicologically active metabolites at target sites in humans and in rodent test species. |
| The active GSTT1 enzyme in humans was associated with increased kidney cancer risk, whereas the lack of active enzyme was associated with no increased risk (Moore et al. 2010). | |
| TCE is carcinogenic in rodents; cancer types with increased incidences include kidney, liver, and lymphohematopoietic cancers. | |
| A mutagenic mode of action is considered operative for TCE-induced kidney tumors, based on mutagenicity of GSH-conjugation metabolites and the toxicokinetic availability of these metabolites to the target tissue. | |
| Modes of action are not established for other rodent cancer findings; human relevance is not precluded by any hypothesized modes of action due to inadequate support. | |
| NHL, non-Hodgkin lymphoma. Data from U.S. EPA (2011d). | |
The evidence on carcinogenicity from epidemiologic studies of TCE exposure was strong for NHL, although less convincing than for kidney cancer (U.S. EPA 2011d). Of the 17 studies that met the criteria for meta-analysis inclusion, most observed increased RR estimates (Figure 2A). The increased RRm estimate observed in the meta-analysis of NHL and overall TCE exposure was robust because it was not sensitive to different study or RR inputs. However, some heterogeneity among studies was observed, although it was not statistically significant. There was also some evidence of potential publication bias. An exposure–response gradient is supported by observations of a greater RRm estimate with the highest exposure groups (Figure 2B) and of a statistically significant trend between TCE exposure and NHL in a high-quality epidemiologic study (Purdue et al. 2011).
The epidemiologic evidence was more limited for liver cancer, where only cohort studies with small numbers of cases were available (U.S. EPA 2011d). Of the nine studies that met the criteria for meta-analysis inclusion, most reported increased RR estimates (Figure 2A). The U.S. EPA (2011d) observed a statistically significantly increased RRm estimate in their meta-analysis of liver cancer and overall TCE exposure, but the statistical significance depended on the large study by Raaschou-Nielsen et al. (2003). There was no evidence of heterogeneity or publication bias. However, the data available did not support an exposure–response gradient because the RRm estimate for the highest exposure groups was lower than that for overall exposure (Figure 2B) and because none of the available studies reported a statistically significant trend between TCE exposure and liver cancer.
Experimental animal studies, analysis of mode of action, and toxicokinetic considerations. There is clear evidence of TCE carcinogenicity in rodents. Particularly notable is the site-concordant finding of TCE-induced kidney tumors in multiple strains and both sexes of rats exposed by inhalation or gavage [Maltoni et al. 1986; National Toxicology Program (NTP) 1988, 1990]. Although the increased incidences were low, they were sometimes statistically significant and were considered biologically significant in light of the very low historical incidences of renal tumors in control rats in various laboratories. There is also site concordance for liver tumors, which were reported in both Swiss and B6C3F1 mice (strains with lower and higher background rates of this tumor, respectively), and in both sexes in the latter strain (Maltoni et al. 1986; National Cancer Institute 1976; NTP 1990). The evidence was more limited for TCE-induced lymphohematopoietic cancers in rats and mice (Henschler et al. 1980; Maltoni et al. 1986; NTP 1988, 1990). TCE inhalation bioassays have demonstrated a statistically significant increase in pulmonary tumors in mice (Fukuda et al. 1983; Maltoni et al. 1986) but not other species [i.e., rats and hamsters (Fukuda et al. 1983; Henschler et al. 1980; Maltoni et al. 1986)]. Finally, testicular (interstitial cell and Leydig cell) tumors were significantly increased in Sprague-Dawley rats exposed via inhalation (Maltoni et al. 1986) and Marshall rats exposed via gavage (NTP 1988). In three other tested rat strains, ACI, August, and F344/N, a high (> 75%) control rate of testicular tumors limited the ability to detect a treatment effect, although a positive trend was reported in ACI rats (NTP 1988, 1990). Overall, the rodent cancer data add substantial biological plausibility for TCE carcinogenicity in humans, particularly when combined with the mechanistic data findings.
Table 2 summarizes hypothesized modes of action and mechanistic data informative to the evaluation of TCE’s carcinogenic mode of action for liver, kidney, and other tumors. Mode-of-action analyses can inform judgments regarding the human relevance of animal bioassay results and aid in identifying particularly susceptible populations or life stages (U.S. EPA 2005). For kidney carcinogenicity, the U.S. EPA (2011d) concluded that a mutagenic mode of action is operative for TCE, providing further biological plausibility for the epidemiologic findings of TCE-induced kidney cancer. The identification of the mutagenic metabolites as being derived from the GSH conjugation pathway further suggests increased susceptibility in populations with greater metabolism through this pathway. Consistent with this hypothesis, Moore et al. (2010) found a statistically significant association among TCE-exposed persons with an active GSTT1 (glutathione-S-transferase theta-1) enzyme [odds ratio (OR) = 1.88; 95% CI: 1.06, 3.33], but not among those with no GSTT1 activity (OR = 0.93; 95% CI: 0.35, 2.44). Although data are lacking on early-life susceptibility to TCE carcinogenicity, the analysis by Barton et al. (2005) suggested increased susceptibility to cancer from early-life exposures, particularly for chemicals acting through a mutagenic mode of action. For other end points, there are inadequate data to support a particular hypothesized mode of action.
Table 2.
Selected key mode-of-action hypotheses and support.
| End point/hypothesized mode of action | Summary of weight of evidence | ||
|---|---|---|---|
| Kidney tumors | |||
| Mutagenicity | Data sufficient to conclude a mutagenic mode of action is operative. | ||
| GSH conjugation–derived metabolites are produced in the kidney. | Studies demonstrate TCE metabolism via GSH conjugation pathway; availability of metabolites to the kidney in laboratory animals and humans. | ||
| Metabolites directly induce mutations in kidney cells, advancing acquisition of critical traits contributing to carcinogenesis. | Predominance of positive genotoxicity data for GSH pathway metabolites in experimental systems. | ||
| Cytotoxicity and regenerative proliferation | Data consistent with cytotoxicity contributing to carcinogenesis in rodents, but the evidence is not as strong as that for a mutagenic mode of action. | ||
| GSH conjugation–derived metabolites are produced in kidney. | Studies demonstrate TCE metabolism via GSH conjugation pathway; availability of metabolites to the kidney in humans and laboratory animals. | ||
| Metabolites directly induce death in kidney cells (cytotoxicity). | Studies demonstrating TCE-induced rare form of nephrotoxicity in laboratory animals; similarity of renal tubular effects induced by TCE and its GSH metabolites. However, cytopathology involves changes in cell and nuclear sizes. | ||
| Compensatory cell proliferation occurs to repair damage. | Data linking TCE-induction of proliferation and clonal expansion are lacking. | ||
| Clonal expansion of initiated cells occurs, leading to cancer. | |||
| Liver tumors | |||
| Mutagenicity | Data are inadequate to support a mutagenic mode of action | ||
| Oxidation-pathway–derived metabolites are produced in and/or distributed to the liver. | Studies demonstrate TCE metabolism via oxidative pathway: availability of numerous metabolites to the liver. | ||
| Metabolites directly induce mutations in liver, advancing acquisition of critical traits contributing to carcinogenesis. | Strong data for mutagenic potential is CH, but difficult to assess the contributions from CH along with genotoxic and non-genotoxic effects of other oxidative metabolites. | ||
| PPARα activation | Data are inadequate to support a PPARα activation mode of action. | ||
| Oxidation-pathway–derived PPAR agonist metabolites (TCA and/or DCA) are produced in and/or distributed to the liver. | Studies demonstrate TCE metabolism via oxidative pathway: availability of some metabolites that are PPAR agonists to the liver. | ||
| Metabolites activate PPARα in the liver. | Studies demonstrating activation of hepatic PPARα in rodents exposed to TCE and TCA. | ||
| Alteration of cell proliferation and apoptosis occurs. | However, inadequate evidence that PPARα is necessary for liver tumors induced by TCE or that hypothesized key events are collectively sufficient for carcinogenesis. | ||
| Clonal expansion of initiated cells occurs, leading to cancer. | |||
| Other end points and/or modes of action | |||
| Inadequate data to support one or more of the following: | |||
| An identified sequence of key events. | |||
| TCE or metabolites induce key events. | |||
| Key events are individually necessary for inducing the end point. | |||
| Key events are collectively sufficient for inducing the end point. | |||
| Abbreviations: CH, chloral hydrate; DCA, dichloroacetic acid; PPARα, peroxisome proliferator activated receptor α; TCA, trichloroacetic acid. Data from U.S. EPA (2011d). | |||
The evaluation of TCE carcinogenicity (U.S. EPA 2011d) also considered toxicokinetic data on TCE and metabolites, which are consistent with qualitatively similar absorption, distribution, metabolism, and excretion across species and routes of exposure (Lash et al. 2000a). Mice, rats, and humans all metabolize TCE via the pathways illustrated in Figure 1. Thus, toxicokinetic data support the biological plausibility of TCE carcinogenicity in humans because humans and experimental animals have similar mixtures of TCE and metabolites in target tissues.
Another issue informed by toxicokinetic data is whether TCE carcinogenicity depends on route of exposure, given that the vast majority of the available epidemiologic data are from inhalation exposures to TCE. Because TCE is systemically distributed and undergoes systemic metabolism from all routes of exposure, there is no reason to expect that cancers such as kidney cancer, NHL, or liver cancer, which originate in separate tissues, would be dependent on route of exposure. Also, TCE-induced tumors have been reported in rodents by both the oral and inhalation routes (Maltoni et al. 1986; NTP 1988, 1990). Therefore, conclusions regarding TCE carcinogenicity would apply equally to any exposure route.
Conclusions as to carcinogenic hazard. Supported by the analyses described above and following the U.S. EPA’s Guidelines for Carcinogen Risk Assessment (U.S. EPA 2005), TCE is characterized as “carcinogenic to humans” by all routes of exposure (U.S. EPA 2011d). This conclusion was based primarily on convincing evidence of a causal association between TCE exposure and kidney cancer in humans. The epidemiologic evidence is strong for NHL, although less convincing than for kidney cancer. Issues increasing the uncertainty in the NHL association include study heterogeneity, potential publication bias, and less evidence for an exposure–response gradient. The epidemiologic evidence was more limited for liver cancer, where only cohort studies with small numbers of cases were available. Finally, animal bioassay, mechanistic, and toxicokinetic data provide further corroboration and biological plausibility to the epidemiologic findings, thus supporting a causal link between TCE exposure and cancer (Table 1).
Noncancer Toxicity
As part of its evaluation of TCE noncancer toxicity, the U.S. EPA analyzed the available experimental animal, human epidemiologic, and mechanistic studies of TCE. A summary of the relevant studies for each end point is available in Supplemental Material, Table S1 (http://dx.doi.org/10.1289/ehp.1205879). Below we discuss the data pertaining to immunotoxicity and developmental cardiac toxicity, for which there are substantial new experimental and epidemiologic studies (U.S. EPA 2011d), and about which scientific issues have been raised by reviewers or comments. We also provide an overall summary of the hazard conclusions for noncancer toxicity.
Immunotoxicity. As recently reviewed by Cooper et al. (2009) and documented in the TCE assessment (U.S. EPA 2011d), the human and laboratory animal studies of TCE and immune-related effects provide strong evidence that TCE exposure increases the risk of autoimmune disease and a specific type of generalized hypersensitivity syndrome. In addition to the epidemiologic studies of specific diseases (e.g., systemic sclerosis), changes in cytokine levels reflecting an inflammatory immune response have been reported in relation to TCE exposure in occupational (Iavicoli et al. 2005) and residential (i.e., infants exposed to TCE in indoor air) (Lehmann et al. 2001, 2002) settings. Also, many case reports have associated a severe hypersensitivity skin disorder, distinct from contact dermatitis and often accompanied by hepatitis, with occupational TCE exposure, with prevalences as high as 13% of workers in the same location (Kamijima et al. 2007, 2008).
Human evidence for autoimmune-related effects is supported by experimental animal studies. Numerous studies have demonstrated TCE-induced progressive, accelerated autoimmune responses in autoimmune-prone mice (reviewed by Cooper et al. 2009). After shorter exposure periods, changes in cytokine levels appear similar to those reported in human studies. Longer exposure periods led to more severe effects, including autoimmune hepatitis, inflammatory skin lesions, and alopecia, that differ from the “normal” expression of autoimmune effects in these mice. TCE-induced autoimmune effects have also been reported in B6C3F1 mice, which are not known to have any particular immune-related susceptibility (Gilkeson et al. 2004; Peden-Adams et al. 2006). A treatment-related increase in delayed hypersensitivity response accompanied by hepatic damage has been observed in guinea pigs following intradermal TCE injection (Tang et al. 2002, 2008), and increased hypersensitivity response was reported in mice exposed via drinking water prenatally and postnatally (gestation day 0 through to 8 weeks of age) (Peden-Adams et al. 2006).
There is less evidence regarding a possible role of TCE exposure in immunosuppression. Immunosuppressive effects have been reported in a number of experimental studies in mice and rats [see Supplemental Material, Table S1 (http://dx.doi.org/10.1289/ehp.1205879)]. Reported effects include reduced responses to bacterial challenge in mice (Aranyi et al. 1986; Selgrade and Gilmour 2010) and decreased numbers of antibody-forming cells in rats and developmentally exposed mice (Peden-Adams et al. 2006; Woolhiser et al. 2006).
Overall, the concordance of human and laboratory animal studies and the spectrum of effects (from biomarkers to frank expressions of disease) strongly support the conclusion that TCE causes immunotoxicity, particularly in the form of autoimmune disease and a specific type of severe hypersensitivity skin disorder, with more limited evidence for immunosuppression. Moreover, these findings lend additional biological plausibility to the association between TCE and NHL, as alterations in immune status are associated with increased risk of NHL (Grulich et al. 2007).
Developmental cardiac toxicity. The TCE data include a number of epidemiologic and animal toxicity studies that indicate TCE-induced developmental toxicity. Congenital malformations, particularly cardiac defects, have been associated with exposures to TCE and/or its metabolites in both humans and experimental animals [for example studies, see Supplemental Material, Table S1 (http://dx.doi.org/10.1289/ehp.1205879)]. Other TCE-related developmental outcomes observed in both humans and experimental animals include embryonic or fetal mortality, prenatal growth inhibition, and neurological and immunological functional deficits. (see Supplemental Material, Table S1).
As noted by the NRC (2006), the cardiac teratogenicity of TCE has been the focus of considerable study and analysis (Bove et al. 2002; Hardin et al. 2005; Johnson et al. 1998b; Watson et al. 2006). Only geography-based epidemiology studies have evaluated whether there is an association between maternal TCE exposure and cardiac defects in offspring [see Supplemental Material, Table S1 (http://dx.doi.org/10.1289/ehp.1205879)], with some of the studies reporting statistically significant elevations in a variety of cardiac defects [Agency for Toxic Substances and Disease Registry (ATSDR) 2006, 2008; Yauck et al. 2004], and others reporting nonstatistically significant elevations in risk (Bove 1996; Bove et al. 1995; Goldberg et al. 1990). Interpretation of these data has been controversial because many of the studies are limited by small numbers of cases, insufficient exposure characterization, chemical coexposures, and other methodological deficiencies. In addition, these studies aggregate a broad array of TCE-associated cardiac malformations and have inadequate statistical power to identify any particular kind(s) of defect that may be more susceptible to induction by TCE. The NRC (2006) noted that the epidemiologic studies—although limited individually—as a whole showed relatively consistent elevations for cardiac malformations with similar relative effect sizes of 2- to 3-fold, some of which were statistically significant, associated with TCE exposure across multiple studies.
The outcomes of studies in rodents exposed to TCE during gestation show an inconsistent pattern. Some studies identified significant treatment-related increases in the overall incidence of cardiac anomalies at environmentally relevant exposure levels (e.g., Johnson et al. 2003, 2005), whereas others reported no excess cardiac abnormalities at much higher dose levels (e.g., Carney et al. 2006; Fisher et al. 2001). Several methodological factors may contribute to differences across study outcomes, such as the route of administration, test substance purity, test species or strain, timing of dosing or fetal evaluation, procedures used in dissecting and examining fetal hearts, statistical approaches applied to data evaluation, and generally uncharacterized interlaboratory variation.
Other available data providing evidence of TCE cardiac teratogenicity come from avian and in vitro mechanistic studies (NRC 2006). For instance, studies in chick embryos reported consistent effects on cardiogenesis (many demonstrating septal and valvular alterations) when TCE was administered during critical stages of heart development (Drake et al. 2006a, 2006b; Loeber et al. 1988; Rufer et al. 2010); these findings are similar to some of the cardiac defects observed in rodent studies following in utero TCE exposures (Johnson et al. 2003). The events of cardiac morphogenesis in birds and mammals are similar; both involving mesenchymal cells that form endocardial cushion tissue with subsequent differentiation into septa and valvular structures in the adult heart (NRC 2006). Thus, cultured embryonic chick atrioventricular canal cushion cells have been used to examine chemically induced disruptions in cardiac morphogenesis. In this model, TCE inhibited endothelial separations and mesenchymal cell formation (Boyer et al. 2000; Mishima et al. 2006) or adhesive properties of endocardial cells (Hoffman et al. 2004), either of which could potentially result in septal or valvular malformations. Other TCE-induced effects that may have morphologic consequences in the developing heart include disruption of endothelial oxide synthetase, which has a role in endothelial cell proliferation (Ou et al. 2003), and interference with proteins involved in intercellular Ca2+ regulation, which may result in altered blood flow (Caldwell PT et al. 2008, 2010; Collier et al. 2003; Selmin et al. 2008).
Overall, the avian and in vitro data substantially increase the biological plausibility for TCE-induced cardiac teratogenesis, and thus strongly support the more limited epidemiologic and in vivo rodent data suggesting that TCE induces cardiac teratogenicity. Moreover, mechanistic data support the possibility that multiple modes of action with different targets within the developing heart may be operant in eliciting cardiac malformations, consistent with the reported association between TCE and overall cardiac malformations in the absence of a strong association with any particular type of defect.
Conclusions as to noncancer hazard. Table 3 summarizes the evidence for TCE noncancer toxicity across target organs and systems (for additional details, see U.S. EPA 2011d). In addition to the immunotoxicity and developmental cardiac toxicity discussed above, there is strong evidence for TCE-induced neurotoxicity, kidney toxicity, liver toxicity, male reproductive toxicity, and several developmental effects in addition to cardiac toxicity. More limited evidence exists for the toxicity of TCE in the respiratory tract and female reproductive system.
Table 3.
Key conclusions for TCE noncancer toxicity.
| Tissue or organ system | Key conclusions as to human health hazard |
|---|---|
| Central nervous system | Strong evidence, based on multiple human and experimental animal studies, that TCE causes |
| Changes in trigeminal nerve function or morphology | |
| Impairment of vestibular function. | |
| Limited evidence, primarily from experimental animal studies, with fewer/more limited human studies, that TCE causes | |
| Delayed motor function, including during neurodevelopment | |
| Changes in auditory, visual, and cognitive function or performance. | |
| Kidney | Strong evidence, based on experimental animal studies, a few human studies, and mechanistic studies, that TCE causes nephrotoxicity, particularly in the form of tubular toxicity. Nephrotoxicity is likely mediated primarily through the TCE GSH conjugation metabolite DCVC. |
| Liver | Limited evidence in humans and strong evidence from experimental animal studies that TCE causes hepatotoxicity but not necrosis. Mice appear to be more sensitive than other experimental species, and hepatotoxicity is likely mediated through oxidative metabolites including, but not exclusively, TCA. |
| Immune system | Strong evidence, based on multiple human and experimental animal studies, that TCE exposure causes |
| Autoimmune disease, including scleroderma | |
| A specific type of generalized hypersensitivity disorder. | |
| Limited evidence, primarily from experimental animal studies, with fewer/more limited human studies, that TCE causes immunosuppression. | |
| Respiratory tract | Suggestive evidence, primarily from short-term experimental animal studies, that TCE causes respiratory tract toxicity, primarily in Clara cells. |
| Reproductive system | Strong evidence, based on multiple human and experimental animal studies, that TCE causes male reproductive toxicity, primarily through effects on the testes, epididymides, sperm, or hormone levels. |
| Suggestive evidence, based on few/limited human and experimental animal studies, that TCE causes female reproductive toxicity. | |
| Development | Strong evidence, based on weakly suggestive epidemiologic studies, limited experimental animal studies, and multiple mechanistic studies, that TCE causes fetal cardiac malformations; limited experimental evidence that oxidative metabolites, such as TCA and/or DCA, cause similar effects. |
| Limited evidence, primarily from experimental animal studies, with weakly suggestive epidemiologic studies, that TCE causes fetal malformations (in addition to cardiac), prenatal losses, decreased growth or birth weight of offspring, and alterations in immune system function. | |
| Abbreviations: DCVC, S-dichlorovinyl-l-cysteine. Data from U.S. EPA (2011d). | |
Summary
TCE is carcinogenic to humans by all routes of exposure and poses a potential human health hazard for noncancer toxicity to the central nervous system, kidney, liver, immune system, male reproductive system, and the developing embryo/fetus. These conclusions are based on analyses of a broad spectrum of information from thousands of scientific studies and input from numerous scientific reviews. In the last decade, substantial new scientific data on the human health effects of TCE have become available. Moreover, methodologic advancements—such as modeling of TCE toxicokinetics, meta-analyses of epidemiologic studies, and analyses of mechanistic and noncancer hazard information—have improved the scientific rigor and transparency of data interpretation. The approaches and conclusions of the U.S. EPA’s analyses (U.S. EPA 2011d) are consistent with the recommendations of the NRC (2006) and were affirmed by independent peer review through the U.S. EPA’s Science Advisory Board (U.S. EPA SAB 2011). In addition, the International Agency for Research on Cancer (IARC) recently upgraded its carcinogenicity classification of TCE to “carcinogenic to humans” (Guha et al. 2012). Finally, studies on the health effects of TCE continue to report findings similar to those described in the U.S. EPA’s assessment, such as kidney carcinogenicity and toxicity (Karami et al. 2012; Vermeulen et al. 2012), immunotoxicity (Hosgood et al. 2011), and developmental cardiac toxicity (Forand et al. 2012).
Supplemental Material
Acknowledgments
This work has benefitted from advice and comments from a number of scientific reviewers—including members of the National Academies of Sciences, National Research Council panel on Trichloroethylene Health Risks, two U.S. EPA Science Advisory Board review panels, and scientists at federal agencies (including the U.S. EPA)—as well as from others who prepared public comments. We also thank P. Anastas, R. Clark, P. Preuss, D. Bussard, V. Cogliano, B. Sonawane, and P. White for providing U.S. EPA management support.
Footnotes
The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. EPA.
The authors declare they have no actual or potential competing financial interests.
References
- Anders MW, Lash L, Dekant W, Elfarra AA, Dohn DR. Biosynthesis and biotransformation of glutathione S-conjugates to toxic metabolites. Crit Rev Toxicol. 1988;18:311–341. doi: 10.3109/10408448809037470. [DOI] [PubMed] [Google Scholar]
- Anttila A, Pukkala E, Sallmen M, Hernberg S, Hemminki K. Cancer incidence among Finnish workers exposed to halogenated hydrocarbons. J Occup Environ Med. 1995;37:797–806. doi: 10.1097/00043764-199507000-00008. [DOI] [PubMed] [Google Scholar]
- Aranyi C, O’Shea W, Graham J, Miller F. The effects of inhalation of organic chemical air contaminants on murine lung host defenses. Fundam Appl Toxicol. 1986;6:713–720. doi: 10.1016/0272-0590(86)90184-3. [DOI] [PubMed] [Google Scholar]
- ATSDR (Agency for Toxic Substances and Disease Registry) Health Consultation: Endicott Area Investigation, Health Statistics Review. Cancer and Birth Outcome Analysis: Endicott Area, Town of Union, Broome County, New York. Atlanta, GA:ATSDR. 2006. Available: http://www.atsdr.cdc.gov/HAC/pha/EndicottAreaInvestigation/EndicottHealthStatsReviewHC052606.pdf [accessed 19 September 2012]
- ATSDR (Agency for Toxic Substances and Disease Registry) Health Consultation: Health Statistics Review Follow-Up. Cancer and Birth Outcome Analysis: Endicott Area Investigation, Endicott Area, Town of Union, Broome County, New York. Atlanta, GA:ATSDR. 2008. Available: http://www.atsdr.cdc.gov/hac/pha//EndicottAreaInvestigationFollowUp/EndicottAreaHC051508.pdf [accessed 19 September 2012]
- Axelson O, Seldén A, Andersson K, Hogstedt C. Updated and expanded Swedish cohort study on trichloroethylene and cancer risk. J Occup Med. 1994;36:556–562. [PubMed] [Google Scholar]
- Bale AS, Barone S, Jr, Scott CS, Cooper GS. A review of potential neurotoxic mechanisms among three chlorinated organic solvents. Toxicol Appl Pharmacol. 2011;255:113–126. doi: 10.1016/j.taap.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Barton HA, Cogliano VJ, Flowers L, Valcovic L, Setzer RW, Woodruff TJ. Assessing susceptibility from early-life exposure to carcinogens. Environ Health Perspect. 2005;113:1125–1133. doi: 10.1289/ehp.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernauer U, Birner G, Dekant W, Henschler D. Biotransformation of trichloroethene: dose-dependent excretion of 2,2,2-trichloro-metabolites and mercapturic acids in rats and humans after inhalation. Arch Toxicol. 1996;70:338–346. doi: 10.1007/s002040050283. [DOI] [PubMed] [Google Scholar]
- Birner G, Vamvakas S, Dekant W, Henschler D. Nephrotoxic and genotoxic N-acetyl-S-dichlorovinyl-l-cysteine is a urinary metabolite after occupational 1,1,2-trichloroethene exposure in humans: Implications for the risk of trichloroethene exposure. Environ Health Perspect. 1993;99:281–284. doi: 10.1289/ehp.9399281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boice JD, Jr, Marano DE, Cohen SS, Mumma MT, Blot WJ, Brill AB, et al. Mortality among Rocketdyne workers who tested rocket engines, 1948–1999. J Occup Environ Med. 2006;48:1070–1092. doi: 10.1097/01.jom.0000240661.33413.b5. [DOI] [PubMed] [Google Scholar]
- Boice JD, Jr, Marano D, Fryzek J, Sadler C, McLaughlin JK. Mortality among aircraft manufacturing workers. Occup Environ Med. 1999;56:581–597. doi: 10.1136/oem.56.9.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois FY. Statistical analysis of Clewell et al. PBPK model of trichloroethylene kinetics. Environ Health Perspect. 2000a;108(suppl 2):307–316. doi: 10.1289/ehp.00108s2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois FY. Statistical analysis of Fisher et al. PBPK model of trichloroethylene kinetics. Environ Health Perspect. 2000b;108(suppl 2):275–282. doi: 10.1289/ehp.00108s2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove F. Public drinking water contamination and birthweight, prematurity, fetal deaths, and birth defects. Toxicol Ind Health. 1996;12:255–266. [PubMed] [Google Scholar]
- Bove F, Fulcomer M, Klotz J, Esmart J, Dufficy E, Savrin J. Public drinking water contamination and birth outcomes. Am J Epidemiol. 1995;141:850–862. doi: 10.1093/oxfordjournals.aje.a117521. [DOI] [PubMed] [Google Scholar]
- Bove F, Shim Y, Zeitz P. Drinking water contaminants and adverse pregnancy outcomes: a review. Environ Health Perspect. 2002;110(suppl 1):61–74. doi: 10.1289/ehp.02110s161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer AS, Finch WT, Runyan RB. Trichloroethylene inhibits development of embryonic heart valve precursors in vitro. Toxicol Sci. 2000;53:109–117. doi: 10.1093/toxsci/53.1.109. [DOI] [PubMed] [Google Scholar]
- Brauch H, Weirich G, Hornauer M, Störkel S, Wöhl T, Brüning T. Trichloroethylene exposure and specific somatic mutations in patients with renal cell carcinoma. J Natl Cancer Inst. 1999;91:854–861. doi: 10.1093/jnci/91.10.854. [DOI] [PubMed] [Google Scholar]
- Brauch H, Weirich G, Klein B, Rabstein S, Bolt HM, Brüning T. VHL mutations in renal cell cancer: does occupational exposure to trichloroethylene make a difference. Toxicol Lett. 2004;151:301–310. doi: 10.1016/j.toxlet.2003.12.074. [DOI] [PubMed] [Google Scholar]
- Brüning T, Pesch B, Wiesenhütter B, Rabstein S, Lammert M, Baumüller A, et al. Renal cell cancer risk and occupational exposure to trichloroethylene: results of a consecutive case-control study in Arnsberg, Germany. Am J Ind Med. 2003;43:274–285. doi: 10.1002/ajim.10185. [DOI] [PubMed] [Google Scholar]
- Brüning T, Weirich G, Hornauer MA, Höfler H, Brauch H. Renal cell carcinomas in trichloroethene (TRI) exposed persons are associated with somatic mutations in the von Hippel-Lindau (VHL) tumour suppressor gene. Arch Toxicol. 1997;71:332–335. doi: 10.1007/s002040050394. [DOI] [PubMed] [Google Scholar]
- Buben J, O’Flaherty E. Delineation of the role of metabolism in the hepatotoxicity of trichloroethylene and perchloroethylene: a dose-effect study. Toxicol Appl Pharmacol. 1985;78:105–122. doi: 10.1016/0041-008x(85)90310-2. [DOI] [PubMed] [Google Scholar]
- Bull RJ. Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ Health Perspect. 2000;108(suppl 2):241–259. doi: 10.1289/ehp.00108s2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Guengerich FP. Mechanisms of aqueous decomposition of trichloroethylene oxide. J Am Chem Soc. 1999;121:11656–11663. [Google Scholar]
- Caldwell JC, Keshava N. Key issues in the modes of action and effects of trichloroethylene metabolites for liver and kidney tumorigenesis. Environ Health Perspect. 2006;114:1457–1463. doi: 10.1289/ehp.8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JC, Keshava N, Evans MV. Difficulty of mode of action determination for trichloroethylene: an example of complex interactions of metabolites and other chemical exposures. Environ Mol Mutagen. 2008;49:142–154. doi: 10.1002/em.20350. [DOI] [PubMed] [Google Scholar]
- Caldwell PT, Manziello A, Howard J, Palbykin B, Runyan RB, Selmin O. Gene expression profiling in the fetal cardiac tissue after folate and low-dose trichloroethylene exposure. Birth Defects Res A Clin Mol Teratol. 2010;88:111–127. doi: 10.1002/bdra.20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell PT, Thorne PA, Johnson PD, Boitano S, Runyan RB, Selmin O. Trichloroethylene disrupts cardiac gene expression and calcium homeostasis in rat myocytes. Toxicol Sci. 2008;104:135–143. doi: 10.1093/toxsci/kfn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney E, Thorsrud B, Dugard P, Zablotny C. Developmental toxicity studies in Crl:CD (SD) rats following inhalation exposure to trichloroethylene and perchloroethylene. Birth Defects Res B Dev Reprod Toxicol. 2006;77:405–412. doi: 10.1002/bdrb.20091. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Fourth National Report on Human Exposure to Chemicals. 2009. Available: http://www.cdc.gov/exposurereport/pdf/FourthReport.pdf [accessed 19 September 2012]
- Charbotel B, Fevotte J, Hours M, Martin JL, Bergeret A. Case–control study on renal cell cancer and occupational exposure to trichloroethylene. Part II: Epidemiological aspects. Ann Occup Hyg. 2006;50:777–787. doi: 10.1093/annhyg/mel039. [DOI] [PubMed] [Google Scholar]
- Chiu WA. Trichloroacetic acid: updated estimates of its bioavailability and its contribution to trichloroethylene-induced mouse hepatomegaly. Toxicology. 2011;285:114–125. doi: 10.1016/j.tox.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Caldwell JC, Keshava N, Scott CS. Key scientific issues in the health risk assessment of trichloroethylene. Environ Health Perspect. 2006a;114:1445–1449. doi: 10.1289/ehp.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, Okino MS, Evans MV. Characterizing uncertainty and population variability in the toxicokinetics of trichloroethylene and metabolites in mice, rats, and humans using an updated database, physiologically based pharmacokinetic (PBPK) model, and Bayesian approach. Toxicol Appl Pharmacol. 2009;241:36–60. doi: 10.1016/j.taap.2009.07.032. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Okino MS, Lipscomb JC, Evans MV. Issues in the pharmacokinetics of trichloroethylene and its metabolites. Environ Health Perspect. 2006b;114:1450–1456. doi: 10.1289/ehp.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell HJ, III, Gentry PR, Covington TR, Gearhart JM. Development of a physiologically based pharmacokinetic model of trichloroethylene and its metabolites for use in risk assessment. Environ Health Perspect. 2000;108(suppl 2):283–305. doi: 10.1289/ehp.00108s2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco P, t’Mannetje A, Fadda D, Melis M, Becker N, de Sanjosé S, et al. 2010Occupational exposure to solvents and risk of lymphoma subtypes: results from the Epilymph case–control study. Occup Environ Med 67341–347. [DOI] [PubMed] [Google Scholar]
- Collier JM, Selmin O, Johnson PD, Runyan RB. Trichloroethylene effects on gene expression during cardiac development. Birth Defects Res A Clin Mol Teratol. 2003;67:488–495. doi: 10.1002/bdra.10073. [DOI] [PubMed] [Google Scholar]
- Cooper GS, Makris SL, Nietert PJ, Jinot J. Evidence of autoimmune-related effects of trichloroethylene exposure from studies in mice and humans. Environ Health Perspect. 2009;117:696–702. doi: 10.1289/ehp.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci M, Cocco P, Chow WH. Gender differences in risk of renal cell carcinoma and occupational exposures to chlorinated aliphatic hydrocarbons. Am J Ind Med. 1999;36:54–59. doi: 10.1002/(sici)1097-0274(199907)36:1<54::aid-ajim8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski S, Hu N, Smith S, Lough J. Cardiogenic effects of trichloroethylene and trichloroacetic acid following exposure during heart specification of avian development. Toxicol Sci. 2006a;94:153–162. doi: 10.1093/toxsci/kfl083. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski S, Lough J, Hu N, Smith S. Trichloroethylene exposure during cardiac valvuloseptal morphogenesis alters cushion formation and cardiac hemodynamics in the avian embryo. Environ Health Perspect. 2006b;114:842–847. doi: 10.1289/ehp.8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MV, Chiu WA, Okino MS, Caldwell JC. Development of an updated PBPK model for trichloroethylene and metabolites in mice, and its application to discern the role of oxidative metabolism in TCE-induced hepatomegaly. Toxicol Appl Pharmacol. 2009;236:329–340. doi: 10.1016/j.taap.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Fisher J. Physiologically based pharmacokinetic models for trichloroethylene and its oxidative metabolites. Environ Health Perspect. 2000;108(suppl 2):265–273. doi: 10.1289/ehp.00108s2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J, Channel S, Eggers J, Johnson P, MacMahon K, Goodyear C, et al. Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development. Int J Toxicol. 2001;20:257–267. doi: 10.1080/109158101753252992. [DOI] [PubMed] [Google Scholar]
- Forand SP, Lewis-Michl EL, Gomez MI. Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State. Environ Health Perspect. 2012;120:616–621. doi: 10.1289/ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Takemoto K, Tsuruta H. Inhalation carcinogenicity of trichloroethylene in mice and rats. Ind Health. 1983;21:243–254. doi: 10.2486/indhealth.21.243. [DOI] [PubMed] [Google Scholar]
- Gilkeson GS, Keil D, Peden-Adams M. Charleston, SC: Medical University of South Carolina Press, 87–98; 2004. Immune effects of trichloroethylene on autoimmune disease in mice. In: Trichloroethylene: The Scientific Basis of Risk Assessment (Mohr LC, Hoel DG, Jollow D, eds) [Google Scholar]
- Goldberg S, Lebowitz M, Graver E, Hicks S. An association of human congenital cardiac malformations and drinking water contaminants. J Am Coll Cardiol. 1990;16:155–164. doi: 10.1016/0735-1097(90)90473-3. [DOI] [PubMed] [Google Scholar]
- Greenland S, Salvan A, Wegman DH, Hallock MF, Smith TJ. A case-control study of cancer mortality at a transformer-assembly facility. Int Arch Occup Environ Health. 1994;66:49–54. doi: 10.1007/BF00386579. [DOI] [PubMed] [Google Scholar]
- Grulich AE, Vajdic CM, Cozen W. Altered immunity as a risk factor for non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16(3):405–408. doi: 10.1158/1055-9965.EPI-06-1070. [DOI] [PubMed] [Google Scholar]
- Guha N, Loomis D, Grosse Y, Lauby-Secretan B, El Ghissassi F, et al. Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 2012;13:1192–1193. doi: 10.1016/s1470-2045(12)70485-0. [DOI] [PubMed] [Google Scholar]
- Hack CE, Chiu WA, Jay Zhao Q, Clewell HJ. Bayesian population analysis of a harmonized physiologically based pharmacokinetic model of trichloroethylene and its metabolites. Regul Toxicol Pharmacol. 2006;46:63–83. doi: 10.1016/j.yrtph.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Hansen J, Raaschou-Nielsen O, Christensen JM, Johansen I, McLaughlin JK, Lipworth L, et al. Cancer incidence among Danish workers exposed to trichloroethylene. J Occup Environ Med. 2001;43:133–139. doi: 10.1097/00043764-200102000-00012. [DOI] [PubMed] [Google Scholar]
- Hardell L, Eriksson M, Degerman A. Exposure to phenoxyacetic acids, chlorophenols, or organic solvents in relation to histopathology, stage, and anatomical localization of non-Hodgkin’s lymphoma. Cancer Res. 1994;54:2386–2389. [PubMed] [Google Scholar]
- Hardin B, Kelman B, Brent R. Trichloroethylene and dichloroethylene: a critical review of teratogenicity. Birth Defects Res A Clin Mol Teratol. 2005;73:931–955. doi: 10.1002/bdra.20192. [DOI] [PubMed] [Google Scholar]
- Henschler D, Romen W, Elsaesser HM, Reichert D, Eder E, Radwan Z. Carcinogenicity study of trichloroethylene by longterm inhalation in three animal species. Arch Toxicol. 1980;43:237–248. doi: 10.1007/BF00366179. [DOI] [PubMed] [Google Scholar]
- Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman S, Mishima N, Krug EL, eds. Charleston, SC: Medical University of South Carolina Press; 2004. An Improved Model for Evaluating Trichloroethylene and Its Metabolites as Cardiac Specific Teratogens. [Google Scholar]
- Hosgood HD, III, Zhang L, Tang X, Vermeulen R, Qiu C, Shen M, et al. 2011Decreased numbers of CD4+ naive and effector memory T cells, and CD8+ naive T cells, are associated with trichloroethylene exposure. Front Oncol 153; doi: 10.3389/fonc.2011.00053[Online 10 January 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavicoli I, Marinaccio A, Carelli G. Effects of occupational trichloroethylene exposure on cytokine levels in workers. J Occup Environ Med. 2005;47:453–457. doi: 10.1097/01.jom.0000161728.23285.66. [DOI] [PubMed] [Google Scholar]
- Johnson P, Dawson B, Goldberg S. Cardiac teratogenicity of trichloroethylene metabolites. J Am Coll Cardiol. 1998a;32:540–545. doi: 10.1016/s0735-1097(98)00232-0. [DOI] [PubMed] [Google Scholar]
- Johnson P, Dawson B, Goldberg S. A review: Trichloroethylene metabolites: potential cardiac teratogens. Environ Health Perspect. 1998b;106(suppl 4):995–999. doi: 10.1289/ehp.98106s4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P, Goldberg S, Mays M, Dawson B. Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environ Health Perspect. 2003;111:289–292. doi: 10.1289/ehp.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P, Goldberg S, Mays M, Dawson B. Environ Health Perspect. 2005;113:A18. doi: 10.1289/ehp.5125. Errata [Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijima M, Hisanaga N, Wang H, Nakajima T. Occupational trichloroethylene exposure as a cause of idiosyncratic generalized skin disorders and accompanying hepatitis similar to drug hypersensitivities. Int Arch Occup Environ Health. 2007;80:357–370. doi: 10.1007/s00420-006-0147-y. [DOI] [PubMed] [Google Scholar]
- Kamijima M, Wang H, Huang H, Li L, Shibata E, Lin B, et al. Trichloroethylene causes generalized hypersensitivity skin disorders complicated by hepatitis. J Occup Health. 2008;50:328–338. doi: 10.1539/joh.l8013. [DOI] [PubMed] [Google Scholar]
- Karami S, Lan Q, Rothman N, Stewart PA, Lee KM, Vermeulen R, et al. Occupational trichloroethylene exposure and kidney cancer risk: a meta-analysis. Occup Environ Med. 2012;69:858–867. doi: 10.1136/oemed-2012-100932. [DOI] [PubMed] [Google Scholar]
- Keshava N, Caldwell J. Key issues in the role of peroxisome proliferator-activated receptor agonism and cell signaling in trichloroethylene toxicity. Environ Health Perspect. 2006;114:1464–1470. doi: 10.1289/ehp.8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause RJ, Lash LH, Elfarra AA. Human kidney flavin-containing monooxygenases and their potential roles in cysteine S-conjugate metabolism and nephrotoxicity. J Pharmacol Exp Ther. 2003;304:185–191. doi: 10.1124/jpet.102.042911. [DOI] [PubMed] [Google Scholar]
- Lash LH, Fisher JW, Lipscomb JC, Parker JC. Metabolism of trichloroethylene. Environ Health Perspect. 2000a;108:177–200. doi: 10.1289/ehp.00108s2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Parker JC, Scott CS. Modes of action of trichloroethylene for kidney tumorigenesis. Environ Health Perspect. 2000b;108(suppl 2):225–240. doi: 10.1289/ehp.00108s2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Hueni SE, Krause RJ, Elfarra AA. Roles of necrosis, apoptosis, and mitochondrial dysfunction in S-(1,2-dichlorovinyl)-l-cysteine sulfoxide-induced cytotoxicity in primary cultures of human renal proximal tubular cells. J Pharmacol Exp Ther. 2003;305:1163–1172. doi: 10.1124/jpet.102.046185. [DOI] [PubMed] [Google Scholar]
- Lehmann I, Rehwagen M, Diez U, Seiffart A, Rolle-Kampczyk U, Richter M, et al. Enhanced in vivo IgE production and T cell polarization toward the type 2 phenotype in association with indoor exposure to VOC: results of the LARS study. Int J Hyg Environ Health. 2001;204:211–221. doi: 10.1078/1438-4639-00100. [DOI] [PubMed] [Google Scholar]
- Lehmann I, Thoelke A, Rehwagen M, Rolle-Kampczyk U, Schlink U, Schulz R, et al. The influence of maternal exposure to volatile organic compounds on the cytokine secretion profile of neonatal T cells. Environ Toxicol. 2002;17:203–210. doi: 10.1002/tox.10055. [DOI] [PubMed] [Google Scholar]
- Loeber C, Hendrix M, Diez De Pinos S, Goldberg S. Trichloroethylene: a cardiac teratogen in developing chick embryos. Pediatr Res. 1988;24:740–744. doi: 10.1203/00006450-198812000-00018. [DOI] [PubMed] [Google Scholar]
- Maltoni C, Lefemine G, Cotti G. Princeton, NJ: Princeton Scientific Publishing; 1986. Experimental Research on Trichloroethylene Carcinogenesis. [Google Scholar]
- Miligi L, Costantini AS, Benvenuti A, Kriebel D, Bolejack V, Tumino R, et al. Occupational exposure to solvents and the risk of lymphomas. Epidemiology. 2006;17:552–561. doi: 10.1097/01.ede.0000231279.30988.4d. [DOI] [PubMed] [Google Scholar]
- Mishima N, Hoffman S, Hill E, Krug E. Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation. Birth Defects Res A Clin Mol Teratol. 2006;76:517–527. doi: 10.1002/bdra.20283. [DOI] [PubMed] [Google Scholar]
- Moore LE, Boffetta P, Karami S, Brennan P, Stewart PS, Hung R, et al. Occupational trichloroethylene exposure and renal carcinoma risk: evidence of genetic susceptibility by reductive metabolism gene variants. Cancer Res. 2010;70:6527–6536. doi: 10.1158/0008-5472.CAN-09-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RW, Kelsh MA, Zhao K, Heringer S. Mortality of aerospace workers exposed to trichloroethylene. Epidemiology. 1998;9:424–431. [PubMed] [Google Scholar]
- National Cancer Institute. Carcinogenesis Bioassay of Trichloroethylene. TR 002. Bethesda, MD:National Cancer Institute. 1976. Available: http://ntp.niehs.nih.gov/ntp/htdocs/LT_rpts/tr002.pdf [accessed 19 September 2012] [PubMed]
- Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström M, Hardell L, Magnuson A, Hagberg H, Rask-Andersen A. Occupational exposures, animal exposure and smoking as risk factors for hairy cell leukaemia evaluated in a case-control study. Br J Cancer. 1998;77:2048–2052. doi: 10.1038/bjc.1998.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC (National Research Council) Assessing the Human Health Risks of Trichloroethylene: Key Scientific Issues. Washington, DC:National Academies Press. 2006. Available: http://www.nap.edu/catalog.php?record_id=11707 [accessed 19 September 2012]
- NTP (National Toxicology Program) Toxicology and Carcinogenesis Studies of Trichloroethylene (CAS No. 79-01-6) in Four Strains of Rats (Aci, August, Marshall, Osborne-Mendel) (Gavage Studies). TR 273. Research Triangle Park, NC:NTP. 1988. Available: http://ntp.niehs.nih.gov/ntp/htdocs/LT_rpts/tr273.pdf [accessed 19 September 2012] [PubMed]
- NTP (National Toxicology Program) Carcinogenesis Studies of Trichloroethylene (without Epichlorohydrin) (CASs No. 79-01-6) in F344/N Rats and B6C3F1 Mice (Gavage Studies). TR 243. Research Triangle Park, NC:NTP. 1990. Available: http://ntp.niehs.nih.gov/ntp/htdocs/LT_rpts/tr243.pdf [accessed 19 September 2012] [PubMed]
- Odum J, Foster J, Green T. A mechanism for the development of Clara cell lesions in the mouse lung after exposure to trichloroethylene. Chem Biol Interact. 1992;83:135–153. doi: 10.1016/0009-2797(92)90042-j. [DOI] [PubMed] [Google Scholar]
- Ou J, Ou Z, McCarver D, Hines R, Oldham K, Ackerman A, et al. Trichloroethylene decreases heat shock protein 90 interactions with endothelial nitric oxide synthase: implications for endothelial cell proliferation. Toxicol Sci. 2003;73:90–97. doi: 10.1093/toxsci/kfg062. [DOI] [PubMed] [Google Scholar]
- Peden-Adams M, Eudaly J, Heesemann L, Smythe J, Miller J, Gilkeson G, et al. 2006. Developmental immunotoxicity of trichloroethylene (TCE): studies in B6C3F1 mice. J Environ Sci Health A Tox Hazard Subst Environ Eng 41:249–271. [DOI] [PubMed]
- Persson B, Fredrikson M. Some risk factors for non-Hodgkin’s lymphoma. Int J Occup Med Environ Health. 1999;12:135–142. [PubMed] [Google Scholar]
- Pesch B, Haerting J, Ranft U, Klimpel A, Oelschlägel B, Schill W. Occupational risk factors for urothelial carcinoma: agent-specific results from a case-control study in Germany. Int J Epidemiol. 2000;29:238–247. doi: 10.1093/ije/29.2.238. [DOI] [PubMed] [Google Scholar]
- Purdue MP, Bakke B, Stewart P, De Roos AJ, Schenk M, Lynch CF, et al. A case-control study of occupational exposure to trichloroethylene and non-Hodgkin lymphoma. Environ Health Perspect. 2011;119:232–238. doi: 10.1289/ehp.1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaschou-Nielsen O, Hansen J, McLaughlin JK, Kolstad H, Christensen JM, Tarone RE, et al. Cancer risk among workers at Danish companies using trichloroethylene: a cohort study. Am J Epidemiol. 2003;158:1182–1192. doi: 10.1093/aje/kwg282. [DOI] [PubMed] [Google Scholar]
- Radican L, Blair A, Stewart P, Wartenberg D. Mortality of aircraft maintenance workers exposed to trichloroethylene and other hydrocarbons and chemicals: extended follow-up. J Occup Environ Med. 2008;50:1306–1319. doi: 10.1097/JOM.0b013e3181845f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer E, Hacker T, Flentke G, Drake V, Brody M, Lough J, et al. Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene. Toxicol Sci. 2010;113:444–452. doi: 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml P, Zhaou M, Richter J, Brüning T, Pommer M, Sauter G, et al. Analysis of kidney tumors in trichloroethylene exposed workers by comparative genomic hybridization and DNA sequence analysis. Verh Dtsch Ges Pathol. 1999;83:218–224. [in German] [PubMed] [Google Scholar]
- Scott CS, Chiu WA. Trichloroethylene cancer epidemiology: a consideration of select issues. Environ Health Perspect. 2006;114:1471–1478. doi: 10.1289/ehp.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CS, Jinot J. Trichloroethylene and cancer: systematic and quantitative review of epidemiologic evidence for identifying hazards. Int J Environ Res Public Health. 2011;8:4238–4272. doi: 10.3390/ijerph8114238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selgrade M, Gilmour M. Suppression of pulmonary host defenses and enhanced susceptibility to respiratory bacterial infection in mice following inhalation exposure to trichloroethylene and chloroform. J Immunotoxicol. 2010;7:350–356. doi: 10.3109/1547691X.2010.520139. [DOI] [PubMed] [Google Scholar]
- Selmin OI, Thorne PA, Caldwell PT, Taylor MR. Trichloroethylene and trichloroacetic acid regulate calcium signaling pathways in murine embryonal carcinoma cells p19. Cardiovasc Toxicol. 2008;8:47–56. doi: 10.1007/s12012-008-9014-2. [DOI] [PubMed] [Google Scholar]
- Siemiatycki J. 1991. Risk Factors for Cancer in the Workplace. Boca Raton, FL:CRC Press. [Google Scholar]
- Tang X, Li L, Huang J, Deng Y. Guinea pig maximization test for trichloroethylene and its metabolites. Biomed Environ Sci. 2002;15:113–118. [PubMed] [Google Scholar]
- Tang X, Que B, Song X, Li S, Yang X, Wang H, et al. Characterization of liver injury associated with hypersensitive skin reactions induced by trichloroethylene in the guinea pig maximization test. J Occup Health. 2008;50:114–121. doi: 10.1539/joh.l7114. [DOI] [PubMed] [Google Scholar]
- U.S. EPA (U.S. Environmental Protection Agency) Guidelines for Carcinogen Risk Assessment (2005). EPA/630/P-03/001F. Washington, DC:U.S. EPA. 2005. Available: http://www.epa.gov/cancerguidelines/ [accessed 19 September 2012]
- U.S. EPA (U.S. Environmental Protection Agency) Draft Toxicological Review of Trichloroethylene: in Support of the Summary Information in the Integrated Risk Information System (IRIS). Docket ID: EPA-hq-ord-2009-0791. 2009a. Available: http://www.regulations.gov/#!docketDetail;D=EPA-HQ-ORD-2009-0791 [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) IRIS Toxicological Review of Trichloroethylene (Interagency Science Consultation Draft). 2009b. Available: http://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=22536 [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) A New Approach to Protecting Drinking Water and Public Health. EPA 815F10001. Washington, DC:U.S. EPA Office of Water. 2010. Available: http://water.epa.gov/lawsregs/rulesregs/sdwa/dwstrategy/upload/Drinking_Water_Strategyfs.pdf [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) 2011 Edition of the Drinking Water Standards and Health Advisories. EPA 820-R-11-002. Washington, DC:U.S. EPA Office of Water. 2011a. Available: http://water.epa.gov/action/advisories/drinking/upload/dwstandards2011.pdf [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) IRIS Toxicological Review of Trichloroethylene (Interagency Science Discussion Draft). 2011b. Available: http://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=237625 [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) News Release: EPA Releases Final Health Assessment for TCE. 2011c. Available: http://yosemite.epa.gov/opa/admpress.nsf/03dd877d6f1726c28525735900404443/b8d0e4d8489ad991852579190058d6c3!OpenDocument [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) Trichloroethylene Toxicological Review and Appendices. 2011d. Available: http://www.epa.gov/iris/supdocs/0199index.html [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) TSCA Work Plan Chemicals: Methods Document. Washington, DC:U.S. EPA Office of Pollution Prevention and Toxics. 2012a. Available: http://www.epa.gov/oppt/existingchemicals/pubs/wpmethods.pdf [accessed 24 May 2012]
- U.S. EPA (U.S. Environmental Protection Agency) TRI Explorer: Release Trends Report. 2012b. Available: http://iaspub.epa.gov/triexplorer/tri_release.trends [accessed 23 July 2012]
- U.S. EPA SAB (U.S. Environmental Protection Agency Science Advisory Board) Review of Draft Trichloroethylene Health Risk Assessment: Synthesis and Characterization: An EPA Science Advisory Board Report. Washington, DC:U.S. EPA. 2002. Available: http://www.epa.gov/sab/pdf/ehc03002.pdf [accessed 17 January 2013]
- U.S. EPA SAB (U.S. Environmental Protection Agency Science Advisory Board) Review of EPA’s Draft Assessment Entitled “Toxicological Review of Trichloroethylene” (October 2009). EPA-SAB-11-002. Washington, DC:U.S. EPA. 2011. Available: http://yosemite.epa.gov/sab/sabproduct.nsf/B73D5D39A8F184BD85257817004A1988/$File/EPA-SAB-11-002-unsigned.pdf [accessed 19 September 2012]
- Vermeulen R, Zhang L, Spierenburg A, Tang X, Bonventre JV, Reiss B, et al. Elevated urinary levels of kidney injury molecule-1 among Chinese factory workers exposed to trichloroethylene. Carcinogenesis. 2012;33:1538–1541. doi: 10.1093/carcin/bgs191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang Y, Lan Q, Holford TR, Leaderer B, Zahm SH, et al. Occupational exposure to solvents and risk of non-Hodgkin lymphoma in Connecticut women. Am J Epidemiol. 2009;169:176–185. doi: 10.1093/aje/kwn300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Jacobson CF, Williams AL, Howard WB, DeSesso JM. Trichloroethylene-contaminated drinking water and congenital heart defects: a critical analysis of the literature. Reprod Toxicol. 2006;21:117–147. doi: 10.1016/j.reprotox.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Werner M, Birner G, Dekant W. The role of cytochrome p4503a1/2 in the sex-specific sulfoxidation of the hexachlorobutadiene metabolite, N-acetyl-S-(pentachlorobutadienyl)-l-cysteine in rats. Drug Metab Dispos. 1995a;23:861–868. [PubMed] [Google Scholar]
- Werner M, Guo Z, Birner G, Dekant W, Guengerich FP. The sulfoxidation of the hexachlorobutadiene metabolite N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-l-cysteine is catalyzed by human cytochrome p450 3A enzymes. Chem Res Toxicol. 1995b;8:917–923. doi: 10.1021/tx00049a004. [DOI] [PubMed] [Google Scholar]
- Woolhiser M, Krieger S, Thomas J, Hotchkiss J. Midland, MI: Dow Chemical Company; 2006. Trichloroethylene (TCE): Immunotoxicity Potential in CD Rats following a 4-week Vapor Inhalation Exposure. [Google Scholar]
- Wu C, Schaum J. Exposure assessment of trichloroethylene. Environ Health Perspect. 2000;108(suppl 2):359–363. doi: 10.1289/ehp.00108s2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauck J, Malloy M, Blair K, Simpson P, McCarver D. Proximity of residence to trichloroethylene-emitting sites and increased risk of offspring congenital heart defects among older women. Birth Defects Res A Clin Mol Teratol. 2004;70:808–814. doi: 10.1002/bdra.20060. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Krishnadasan A, Kennedy N, Morgenstern H, Ritz B. Estimated effects of solvents and mineral oils on cancer incidence and mortality in a cohort of aerospace workers. Am J Ind Med. 2005;48:249–258. doi: 10.1002/ajim.20216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.