

Abstract
Successful development of a multicellular organism depends on the finely tuned orchestration of cell proliferation, differentiation and apoptosis from embryogenesis through adulthood. The MYB-gene family encodes sequence-specific DNA-binding transcription factors that have been implicated in the regulation of both normal and neoplastic growth. The Drosophila Myb protein, DMyb (and vertebrate B-Myb protein), has been shown to be part of the dREAM/MMB complex, a large multi-subunit complex, which in addition to four Myb-interacting proteins including Mip130, contains repressive E2F and pRB proteins. This complex has been implicated in the regulation of DNA replication within the context of chorion gene amplification and transcriptional regulation of a wide array of genes. Detailed phenotypic analysis of mutations in the Drosophila myb gene, Dm myb, has revealed a previously undiscovered function for the dREAM/MMB complex in regulating programmed cell death (PCD). In cooperation with the pro-apoptotic protein Grim and dREAM/MMB, DMyb promotes the PCD of specified sensory organ precursor daughter cells in at least two different settings in the peripheral nervous system: the pIIIb precursor of the neuron and sheath cells in the posterior wing margin and the glial cell in the thoracic microchaete lineage. Unlike previously analyzed settings, in which the main role of DMyb has been to antagonize the activities of other dREAM/MMB complex members, it appears to be the critical effector in promoting PCD. The finding that Dm myb and grim are both involved in regulating PCD in two distinct settings suggests that these two genes may often work together to mediate PCD.
Keywords: Drosophila melanogaster, myb, apoptosis, programmed cell death, sensory organ, cell lineage
INTRODUCTION
Apoptosis plays a fundamental role in the development of all multicellular organisms. This process of programmed cell death (PCD) is required for eliminating excess or mis-specified cells, refining morphogenesis, and ensuring viability (reviewed in Conradt, 2009; Jacobson et al., 1997). Developmentally programmed apoptosis in Drosophila requires the pro-apoptotic RHG proteins, encoded by the reaper (rpr), head involution defective (hid), and grim genes, which are clustered within a ~300-kb region on chromosome III (Chen et al., 1996; Grether et al., 1995; White et al., 1994). These genes, along with two more recently identified family members, sickle (skl) and Jafrac2 (Srinivasula et al., 2002; Tenev et al., 2002; Wing et al., 2002), are differentially expressed and function as antagonists to DIAP1, the most central member of the inhibitor of apoptosis (IAP) protein family in Drosophila, culminating in caspase-dependent apoptosis (reviewed in Orme and Meier, 2009).
The requirement for RHG proteins during development was first revealed by the finding that embryos lacking rpr, hid, and grim, all three of which are deleted in the Df(3L)H99 chromosome (henceforth referred to as H99), die with excess cells due to lack of developmental apoptosis (White et al., 1994). These pro-apoptotic proteins function in a semi-redundant manner, at least during metamorphosis and imaginal disc development (Nicholson et al., 2009; Robinow et al., 1997; Yin and Thummel, 2004). However, each RHG protein has also been shown to have discreet functions, with hid being involved mainly in pruning excess cells in embryos and imaginal discs (de la Cova et al., 2004; Kurada and White, 1998; Zhou et al., 1997), and rpr being responsible for neural refinement during larval development (Peterson et al., 2002).
Studies on the function of grim have been limited by the lack of loss-of-function mutations. However, recent generation and analysis of a grim-null line has revealed a key role for grim in the apoptosis of thoracic microchaete glial cells (Wu et al., 2010). Microchaete differentiation follows the canonical pattern of sensory organ development in which one sensory organ precursor (SOP) cell divides asymmetrically to give rise to two daughter cells, pIIa and pIIb, which in turn also asymmetrically divide and generate the socket, shaft, sheath, neuron, and glial cells (Gho et al., 1999; Hartenstein and Posakony, 1989; Reddy and Rodrigues, 1999; and see Fig. 3A). However, it was subsequently discovered that in the thoracic microchaete lineage, glial cells undergo apoptosis (PCD) after the division of the pIIb cell, or around 23 hours after puparium formation (APF) (Fichelson and Gho, 2003). The non-innervated mechanoreceptors that are present in the posterior wing margin (PWM) of adult flies provide another example in which modified sensory organs appear to be generated via PCD (Lai and Orgogozo, 2004). In the PWM, asymmetric cell divisions begin at 11 hours APF, and by 16–20 hours APF, the presumptive socket and shaft cells have been specified (Hartenstein and Posakony, 1989; Takemura and Adachi-Yamada, 2011). The lack of neurons and sheath cells in the PWM bristles gave rise to the hypothesis that their precursor cells undergo lineage-specific apoptosis, a model strongly supported by the presence of neurons when apoptosis is blocked via ectopic expression of the caspase inhibitor p35 (Blair, 1992; Jafar-Nejad et al., 2006; Lai and Orgogozo, 2004).
Figure 3. Each ectopic PWM neuron is always accompanied by a sheath cell.

(A) A schematic of the canonical lineage specification process involving asymmetric cell divisions from one sensory organ precursor (pI) to give rise to specialized daughter cells. Variations of the canonical lineage generate diverse sensory organ types of the PNS (see Fig.8J and (Lai and Orgogozo, 2004). (B–C″) Shown are PWM of 24–28-hr APF (25°C) pupae labeled with anti-Elav (red) and anti-D-Pax2, which labels the sheath and shaft cells (green). Each ectopic neuron (red) at the PWM is accompanied by a sheath and a shaft cell (green) in both w; H99/+ (B-B″) and w myb2; H99/+ (C-C″). A few examples of this correlation are indicated as follows: sheath cells (arrowheads), corresponding neurons (arrows); shaft cells are marked with asterisks. Scale bar in B is 0.02 mm. All images are shown at the same magnification.
Throughout organismal development, cell proliferation, differentiation and apoptosis are finely orchestrated. The MYB-gene family encodes sequence-specific DNA binding transcription factors that play important roles in these developmental processes (reviewed in Oh and Reddy, 1999; Katzen, 2004). The sole myb gene in Drosophila, Dm myb, encodes DMyb, which is related to the three closely related members of the MYB family in vertebrates (A-Myb, B-Myb and c-Myb; aka MYBL1, MYBL2 and MYB, respectively) and has been shown to regulate cell proliferation in many tissues throughout development (Fitzpatrick et al., 2002; Fung et al., 2002, 2003; Katzen et al., 1998; Manak et al., 2002; Manak et al., 2007; Okada et al., 2002). DMyb has been found to be a member of a multi-subunit complex, dubbed the Myb complex, with four other proteins designated as Myb-interacting proteins (Mips) (Beall et al., 2004). The Myb complex was originally implicated in the repression and regulated derepression of a specialized form of DNA replication (chorion gene amplification) and transcription (Beall et al., 2004; Korenjak et al., 2004; Lewis et al., 2004), but has more recently been shown to have activating roles as well (Georlette et al., 2007; Wen et al., 2008). With respect to some repressive activities of the Myb complex, DMyb was shown to be a nonessential component of the complex, while another member of the complex, Mip130, has been shown to be critical for both repression and stability of the complex (Beall et al., 2004; Korenjak et al., 2004; Lewis et al., 2004). Thus, the absence of Mip130 leads to destabilization of the complex and loss of repression.
Subsequently, it was discovered that the Myb complex could be purified as part of a larger protein complex, the dREAM/MMB (Drosophila RBF, E2F2, and Myb/Myb-MuvB) complex, which in addition to the Myb complex proteins, invariably contains Drosophila’s repressive E2F and pRB proteins: dE2F2 and RBF1 or RBF2 (Korenjak et al., 2004; Lewis et al., 2004). The dREAM/MMB complex has been implicated in transcriptional repression of the developmentally regulated E group of dE2F target genes (Korenjak et al., 2004). However, a comprehensive analysis of the complex components in tissue culture cells has implicated many of the components in transcriptional activation as well (Georlette et al., 2007).
Studies on vertebrate MYB family proteins in apoptosis have implicated these proteins in both pro- and anti-apoptotic roles (reviewed in Greene et al., 2007; Ramsay and Gonda, 2008). In Drosophila, overexpression of an “activated” version of DMyb in the imaginal discs led to increased levels of apoptosis, although it was not determined whether this was a direct effect of increased DMyb activity or an indirect consequence of excess proliferation (Fitzpatrick et al., 2002; Okada et al., 2002). The possibility of DMyb having a direct role in apoptosis, however, is supported by its identification as a mild effector in response to apoptotic stimuli in an RNAi-based screen using S2R+ cells (Chew et al., 2009).
Here, we demonstrate a specific requirement for DMyb in the appropriate development of a subset of the diverse organs generated by SOPs of the peripheral nervous system (PNS). In cooperation with Grim and the dREAM/MMB complex, DMyb promotes the programmed cell death of specified SOP daughter cells in these developmental settings. In myb-null wings, neurons developed inappropriately at the posterior margin due to a defect in developmentally specified apoptosis. Although genetic interaction studies indicated that the pro-apoptotic genes reaper and hid contributed weakly to this phenotype, grim interacted strongly with myb and was found to be the major effector of this lineage-specific developmental PCD at the posterior wing margin. myb was also shown to participate in the grim-mediated glial cell death that occurs in the thoracic microchaete lineage, suggesting that these two genes may often work together to mediate PCD. In addition, our wing data provide strong support for the hypothesis that at the PWM, it is the neural precursor pIIIb cell that undergoes apoptosis, rather than the pIIb cell or the neurons and sheath cells themselves. Surprisingly, unlike other settings, we found that the Mip130 protein does not play an active role in this developmental process, but instead contributes solely through its stabilization of DMyb and the Myb complex.
MATERIALS AND METHODS
Drosophila strains
The following Drosophila strains were used in this study: white (referred to as w), w myb1, w myb2 and Dp(1;Y)shi+1 (carries a wild-type copy of Dm myb and is referred to as YDup) have been described previously (Katzen and Bishop, 1996); mybMH107/FM7, Act-GFP [(Manak et al., 2002), from J. Lipsick]; mip1301-36/FM7a and w mip1301-36 mybMH107/FM7, Kr-Gal4, UAS-GFP(S65T) [(Beall et al., 2004), from M. Botchan]; e2f2c03344/CyO [(Thibault et al., 2004), from the Exelixis/Harvard collection]; w1118 Ubi-GFP(S65T) RPS5a2 FRT18A/FM7a (referred to as Minute FRT18A), w FRT18A piMyc; MKRS hs-FLP/TM6, Tb and w FRT18A Ubi-GFP(S65T) piMyc; MKRS hs-FLP/TM6, Tb, all from B. Edgar; P{GawB}bbgC96 [referred to as C96-Gal4, (Gustafson and Boulianne, 1996), from E. Siegfried]; P{UAS-p35.H} (second- and third-chromosome inserts, referred to as UAS-P35, from G. Rubin); Df(3L)H99/TM6B, Df(3L)WRX25/TM3 (White et al., 1994), XR38/TM3, Sb Kr-GFP (Peterson et al., 2002), rpr87/TM2 (Moon et al., 2008), and hid05014/TM6 (Grether et al., 1995), all from K. White; grimA6C/TM3, Sb twi-GFP Ser and UAS-grimRNAi have been described previously (Wu et al., 2010); P{GMR-grim}1 (referred to as GMR-grim, from Bloomington Stock Center); w mip1301-36 myb2/FM7, w mip1301-36 FRT18A/FM7, w mip1301-36 mybMH107 FRT18A/FM7, and (y) w mybMH107 FRT18A/FM7; MKRS hs-FLP/TM6, Tb were generated from the fly lines listed above for this study. Detailed descriptions of all alleles, transgenic lines and aberrations can be found on Flybase at http://flybase.org/.
Mosaic analysis
Mitotic recombination was induced using the FLP/FRT method (Xu and Rubin, 1993). Animals were raised and staged at 24°C. Relevant myb and mip130 lines were recombined onto the FRT18A chromosome by crossing each to the w1118 piMyc FRT18A line (Bloomington Stock Center). To generate mip1301-36 and mip1301-36 mybMH107 clones, w mip1301-36 FRT18A males or w mip1301-36 mybMH107 FRT18A/YDup males were crossed to w FRT18A Ubi-GFP piMyc; MKRS hs-FLP/TM6 Tb females. We found that mybMH107 clones were small and did not compete/survive well with neighboring cells. So to generate clones in a Minute FRT18A background, w mybMH107 FRT18A/YDup; MKRS hs-FLP/TM6 Tb males or w FRT18A piMyc; MKRS hs-FLP/TM6 Tb males were crossed to Minute FRT18A females. At 72 hours after egg deposition, the progeny were heat shocked for one hour at 37°C. Desired female prepupae that were GFP-positive and non-Tb were picked 4–5 or 5–6 (Minute background) days after the heat-shock treatment.
Tissue preparation and immunohistochemistry
Animals were raised and staged at 24°C unless specified otherwise. Adult wing dissection was performed in 100% isopropanol. The wings were then mounted in Euparal mounting medium (ASCO Laboratories).
For immunostaining pupal wings, white prepupae [0 hours after puparium formation (APF)] were picked and aged for 24–28 hours APF at 24°C or to 72 hours APF at 18°C, and then fixed overnight in 4% formaldehyde/PBS at 4°C. For pupal nota, white prepupae were aged for 27–28 hours APF at 24°C and then fixed in 4% paraformaldehyde/PBS with 0.1% Triton-X for 30 minutes at room temperature. For immunostaining of larval tissue, wandering third-instar larvae were dissected and fixed in 4% paraformaldehyde/PBS with 0.1% Triton-X for 30 minutes at room temperature. Immunostaining was performed as described previously (Fitzpatrick et al., 2002) using the following primary antibodies: mouse anti-22C10 [1:100; Developmental Studies Hybridoma Bank (DSHB)] (Zipursky et al., 1984), mouse anti-Elav (1:100; DSHB) and rat anti-Elav (1:200; DSHB) (Robinow and White, 1991), rabbit anti-DPax2 (1:100; gift from M. Noll) (Kavaler et al., 1999), goat anti-DMyb (dN-17, lot #A2804, 1:50; Santa Cruz Biotechnology) (Scaria et al., 2008), mouse anti-Repo (1:200; DSHB) (Alfonso and Jones, 2002). To detect apoptotic cells or Grim protein expression, wings were dissected from pupae at 20–24 hr APF and stained with rabbit anti-Cleaved Caspase3 (1:150; Cell Signaling Technology) or goat anti-Grim (dN-16, lot #A222, 1:50; Santa Cruz Biotechnology), respectively. Secondary antibodies conjugated to FITC, Texas Red, or Cy5 (Jackson ImmunoResearch) were used at 1:200 dilutions. Samples were mounted in Vectashield (Vector Labs) and imaged using a Zeiss Axiovert 200M microscope; confocal images were obtained with the LSM 5 Pascal system. All images were processed with Adobe Photoshop 10.0.1.
Quantification of cell survival
To quantify ectopic neurons in the wing, Elav-positive cells along the posterior wing margin were counted from 10–15 mounted pupal wings dissected from independent pupae of each of the indicated genotypes. Quantitative assessment of glial cell apoptosis in the notum was performed by counting the number of Repo-positive glial cells associated with the Elav-positive neurons as a percentage of the total number of Elav-positive neurons for each genotype.
RESULTS
Loss of DMyb leads to inappropriate neuronal development at the PWM
Using temperature-sensitive, hypomorphic mutant alleles, we have previously shown that decreased DMyb function leads to a reduction in the number of wing hairs due to the majority of cells being arrested before the last round of mitosis, and instead undergoing endoreplication (Katzen et al., 1998). These mutants also exhibited larger nuclei, defects in wing hair orientation, and thicker wing veins. Surprisingly however, there was no obvious effect on the number of bristles along the wing margin. To further understand the role of DMyb in wing development, a null mutation that is inviable as a homozygote, mybMH107 (Manak et al., 2002), was used to generate loss-of-function clones in the wing. Clonal analyses revealed that in addition to wing hair defects that were similar to those shown by the hypomorphs, albeit more extreme, the null clones unexpectedly exhibited novel phenotypic abnormalities in the bristles along the wing margin. In adult wings, mechanosensory and chemosensory bristles, which are generated from SOP cells, are stereotypically arranged along the anterior wing margin (AWM; Fig. 1A), while slender, non-innervated bristles, which are also derived from the SOP cells, line the posterior margin (PWM; Fig. 1C) (Hartenstein and Posakony, 1989). mybMH107 clones (marked by yellow) exhibited both loss and gain of bristles at the anterior and posterior wing margins, and some of the mutant bristles were noticeably thicker than their wild-type counterparts (Fig. 1B,D). Although the loss of bristles could be a consequence of cell loss due to more severe defects in the proliferation of wing cells, it is unlikely that these mitotic defects could cause the gain and change of bristles.
Figure 1. Drosophila myb plays a role in sensory organ development in the wing.

(A–D) Shown are adult wings from y mybMH107 FRT/myb+ Ubi-GFP Minute FRT; hs-FLP/+ flies. Control wings, which do not contain any clones, display the normal array of mechanosensory (ms) and chemosensory (cs) bristles along the AWM (A) and slender, non-innervated (ni) bristles along the PWM (C). mybMH107 clones (marked with yellow) at the AWM (B) and PWM (D) exhibit thicker bristles (black arrows), loss of bristles (asterisk), gain of bristles (open arrowheads), and split bristles (black arrowheads). (E–F″) Shown are pupal wings dissected 24–28 hours APF from mybMH107 FRT/myb+ Ubi-GFP Minute FRT; hs-FLP/+ animals and stained with anti-22C10 (red; E,F) and anti-Elav (red in merges; E′-E″; F′-F″). In control wings without clones as shown by GFP fluorescence (green) throughout the wing (E-E″), neurons are not present at the PWM. In contrast, ectopic neurons were invariably present in mutant clones (marked with lack of GFP) that straddle the PWM (arrow in F, F′ and F″). In this and all subsequent figures, pupal wings are presented in the same orientation, and white arrowheads mark the distal tip of longitudinal vein L3. The orientation of the AWM and PWM are indicated in panel E (yellow arrows). Scale bars in A and E are 0.05 mm, A–D and E-F″ are shown at the same magnifications.
The bristle defects led us to examine developing wings at 24 hours APF, a time point when the cell division cycle has ended and bristle development can be followed using antibodies that recognize the different cell types that make up each bristle. Specifically, we used Elav antibody (Robinow and White, 1991), which stains the neurons, and the 22C10 antibody (Zipursky et al., 1984), which stains the axons extending from the neurons and the shaft cells of the innervated sensory bristles in the wing. As expected, staining of 24–28 hour APF wings with DAPI and 22C10 showed larger and less organized cells in the clones (see Fig. S1 in supplementary material), reminiscent of the phenotypes of myb hypomorphs (Katzen et al., 1998). Our attempts to discern the basis of the bristle defects in mutant myb tissues was hampered by the disorganization of the cells (and bristle positioning) along with the multitude of sensory organ cells at the AWM. The AWM contains two rows of mechanosensory bristles and one row of chemosensory bristles, with each chemosensory bristle being associated with five neurons (Hartenstein and Posakony, 1989).
However, we also made an unexpected observation. In wild-type and control wings that are heterozygous for the mybMH107allele, neuronal innervation of sensory bristles is restricted to the anterior compartment (Palka et al., 1983; and see Fig. 1E-E″). In contrast, labeling with Elav and 22C10 antibodies revealed that mybMH107 clones exhibit ectopic neuronal development at the PWM (ca. 40 neurons; Fig. 1F-F″). Since there have been reports of abnormalities with the FRT insert at 18A in some contexts (Ebacher, 2002), and the clones were generated in a Minute background to provide myb−/− cells with a relative growth advantage, we examined PWM myb+/+ clones made in the Minute background using the starting FRT line (FRT18A) from which the myb FRT line was generated as a control. We did not detect any ectopic neurons in these clones, indicating that neither the Minute background nor the FRT18A insert had any influence on this phenotype (Fig. S2). Therefore, we conclude that the ectopic-neuron phenotype was specific to loss of Dm myb. This ectopic-neuron phenotype was unexpected since mutations in myb have previously been shown to result in slower rates of proliferation, leading to a reduction in the final number of cells (Fung et al., 2002; Katzen et al., 1998). Consequently, the presence of neurons at the PWM, reflecting the presence of ectopic or mis-specified cells, suggested to us that this phenotype might be independent of DMyb’s role in cell cycle.
DMyb is required for neuronal apoptosis at the PWM and synergizes with the apoptotic pathway
Our discovery of neurons at the PWM of developing wings was not the first such observation. The SOPs of non-innervated bristles at the PWM appear and divide asymmetrically by 11 hours APF, and their descendants, the socket and shaft cells, are specified by 16–20 hours APF (Hartenstein and Posakony, 1989). Jafar-Nejad and colleagues (2006) reported the appearance of neurons along the PWM when the baculovirus p35 caspase-inhibitor protein was ectopically expressed along the wing margin via the C96-Gal4 driver. This finding agrees with the previous reports that in wild-type wings, lack of neurons in the bristles along the PWM is caused by a lineage-specific apoptosis of the neurons or their precursor cells, the pIIb or pIIIb cells (Blair, 1992; Lai and Orgogozo, 2004; and see Fig. 3A). This would then be an example of developmentally targeted PCD to ensure that the posterior bristles are not innervated, even though the same lineage pathway to generate the different cell types of the sensory organs was initiated at both the AWM and PWM.
The survival of neurons in myb-null clones at the PWM suggested that loss of DMyb function led to suppression of apoptosis in the neural lineage of developing bristles. To determine whether the suppression of apoptosis in myb mutants is specific to the PCD at the PWM, we examined other cells in mybMH107 clones to assess whether general apoptosis in the developing wing could be suppressed by loss of DMyb. Double labeling of pupal wings with cleaved Caspase-3 and Elav antibodies revealed that in control wings, cells along the wing margins are undergoing apoptosis, with scattered apoptosis also occurring throughout the wing blade (Fig. S3A), as has been reported in wild-type 20–24-hour APF wings (Milan et al., 1997; Takemura and Adachi-Yamada, 2011). In mybMH107 clones within the wings, we found that loss of DMyb actually resulted in increased, rather than decreased, levels of apoptosis, presumably due to cell division defects (Fig. S3B″). These results demonstrate that loss of Dm myb function does not suppress general apoptosis of non-sensory cells in the wing, thereby indicating that the DMyb protein is actively involved in promoting the developmentally specified neural apoptosis at the PWM.
To further investigate the role of DMyb in this process, we proceeded to examine the genetic interaction between Dm myb and the apoptotic pathway by introducing one copy of the H99 deficiency chromosome, which deletes one copy of reaper, hid and grim, to mildly down-regulate the apoptotic response (Abbott and Lengyel, 1991; Chen et al., 1996; White et al., 1994). In this study, the temperature-sensitive (ts) mutant myb2 allele was used at 18°C, since at this temperature, this hypomorphic allele exhibits minimal cell-cycle defects (Katzen and Bishop, 1996; Katzen et al., 1998). Ectopic neurons were never observed at the PWM of control wings but were detected at low incidence in myb2 wings (ca. 3 neurons per wing), and at a modestly higher level in pupal wings that were heterozygous for the H99 deficiency but wild type for myb (ca. 12 neurons) (Fig. 2A–C). In contrast, when H99 was introduced into the myb2 background, the anti-apoptotic effects synergized so that large numbers of ectopic neurons were consistently observed at the PWM (ca. 111 neurons; Fig. 2D). This result is similar to the PWM phenotype resulting from when a single copy of UAS-p35 is driven by C96-Gal4 throughout the wing margin (Fig. 2E). In addition, labeling with the 22C10 antibody indicated that the ectopic neurons observed in w;H99/+ and myb2;H99/+ wings are functional since they sent out axons, with the latter generating a thicker axon bundle due to the presence of more neurons (Fig. 2C′-C″,D′-D″). The average number of ectopic neurons, which reflects the level of suppression of developmentally programmed apoptosis along the PWM, is depicted in Figure 2F. The synergy between myb2 and H99 was even more pronounced at 24°C, a temperature at which myb2 does exhibit some cell cycle defects. While there were still no ectopic neurons in control wings, there were more ectopic neurons at 24°C than at 18°C in the presence of myb2 and H99, both independently and in combination (Fig. 4). Notably, myb1, a second temperature-sensitive allele that displays stronger cell-cycle defects than myb2 in the wing (Katzen et al., 1998), exhibited a more modest interaction with H99 than myb2 did (Fig. S4). Since the genetic interaction between myb2 and H99 was observed at a temperature with minimal myb-associated cell cycle defects (Fig. 2F), we concluded that the ectopic-neuron phenotype was largely independent of perturbations in the cell cycle.
Figure 2. H99 synergizes with myb2 in suppressing neural apoptosis at the PWM.
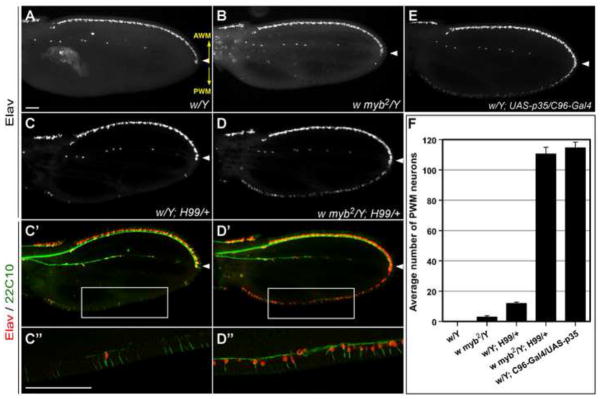
Shown are developing wings dissected 72 hours APF (18°C) from pupae of the indicated genotypes, which were then stained with Elav (red in merges) and 22C10 (green in merges) antibodies. No neurons were detected by Elav staining at the PWM of white (w) control wings (A). A few neurons were detected at the PWM of w myb2 wings (B), and a modestly higher level was present at the PWM of w; H99/+ wings (C). In contrast, a continuous row of ectopic neurons along the entire PWM were detected in w myb2; H99/+ wings (D), reaching levels that phenocopy w; C96-Gal4/UAS-p35 wings (E). 22C10-labeling revealed axonal projections from the ectopic PWM neurons and their shaft cells (C′-C″; D′-D″). C″ and D″ are magnifications of boxed areas in C′ and D′, respectively. Scale bars in A and C″ are 0.05 mm; A-D′, E and C″-D″ are shown at the same magnifications. The orientation of the AWM and PWM are indicated in panel A (yellow arrows). (F) Quantification of the number of neurons at the PWM from 10–15 independent pupal wings of each genotype indicated. Error bars in this and all other graphs represent s.e.m. Only males are shown; the data from females is similar, albeit with slightly higher numbers for the mutant genotypes.
Figure 4. Loss of mip130 enhances myb2 phenotype at the PWM.

As illustrated by the graph, a few neurons were present at the PWM of myb2 and mip1301-36 wings, and more were present in double-mutant wings (note that in this and subsequent graph labels, mip1301-36 is abbreviated as mip130). Independently, myb2 and mip1301-36 appeared to synergize with H99 to similar extents. The synergistic effects were stronger when both the mip1301-36 and myb2 mutations were combined with H99. Quantification was performed on wings dissected at 24–28 hours APF. Note that for the genotypes that are also shown in Figure 2F, the increase in the numbers of PWM neurons shown here is due to a difference in the temperatures at which the animals were cultured: 18°C in Figure 2F versus 25°C in this graph.
To discern the basis of ectopic neurons at the PWM, we examined whether these neurons might be generated at the expense of their sister cells, the sheath, socket, or shaft cells (i.e. due to mis-specification). The 22C10 labeling of the myb-mutant wings already indicated that each ectopic neuron was associated with a shaft cell (Fig. 2D″). To investigate this issue further, an antibody against D-Pax2, which labels the shaft and sheath cells (Kavaler et al., 1999), was used (Fig. 3B′,C′). Double labeling of w;H99/+ and myb2;H99/+ wings with Elav and D-Pax2 demonstrated that in addition to a shaft cell, each ectopic neuron at the PWM was also associated with a sheath cell (Fig. 3B″,C″). Since ectopic neuron and sheath cells are always detected together, it is highly unlikely that these cells are being protected from apoptosis independently. Therefore, this finding supports the hypothesis that at the PWM, the program to make all of the specialized cells that compose sensory bristles is initiated, but that one of the precursor cells (pIIb or pIIIb), as opposed to the neuron and sheath cells themselves, then undergoes apoptosis. Taken together, these data indicate that DMyb works in cooperation with the apoptotic pathway to ensure the developmentally specified apoptosis of the neuronal precursor cells at the PWM.
As a component of the Myb complex, DMyb plays an active role in the PCD of neuronal precursors at the PWM
Initial studies of the multi-subunit Myb complex suggested that the primary function of DMyb is to antagonize the repression of replication or transcription exerted by other components of the Myb complex, and that therefore, in the absence of the Myb complex, DMyb function is no longer required. Support for this hypothesis comes from the finding that while flies homozygous for a null mutation in Dm myb are inviable, flies that are homozygous for null mutations in both Dm myb and mip130 can survive into adulthood (Beall et al., 2004; Manak et al., 2002). We also found that in the context of the adult wing, the wing-hair phenotype observed in myb mutants, which we have previously reported to be a consequence of cell division defects (Katzen et al., 1998), was suppressed in flies that were doubly homozygous for mutations in both myb and mip130 (Fig. S5), which is in agreement with the model that the two proteins are working antagonistically within the complex, and that DMyb is only required to counteract the activity of the other complex members. However, results from microarray expression analysis of cultured Drosophila cells that had been depleted of individual Myb complex components using RNA interference are suggestive of a more complex interaction between the complex members (Georlette et al., 2007). Therefore, we decided to investigate whether mip130 plays a role in promoting neural apoptosis at the PWM, and if so, determine its interaction with Dm myb in this process.
Elav staining of pupal wings dissected from mip1301-36 mutants (a knockout mutation in mip130) (Beall et al., 2004) revealed that loss of Mip130 by itself had a mild effect on the suppression of apoptosis, which was similar to the myb2 phenotype (Fig. 4). In mip1301-36 myb2 double mutants, the phenotype was enhanced, suggesting that contrary to their antagonistic interaction during the cell division cycle, DMyb and Mip130 might work cooperatively to ensure that the developmentally specified neural apoptosis occurs appropriately at the PWM (Fig. 4). Combining myb2 or mip1301-36, individually or in combination, with the H99 deficiency led to potent phenotypic enhancement, suggesting that there are synergistic interactions between the Myb complex genes and the pro-apoptotic genes uncovered by the H99 deficiency chromosome (Fig. 4).
As noted above, it has been shown previously that at least in some settings, the Myb complex is destabilized and DMyb levels are reduced in the absence of Mip130 (Beall et al., 2004; Georlette et al., 2007). We have found this to be true for many, but not all tissues (J. McEllin, G. Scaria, manuscript in preparation; ALK, unpublished observations). Therefore, we decided to investigate whether the absence of Mip130 affects DMyb levels in the pupal wing, which may be part of the basis for the genetic interaction between the myb and mip130 mutants in the PWM. In order to be able to accurately compare the relative levels of DMyb protein in wing cells of different genotypes, we generated clones of mip1301-36 mutant cells within heterozygous animals (Fig. 5B). When pupal wings were stained with DMyb antibodies that have previously been shown to be specific for the DMyb protein via the absence of signal in null myb clones (Scaria et al., 2008), relatively uniform levels of DMyb were observed throughout control wings (heterozygous for mip1301-36 and without any generated clones; Fig. 5A-A′), whereas DMyb levels were significantly lower in cells that were homozygous for the mip1301-36 mutation, and higher in the twin spot cells that were homozygous for the wild-type allele of mip130 (Fig. 5B-B′).
Figure 5. The role of Mip130 protein and the Myb complex in neural apoptosis at the PWM is executed through DMyb.

(A–B″) Shown are pupal wings dissected 24-28 hours APF from mip1301-36 FRT/Ubi-GFP FRT; hs-FLP/+ animals and stained with anti-DMyb. DMyb was expressed at relatively uniform levels throughout the control wing, which did not contain any clones (A,A′). In wings bearing mip1301-36 clones, the levels of DMyb were inversely proportional to those of Mip130 (B,B′; clone borders are delineated with dashed lines). B″ shows a higher magnification of the boxed area in B′, with (+/+) and (−/−) indicating the homozygous wild-type and mutant regions of the twin spot, respectively, and (+/−) indicating the mip130-heterozygous cell population in the wing. Scale bars in A and B″ are 0.05 mm; A-B′ are shown at the same magnification. (C-D′) Comparison of mutant mybMH107 (C,C′) and mip1301-36 mybMH107 (D,D′) clones (indicated by lack of GFP) in wings dissected 24–28 hours APF from mybMH107 FRT/myb+ Ubi-GFP Minute FRT; hs-FLP/+animals (C,C′) and mip1301-36 mybMH107 FRT/Ubi-GFP FRT; hs-FLP/+ animals (D,D′). C′ and D′ are higher magnification images of boxed areas in C and D, respectively. This analysis demonstrated that in myb-null clones, lack of mip130 had no effect on the number of surviving neurons (red), although it did suppress the larger cell size phenotype associated with myb cell-cycle and endoreduplication defects. Scale bars in C and C′ are 0.05 mm; C, D and C′, D′ are shown at the same magnification. (E) Quantitative comparison of the number of neurons at the PWM of mybMH107 clones and mip1301-36 mybMH107 animals. As a reference, the number of neurons at the PWM of mip1301-36 wings is also shown. To quantify neurons at the PWM of the mybMH107 FRT animals, only wings bearing myb clones throughout the entire posterior wing compartment were used. Wings were dissected from 24–28-hr APF pupae.
Based on these results, we hypothesized that among the Myb complex proteins, DMyb plays the critical role in promoting programmed neural apoptosis in the PWM, and that the effects of mutations in mip1301-36 on this phenotype (in mip1301-36 and mip1301-36 myb2 mutant wings) is a consequence of the destabilization of the DMyb protein. This hypothesis is testable since it predicts that the absence of Mip130 should not have any effect on the neuronal phenotype in the context of a null mutation for Dm myb. Since mybMH107 homozygotes are inviable, we compared mybMH107 single-mutant and mip1301-36 mybMH107 double-mutant clones for the formation of ectopic neurons within a given area of the PWM. Visual inspection of the clones revealed no apparent difference (Fig. 5C-D′), an observation that was confirmed by quantitative analysis of the number of ectopic neurons in the PWM of mip1301-36 mybMH107 pupal wings and mybMH107 clones that occupied the entire posterior compartment of the wing (Fig. 5E). Furthermore, we found that introducing one copy of a transgene that carried a wild-type genomic copy of myb into the mip1301-36 mybMH107 background reverted the PWM phenotype to a nearly wild-type level (average of less than one neuron/wing).
These results provide strong support for the hypothesis that Mip130 plays no active role in the neural apoptosis that normally occurs in the PWM, and that ectopic neurons detected in mip130 mutant wings were not a direct result of the mutation itself, but instead a consequence of DMyb destabilization. Furthermore, the finding that the mip130 mutation was able to suppress the myb wing-hair phenotype that results from cell cycle defects in the body of the wing, but not the ectopic-neuron phenotype at the PWM, confirms that the survival of neurons in myb-mutant wings is independent of the role of Dm myb in the cell cycle. Therefore, while the primary role of DMyb in chorion gene amplification and cell division may be to act as an antagonist of other Myb complex members, it appears to play a more active role in promoting developmentally programmed neural apoptosis at the PWM.
DMyb works together with dE2F2 in ensuring appropriate neural apoptosis at the wing margin
As noted in the Introduction, Myb complex proteins were also shown to be part of the larger dREAM/MMB complex, which also contains Drosophila’s repressive E2F and pRb proteins (Korenjak et al., 2004; Lewis et al., 2004). In genetic modifier studies, we found a strong interaction between myb and de2f2 mutants, such that such that a heterozygous de2f2 mutant allele in a myb background could effectively suppress the myb wing-hair defects (Fig. S5). As this was a more potent interaction than had been seen with a mutation in mip130, which only suppresses the wing-hair phenotype when homozygous, we decided to examine the role of dE2F2 on neuronal apoptosis at the PWM.
We found that when de2f2 levels were reduced by approximately 50% in e2f2C03344/+ heterozygotes that were otherwise wild type, there was no effect on neuronal survival at the PWM, and only a mild effect was observed in e2f2C03344 homozygotes (Fig 6A,G). However, a role for the dE2F2 protein in this process was indicated by the finding that when de2f2 levels were reduced in an H99 background, the number of ectopic neurons at the PWM almost doubled relative to H99 alone (Fig. 6B,G). Therefore, we decided to examine the effects of reducing de2f2 levels within the context of Myb complex gene mutations. In the background of the ts myb2 mutation, the number of surviving neurons increased by greater than threefold (from an average of 3 to 11) when de2f2 levels were reduced (Fig. 6G). To examine the effects of reduced de2f2 levels when DMyb (and Myb complex activity) is entirely absent, we used the mip1301-36 mybMH107 double mutant, since loss of Mip130 is able to rescue mybMH107 lethality but does not affect the mutant myb phenotype at the PWM. We found that the survival of neurons at the PWM in mip1301-36 mybMH107 double mutants was enhanced and approximately doubled by reducing de2f2 levels (Fig. 6E,G; note that the number of neurons detected in the double mutants in these experiments differs from that shown in Fig. 5E because of a difference in genders). Furthermore, the strong synergy that occurs between the mip1301-36 mybMH107 double mutant and H99 was even further enhanced by reducing de2f2 levels (Fig. 6D,F,G), with the average number of neurons approaching the theoretical maximum of approximately 230 (based upon counting the number of PWM bristles in adult male flies). These results indicate that dE2F2 plays a role in the programmed cell death of neurons at the PWM that is independent of DMyb, and that the two proteins of the dREAM/MMB complex cooperate to ensure that the developmentally programmed neural apoptosis is achieved at the PWM.
Figure 6. Reduced levels of the dREAM component dE2F2 enhanced the effect of myb mutations and H99 at the PWM.

No ectopic neurons were detected at the PWM when E2F2 levels were reduced in w/Y; e2f2C03344/+ heterozygotes (A), but in an H99 background (B), the number of surviving PWM neurons was approximately doubled over that of H99 alone (compare to Fig. 2C and see panel G in this figure). In addition, the number of ectopic PWM neurons resulting from the complete loss of DMyb in mip1301-36 mybMH107 mutants (C) was approximately doubled when one copy of e2f2C03344 was introduced (E,G). Note that the mip1301-36 mybMH107 double mutant was used to avoid having to generate clones, but as shown in Figure 5, the phenotype is identical to that of a complete loss of DMyb in mybMH107 clones. As expected, the H99 deletion synergized with the mip1301-36 mybMH107 double mutant (D), but unexpectedly, the introduction of one copy of the e2f2C03344 allele was able to further enhance the phenotype (F,G), resulting in a continuous row of ectopic neurons throughout the PWM. Scale bar in A is 0.05 mm; all images are shown at the same magnification. (G) Quantification of the number of neurons at the PWM from pupal wings of indicated genotypes dissected at 24–28 hr APF. In the image and graph labels, e2f2C03344 is abbreviated as e2f2.
Dm myb works with the pro-apoptotic gene grim to promote neural apoptosis at the PWM
Neural apoptosis at the PWM has not been previously analyzed in detail. The strong synergy in suppression of neural apoptosis at the PWM that was observed between mutations in members of the dREAM/MMB complex and the H99 deficiency chromosome indicated that one or more of the pro-apoptotic genes that are deleted in H99 (reaper, grim, hid) might be specifically required for apoptosis of these cells. Therefore, we decided to dissect this interaction further by testing for interactions between the myb2 mutation and either overlapping deletions or individual mutations in the pro-apoptotic genes.
In a myb+ background, introduction of one copy of a loss-of-function mutation in either rpr (Moon et al., 2008) or hid (Grether et al., 1995) had no effect on neural apoptosis at the PWM (Fig. 7A), and in a myb2 background, only a modest enhancement of the myb2 phenotype was observed (Fig. 7B). Likewise, little effect was seen with the Df(3L)XR38 chromosome (not shown), which deletes rpr and sickle, another neighboring pro-apoptotic gene that is not uncovered by H99 (Peterson et al., 2002; Wing et al., 2002). In contrast, a strong interaction was observed between myb2 and Df(3L)X25, which deletes the hid and grim genes (Chen et al., 1996; White et al., 1994) (Fig. 7B). Together, these data suggested that the grim gene was likely to be the critical pro-apoptotic gene for this process. Indeed, when we examined the effects of a newly isolated mutation in this gene, grimA6C (Wu et al., 2010), as a heterozygote in a myb+ background, mild suppression of apoptosis at the PWM was observed (Fig. 7A,C); and when the heterozygous grimA6C was introduced into the myb2 background, a synergistic interaction that was almost as strong as that of myb2 and H99 was observed (Fig. 7B,E). This synergistic interaction was further enhanced when DMyb was eliminated and the Myb complex was destabilized in mip1301-36 mybMH107; grimA6C/+ mutants (Fig. S4). Furthermore, neural apoptosis was also strongly suppressed at the PWM of grimA6C homozygotes (Fig. 7A,F), indicating that grim is the main cell death gene within the H99 interval that is involved in this process. We obtained similar results using the UAS-grimRNAi line (Wu et al., 2010) to reduce grim levels in the PWM of wild-type and myb mutant wings (Fig. S4).
Figure 7. Among the pro-apoptotic genes, grim is the major mediator of neural apoptosis at the PWM.

(A–B) Graphs showing the effect of reducing the levels of pro-apoptotic genes rpr, hid, and grim, individually or in combination, in white control (A) and myb2 (B) backgrounds. Quantification was performed on pupal wing samples of the indicated genotypes dissected at 24–28 hr APF. (C–F) Shown are wings immunostained with anti-Elav that illustrate the myb-grim interaction. Only a few neurons (<5) were observed at the PWM of w; grimA6C/+ (C) and w myb2 wings (D), but more than 80 were present at the PWM of w myb2; grimA6C/+ wings, indicating a synergistic interaction (E). Complete absence of grim in the white background leads to a strong ectopic-neuron phenotype at the PWM (F), which is even further enhanced in a myb2 background (compare genotypes at the far right in panels A and B). Scale bar in C is 0.05 mm; all images are shown at the same magnification.
To test whether DMyb might regulate grim expression in the posterior wing margin, we performed quantitative real-time PCR analysis on whole pupal wing samples, but did not detect any differences between relevant samples (not shown). Since the cells of interest (descendants of the SOP in the PWM) represent a very small proportion of the cells in pupal wings, we then examined the levels of Grim expression in wild-type and mip1301-36 mybMH107 double mutant tissues using a polyclonal antibody directed against the N-terminus of Drosophila Grim (Santa Cruz Biotechnology). We found that Grim signals could be detected in GMR-grim larval eye discs and white pupal wings, but was undetectable in eye discs and pupal wings that were homozygous null for grim (Fig. S6A-D′). Clonal analysis subsequently revealed that Grim expression levels in mip1301-36 mybMH107 double mutant cells were not significantly different from those in wild-type cells (Fig. S6E-E′). Taken together, these results indicate that although DMyb and Grim work together to promote developmentally programmed neural apoptosis at the PWM, the mechanism is not via DMyb regulating grim expression levels. There are also indications that rpr and hid may cooperate with grim to some extent in promoting this PCD, including the findings that: compared to in the wild-type background, more neurons survive when grim is totally eliminated in a myb2 background; total elimination of rpr significantly enhances the myb2 PWM phenotype; and deficiency chromosomes which delete rpr, hid and grim or hid and grim alone (H99 and X25, respectively) enhance the myb2 PWM phenotype more than grim alone (Fig. 7A,B).
Dm myb also interacts with grim in glial cell apoptosis in the thoracic microchaete lineage
Since grim has been shown to play a key role in the apoptosis of microchaete glial cells in the thorax (Wu et al., 2010), we decided to examine whether Dm myb works together with grim in this setting as well as in the PWM. Using an antibody against Repo, which detects glial cells (Halter et al., 1995), we found that glial cell survival levels were comparable in wild-type, myb2, and heterozygous grimA6C pupal nota at 27-28 hours APF (Fig. 8A–C,E). However, glial cell survival was enhanced in myb2; grimA6C/+ nota (Fig. 8D,E), indicating that DMyb participates in grim-mediated apoptosis of the microchaete glial cells in the thorax. Since glial cells are targeted for PCD in the thoracic microchaete, and we had examined only neuron and sheath cells in the PWM, we returned to the wing to assess the status of glial cells and determine whether myb and grim might be involved in promoting their apoptosis during wing development. In wild-type pupal wings, glial cells were only present along the AWM and the presumptive L3 longitudinal vein (Fig. 8F). However, the presence of glial cells at the PWM could be detected when p35 was overexpressed along the wing margin (Fig. 8G), indicating that similarly to neurons and sheath cells, glial cells are initially generated at the PWM, but that they are then eliminated via PCD. Surprisingly, however, reducing grim and myb levels individually or in combination did not promote ectopic glial cell survival at the PWM, indicating that neither grim nor myb plays an essential role in the apoptosis of glial cells in the wing (Fig. 8H,I). Our data therefore suggest that at least two distinct mechanisms are used to regulate the PCD of glial cells in Drosophila: one of which is mediated by grim and myb, as has been shown in the thoracic microchaete, and another that is independent of both grim and myb, as exemplified by the glial cells in the PWM.
Figure 8. DMyb participates in more than one grim-mediated cell death process in the PNS.

(A–D) Shown are portions of nota dissected from male pupae and immunostained with anti-Elav to label the neurons (red), and anti-Repo to label glial cells (green). In the fields shown at 27–28 hours APF, white (A), myb2 (B), and heterozygous grimA6C (C) nota exhibit only a few surviving glial cells relative to the number of neurons, whereas w myb2; grimA6C/+ nota from the same developmental time point display a much higher rate of glial cell survival (D). Scale bar in A is 0.02 mm; images in A–D are shown at the same magnification. (E) Quantification of surviving glial cells in the nota, calculated as a percentage of the number of neurons, demonstrates that significantly more glial cells survive in w myb2; grimA6C/+ and grimA6C homozygous nota than in the other genotypes. The number of neurons counted were as follows: w/Y (n=329; from 6 independent nota), w myb2/Y (n=302; 7), w/Y; grimA6C/+ (n=196; 6), w myb2/Y; grimA6C/+ (n=229; 7), w/Y; grimA6C (n=302; 5). (F–I) Shown are wings which demonstrate that glial cells can be detected at the PWM when apoptosis is inhibited, but that myb and grim are not the critical regulators of their apoptosis. Wings were dissected from male pupae of the indicated genotypes at 24–28 hours APF and stained with anti-Elav (red) and anti-Repo (green). In white control wings, neurons (red) and glial cells (green) were only present along the AWM and the presumptive L3 vein, while the PWM was devoid of any neural cells (F). In contrast, the PWM of w; UAS-p35; C96-Gal4 wings were lined with neurons and glial cells (G). However, although both w myb2/Y; grimA6C/+ and w/Y; grimA6C wings exhibited ectopic neurons at the PWM, ectopic glial cells were not detected at the PWM of either genotype (H–I). G′-I′ are higher magnification images of selected areas on the PWM of G–I, respectively. Scale bars in F and G′ are 0.05 mm; F–I and G′-I′ are shown at the same magnification. (J) Proposed model of the stages at which DMyb plays a role in the developmentally specified apoptosis of SOP descendants in certain PNS lineages. Red X’s indicate apoptosis regulated by Dm myb and grim, whereas the green X indicates apoptosis regulated by a yet unknown mechanism.
DISCUSSION
DMyb promotes programmed neural cell death in the PWM, a function that is independent of its role in regulating cell proliferation
Developmentally specified apoptosis is required to shape the neural pattern of sensory bristles along the wing margin. Jafar-Nejad and colleagues (2006) observed that blocking apoptosis along the wing margin leads to ectopic neurons at the PWM, demonstrating that these neurons or their precursors are normally fated to die during early pupal development. In this study, we show that DMyb, as a member of the dREAM/MMB complex, is required to ensure that this developmental event occurs, leading to a neural pattern that is restricted to the anterior compartment of the wing.
While DMyb has been largely implicated in the regulation of cell cycle progression (Fitzpatrick et al., 2002; Fung et al., 2002, 2003; Katzen et al., 1998; Manak et al., 2002; Manak et al., 2007; Okada et al., 2002), several observations indicate that its function in regulating PCD at the wing margin is independent of its role in the cell cycle. For example, the PWM phenotype is undiminished in experimental settings that minimize cell cycle defects (i.e. myb2 at 18°C and in mip130 myb double-mutant animals), and it is weaker in a myb1 background than a myb2 background, even though the myb1 allele exhibits stronger cell division defects (Katzen et al., 1998). These findings suggest that these two phenotypes and their corresponding functions are separable. Furthermore, since non-sensory pupal wing cells in myb mutants do undergo apoptosis, the requirement for DMyb function in apoptosis appears to be specific to the developmentally programmed death of neural cells at the PWM.
The role of the Myb complex members DMyb and Mip130 in neural apoptosis at the PWM
The existence of the Myb complex was discovered in a very different developmental setting, that of chorion gene amplification within somatic follicle cells of the ovary, and the functional relationship between DMyb and Mip130 was also initially studied within this context (Beall et al., 2004; Beall et al., 2002). In this setting, Mip130 was shown to be critical for stabilizing the Myb complex and for repressing the initiation of DNA replication, while DMyb was implicated as the antagonist of the repressive activity, thereby allowing replication to initiate at appropriate origins so that the clusters of the chorion genes could be amplified. This finding suggests that at least in this setting, the sole function of DMyb is to antagonize the repression exerted by Mip130 and other components of the Myb complex. Evidence that this type of interplay between DMyb and Mip130 is not limited to chorion gene amplification is provided by the findings that elimination of mip130 (via mutation) can rescue both the inviability of myb null mutants (Beall et al., 2004) as well as their the cell cycle-related wing defects (Fig. S5); and that unlike other members of the dREAM/MMB complex, DMyb is not required to repress the E group of dE2F-regulated genes in cultured Drosophila cells (Korenjak et al., 2004; Lewis et al., 2004). Together, these data indicate that in several different settings, the requirement for DMyb function can be abrogated by the removal of Mip130, which destabilizes the Myb complex and eliminates its repressive activities.
Our studies reveal a different type of functional interplay between DMyb and Mip130 in their regulation of developmentally specified neural apoptosis in the PWM. Based on the data that the absence of Mip130 results in a mild PWM phenotype in a wild-type myb background, an enhanced phenotype in a hypomorphic-mutant myb background, and no additional effect in a myb-null background, we conclude that the role of Mip130 in this context is solely to stabilize the DMyb protein. Therefore, this analysis provides the first example of a physiological process regulated by the Myb complex in which DMyb plays the critical role while still being dependent on the presence of other components of the complex.
The pro-apoptotic role of dE2F2 and the dREAM/MMB complex
In addition to the four tightly associated Mips, the Myb complex is associated with the larger dREAM/MMB complex, which also contains Drosophila’s repressive E2F/pRB proteins (Korenjak et al., 2004; Lewis et al., 2004). The presence of DMyb and dE2F2 within the same protein complex suggests that they may be involved in regulating at least a subset of the same target genes. Georlette and colleagues (2007) carried out a comprehensive, genome-wide gene expression analysis of the effects of depleting individual dREAM/MMB complex components from Drosophila cultured cells. Although this analysis implicates DMyb in both positive and negative regulation of gene expression, there is virtually no overlap of genes that are negatively regulated by both DMyb and dE2F2. In contrast, there are numerous examples of genes that are negatively regulated by dE2F2 and positively regulated by DMyb. This data, along with evidence from other investigators that dE2F1 can act to promote apoptosis (Asano et al., 1996; Du et al., 1996; Moon et al., 2008; Moon et al., 2005), whereas dE2F2 acts as an anti-apoptotic factor (Wichmann et al., 2010), indicate that similarly to what has been suggested for DMyb and Mip130, DMyb and dE2F2 may function as antagonists within the dREAM/MMB complex. In support of this hypothesis, we found that reducing dE2F2 levels in a mutant myb background suppresses the mutant myb cell cycle-related wing phenotype.
Therefore, within the context of the developmentally programmed neural apoptosis that occurs in the PWM, we had expected to observe either an antagonistic genetic interaction between Dm myb and de2f2 or no interaction at all. Instead, we found that while reduction of dE2F2 alone does not affect neural apoptosis within the PWM (and complete elimination has only a mild effect), decreased levels of dE2F2 do enhance the neural survival phenotype in the PWM when combined with either the H99 chromosome or a mutant mip130 myb background, or both together. Differently from the situation with mip130, reduced levels of dE2F2 are able to enhance the PWM phenotype in animals that are doubly mutant for null alleles of both myb and mip130. This enhancement is even more dramatic in the presence of the H99 chromosome where an average of 191 neurons survived, the highest number observed in these experiments. These results suggest that dE2F2 plays an active role in promoting apoptosis of neural cells in the PWM, and provides the first example in which dE2F2, DMyb and Mip130 all act in concert to promote the same outcome, in this case, the developmentally specified death of neural cells in the PWM. The finding that dE2F2 plays a pro-apoptotic, rather than an anti-apoptotic role in this process exemplifies the diverse and expanding roles played by E2F proteins in different cell types and developmental settings.
Lineage-specific apoptosis of the PWM neuronal precursor cells
In general, sensory organs are composed of five cell types, including the shaft, socket, neuron, sheath, and glial (often destined to die) cells, with the first two forming the visible portions of the bristles (reviewed in Lai and Orgogozo, 2004). These cells are generated through sequential asymmetric cell divisions from a single SOP cell (Fig. 3A). PWM bristles in wild-type animals represent a modified class of sensory organs, being comprised of only shaft and socket cells and are therefore non-innervated. The finding that blocking apoptosis along the wing margin leads to the presence of ectopic neurons at the PWM confirms that the variant composition of the PWM bristles is not achieved through a change in the cell division pattern of the SOP, but instead via PCD of either the neurons or their precursor cells (pIIb or pIIIb) (Jafar-Nejad et al., 2006). Our analysis of the fate of these cells in mutant alleles of multiple genes encoding components of the dREAM/MMB complex and the apoptotic pathway have allowed us to corroborate and expand upon these conclusions. Using the D-Pax2 antibody, we have been able to confirm that PWM bristles do not normally contain sheath cells. More importantly, we found that in mutant animals with compromised neural apoptosis, each time a surviving PWM neuron is present (and only when such a neuron is present), it is accompanied by its sister sheath cell, and that this is the case even in genotypes with relatively few surviving neurons. Since it is unlikely that sister neuron and sheath cells would always survive together if they were apoptosing independently, this finding strongly suggests that a precursor cell is being targeted for death.
Examination of glial cell fate in the PWM provides further insights. The presence of glial cells in the PWM when the p35 protein is ectopically expressed demonstrates that the absence of these cells in wild-type animals is due to apoptosis. However, unlike the PWM neuron and sheath cells or the microchaete glial cells, glial cells at the PWM do not survive in any of the dREAM/MMB and/or RHG mutant combinations tested, indicating that glial cells undergo apoptosis by a dREAM/MMB-independent mechanism. Thus, two distinct mechanisms lead to glial cell death in two different developmental settings. In addition, the presence of a developmental program to generate glial cells at the PWM indicates that one extra cell division cycle occurs after the division of the pIIb and before the ultimate specification of the neuron and sheath cells. This supports the hypothesis that the development of PWM bristles follows the same lineage as that of the thoracic microchaete, in which the pIIb cell produces a glial cell and a pIIIb cell, which in turn gives rise to the neuron and sheath cell (Fig. 8J). Furthermore, since glial cells die when neurons and sheath cells survive at the PWM, dREAM/MMB-promoted apoptosis in this setting cannot be directed at the pIIb precursor which gives rise to all three cell types, and is therefore most likely to be targeting the pIIIb precursor cell (Fig. 8J).
The pro-apoptotic role of the dREAM/MMB complex appears to be specific to grim-mediated cell death
Our data demonstrate that grim is the critical RHG gene involved in mediating the dREAM/MMB signal to initiate developmentally specified neural apoptosis at the PWM. Until recently, of the three RHG genes deleted in the H99 chromosome, only rpr and hid had been shown to be important for PCD in specific tissues during development (Grether et al., 1995; Peterson et al., 2002; Yin and Thummel, 2004; Yu et al., 2002; Zhou et al., 1997). However, grim has now been shown to be required for the PCD of glial cells in thoracic microchaete (Wu et al., 2010). Our study provides a second example in which grim plays the critical role in promoting the apoptosis of a specific SOP daughter cell and reveals that Dm myb plays a role in both of the grim-mediated cell death processes. In examining the specificity of the myb-grim linkage in promoting apoptosis, we also looked at whether either of these gene plays a role in promoting the PCD of neuroblasts in abdominal neuromeres, a process in which reaper has been previously implicated (Peterson et al., 2002). We were unable to find any indication that myb or grim, individually or in combination with H99, play a role in regulating the apoptosis of these neuroblasts (not shown), strengthening the hypothesis that the interaction between DMyb and the apoptotic machinery may be specific to grim and possibly to developmentally specified apoptosis in the PNS.
Given the transcriptional regulatory roles of DMyb and the dREAM/MMB complex, the most obvious model to explain the interplay between myb and grim is that as a component of the dREAM/MMB complex, DMyb positively regulates the expression of grim. However, no differences were detected between the relevant genotypes when quantitative real-time PCR analysis was performed on whole pupal wing and notum samples (not shown). Since we believe that the unique interaction between myb and grim only occurs in a subset of SOP daughter cells, which are a small fraction (<1%) of the whole wing and thoracic tissues, no conclusions could be drawn from this negative results. However, we also did not detect any significant difference in Grim protein levels between wild-type and mip1301-36 mybMH107 double mutant cells (Fig. S6), suggesting that grim expression is not regulated by DMyb. These findings are in agreement with results from Drosophila Kc cells, in which none of the dREAM/MMB complex members were found to be bound in the vicinity of the grim promoter region, and grim was not identified as a target of any of the dREAM/MMB complex members in RNAi knock-down studies (Georlette et al., 2007).
CONCLUSION
As discussed in the Introduction, both pro- and anti-apoptotic roles have been ascribed to mammalian and Drosophila myb genes, but as these effects have generally been observed in settings with abnormally elevated levels of Myb proteins or activity, which also have associated effects in the cell cycle and/or cellular proliferation, it has been difficult to distinguish between direct and indirect effects. The data presented here demonstrate that Drosophila myb plays a critical role in promoting highly tissue-specific and developmentally specified examples of PCD in two settings, that this apoptotic event is separable from any other effects that Dm myb has on the cell cycle, and that this process is mediated by the pro-apoptotic grim gene. These studies reveal a previously undiscovered function for the dREAM/MMB complex, that of regulating PCD. We posit that these first two examples will herald the discovery of additional settings in Drosophila and other organisms in which Myb and the dREAM/MMB complex regulate developmentally specified cell death.
Supplementary Material
HIGHLIGHTS.
DMyb promotes lineage-specific apoptosis of neural precursor cells at the PWM
This is a novel function of DMyb that is independent of its roles in the cell cycle
dREAM/MMB complex member dE2F2 is involved in this process, while Mip130 is not
Pro-apoptotic role of DMyb in lineage-specific apoptosis is mediated by Grim
DMyb participates in more than one grim-mediated cell death processes in the PNS
Acknowledgments
We thank J. Abrams, M. Botchan, S. Bray, B. Edgar, J. Lipsick, M. Noll, G. Rubin, E. Siegfried, K. White, and the Bloomington Stock Center for reagents; and J. McEllin, T. Orenic, M. Frolov, and members of the Frolov lab for valuable discussion and ideas. The 22C10, Elav, and Repo monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. Work reported here was supported by a National Institutes of Health Grant to A.L.K. (GM68961). MKR was partially supported by a University Fellowship from the University of Illinois and by the Department of Biochemistry and Molecular Genetics, University of Illinois at Chicago; CBB was partially supported by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott MK, Lengyel JA. Embryonic head involution and rotation of male terminalia require the Drosophila locus head involution defective. Genetics. 1991;129:783–789. doi: 10.1093/genetics/129.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso TB, Jones BW. gcm2 promotes glial cell differentiation and is required with glial cells missing for macrophage development in Drosophila. Dev Biol. 2002;248:369–383. doi: 10.1006/dbio.2002.0740. [DOI] [PubMed] [Google Scholar]
- Asano M, Nevins JR, Wharton RP. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev. 1996;10:1422–1432. doi: 10.1101/gad.10.11.1422. [DOI] [PubMed] [Google Scholar]
- Beall EL, Bell M, Georlette D, Botchan MR. Dm-myb mutant lethality in Drosophila is dependent upon mip130: positive and negative regulation of DNA replication. Genes Dev. 2004;18:1667–1680. doi: 10.1101/gad.1206604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature. 2002;420:833–837. doi: 10.1038/nature01228. [DOI] [PubMed] [Google Scholar]
- Blair SS. Shaggy (zeste-white 3) and the formation of supernumerary bristle precursors in the developing wing blade of Drosophila. Dev Biol. 1992;152:263–278. doi: 10.1016/0012-1606(92)90134-3. [DOI] [PubMed] [Google Scholar]
- Chen P, Nordstrom W, Gish B, Abrams JM. grim, a novel cell death gene in Drosophila. Genes Dev. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- Chew SK, Chen P, Link N, Galindo KA, Pogue K, Abrams JM. Genome-wide silencing in Drosophila captures conserved apoptotic effectors. Nature. 2009;460:123–127. doi: 10.1038/nature08087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. 2009;43:493–523. doi: 10.1146/annurev.genet.42.110807.091533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- Du W, Xie JE, Dyson N. Ectopic expression of dE2F and dDP induces cell proliferation and death in the Drosophila eye. EMBO J. 1996;15:3684–3692. [PMC free article] [PubMed] [Google Scholar]
- Ebacher D. Personal communication to FlyBase. 2002 < http://flybase.org/reports/FBrf0150864.html>.
- Fichelson P, Gho M. The glial cell undergoes apoptosis in the microchaete lineage of Drosophila. Development. 2003;130:123–133. doi: 10.1242/dev.00198. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick CA, Sharkov NV, Ramsay G, Katzen AL. Drosophila myb exerts opposing effects on S phase, promoting proliferation and suppressing endoreduplication. Development. 2002;129:4497–4507. doi: 10.1242/dev.129.19.4497. [DOI] [PubMed] [Google Scholar]
- Fung SM, Ramsay G, Katzen AL. Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development. 2002;129:347–359. doi: 10.1242/dev.129.2.347. [DOI] [PubMed] [Google Scholar]
- Fung SM, Ramsay G, Katzen AL. MYB and CBP: physiological relevance of a biochemical interaction. Mech Dev. 2003;120:711–720. doi: 10.1016/s0925-4773(03)00044-3. [DOI] [PubMed] [Google Scholar]
- Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, Beall EL, Bell SP, Speed T, Manak JR, Botchan MR. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev. 2007;21:2880–2896. doi: 10.1101/gad.1600107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gho M, Bellaiche Y, Schweisguth F. Revisiting the Drosophila microchaete lineage: a novel intrinsically asymmetric cell division generates a glial cell. Development. 1999;126:3573–3584. doi: 10.1242/dev.126.16.3573. [DOI] [PubMed] [Google Scholar]
- Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta. 2007;1772:392–401. doi: 10.1016/j.bbadis.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Gustafson K, Boulianne GL. Distinct expression patterns detected within individual tissues by the GAL4 enhancer trap technique. Genome. 1996;39:174–182. doi: 10.1139/g96-023. [DOI] [PubMed] [Google Scholar]
- Halter DA, Urban J, Rickert C, Ner SS, Ito K, Travers AA, Technau GM. The homeobox gene repo is required for the differentiation and maintenance of glia function in the embryonic nervous system of Drosophila melanogaster. Development. 1995;121:317–332. doi: 10.1242/dev.121.2.317. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Posakony JW. Development of adult sensilla on the wing and notum of Drosophila melanogaster. Development. 1989;107:389–405. doi: 10.1242/dev.107.2.389. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Tien AC, Acar M, Bellen HJ. Senseless and Daughterless confer neuronal identity to epithelial cells in the Drosophila wing margin. Development. 2006;133:1683–1692. doi: 10.1242/dev.02338. [DOI] [PubMed] [Google Scholar]
- Katzen AL, Bishop JM. myb provides an essential function during Drosophila development. Proc Natl Acad Sci U S A. 1996;93:13955–13960. doi: 10.1073/pnas.93.24.13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen AL, Jackson J, Harmon BP, Fung SM, Ramsay G, Bishop JM. Drosophila myb is required for the G2/M transition and maintenance of diploidy. Genes Dev. 1998;12:831–843. doi: 10.1101/gad.12.6.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen AL. Drosophila Myb: lessons for the understanding of vertebrate Myb proteins. In: Frampton J, editor. Myb Transcription Factors: Their Role in Growth, Differentiation and Disease, Proteins and Cell Regulation. Vol. 2. Kluwer Academic Publishers B.V., Dordrecht; The Netherlands: 2004. pp. 36–64. [Google Scholar]
- Kavaler J, Fu W, Duan H, Noll M, Posakony JW. An essential role for the Drosophila Pax2 homolog in the differentiation of adult sensory organs. Development. 1999;126:2261–2272. doi: 10.1242/dev.126.10.2261. [DOI] [PubMed] [Google Scholar]
- Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell. 2004;119:181–193. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Kurada P, White K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell. 1998;95:319–329. doi: 10.1016/s0092-8674(00)81764-x. [DOI] [PubMed] [Google Scholar]
- Lai EC, Orgogozo V. A hidden program in Drosophila peripheral neurogenesis revealed: fundamental principles underlying sensory organ diversity. Dev Biol. 2004;269:1–17. doi: 10.1016/j.ydbio.2004.01.032. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Beall EL, Fleischer TC, Georlette D, Link AJ, Botchan MR. Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev. 2004;18:2929–2940. doi: 10.1101/gad.1255204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manak JR, Mitiku N, Lipsick JS. Mutation of the Drosophila homologue of the Myb protooncogene causes genomic instability. Proc Natl Acad Sci U S A. 2002;99:7438–7443. doi: 10.1073/pnas.122231599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manak JR, Wen H, Van T, Andrejka L, Lipsick JS. Loss of Drosophila Myb interrupts the progression of chromosome condensation. Nat Cell Biol. 2007;9:581–587. doi: 10.1038/ncb1580. [DOI] [PubMed] [Google Scholar]
- Milan M, Campuzano S, Garcia-Bellido A. Developmental parameters of cell death in the wing disc of Drosophila. Proc Natl Acad Sci U S A. 1997;94:5691–5696. doi: 10.1073/pnas.94.11.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon NS, Di Stefano L, Morris EJ, Patel R, White K, Dyson NJ. E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet. 2008;4:e1000153. doi: 10.1371/journal.pgen.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon NS, Frolov MV, Kwon EJ, Di Stefano L, Dimova DK, Morris EJ, Taylor-Harding B, White K, Dyson NJ. Drosophila E2F1 has context-specific pro- and antiapoptotic properties during development. Dev Cell. 2005;9:463–475. doi: 10.1016/j.devcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Nicholson SC, Gilbert MM, Nicolay BN, Frolov MV, Moberg KH. The archipelago tumor suppressor gene limits rb/e2f-regulated apoptosis in developing Drosophila tissues. Curr Biol. 2009;19:1503–1510. doi: 10.1016/j.cub.2009.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Okada M, Akimaru H, Hou DX, Takahashi T, Ishii S. Myb controls G(2)/M progression by inducing cyclin B expression in the Drosophila eye imaginal disc. EMBO J. 2002;21:675–684. doi: 10.1093/emboj/21.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme M, Meier P. Inhibitor of apoptosis proteins in Drosophila: gatekeepers of death. Apoptosis. 2009;14:950–960. doi: 10.1007/s10495-009-0358-2. [DOI] [PubMed] [Google Scholar]
- Palka J, Schubiger M, Ellison RL. The polarity of axon growth in the wings of Drosophila melanogaster. Dev Biol. 1983;98:481–492. doi: 10.1016/0012-1606(83)90377-9. [DOI] [PubMed] [Google Scholar]
- Peterson C, Carney GE, Taylor BJ, White K. reaper is required for neuroblast apoptosis during Drosophila development. Development. 2002;129:1467–1476. doi: 10.1242/dev.129.6.1467. [DOI] [PubMed] [Google Scholar]
- Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8:523–534. doi: 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- Reddy GV, Rodrigues V. A glial cell arises from an additional division within the mechanosensory lineage during development of the microchaete on the Drosophila notum. Development. 1999;126:4617–4622. doi: 10.1242/dev.126.20.4617. [DOI] [PubMed] [Google Scholar]
- Robinow S, Draizen TA, Truman JW. Genes that induce apoptosis: transcriptional regulation in identified, doomed neurons of the Drosophila CNS. Dev Biol. 1997;190:206–213. doi: 10.1006/dbio.1997.8696. [DOI] [PubMed] [Google Scholar]
- Robinow S, White K. Characterization and spatial distribution of the ELAV protein during Drosophila melanogaster development. J Neurobiol. 1991;22:443–461. doi: 10.1002/neu.480220503. [DOI] [PubMed] [Google Scholar]
- Scaria GS, Ramsay G, Katzen AL. Two components of the Myb complex, DMyb and Mip130, are specifically associated with euchromatin and degraded during prometaphase throughout development. Mech Dev. 2008;125:646–661. doi: 10.1016/j.mod.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Datta P, Kobayashi M, Wu JW, Fujioka M, Hegde R, Zhang Z, Mukattash R, Fernandes-Alnemri T, Shi Y, Jaynes JB, Alnemri ES. sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr Biol. 2002;12:125–130. doi: 10.1016/s0960-9822(01)00657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M, Adachi-Yamada T. Cell death and selective adhesion reorganize the dorsoventral boundary for zigzag patterning of Drosophila wing margin hairs. Dev Biol. 2011;357:336–346. doi: 10.1016/j.ydbio.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Tenev T, Zachariou A, Wilson R, Paul A, Meier P. Jafrac2 is an IAP antagonist that promotes cell death by liberating Dronc from DIAP1. EMBO J. 2002;21:5118–5129. doi: 10.1093/emboj/cdf530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, Buchholz R, Demsky M, Fawcett R, Francis-Lang HL, Ryner L, Cheung LM, Chong A, Erickson C, Fisher WW, Greer K, Hartouni SR, Howie E, Jakkula L, Joo D, Killpack K, Laufer A, Mazzotta J, Smith RD, Stevens LM, Stuber C, Tan LR, Ventura R, Woo A, Zakrajsek I, Zhao L, Chen F, Swimmer C, Kopczynski C, Duyk G, Winberg ML, Margolis J. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Wen H, Andrejka L, Ashton J, Karess R, Lipsick JS. Epigenetic regulation of gene expression by Drosophila Myb and E2F2-RBF via the Myb-MuvB/dREAM complex. Genes Dev. 2008;22:601–614. doi: 10.1101/gad.1626308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- Wichmann A, Uyetake L, Su TT. E2F1 and E2F2 have opposite effects on radiation-induced p53-independent apoptosis in Drosophila. Dev Biol. 2010;346:80–89. doi: 10.1016/j.ydbio.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing JP, Karres JS, Ogdahl JL, Zhou L, Schwartz LM, Nambu JR. Drosophila sickle is a novel grim-reaper cell death activator. Curr Biol. 2002;12:131–135. doi: 10.1016/s0960-9822(01)00664-9. [DOI] [PubMed] [Google Scholar]
- Wu JN, Nguyen N, Aghazarian M, Tan Y, Sevrioukov EA, Mabuchi M, Tang W, Monserrate JP, White K, Brachmann CB. grim promotes programmed cell death of Drosophila microchaete glial cells. Mech Dev. 2010;127:407–417. doi: 10.1016/j.mod.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yin VP, Thummel CS. A balance between the diap1 death inhibitor and reaper and hid death inducers controls steroid-triggered cell death in Drosophila. Proc Natl Acad Sci U S A. 2004;101:8022–8027. doi: 10.1073/pnas.0402647101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SY, Yoo SJ, Yang L, Zapata C, Srinivasan A, Hay BA, Baker NE. A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development. 2002;129:3269–3278. doi: 10.1242/dev.129.13.3269. [DOI] [PubMed] [Google Scholar]
- Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, Nambu JR. Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci U S A. 1997;94:5131–5136. doi: 10.1073/pnas.94.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Venkatesh TR, Teplow DB, Benzer S. Neuronal development in the Drosophila retina: monoclonal antibodies as molecular probes. Cell. 1984;36:15–26. doi: 10.1016/0092-8674(84)90069-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.