

Abstract
Prosaposin is a precursor of saposins A, B, C, and D. Saposins are indispensable for lysosomal hydrolysis of sphingolipids. The notion that prosaposin itself is likely involved in brain development led us to generate an anti-mouse prosaposin-specific antibody that do not cross-react with any of the processed saposins. We have used it to study expression of prosaposin in the brain of wild-type (WT) and saposin D knockout mice (Sap-D−/−). Immunoblot studies indicated that prosaposin, already abundant in the brain of WT, was dramatically increased in Sap-D−/−. By immunohistochemistry, the brain of WT was rich in prosaposin in hippocampal CA3 pyramidal neurons, tufted cells and mitral cells in olfactory bulb, and cerebellar Purkinje cells. In Sap-D−/−, immunoreactivity of prosaposin was increased in these neurons, most notably in the CA3 pyramidal neurons which contained prosaposin immuno-positive inclusion bodies in the endoplasmic reticulum. Further characterization of these prosaposin-rich neurons may provide new insights into the physiological functions of prosaposin in the nervous system.
Keywords: Sphingolipid activator protein, saposins, neurotrophic factor, hippocampus, olfactory bulb, cerebellar Purkinje cell
Introduction
Prosaposin is a highly conserved glycoprotein of about 70 kDa and is the precursor of four saposins A, B, C, and D. Prosaposin (Psap) gene encodes saposins A–D in tandem with N- and C-terminal and inter-saposin sequences.1),2) Prosaposin is synthesized in the endoplasmic reticulum (ER) and transported through the Golgi apparatus. Major fractions of prosaposin are intracellularly targeted to the lysosomes via the mannose-6-phosphate receptor (M6PR) or by sortilin, where it is sequentially processed to individual saposins by proteolytic digestion. The remainder of prosaposin is secreted to the extracellular space and re-enters the cell by endocytosis through M6PR, a low-density lipoprotein receptor-related protein (LRP), or mannose receptors.3)–6)
The mature four saposins A–D are heat-stable glycoproteins of approximately 80 amino acids and 15 kDa.1),2) The most notable in vivo function of each saposin is to facilitate degradation of some sphingolipids in the lysosome as a cofactor of hydrolases. It has been clarified definitively that the deficiency of each saposin causes a lysosomal storage disease similar to that produced by the absence of the corresponding hydrolase both in humans and the mouse.1),2),7)–9) On the other hand, there is growing evidence showing that saposins have membrane-binding and lipid transport properties other than their basic function as enzyme cofactors.2)
It has been known that secreted prosaposin is present at relatively high concentrations in body fluids such as milk, cerebrospinal fluid, semen, bile, and pancreatic juice.10),11) In contrast to the ubiquitous expression of mature saposins in tissues, unprocessed prosaposin has been detected mainly in the brain, heart, and muscle tissues.12)–14) These observations support often-stated notion that prosaposin may possess its own physiological functions other than being a mere precursor of saposins. For example, the function of secreted prosaposin as a neurotrophic factor has long been proposed in the nervous system15),16) and as a spermatogenesis related factor in the reproductive organs.17),18) Recently, in the prostate cancers, the new role of secreted prosaposin to control the tumor metastasis has been reported.19),20) However, the specific in vivo functions of prosaposin in the nervous system and other organs are still not well understood.
In the present work, in order to clarify the possible physiological role of unprocessed prosaposin itself, we have generated an anti-mouse prosaposin-specific antibody that does not cross-react with any processed saposins. Using this antibody, we investigated the expression pattern of prosaposin in the tissues of wild-type mice and saposin D knockout (Sap-D−/−) mice9) by immunoblot and immunohistochemical analyses.
Materials and methods
Antibody
Polyclonal antibody against mouse prosaposin was generated by immunizing rabbits with three synthetic oligopeptides corresponding to 178-LYPQDHPRSQPQPKAN-193, 299-EMMDPYEQNLVQAH-312, and 413-KEPTPPKQPAQPKQSALP-430. These oligopeptides were selected from the mouse Psap sequences (GenBank accession number: U-57999) such that do not encode any saposins. These synthetic peptides with an additional cysteine residue at the N-terminal were conjugated to keyhole limpet hemocyanin. The antiserum was passed through a Sepharose column with bound antigen peptides and the bound antibody eluted with 0.1 M glycine (pH 2.7), and neutralized immediately with 1 M Tris-HCl (pH8.0). The rabbit antiserum against recombinant mouse saposin D was a gift from the laboratory of Dr. G. Grabowski.21)
Animals
The prosaposin, saposin A, and saposin D knockout mice (Psap−/−, Sap-A−/−, and Sap-D−/−) were originally generated in our laboratory by the gene targeting technology.7),9),22)Twitcher mouse, which is the naturally occurring mouse model of human Krabbe disease due to genetic galactosylceramidase deficiency, was originally purchased from the Jackson Laboratory (Bar Harbor, ME). The Psap−/−, Sap-A−/−, and Sap-D−/− mice were back-crossed to the C57BL/6J strain more than 10 generations so that the genetic background of all mice used in this study was C57BL/6J. A colony was maintained in the facility for the experimental animals at the Tokai University and all experimental procedures were approved by the institutional review committee.
Immunoblot analyses
Mouse tissues (brain, liver, kidney, testis, intestine, and placenta) from each genotype were dissected freshly and homogenized with a Potter-Elvehjem homogenizer. For cell fractionation, tissues were homogenized in an ice-cold 10 mM Tris-HCl (pH 7.0) containing 0.25 M sucrose, 1 mM ethylenediaminetetraacetic acid (EDTA) and 1.5% protease inhibitor cocktail (Sigma, St. Louis, MO). The homogenate was centrifuged at 10,000 × g for 15 minutes at 4 °C and the supernate was centrifuged at 100,000 × g for 1 hour at 4 °C. The separated pellet (membrane fraction) was suspended in a buffer of the same volume as the supernate. The supernate was designated as the cytosolic fraction. The solubilized proteins (50 μg) were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on 10–15% polyacrylamide gels (Daiichi Pure Chemicals, Tokyo) and transferred to polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ). The membranes were incubated with a 1% blocking solution (BM Chemiluminescence Western Blotting Kit (Mouse/Rabbit); Roche Molecular Biochemicals, Mannheim, Germany) in TBS buffer (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl) for 16 hours at 4 °C, then incubated with either the rabbit polyclonal anti-mouse prosaposin antibody (3.74 μg/ml), anti-saposin D antiserum diluted 1:100, or anti-mouse prosaposin antibody pre-absorbed by a 10 mg/ml solution of the three antigen peptides for 2 hour at room temperature. The membrane was then washed with the TBS-T (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1% Tween-20) buffer and incubated with peroxidase-labeled anti-rabbit IgG (40 mU/ml; Roche Molecular Biochemicals) for 1 hour at room temperature, followed by treatment with the detection solution of a BM chemiluminescence kit (Roche Molecular Biochemicals). The immunoreactive protein bands were visualized with a LAS-1000 Plus luminescence image analyzer (Fujifilm, Tokyo).
Mouse brain lysates (125 μg) were denatured in 1 × glycoprotein denaturing buffer with 0.2 M 2-mercaptoethanol at 100 °C for 3 min. The deglycosylation was carried by glycopeptidase F (1 mU) at 37 °C for 16 hours following manufacture’s instruction (Takara Bio Inc., Shiga). The reaction mixtures were analyzed by SDS-PAGE (50 μg protein/lane, 10% polyacrylamide gel) and probed by anti-mouse prosaposin antibody.
Real-time quantitative reverse transcrip-tase-PCR
Adult mouse brain tissues (n = 4) were prepared from 80-day old male mice of wild-type and Sap-D−/− mice. The fetal mouse brain and placental tissues were prepared from the pregnant female mice of wild-type and Sap-D−/− mice (n = 4). Isolated tissues were immediately frozen in liquid nitrogen and stored at −80 °C until use. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The total RNA content was quantified by UV-spectrophotometry and 10 μg of RNA was used for analysis. The TaqMan Reverse Transcription Reagent and TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) were used for RT-PCR. Mouse prosaposin RNA was quantified using AB 7500 Real time PCR System (Applied Biosystems). The oligonucleotide primers and FAM-labeled TaqMan probe for mouse prosaposin were purchased from Applied Biosystems (TaqMan® Gene Expression Assay). The mouse glyceraldehydes-3-phosphate dehydrogenase (GAPDH) gene was used as an endogeneous control to normalize the amount of total RNA in each sample. The oligonucleotide primers and VIC® labeled TaqMan probe for GAPDH were purchased from Applied Biosystems (TaqMan Rodent GAPDH Control Reagent VIC® Probe). The prosaposin expression levels were evaluated by Relative Quantification (RQ) using SDS v1.3 Software (Applied Biosystems). The relative expression ratio of prosaposin to GAPDH of each sample was calculated by 2–ΔCT (CT; Threshold cycle).
Immunohistochemical study
Tissue preparation
Three wild-type and Sap-D−/− male mice at 5–6 months-old were used for immunohistochemical study as previously described.23) The mice were anesthetized with ether and perfused through the left ventricle of the heart briefly with physiological saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB) (pH 7.4) and immersed in the same fixative at 4 °C overnight. Then the brain was dissected and processed for sectioning. For free-floating immunohistochemical studies, the brain was sliced in the coronal and parasagittal directions at 50 μm thickness with a Microslicer (DTK-1000, Dosaka Co., Ltd., Kyoto). For cryosections, after rinsing the brain tissues in phosphate buffered saline (PBS), it was sequentially immersed in 10%, 20% and finally 30% sucrose in PBS overnight at 4 °C. The brain was sectioned, placed in the O.C.T compound (Sakura Finetek U.S.A., Inc., Torrance, CA) and frozen on the dry ice. For sectioning, the samples were cut to 6 μm thickness with a cryostat microtome (Leica) and thaw-mounted on Matsunami adhesive silane (MAS)-coated glass slides (MATSUNAMI, Osaka). The mounted sections were air-dried for 1 hour.
Confocal microscopic immunohistochemical analyses
50 μm thick microsliced sections were incubated with 1% bovine serum albumin/phosphate-buffered saline with 0.3% Triton X-100 for 1 hour to block and increase the penetration of antibodies. They were then incubated in the primary antibodies by free-floating methods at 20 °C for overnight, and in the second antibodies for 2 hours as previously described. 23) The anti-mouse prosaposin antibody was used at a concentration of 3.74 μg/ml. Other primary antibodies used for the study were anti-calbindin D-28 k antibody [mouse monoclonal: Swant (Bellinzona, Switzerland)] and anti-parvalbumin antibody (mouse monoclonal: Swant). After incubation in mixtures of the primary antibodies, the sections were then incubated in mixtures of the species-specific secondary antibodies labeled with fluorescein isothiocyanate (FITC), Cy-3, or Cy-5 (Jakson ImmunoResearch Laboratories, Inc. West Grove, PA). All the secondary antibodies were used at 1:200 dilution. Hoechst 33258 (Dojindo, Tokyo) stain was used for DNA staining. Immunostained sections were mounted in Vectashield (Vector Laboratory, Burlingame, CA) and examined with a confocal laser scanning microscope (LSM510, Zeiss, Oberkochen, Germany). As a negative control for non-specific staining, additional sections were subjected to the immunostaining procedure with anti-mouse prosaposin antibody preabsorbed by the three antigen peptides. The experiments were repeated 3 times using three different sections from different mice.
Immunoelectron microscopic analyses
The 6 μ-thick cryosections were pretreated by 3% H2O2 in distilled water for 30 minutes to reduce the endogenous peroxidase activities. Sections were incubated with 1% BSA/PBS for 1 hour. They were incubated with rabbit anti-mouse prosaposin antibody at 20 °C overnight and then with the 1:200 diluted HRP-conjugated goat anti-rabbit IgG, F(ab’)2 specific antibodies (Jackson ImmunoResearch Laboratories, Inc.) at room temperature for 2 hours. After rinsing in PBS, they were fixed with 1% glutaraldehyde in 0.1 M PB for 10 minutes. Immunoreactivity was visualized by diaminobenzidine (DAB). Subsequently, sections were treated with 2% osmium tetroxide in 0.05 M PB for 1 hour. After dehydration in a series of graded ethanol, they were embedded in epoxy resin using the inverted gelatin capsule method. Ultrathin sections at 70-nm thickness were cut on an ultramicrotome (LKB 2800, Reichert-Jung, Wien, Austria). Sections were stained by a lead solution to identify the localization of DAB reaction products, and examined at an accelerating voltage at 80 kV by a transmission electron microscope (JEM-1200 ExII, JEOL, Tokyo) as previously described.24)
Results
Anti-mouse prosaposin antibody specifically recognized prosaposin
The specificity of the anti-mouse prosaposin antibody was demonstrated by immunoblot analyses of mouse whole brain homogenates. In the 15% gel SDS-PAGE, anti-prosaposin antibody recognized a major band with molecular weight about 70 kDa corresponding to the size of prosaposin, but no bands were detected at around 15 kDa corresponding to the size of mature saposins and of other partially processed forms at around 30 kDa or 45 kDa (Fig. 1A). The 70 kDa band was not detected in the brain of Psap−/− mice either by the anti-mouse prosaposin antibody or the anti-saposin D antibody, confirming that this 70 kDa band detected by the anti-prosaposin antibody was prosaposin (Fig. 1A). On the other hand, anti-saposin D antibody recognized not only the 70 kDa band but also the 15 kDa band, which was completely absent in Sap-D−/− mice, but clearly detected in Sap-A−/− mice (Fig. 1A). These results not only confirmed the absence of saposin D protein in Sap-D−/− mice but also the retention of saposin D protein in Sap-A−/− mice supporting the specific destruction of each saposin in our targeting strategy used previously to generate specific saposin knockout mice.7),9) Unexpectedly, the intensity of the 70 kDa band was dramatically increased in Sap-D−/− mice and it was also increased in Sap-A−/− and Twitcher mice to a lesser degree (Fig. 1A). In 10% SDS-PAGE, anti-mouse prosaposin antibody recognized additional band of about 60 kDa. Both 70 and 60 kDa bands were not detected in prosaposin knockout mouse (Fig. 1B). To check whether the difference of two bands of 70 kDa and 60 kDa were due to the glycosylation of prosaposin, brain lysates from wild-type and Sap-D−/− mice were treated with glycopeptidase F, which cleaves N-glycans from glycoproteins, and visualized by immunoblot using anti-mouse prosaposin antibody (Fig. 1B). Both in the wild-type and Sap-D−/− mice, the major portion of 70 kDa band was glycopeptidase F sensitive and shifted to the 60 kDa band whereas the 60 kDa band showed no changes in its size. These results indicate that the 70 kDa bands were N-glycosylated prosaposin and that the 60 kDa bands were prosaposin without N-glycosylation. Therefore, it could be concluded that the glycosylated prosaposin was dramatically increased in Sap-D−/− mice. By the cell fractionation experiment, prosaposin was detected both in the membrane fraction and cytosole fraction as expected from the presence of both secreted and endocytosed forms of prosaposin. Both 70 kDa and 60 kDa bands were significantly reduced with antibody preabsorbed by three antigen peptides, confirming that these two bands were specific to the antigenic peptides (Fig. 1C). Furthermore, although the reactivity was much weak, this anti-mouse prosaposin antibody also recognized the human prosaposin in peripheral leukocytes and fibroblasts (data not shown).
Fig. 1.
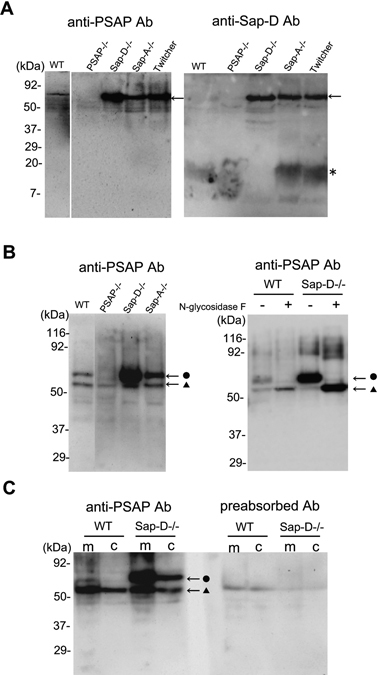
Immunoblot analyses of mouse brain homogenates with the anti-mouse prosaposin antibody. A: Tissue lysates from mouse brain of wild-type, Psap−/−, Sap-D−/−, Sap-A−/−, and Twitcher mice were separated by SDS-PAGE (15% gel) and probed with anti-mouse prosaposin antibody and anti-mouse saposin D antibody, respectively. Anti-mouse prosaposin antibody recognized only a band of approximately 70 kDa (arrow) corresponding to the molecular weight of prosaposin, but not a band of approximately 15 kDa (*), corresponding to the mature saposins in wild-type mice. This 70 kDa band was completely absent in Psap−/− mice and was increased in the Sap-D−/−, Sap-A−/−, and Twitcher mice. In contrast, anti-mouse saposin D antibody recognized not only the 70 kDa band but also the 15 kDa band, which was completely absent in Sap-D−/− mice and clearly detected in Sap-A−/− mice. B: In 10% gel, in addition to the 70 kDa band (•), anti-mouse prosaposin antibody recognized 60 kDa band (▴). The intensity of 70 kDa band was dramatically increased in Sap-D−/− mice followed by Sap-A−/− mice. Both bands were not detected in Psap−/− mice. By the glycopeptidase F treatment, in both the wild-type and Sap-D−/− mice, the majority of 70 kDa band was shifted to 60 kDa band. C: Both 70 kDa and 60 kDa bands were detected both in the membrane (m) and cytosolic (c) fraction and were significantly reduced with antibody preabsorbed by the three antigen peptides.
Prosaposin expression was dominant in the brain and glycosylated prosaposin was increased in Sap-D−/− mice
Tissue distribution of prosaposin in wild-type and Sap-D−/− mice was examined in adult and fetal tissues. In wild-type mice, prosaposin was expressed mainly in the brain followed by the kidney (Fig. 2A). In Sap-D−/− mice, in both adult and fetus, the remarkable increase of prosaposin with molecular weight of 70 kDa was demonstrated in all tissues examined, especially in the brain and the placenta (Fig. 2A). When we have done the immunoblot of fetal tissues from a Sap-D+/− crossing, the protein levels of prosaposin were elevated in the order of Sap-D−/−, Sap-D+/−, Sap-D+/+ both in the brain and placenta, suggesting that the increase of prosaposin in Sap-D−/− mice might be due to the mutant prosaposin protein (Fig. 2B). In Twitcher mice, the protein levels of prosaposin were not elevated in the fetal tissues (data not shown), suggesting that the elevation of prosaposin in the brain of symptomatic Twitcher mice (30 days-old) (Fig. 1A) was due to the secondary response to the pathology.25),26)
Fig. 2.
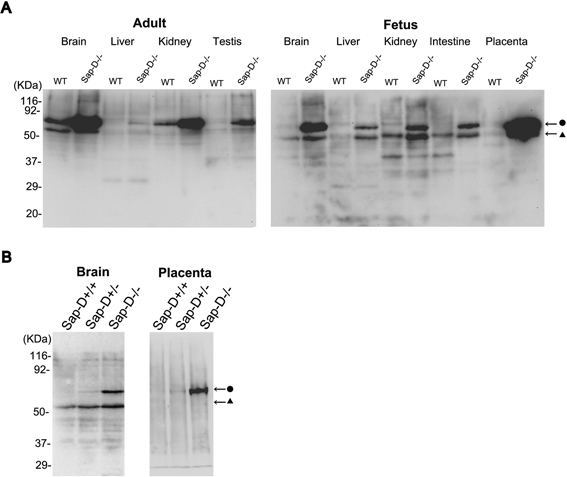
Tissue distribution of prosaposin and its elevation in Sap-D−/− mice. A: In tissues of wild-type mice, prosaposin was expressed mainly in the brain followed by the kidney. In Sap-D−/− mice, the protein levels of prosaposin, mainly of the 70 kDa size, were increased in all tissues examined both in adults and fetuses, most dramatically in the brain and placenta. B: The protein levels of prosaposin were already elevated fetuses of the Sap-D−/− mice both in the brain and placenta, compared to wild-type or heterozygous littermates.
No increase of prosaposin RNA level in Sap-D−/− mice
To address the question of whether the accumulation of prosaposin protein in the tissues of Sap-D−/− mice is due to the transcriptional regulation, we analyzed the mRNA levels of prosaposin in the brain and placenta of wild-type and Sap-D−/− mice. Although it is known that the mRNA that encodes proteins of the glycosphingolipid catabolism is often upregulated,25) no increase was detected in the mRNA levels of prosaposin either in adult or fetal tissues between wild-type and Sap-D−/− mice (Fig. 3). These results indicated that there is no transcriptional upregulation of prosaposin in the tissues of Sap-D−/− mice.
Fig. 3.

qRT-PCR analyses of prosaposin RNA in wild-type and Sap-D−/− mice. qRT-PCR analyses showed no significant difference in prosaposin mRNA levels between wild-type and Sap-D−/− mice, both in the brain and placenta. Prosaposin mRNA amounts were normalized to GAPDH mRNA for each sample. The data were presented as means ± S.E. *: P < 0.05 compared to the value of wild-type mice.
Immunohistochemical localization of prosaposin in the brain of wild-type and Sap-D−/− mice
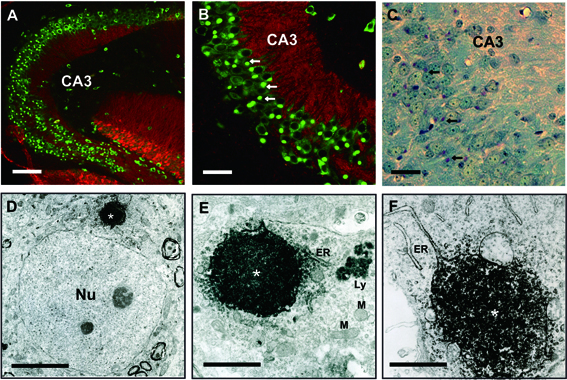
We next examined the in situ expression of prosaposin in the brain of wild-type and Sap-D−/− mice by immunohistochemical analyses using the anti-mouse prosaposin antibody and anti-mouse calbindin D-28K antibody. No immunoreactivity was observed by the incubation with pre-immune serum or with antibody preabsorbed with excess antigen peptides supporting the reliability and specificity of the anti-mouse prosaposin antibody (data not shown). In the brain of wild-type mice, the immunoreactivity of prosaposin was relatively weak, but regionally observed in the cytoplasm of certain types of neurons. In the hippocampus, pyramidal neurons especially in the CA3 area were prosaposin immune-positive (Fig. 4A). In the olfactory bulb, the immunoreactivity of prosaposin was clearly observed in tufted cells and mitral cells (Fig. 4B). In the cerebral cortex, neurons in layer II to VI were weakly prosaposin immune-positive (Fig. 4C). In the cerebellum, Purkinje cell bodies and granular cell layer were prosaposin immune-positive (Fig. 4D).
Fig. 4.

Immunohistochemical localization of prosaposin in the brain of wild-type and Sap-D−/− mice. Confocal microscopic analyses using anti-mouse prosaposin antibody (green) and calbindin (red) in wild-type (A–D) and Sap-D−/− mice (E–H). Overall, immunoreactivity of prosaposin was weak in wild-type mice and was dramatically increased in the brain of Sap-D−/− mice. In the hippocampus (A, E), the cytoplasm of pyramidal neurons, especially of those in the hippocampal CA3 area (arrow), was prosaposin immunoreactive. In Sap-D−/− mice, it was dramatically increased in the pyramidal neurons, especially in the CA3 area. In the CA3 pyramidal neurons, the prosaposin immunoreactive inclusion bodies were observed. In the olfactory bulb (B, F), the immunoreactivity of prosaposin was prominently observed in tufted cells (arrow) and mitral cells (arrow head) in wild-type and Sap-D−/− mice. In the cerebral cortex (C, G), neurons in layer II to VI were weakly prosaposin immunoreactive in wild-type mice, but the neurons of layer III and V in Sap-D−/− mice were strongly positive. In the cerebellum (C, H), Purkinje cells (arrow) and granular layer were immune-positive in wild-type and Sap-D−/− mice. GL: glomerular layer, MCL: mitral cell layer, GRL: granular cell layer, EPL: external plexiformlayer, ML: molecular layer, P: Purkinje cell layer, G: glanular cell layer. WM: white matter. The lines indicate scales of 100 μm.
In the brain of Sap-D−/− mice, over all, the immunoreactivity of prosaposin was increased and clearly detected even at 40 days, when Sap-D−/− mice do not yet show any clinical, pathological, and biochemical changes. In the hippocampus of Sap-D−/− mice, immunoreactivity of prosaposin was dramatically increased in the pyramidal neurons, especially in the CA3 area (Fig. 4E). In the CA3 pyramidal neurons, the prosaposin immune-positive perinuclear inclusion bodies were detected (Fig. 5A, B). These were periodic acid-Schiff (PAS) positive in paraffin sections suggesting that these were glycoproteins (Fig. 5C). These inclusion bodies were rarely observed in other pyramidal neurons in CA1 or CA2 area (Fig. 5A, B). In the olfactory bulb, the immunoreactivity of prosaposin was clearly observed in tufted cells and mitral cells (Fig. 4F). In the cerebral cortex, neurons in layer II to VI were strongly prosaposin immunoreactive and some neurons in Layer III and V had prosaposin immune-positive granular staining (Fig. 5G). In the cerebellum, Purkinje cells and the granular cell layer were strongly prosaposin immune-positive (Fig. 5H). These results suggest that there may be a neuron type-specific regulation of prosaposin expression in the central nervous system.
Fig. 5.
The prosaposin immuno-positive inclusion bodies in the CA3 pyramidal neuron of Sap-D−/− mice were localized in the ER. In the hippocampus of Sap-D−/− mice, prosaposin immuno-positive inclusion bodies were characteristically observed in the CA3 pyramidal neuron (A, B), corresponding to the periodic acid-Schiff (PAS) positive materials in paraffin embedded sections (arrows). In the immune-electronmicroscopic analyses (D–F), electron dense prosaposin immuno-positive reactions were observed (*) in the ER, and some ER showed balloon-like enlargements due to the retention of the prosaposin immunoreactive products (*) in E and F. This finding suggests impaired intracellular transport of prosaposin in the Sap-D−/− mice. ER: endoplasmic reticulum, Ly: lysosome, M: mitochondria, Nu: nucleus. The lines indicate scales of 50 μm in A, 25 μm in B, C, 2 μm in D, 1 μm in E, F.
Prosaposin were abnormally trapped in the ER of Sap-D−/− mice
To define the cellular localization of prosaposin immune-positive inclusion bodies in the hippocampal CA3 pyramidal neurons of Sap-D−/− mice, the immunoelectron microscopic analyses were carried out. In the hippocampal CA3 pyramidal neurons, prosaposin immuno-positive products were observed in the ER, but not in the Golgi apparatus (Fig. 5D, E). In most of the CA3 pyramidal neurons, balloon-like ER with the prosaposin immune-positive products were observed, which may correspond to the inclusion bodies (Fig. 5F). By the confocal microscopic analyses, these inclusion bodies were colocalized with the ER marker protein of BiP/GRP78 (data not shown). These findings suggested that the processing of prosaposin in the ER to Golgi apparatus were impaired in Sap-D−/− mice.
Discussion
Because the prosaposin is proteolytically processed to four saposins efficiently within the acidic compartments,1),2) it can be assumed that, the unprocessed prosaposin play no role in the membrane digestion in the lysosome. If the prosaposin functions just as the precursor of four saposins, why the unprocessed prosaposin is secreted or rich in the nervous system? There are reports that the secreted form of prosaposin could act as a glycolipid transfer protein, since several gangliosides were found to bind with high affinity to prosaposin.27),28) It has also been proposed that the prosaposin has the function to promote neurite outgrowth and nerve regeneration, and prevent cell death, when supplied exogeneously. 15),29)–31) Furthermore, the expression of prosaposin markedly increases in response to brain injury such as ischemia, or sciatic nerve crush injury.32),33) Several reports have identified the saposin C domain of prosaposin responsible for its in vitro and in vivo neurotrophic activity.16),34)
There is no doubt that inherited deficiency of prosaposin can be an indispensable tool to elucidate the function of prosaposin itself.1) In humans, four different genotypes (all homozygous) that lead to a complete loss of functional prosaposin and consequently of all four saposins have been reported to date.35)–39) In all cases so far studied in detail, patients showed a rapidly progressive, severe neurological disease with an accumulation of multiple sphingolipids in the brain and other organs leading to early death. A mouse model of prosaposin deficiency (Psap−/−) we generated earlier showed clinical, pathological and biochemical abnormalities similar to those of human patients.22),40) A significant number of Psap−/− mice die either in utero or within 1–2 days after birth, and most of those which survive these periods do not live beyond 30 days of life. If the prosaposin itself has neurotrophic activities, human patients of prosaposin deficiency or Psap−/− mice would be expected to present neuropathological changes in the organization of cerebral architecture or the vulnerability to the brain or nerve injury. However, the neuropathological phenotypes in the human and mouse prosaposin deficiencies are too complex due to the combined deficiency of all four saposins to dissect out the function of prosaposin itself.41)
In the present work, we generated an anti-mouse prosaposin-specific antibody by immunizing the rabbit with three oligopeptides, selected from the areas of the prosaposin sequence outside of the saposin-encoding domains. The aim of this work is to have the antibody that can overcome the problem of cross-reactivity with mature saposins we encountered when we used the anti-saposin antibodies to detect the in situ expression of unprocessed prosaposin by the immunohistochemical or immunocytochemical analyses.42),43) By the immunoblot analyses using this antibody, this antibody was confirmed to recognize only unprocessed prosaposin but not to cross-react with any saposins. The tissue distribution of prosaposin in wild-type mice confirmed the previous reports showing the abundance of prosaposin in the brain.12),13) Eventually, we found the dramatic increase of prosaposin, especially its glycosylated form in all the tissues examined in Sap-D−/− mice, most prominently in the brain and placenta. The Sap-D−/− mice were generated by introduction of the C509S mutation into the saposin D domain of Psap gene, which disrupts one of the three disulfide bonds of saposin D. The Sap-D−/− mice accumulate ceramides with hydroxylated fatty acids mainly in the brain and in the kidney.9) Since the mRNA levels of prosaposin were not elevated in any tissues from Sap-D−/− mice, and since the anti-prosaposin immune-positive inclusion bodies were detected in the ER, it was speculated that the C509S mutation in the saposin D domain might result in the misfolding of prosaposin and lead to partial failure of lysosomal targeting and ER trapping of prosaposin. It is also possible that the ER quality control system of misfolded proteins via cytoplasmic peptide: N-glycanase (PNGase) and ER-associated degradation (ERAD) is impaired in Sap-D−/− mice, which might lead to the accumulation of misfolded prosaposin in the ER.44) On the other hand, there is a report showing that C-terminus of prosaposin and saposin D domain may be required for lysosomal targeting.4) The combined deficiency of saposins C and D mouse model also showed the ER trapping of prosaposin.45) The ER-trapped prosaposin in Sap-D−/− mice may suggest the importance of saposin D domain for the proper transport of prosaposin from the ER to Golgi apparatus and to its final destination of the lysosome. A few important points remain for future studies; (1) the findings of a major portion of the mutated prosaposin being trapped in ER and the major portion of the accumulating prosaposin in the Sap-D−/− mice being in the glycosylated form appears to be contradictory in view of the known intracellular glycosylation process of newly synthesized glycoproteins or ER quality control process for misfolded proteins, (2) the ER-trapped prosaposin may trigger the ER stress together with activation of the unfolded protein response, which cause neuronal cell death, and (3) altered processing of prosaposin could cause the decrease of secreted prosaposin. Cells from Sap-D−/− mice could be useful to study the mechanism of intracellular processing of prosaposin. Interestingly, the recent report showed that the secretion and maturation of prosaposin was blocked in the embryonic neural progenitor cells.46) It indicates the importance of temporal and spatial post-translational regulation of prosaposin in the neurons.
By the immunohistochemical analyses using this anti-mouse prosaposin antibody, we investigated the in situ expression pattern of unprocessed prosaposin in the brain and revealed the regional expression of prosaposin in wild-type and Sap-D−/− mice. Based on the immunoblot findings that showed accumulation of prosaposin in the brain of presymptomatic Sap-D−/− mice, it is expected that cells which produce more prosaposin in the normal state show more intense prosaposin immunoreactivity in Sap-D−/− mice. We found it is indeed the case. In the hippocampus, pyramidal neurons in the CA3 region express more prosaposin than CA1 region. Interestingly, only the CA3 pyramidal neurons of Sap-D−/−mice showed characteristic prosaposin immune-positive inclusion bodies suggesting the regional function of prosaposin in these cells. The pyramidal cell layer of the hippocampus has been divided into three regions designated CA1, CA2, and CA3 based on the size and appearance of the neurons. In addition to the morphological differences, there is a clear-cut connectional differences. The CA3 pyramidal neurons receive mossy fiber inputs from the dentate gyrus and the CA2 and CA1 pyramidal neurons do not. The CA3 pyramidal cells project to other levels of CA3 as well as to CA1.47) There is a report showing that stress transiently reduces hippocampal prosaposin levels, not via an increase of glucocorticoid hormone. 48) In the olfactory bulb, mitral cells and tufted cells express more prosaposin than the other type of cells. Both neurons carry the output from the olfactory bulb projecting axons to the olfactory cortex.49) In the cerebellum, Purkinje cells most abundantly express prosaposin. Purkinje cells are some of the largest neurons and a class of GABAergic neurons. They are the single output system of the cerebellar cortex, sending long inhibitory projections to the deep cerebellar nuclei.50) Purkinje cells are affected in a variety of neurodegenerative diseases in humans and in spontaneous or experimental mouse mutants. Purkinje cells degeneration is also a conspicuous finding in Sap-D−/−, combined deficiency of saposins C and D, and other prosaposin related transgenic mice, suggesting the important role of prosaposin in the Purkinje cells.9),34),45) One of the common features of these neurons seems that they are relatively large, long projecting principal neurons. Further characterization of these neurons rich in prosaposin, may provide new insights into the physiological and pathophysiological functions of prosaposin itself. We conclude that this novel prosaposin specific antibody could be the useful tool to reveal the in vivo function of prosaposin itself not only in the central nervous system, but also in the other organs.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Sciences, Grant-in-aid for Scientific Research (18591169), a grant for Research for Intractable Diseases from the Japanese Ministry of Health, Welfare and Labor. J.M. was supported in part by a grant for Hi-Tech Research from Tokai University, Japan.
References
- 1).Sandhoff, K., Kolter, T. and Harzer, K. (2001) Sphingolipid activator proteins. InThe Metabolic and Molecular Basis of Inherited Disease (eds. Scriver, C. R., Beaudet, A. L., Sly, W. S., Valle, D., Childs, B. and Vogelstein, B.). McGraw-Hill, New York, pp. 3371–3388 [Google Scholar]
- 2).Kolter, T. and Sandhoff, K. (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 21, 81–103 [DOI] [PubMed] [Google Scholar]
- 3).Vielhaber, G., Hurwitz, R. and Sandhoff, K. (1996) Biosynthesis, processing, and targeting of sphingolipid activator protein (SAP) precursor in cultured human fibroblasts. Mannose 6-phosphate receptor-independent endocytosis of SAP precursor. J. Biol. Chem. 271, 32438–32446 [DOI] [PubMed] [Google Scholar]
- 4).Lefrancois, S., Zeng, J. B., Hassan, A. J., Canuel, M. and Morales, C. R. (2003) The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 22, 6430–6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Zeng, J., Racicott, J. and Morales, C. R. (2009) The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp. Cell. Res. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 6).Kiss, R. S., Ma, Z., Nakada-Tsukui, K., Brugnera, E., Vassiliou, G., McBride, H. M.et al. (2006) The lipoprotein receptor-related protein-1 (LRP) adapter protein GULP mediates trafficking of the LRP ligand prosaposin, leading to sphingolipid and free cholesterol accumulation in late endosomes and impaired efflux. J. Biol. Chem. 281, 12081–12092 [DOI] [PubMed] [Google Scholar]
- 7).Matsuda, J., Vanier, M. T., Saito, Y., Tohyama, J. and Suzuki, K. (2001) A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 10, 1191–1199 [DOI] [PubMed] [Google Scholar]
- 8).Spiegel, R., Bach, G., Sury, V., Mengistu, G., Meidan, B., Shalev, S.et al. (2005) A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol. Genet. Metab. 84, 160–166 [DOI] [PubMed] [Google Scholar]
- 9).Matsuda, J., Kido, M., Tadano-Aritomi, K., Ishizuka, I., Tominaga, K., Toida, K.et al. (2004) Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum. Mol. Genet. 13, 2709–2723 [DOI] [PubMed] [Google Scholar]
- 10).Hineno, T., Sano, A., Kondoh, K., Ueno, S., Kakimoto, Y. and Yoshida, K. (1991) Secretion of sphingolipid hydrolase activator precursor, prosaposin. Biochem. Biophys. Res. Commun. 176, 668–674 [DOI] [PubMed] [Google Scholar]
- 11).Kondoh, K., Hineno, T., Sano, A. and Kakimoto, Y. (1991) Isolation and characterization of prosaposin from human milk. Biochem. Biophys. Res. Commun. 181, 286–292 [DOI] [PubMed] [Google Scholar]
- 12).Kreda, S. M., Fujita, N. and Suzuki, K. (1994) Expression of sphingolipid activator protein gene in brain and systemic organs of developing mice. Dev. Neurosci. 16, 90–99 [DOI] [PubMed] [Google Scholar]
- 13).Sun, Y., Witte, D. P. and Grabowski, G. A. (1994) Developmental and tissue-specific expression of prosaposin mRNA in murine tissues. Am. J. Pathol. 145, 1390–1398 [PMC free article] [PubMed] [Google Scholar]
- 14).Sun, Y., Jin, P., Witte, D. P. and Grabowski, G. A. (2000) Prosaposin: promoter analysis and central-nervous-system-preferential elements for expression in vivo. Biochem. J. 352, 549–556 [PMC free article] [PubMed] [Google Scholar]
- 15).O’Brien, J. S., Carson, G. S., Seo, H.-C., Hiraiwa, M. and Kishimoto, Y. (1994) Identification of prosaposin as a neurotrophic factor. Proc. Natl. Acad. Sci. USA 91, 9593–9596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).O’Brien, J. S., Carson, G. S., Seo, H.-C., Hiraiwa, M., Weiler, S., Tomich, J. M.et al. (1995) Identification of the neurotrophic factor sequence of prosaposin. FASEB J. 9, 681–685 [DOI] [PubMed] [Google Scholar]
- 17).Collard, M. W., Sylvester, S. R., Tsuruta, J. K. and Griswold, M. D. (1988) Biosynthesis and molecular cloning of sulfated glycoprotein 1 secreted by rat Sertoli cells: sequence similarity with the 70-kilo-dalton precursor to sulfatide/GM1 activator. Biochemistry 27, 4557–4564 [DOI] [PubMed] [Google Scholar]
- 18).Morales, C. R., Zhao, Q., Lefrancois, S. and Ham, D. (2000) Role of prosaposin in the male reproductive system: effect of prosaposin inactivation on the testis, epididymis, prostate, and seminal vesicles. Arch. Androl. 44, 173–186 [DOI] [PubMed] [Google Scholar]
- 19).Koochekpour, S., Zhuang, Y. J., Beroukhim, R., Hsieh, C. L., Hofer, M. D., Zhau, H. E.et al. (2005) Amplification and overexpression of prosaposin in prostate cancer. Genes Chromosomes Cancer 44, 351–364 [DOI] [PubMed] [Google Scholar]
- 20).Kang, S. Y., Halvorsen, O. J., Gravdal, K., Bhattacharya, N., Lee, J. M., Liu, N. W.et al. (2009) Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and Tsp-1. Proc. Natl. Acad. Sci. USA 106, 12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Qi, X., Leonova, T. and Grabowski, G. A. (1994) Functional human saposins expressed in Escherichia coli. Evidence for binding and activation properties of saposins C with acid beta-glucosidase. J. Biol. Chem. 269, 16746–16753 [PubMed] [Google Scholar]
- 22).Fujita, N., Suzuki, K., Vanier, M. T., Popko, B., Maeda, N., Klein, A.et al. (1996) Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 5, 711–725 [DOI] [PubMed] [Google Scholar]
- 23).Tominaga, K., Matsuda, J., Kido, M., Naito, E., Yokota, I., Toida, K.et al. (2004) Genetic background markedly influences vulnerability of the hippocampal neuronal organization in the “twitcher” mouse model of globoid cell leukodystrophy. J. Neurosci. Res. 77, 507–516 [DOI] [PubMed] [Google Scholar]
- 24).Tamaki, T., Uchiyama, Y., Okada, Y., Ishikawa, T., Sato, M., Akatsuka, A.et al. (2005) Functional recovery of damaged skeletal muscle through synchronized vasculogenesis, myogenesis, and neurogenesis by muscle-derived stem cells. Circulation 112, 2857–2866 [DOI] [PubMed] [Google Scholar]
- 25).Potratz, A., Hutler, S., Bierfreund, U., Proia, R. L., Suzuki, K. and Sandhoff, K. (2000) Quantification of mRNAs encoding proteins of the glycosphingolipid catabolism in mouse models of GM2 gangliosidoses and sphingolipid activator protein precursor (prosaposin) deficiency. Biochim. Biophys. Acta. Mol. Basis Dis. 1502, 391–397 [DOI] [PubMed] [Google Scholar]
- 26).Morimoto, S., Yamamoto, Y., O’Brien, J. S. and Kishimoto, Y. (1990) Distribution of saposin proteins (sphingolipid activator proteins) in lysosomal storage and other diseases. Proc. Natl. Acad. Sci. USA 87, 3493–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Hiraiwa, M., Soeda, S., Kishimoto, Y. and O’Brien, J. S. (1992) Binding and transport of gangliosides by prosaposin. Proc. Natl. Acad. Sci. USA 89, 11254–11258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Misasi, R., Sorice, M., Carson, G. S., Griggi, T., Lenti, L., Pontieri, G. M.et al. (1996) Prosaposin and prosaptide, a peptide from prosaposin, induce an increase in ganglioside content on NS20Y neuroblastoma cells. Glycoconj. J. 13, 195–202 [DOI] [PubMed] [Google Scholar]
- 29).Kotani, Y., Matsuda, S., Sakanaka, M., Kondoh, K., Ueno, S. and Sano, A. (1996) Prosaposin facilitates sciatic nerve regeneration in vivo. J. Neurochem. 66, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 30).Tsuboi, K., Hiraiwa, M. and O’Brien, J. S. (1998) Prosaposin prevents programmed cell death of rat cerebellar granule neurons in culture. Dev. Brain Res. 110, 249–255 [DOI] [PubMed] [Google Scholar]
- 31).Calcutt, N. A., Campana, W. M., Eskeland, N. L., Mohiuddin, L., Dines, K. C., Mizisin, A. P.et al. (1999) Prosaposin gene expression and the efficacy of a prosaposin-derived peptide in preventing structural and functional disorders of peripheral nerve in diabetic rats. J. Neuropathol. Exp. Neurol. 58, 628–636 [DOI] [PubMed] [Google Scholar]
- 32).Sano, A., Matsuda, S., Wen, T.-C., Kotani, Y., Kondoh, K., Ueno, S.et al. (1994) Protection by prosaposin against ischemia-induced learning disability and neuronal loss. Biochem. Biophys. Res. Commun. 204, 994–1000 [DOI] [PubMed] [Google Scholar]
- 33).Unuma, K., Chen, J., Saito, S., Kobayashi, N., Sato, K., Saito, K.et al. (2005) Changes in expression of prosaposin in the rat facial nerve nucleus after facial nerve transection. Neurosci. Res. 52, 220–227 [DOI] [PubMed] [Google Scholar]
- 34).Sun, Y., Qi, X., Witte, D. P., Ponce, E., Kondoh, K., Quinn, B.et al. (2002) Prosaposin: threshold rescue and analysis of the “neuritogenic” region in transgenic mice. Mol. Genet. Metab. 76, 271–286 [DOI] [PubMed] [Google Scholar]
- 35).Harzer, K., Paton, B. C., Poulos, A., Kustermann-Kuhn, B., Roggendorf, W., Grisar, T.et al. (1989) Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur. J. Pediatr. 149, 31–39 [DOI] [PubMed] [Google Scholar]
- 36).Hulkova, H., Cervenkova, M., Ledvinova, J., Tochackova, M., Hrebicek, M., Poupetova, H.et al. (2001) A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum. Mol. Genet. 10, 927–940 [DOI] [PubMed] [Google Scholar]
- 37).Millat, G., Verot, L., Rodriguez-Lafrasse, C., Di-Marco, J. N., Rimet, Y., Poujol, Aet al. (2003) Fourth Reported 620 American Journal of Medical Genetics Part A Family with Prosaposin Deficiency. InBook of Abstracts 14th ESGLD Workshop (eds. Elleder, M., Ledvinová, J., Hřebíček, M., Poupetová, H. and Kožich, V.). Podebrady/Prague: Guarant Ltd, Prague, Czech, p 81 [Google Scholar]
- 38).Elleder, M., Jerabkova, M., Befekadu, A., Hrebicek, M., Berna, L., Ledvinova, J.et al. (2005) Prosaposin deficiency – a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipid storage disease. Report of a further patient. Neuropediatrics 36, 171–180 [DOI] [PubMed] [Google Scholar]
- 39).Kuchař, L., Ledvinová, J., Hřrebíček, M., Myšková, H., Dvořáková, L., Berná, L.et al. (2009) Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy): Report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations. Am. J. Med. Genet. Part A 149A, 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Oya, Y., Nakayasu, H., Fujita, N. and Suzuki, K. (1998) Pathological study of mice with total deficiency of sphingolipid activator proteins (SAP knockout mice). Acta Neuropathol. (Berl.) 96, 29–40 [DOI] [PubMed] [Google Scholar]
- 41).Sikora, J., Harzer, K. and Elleder, M. (2007) Neurolysosomal pathology in human prosaposin deficiency suggests essential neurotrophic function of prosaposin. Acta Neuropathol. (Berl). 113, 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Kondoh, K., Sano, A., Kakimoto, Y., Matsuda, S. and Sakanaka, M. (1993) Distribution of prosaposin-like immunoreactivity in rat brain. J. Comp. Neurol. 334, 590–602 [DOI] [PubMed] [Google Scholar]
- 43).Hosoda, Y., Miyawaki, K., Saito, S., Chen, J., Bing, X., Terashita, T.et al. (2007) Distribution of prosaposin in the rat nervous system. Cell Tissue Res. 330, 197–207 [DOI] [PubMed] [Google Scholar]
- 44).Helenius, A. and Aebi, M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 45).Sun, Y., Witte, D. P., Zamzow, M., Ran, H., Quinn, B., Matsuda, J.et al. (2007) Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and α-hydroxy ceramide accumulation, and altered prosaposin trafficking. Hum. Mol. Genet. 16, 957–971 [DOI] [PubMed] [Google Scholar]
- 46).Salvioli, R., Ricci-Vitiani, L., Tatti, M., Scarpa, S., De Maria, R. and Vaccaro, A. M. (2008) The secretion and maturation of prosaposin and procathepsin D are blocked in embryonic neural progenitor cells. Biochim. Biophys. Acta 1783, 1480–1489 [DOI] [PubMed] [Google Scholar]
- 47).Johnston, D. and Amaral, D. G. (2004) Hippocampus. InThe Synaptic Organization of the Brain, 5th edition (eds. Shepherd, G. M.). Oxford University Press, USA, pp. 455–498 [Google Scholar]
- 48).Scaccianoce, S., Mattei, V., Del Bianco, P., Gizzi, C., Sorice, M., Hiraiwa, M.et al. (2004) Hippocampal prosaposin changes during stress: a glucocorticoid-independent event. Hippocampus 14, 275–280 [DOI] [PubMed] [Google Scholar]
- 49).Shepherd, G. M. D., Chen, W. R. G. and Greer, C. A. (2004) Olfactory bulb. InThe Synaptic Organization of the Brain, 5th edition (eds. Shepherd, G. M.). Oxford University Press, USA, pp. 165–216 [Google Scholar]
- 50).Llinás, R. R., Walton, K. D. and Lang, E. J. (2004) Cerebellum. InThe Synaptic Organization of the Brain, 5th edition (eds. Shepherd, G. M.). Oxford University Press, USA, pp. 271–309 [Google Scholar]