

Abstract
To combine benefits stemming from the high nucleophilicity of piperidine and the flexibility afforded by aliphatic triamine linkers, a trimethylene-dipiperidine linker has been used to synthesize triazine dendrimers using a divergent route. The cyclic, secondary amine of the linker reacts with monochlorotriazine monomer units, 1, leading to a dendrimer growth strategy that requires two-steps-per-generation. This strategy reduces the number of steps required for synthesis by 50%. The new linker also reduces complexity in the NMR spectra because rotational isomerism observed in linkers with primary amines is not present. In addition, the final products contain no interior hydrogen-bond donating groups. The high solubility observed in organic solvents for protected dendrimers is attributed to this factor and the inherent flexibility provided by the linker. Gas phase simulation suggests that globular structure emerges after generation three, wherein the core of the dendrimer is effectively shielded from solvent.
Introduction
Interest in dendrimers stems from their unique branched structure, opportunities to exploit multivalency, and well-defined chemical composition. The size and shape of dendrimers allows them to function as protein mimics capable of drug delivery,1 enzyme-like catalysis,2 gene therapy,3 and immunodiagnostics.4 One such class of dendrimers is the triazine-based structures. The primary methods of synthesis for these compounds proceed by either cycloaddition or nucleophilic aromatic substitution (SNAr).5 Despite the synthetic importance of the cycloaddition route, the method is limited to the formation of only symmetrically substituted structures.6
Unlike cycloaddition reactions, SNAr can afford products with rich compositional diversity. The potential use of these triazine dendrimers in both medicine and materials science derives from the formation of unsymmetrically substituted products. Such polyfunctional peripheries could find a role in a number of areas, including the delivery of chemotherapeutic agents,7 sequestration,8 and gene transfection.9
The ease with which these compounds can be synthesized will ultimately impact utility. We have invested significant energies in improving synthetic strategies based on both route and linking groups connecting triazines. The choice of linking diamines has evolved over time. Early efforts utilized p-aminobenzylamine,10 but oxidation and sluggish reaction led us to move to piperazine,11 then aminomethyl-piperidine.12 To facilitate our search for diamine linkers, the relative reactivity of amines were measured using competition reactions with a monochlorotriazine.13 The data are summarized in Figure 1.
Figure 1.
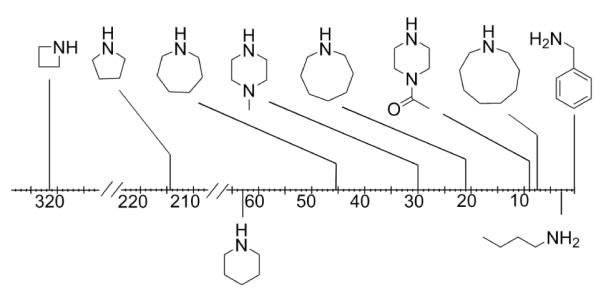
Relative reactivity of amines toward a monochlorotriazine.
By combining two amines with markedly different reactivities, convergent syntheses could proceed without protecting groups.10-12 With the demand of scale, we switched to divergent routes to these materials. Here, protecting groups are required, but the relative reactivity of the amine is still critical. Monomer 1 and the previous monomer, 2, are shown in Chart 1. Initial success with monomer 2 relied on reacting the primary amines with dichlorotriazines.14 The increased reactivity of dichlorotriazines (over the monochlorotriazines used here) overcomes the sluggish relative reactivity of the primary amine. Use of monomer 2 requires that the remaining chlorine atom be replaced prior to iteration. We commonly used piperidine due to its high reactivity with monochlorotriazines, a value recorded at 63x relative to benzyl amine. This capping step, however, translates into a three-step-per-generation iterative process: monomer addition, capping, and deprotection. In addition, hindered rotation around the exocyclic amine-triazine N-C bond (shown in bold) complicated interpretation of the NMR spectra. The new monomer 1 shows sufficient reactivity to react with monochlorotriazines reducing the number of steps-per-generation to two: monomer addition and deprotection. Due to symmetry, no rotational isomerism is observed in the NMR spectra. Monomer 1 also avoids the installation of hydrogen bond donors while maintaining flexibility in the linking domain. Both of these factors were hypothesized, and ultimately observed, to communicate favorable solubility properties to the targets in a range of organic solvents.
Chart 1.
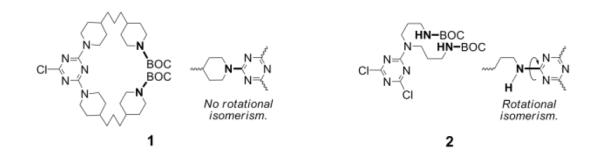
Monomers 1 and 2. The new monomer 1 conveys increased reactivity affording the use of monochlorotriazines and reduces complexity in the NMR spectra due to asymmetry: No rotational isomerism is observed around the N-C bond. The route relies on linking the BOC-protected amine (bold) to the triazine.
Results and Discussion
Synthesis
The synthesis of trimethylene-dipiperidine-linked triazine dendrimers is shown in Scheme 1. The mono-Boc-protected trimethylene-dipiperidine is generated by reaction of the diamine with Boc-anhydride. Following column chromatography, which is necessary to eliminate the bisprotected diamine, the desired product is afforded in approximately 45% yield.
Scheme 1.
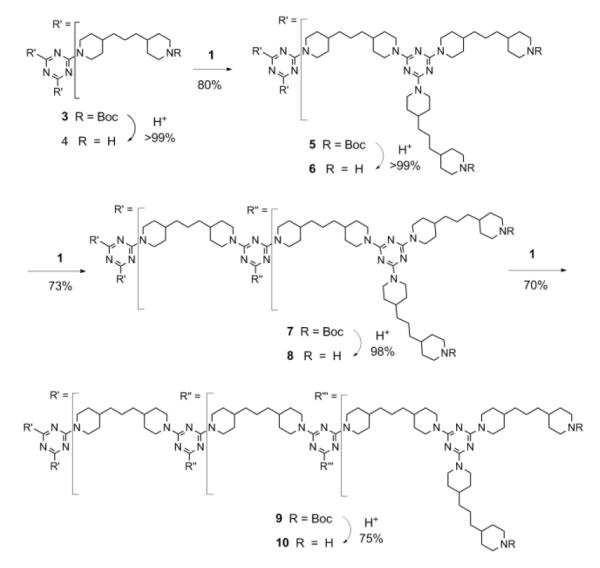
Synthesis of the target dendrimers.
Monomer 1 is obtained upon reaction of the monoprotected amine with cyanuric chloride. Briefly, both cyanuric chloride (0.27 g, 1.49 mmol) and amine (1.02 g, 3.29 mmol) are dissolved in 25 mL CH2Cl2. Diisopropylethylamine (2.9 mL, 16.5 mmol) is added to the amine solution before it is added dropwise to the solution of cyanuric chloride at 0 °C, and after warming to room temperature, the reaction stirs for 12 hr. Subsequently, the reaction is extracted against distilled water, the organic phase is dried, and ultimately is purified by column chromatography using a gradient of ethyl acetate (3% to 10%) in dichloromethane to afford 1 in 93% yield.
Deprotection steps are affected throughout by dissolving the protected material in CH3OH and adding an equal volume of concentrated HCl. After 18 hr, the methanol is removed and the aqueous solution is basified with 5 M NaOH and then extracted with chloroform. After drying the organic phase, evaporation generally provides the desired material in quantitative yields.
Through iterative monomer additions and deprotection steps, generation 1-3 dendrimers are obtained. These additions are typified by the addition of the monomer 1 to 4 to yield 5. Here, 1 (0.37 g, 0.50 mmol) and compound 4 (0.11 g, 0.15 mmol) are each dissolved in 5 mL THF. Diisopropylethylamine (0.9 mL, 5 mmol) is added to the solution of 4. Next, the solution of monomer 1 is added and the reaction mixture is heated to reflux. After 24 hr, the solvent is evaporated, the residue is redissolved in chloroform, and the organic phase is washed three times with distilled water. After evaporation, the solid is purified by column chromatography using a gradient of ethyl acetate (3% to 16%) in dichloromethane to provide the desired material in 80% yield.
Monomer addition yields and reaction times reflect the growing number of reactions required at each generation, from three to six to twelve. Starting from the core, 4, the third generation dendrimer 10 can be afforded in 6 steps in ~30% overall yield. In addition to reducing the number of steps to generation 3 materials from 9 (with monomer 2) to 6, the high solubility of each of the bis-piperizyl-linked dendrimers in a range of organic solvents would predict that higher generation materials are accessible.
Characterization
The characterization of the various generations of these triazine dendrimers has proven to be more straightforward than that of previous targets. Improved solubility increases confidence in adequate representation of all species present when these materials are subjected to NMR spectroscopy or mass spectrometry. The lack of rotational isomerism greatly simplifies interpretation of NMR. In the initial syntheses of triazine dendrimers with primary amine linkers, multiple rotational isomers in the products led to broad signals that could not be readily integrated. In addition, gelation of organic solvents resulted from the hydrogen-bonding facilitated by these diamines.15 The 1H NMR traces for the targets and intermediates are shown in Figure 2. In addition to the iterative appearance and disappearance of BOC groups, the axial and equatorial protons of the piperidine groups can be monitored when attached to a triazine (labeled A and B), substituted with BOC (labeled C and D) or free (labeled C’ and D’).
Figure 2.
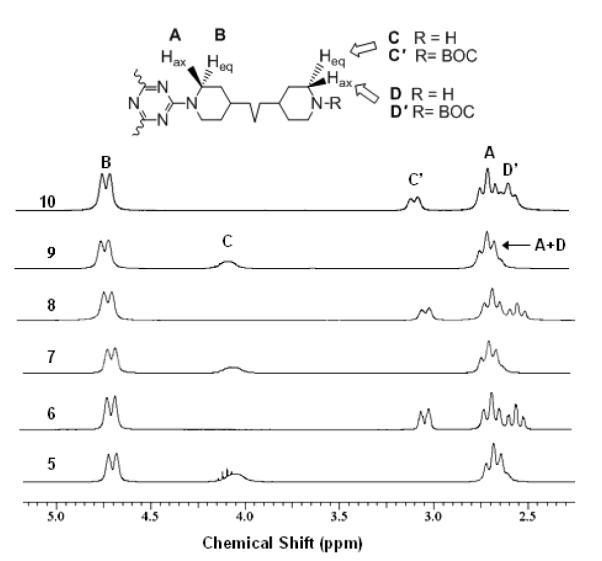
NMR spectra obtained in CDCl3 of the intermediates and targets with labels.
The traces from NMR spectroscopy for these polymeric materials, however, can be misleading. Despite the suggestion of high levels of product purity based on 1H NMR, mass spectrometry suggests that by the third generation, not all twelve substitutions readily go to completion. Mass spectra for both the protected and deprotected materials reveal lines corresponding to 11 of the 12 amines being substituted with monomer. Upon incomplete reaction, differences in relative intensities of these signals in both 9 and 10 do not allow us to quantify the extent of impurity with assumptions on ionization behaviors. NMR also precludes accurate determination. Estimates on the reliability of NMR to detect sideproducts are limited to 1:1 mixtures the desired:undersubstituted material.
Computational models complement our impressions of these materials and their behavior. Figure 3 shows gas phase simulations (front and back) of these materials with surface BOC groups shown in green, the outermost generation in blue, the middle generation in raspberry, the innermost in purple and the core in salmon. Globular structure—wherein the core is sterically forced to the center of the dendrimer—appears to emerge after generation three. Peripheral BOC groups are distributed across the surface, suggesting larger generations may be accessible or that derivatization chemistries would be predicted to be successful assuming that the BOC groups successfully mimic the structure of additional monomer and that hydrogen-bonding interactions between the necessary amine groups of the dendrimers are not limiting. These suppositions are supported in models of the diprotected materials. The significant increase in size from generation 2 to 3 provides visual support for the hypothesis that the 12th substitution reaction might be expected to encounter steric resistance.
Figure 3.
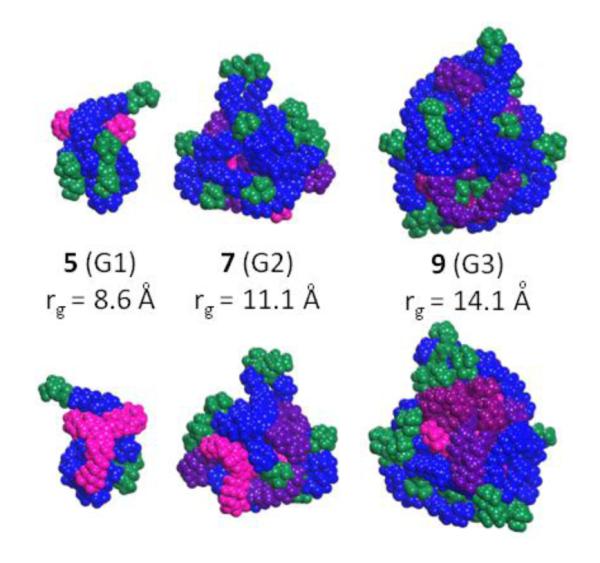
Front/back simulations of the dendrimers with the core in salmon, BOC groups in green, peripheral triazines in blue, generation 1 layer in purple and generation 2 layer in raspberry.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for support (NIGMS 64560).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Spectra (NMR and MS), experimental details and assignments, and gas phase simulations (26 pages).
References
- (1).Svenson S. Eur. J. Pharm. Biopharm. 2008;71:445–462. doi: 10.1016/j.ejpb.2008.09.023. [DOI] [PubMed] [Google Scholar]
- (2).Piotti ME, Rivera F, Jr., Bond R, Hawker CJ, Fréchet JMJ. J. Am. Chem. Soc. 1999;121:9471–9472. [Google Scholar]
- (3).Mintzer MA, Simanek EE. Chem. Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- (4).Singh P. In: Dendrimers and Other Dendritic Polymers. Fréchet JMJ, Tomalia DA, editors. Wiley; Chicester: 2001. pp. 463–484. [Google Scholar]
- (5).Steffensen MB, Hollink E, Kuschel F, Bauer M, Simanek EE. J. Poly. Sci. A. 2006;44:3411–3433. doi: 10.1002/pola.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Bosman AW, Janssen HM, Meijer EW. Chem. Rev. 1999;99:1665–1688. doi: 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]; (b) Fréchet JM. J. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4782–4787. doi: 10.1073/pnas.082013899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tomalia DA, Henderson SA, Diallo MS. Handb. Nanosci., Eng., Technol. (2nd Ed) 2007 24/1-24/47. [Google Scholar]
- (7) (a).Lim J, Guo Y, Rostollan CL, Stanfield J, Hsieh J-T, Sun X, Simanek EE. Mol. Pharmaceutics. 2008;5:540–547. doi: 10.1021/mp8000292. [DOI] [PubMed] [Google Scholar]; (b) Venditto VJ, Allred K, Allred CD, Simanek EE. Chem. Commun. 2009:5541–5542. doi: 10.1039/b911353c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Neitsch SL, McInnes KJ, Senseman SA, White GN, Simanek EE. Chemosphere. 2006;64:704–710. doi: 10.1016/j.chemosphere.2005.11.033. [DOI] [PubMed] [Google Scholar]
- (9) (a).Mintzer MA, Merkel OM, Kissel T, Simanek EE. New J. Chem. 2009;33:1918–1925. doi: 10.1039/b908735d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Merkel OM, Mintzer MA, Sitterberg J, Bakowsky U, Simanek EE, Kissel T. Bioconjugate Chem. 2009;20:1799–1806. doi: 10.1021/bc900243r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).Zhang W, Simanek EE. Org. Lett. 2000;2:843–845. doi: 10.1021/ol005585g. [DOI] [PubMed] [Google Scholar]; (b) Zhang W, Nowlan DT, III, Thomson LM, Lackowski WM, Simanek EE. J. Am. Chem. Soc. 2001;123:8914–8922. doi: 10.1021/ja0041369. [DOI] [PubMed] [Google Scholar]; (c) Zhang W, Tichy SE, Pérez LM, Maria G, Lindahl PA, Simanek EE. J. Am. Chem. Soc. 2003;125:5086–5094. doi: 10.1021/ja0210906. [DOI] [PubMed] [Google Scholar]
- (11).Umali AP, Simanek EE. Org. Lett. 2003;5:1245–1247. doi: 10.1021/ol034161u. [DOI] [PubMed] [Google Scholar]
- (12) (a).Lim J, Simanek EE. Molec. Pharm. 2005;2:273–277. doi: 10.1021/mp050030e. [DOI] [PubMed] [Google Scholar]; (b) Steffensen MB, Simanek EE. Angew. Chem. Int. Ed. 2004;43:5178–5180. doi: 10.1002/anie.200460031. [DOI] [PubMed] [Google Scholar]; (c) Neerman MF, Chen H-T, Parrish AR, Simanek EE. Molec. Pharm. 2004;1:390–393. doi: 10.1021/mp049957p. [DOI] [PubMed] [Google Scholar]
- (13) (a).Steffenson MB, Simanek EE. Org. Lett. 2003;5:2359–2361. doi: 10.1021/ol0347491. [DOI] [PubMed] [Google Scholar]; (b) Moreno KX, Simanek EE. Tet. Lett. 2008;49:1152–1154. doi: 10.1016/j.tetlet.2007.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14) (a).Hollink E, Simanek EE. Org. Lett. 2006;8:2293–2295. doi: 10.1021/ol060559p. [DOI] [PubMed] [Google Scholar]; (b) Crampton H, Hollink E, Perez LM, Simanek EE. New J. Chem. 2007;31:1283–1290. doi: 10.1039/b617875h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang W, Gonzalez SO, Simanek EE. Macromolecules. 2002;35:9015–9021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.