

Abstract
The AMP-activated protein kinase (AMPK) is a key regulator of catabolic versus anabolic processes. Its properties as an energy sensor allow it to couple the energy status of the cell to the metabolic environment. These adaptations not only take place through the acute modulation of key metabolic enzymes via direct phosphorylation, but also through a slower transcriptional adaptative response. The question of how AMPK regulates the expression of a number of gene sets, such as those related to mitochondrial biogenesis, energy production and oxidative protection, is only beginning to be elucidated, and still many questions remain to be answered. In this review we will try to integrate our current knowledge on how AMPK regulates transcription in muscle and liver, which will serve as examples to illustrate the major advances in the field and the key challenges ahead.
Keywords: AMPK, Energy metabolism, PGC-1α, SIRT1
Introduction
One and a half centuries ago, Darwin shocked the world with one of the brightest concepts to ever impact biological sciences, i.e., that the ability of organisms to respond and adapt to environmental challenges has been vital for evolution. To the amazement of the scientific community, this remarkable feat to adapt to environmental changes is consistently found not only in organisms as a whole, but also at the tissue and cellular levels. Given that most biological processes (cell growth, division, movement, etc.) depend on energy consumption, it is not surprising that one outcome of evolution is that cells and organisms can sense energy levels and adapt their energy production to their energy demands.
In order to sustain proper biological functions, ATP levels, the energy currency in cells, are maintained in the low millimolar range, hinting at the existence of molecular mechanisms that keep an appropriate balance between energy-consuming and -producing processes. AMP-activated protein kinase (AMPK), an enzyme that senses AMP levels and that is conserved along the eukaryote kingdom, could be a key molecular player in this adaptation process. This review will focus on mammalian AMPK, but we refer the reader to some recent reviews in order to gain some insight on AMPK homologs in different eukaryotes [1–4].
Deconstructing AMPK: enzyme bricks and regulation of its activity
AMPK is a heterotrimeric enzyme
AMPK is a heterotrimeric Ser/Thr kinase composed of an α, β and γ subunit [3]. There are two different forms of α (α1 and α2) and β (β1 and β2) subunits, while three different γ isoforms (γ1, γ2 and γ3) exist [3]. The α subunits are the catalytic subunits of the functional heterotrimer and contain the Thr172 residue, whose phosphorylation is required for full enzymatic activity [5]. The α subunit partners with the β and γ subunits through its C-terminal region [6]. The β subunit also interacts with both the α and γ subunits, and its mid-region contains an evolutionarily conserved carbohydrate-binding domain, which allows AMPK to interact with glycogen particles [7]. The γ subunits contain one of the critical features of the enzyme, the four tandem repeats known as cystathionine β-synthase (CBS) motifs, which form an interface for interaction with two AMP or ATP molecules in a mutually exclusive way and a third AMP molecule in a non-exchangeable fashion [8]. While the binding of ATP keeps the activity of the enzyme low, the exchange of ATP for AMP is enough to promote a mild, less than fivefold, activation of the kinase through an allosteric mechanism [5]. More importantly, AMP binding renders AMPK a poorer substrate for the α subunit Thr172 phosphatase, which results in increased Thr172 phosphorylation [9]. The combination of the allosteric and phosphorylation effects promoted by AMP leads to a >1,000-fold activation of the enzyme [10]. Due to the reaction catalyzed by adenylate kinase, transforming two ADP molecules into one ATP and one AMP, the AMP/ATP ratio is a very sensitive reflection of metabolic disturbances of the cell [11], and, therefore, transforms AMPK into an exquisite sensor of energy balance.
Regulation of AMPK phosphorylation
As described above, AMPK is maximally active when phosphorylated. Consequently, there has been great interest in identifying the regulators of the phosphorylation state of this enzyme. During the last decade, a number of upstream kinase activities have been identified, and, even though the exact identity of the phosphatase activity remains elusive, it seems to belong to the PP2C family [9, 12]. Among the different kinases proposed to act as AMPKKs, LKB1 and CAMKK are now widely accepted to be key. Others, like transforming growth factor-β-activated kinase 1 (TAK1), can certainly phosphorylate AMPK in vitro [13], but the “in vivo” evidence of their capacity to activate AMPK is still not conclusive. The reasons and scenarios justifying the need for different AMPK upstream kinases are yet to be fully understood.
LKB1/STRAD/MO25
LKB1 is a Ser/Thr kinase that was originally identified as a tumor suppressor mutated in an inherited form of susceptibility to cancer, the Peutz-Jeghers syndrome [14]. LKB1 requires the formation of a heterotrimeric complex with two additional proteins in order to function, Sterile-20-related adaptor (STRAD) and Mouse protein 25 (MO25) [15]. In their absence, LKB1 is weakly active [15]. A number of post-translational modifications can impact LKB1 and potentially modulate its activity [14, 16]. However, most evidence points towards the hypothesis that, in normal physiological settings, the LKB1/STRAD/MO25 complex is a constitutively active kinase [17] and that the regulation of AMPK happens through different accessibility for the phosphatase activity [9]. This particularity might be explained by the fact that the LKB1 complex acts as a master kinase for the 13 members of the entire family of AMPK-related kinases [18], making it necessary to create substrate specificity through additional methods. In this sense, increased AMP only leads to activation of AMPK, and not of the other 12 family members [17]. Studies in the LKB1-deficient mouse have shown that LKB1 is the main AMPK kinase in muscle and liver [19–21]. Muscle-specific LKB-1 KO mice display severely impaired AMPKα2 phosphorylation after stimulation of AMPK with the phamacological AMP-mimetic AICAR (aminoimidazole-4-carboxumide-1-β-d-ribofuranoside) or ex-vivo contraction, demonstrating that LKB1 is the major AMPK kinase in skeletal muscle [19, 21]. In liver, deletion of LKB1 prevented the effects of metformin on AMPK activation and glucose production [20].
CaMKK
Simultaneous work by David Carling and Grahame Hardie’s groups found a second alternative AMPK kinase in brain and LKB1-deficient cells: the Ca2+/calmodulin-dependent kinase kinases (CaMKKs) [22, 23]. Other tissues, like muscle, also express CAMKKα and, not so clearly, CAMKKβ, although at lower levels than brain [24, 25]. The activity of CAMKKs depend on increases in intracellular Ca2+ levels and act on AMPK independently of changes in AMP [10]. It has been hypothesized that CAMKKs could be the main AMPKK during the initial phase of mild-tetanic muscle contraction [26]. Overexpression of CAMKKα or CAMKKβ in muscle is enough to increase AMPK phosphorylation [27], and muscle overload is known to increase AMPK activity in LKB1 knock-out mice, in correlation with an increase in CAMKK expression [25]. However, it must be said that a number of experiments studying the role of CAMKK in muscle have relied on the use of STO-609 as a CAMKK inhibitor, whose specificity is not fully clear [19, 26].
AMPK actions
As mentioned before, AMPK acts as an energy sensor by sensing the AMP/ATP ratio. AMPK activation is generally linked to the stimulation of metabolic responses in order to prevent metabolic and energetic crisis in situations where ATP synthesis is compromised (hypoxia, ischemia, low nutrient availability) or ATP consumption is accelerated. Consequent to this principle, AMPK activation stimulates catabolic processes to generate ATP and inhibits ATP-consuming anabolic processes that are not required for the immediate survival of the cell. Even though this review aims to focus on the transcriptional events regulated by AMPK, it is necessary to understand the acute effects of AMPK activation in order to understand the global physiological actions of AMPK and the implications of its pharmacological activation. Therefore, we will briefly mention the most notable acute effects of AMPK and refer the reader to some recent reviews for more details [3, 28, 29].
One of the immediate whole-body consequences of AMPK activation is an increase in glucose uptake by skeletal muscle through the induction of GLUT4 translocation to the plasma membrane [30]. In fact, AMPK has for a long time been hypothesized as a crucial mediator of the effects of muscle contraction on glucose transport [19, 28, 31]. Muscle contraction activates AMPK as a consequence of ATP depletion [19], and, probably, also through the activation of CAMKK in response to the fluctuations in cytosolic Ca2+ during contraction [26]. The downstream events bridging AMPK activation to GLUT4 translocation are still nebulous. A number of studies have focused their attention on the attractive link provided by TBC1D1 and TBC1D4, two highly related proteins of the same family, that are predominant in glycolytic and oxidative muscle, respectively [32]. TBC1D1 and D4 are Rab GTPase-activating proteins (GAPs), which are believed to slow down or prevent GLUT4 exocytosis by keeping GLUT4-vesicle associated Rab proteins in their GDP-bound form [33]. AMPK phosphorylates TBC1D1 and D4, and this dissociates them from GLUT4 vesicles, allowing GLUT4 translocation [33]. While this conforms an interesting mechanism of action, a number of concerns [28] indicate that there are still many questions open regarding the molecular mechanisms by which AMPK regulates glucose uptake.
Acute activation of AMPK is also associated with decreases in glycogen synthesis rates. This can be achieved through the direct phosphorylation of glycogen synthase on Ser7, which inhibits its activity [34]. The decreased glycogen synthesis rates upon acute AMPK activation are generally coupled to an increase in the glycolytic flux, thanks to the activation of 6-phosphofructo-2-kinase (PFK-2) through direct phosphorylation on Ser466 [35]. PFK-2 catalyzes the synthesis of fructose 2,6-bisphosphate, a potent stimulator of glycolysis. Therefore, activation of AMPK rapidly mobilizes glucose into ATP-generating processes.
AMPK also stimulates fatty acid oxidation as a way to increase energy levels. To achieve this goal, AMPK directly phosphorylates acetyl-coA carboxylase (ACC) 1 and 2 isoforms on Ser79 and Ser212 [36], respectively. ACC is the enzyme that catalyzes the reaction forming malonyl coA from acetyl coA and that constitutes the initial step in lipid synthesis [36]. In addition, malonyl coA is an allosteric inhibitor of CPT1b [37], the protein responsible for fatty acid intake into the mitochondria for β-oxidation. The phosphorylation of ACC by AMPK renders ACC inactive [36], which translates into a decrease in lipid synthesis rates and the relieve of CPT-1b inhibition, leading to increased fatty acid flux into the mitochondria for β-oxidation. This induction of β-oxidation contributes, together with the increased glycolytic rate, to stimulate ATP synthesis in order to meet the energy requirements of the cell.
Also protein metabolism is affected by AMPK activation. Through phosphorylation of TSC2 [38] and raptor [39], AMPK blocks the mTOR pathway, a major controller of protein synthesis and biomass generation. This not only translates into the attenuation of protein biosynthetic processes [40], but also into the induction of protein degradation through autophagy and the ubiquitin-proteasome system [40]. While AMPK activation is generally linked to both degrading processes, this action seems to be largely indirect via mTOR inhibition and, probably, relying on transcriptional events [28].
The importance of different AMPK trimers
The existence of different isoforms for each AMPK subunit highlights the possibility that 12 different combinations of AMPK trimers can exist. To date, however, we know that all combinations are not found in different tissues and, furthermore, that every trimer combination displays a distinct spectrum of biochemical properties.
At the tissue level, AMPK trimer composition is extremely varied. For example, the α1 is the predominant isoform in white adipose tissue, blood cells, smooth muscle, endothelial cells and nerve. In contrast, α2 is the predominant one in tissues such as muscle or heart. Other tissues, like liver, contain both catalytic subunits at similar levels [41]. This tissue-specific pattern is especially clear for the γ3 subunit of AMPK, whose expression is almost restricted to glycolytic skeletal muscle, where it is the predominant γ isoform [42]. A second degree of specificity, yet to be understood, is how a similar subunit repertoire in different tissues does not necessarily lead to equal trimer composition. A clear example of this can be found in the fact that the β1 subunit is the predominant subunit associated to α2 in oxidative muscle, while both β1 and β2 equally bind α2 in glycolytic muscle [43]. An additional layer of complexity is composed by the observation that different trimer compositions can also influence the intracellular localization. Several AMPK subunits (i.e., α2, β2, γ1, γ3) have been found to partly reside in the nuclear compartment [44–46] (see below for discussion), suggesting that they might be involved in the regulation of gene expression.
Most of the studies on AMPK trimer composition have been performed in mouse skeletal muscle, where it has been postulated that distinct trimers might have different biochemical properties. For example, while AMPK activity has long been known to increase in response to muscle contraction [47, 48], recent studies indicate that ex-vivo contraction at different intensities and time periods can promote trimer-specific activation (see [28] for review).
The use of transgenic mice has contributed to the understanding of isoform-specific contributions to general AMPK functions and global metabolism. For example, the AMPKα2 knock-out mice, but not the α1, are insulin resistant, glucose intolerant and resistant to the hypoglycemic action of AICAR [49, 50]. This is a clear indication that the lack of one subunit cannot be compensated by the other by specificity in the localization, the activation mechanism or the functional output.
As of now, it is clear that we are only at the tip of the iceberg on our knowledge of the significance of the different AMPK trimers. However, the fact that the AMPK trimer composition is regulated in a tissue/compartment-specific fashion, that different AMPK trimers can be selectively activated and that different isoforms can affect specific processes clearly indicates that AMPK trimer composition is non-random and aimed to the regulation of specific functions and/or respond to different kinds/intensities of stresses.
Transcriptional actions of AMPK
Nuclear localization of AMPK
The consequences of AMPK activation expand far beyond acute responses. This is due to the ability of AMPK to directly and indirectly regulate transcriptional programs through phosphorylation events. AMPK modulates the transcription of a number of genes that increase ATP production through glycolysis and the use of lipid as a mitochondrial energy source. Studies in yeast described how snf1, the AMPKα subunit yeast homolog, is present in the nucleus and regulates transcription even through the direct phosphorylation of histones [51]. Pioneering studies by Grahame Hardie’s laboratory showed how mammalian AMPK complexes containing the α2 subunit were, at least partly, distributed in the nuclear compartment [44]. This work was further extended by the demonstration that complexes containing the α2 subunit translocate to the nucleus in response to muscle contraction [45] or leptin treatment [52]. This specificity by which AMPKα2 translocates to the nucleus is still largely unknown, but seems to depend on the presence of a nuclear localization signal that is not found in the α1 subunit [52]. A recent report, however, also suggests that α1-trimers might translocate to the nucleus, too [53]. Elucidating how AMPK shuttles in and out of the nucleus warrants future investigation.
By merging the observations that activation of AMPK promotes its nuclear translocation and that AMPK leads to specific changes in gene-expression patterns, it is easy to postulate that AMPK might be targeting nuclear proteins involved in transcriptional regulation. In the chapters below, we will discuss AMPK-regulated gene expression in different tissues, the key transcriptional regulators involved in this process, and how AMPK modulates their activity.
AMPK transcriptional regulation in muscle
Skeletal muscle is the predominant site of post-prandrial glucose uptake and the major affected tissue in insulin-resistant subjects [54]. Upon nutrient scarcity, as occurs during fasting or calorie restriction, the muscle decreases glucose consumption and switches to fatty acid utilization as main energy source [55]. Similarly, with endurance training, skeletal muscle suffers a number of changes, such as fiber-type switch from type IIx to IIa and an increase in mitochondrial biogenesis [56–58], aimed to optimize and enhance energy production. As we will see below, AMPK might act as a key mediator of these adaptations.
Chronic treatment of rodents with AMPK-activating compounds, such as AICAR, β-guanadinopropionic acid (a phosphocreatine depleting agent) or resveratrol, all increase mitochondrial biogenesis in skeletal muscle [59–62]. The actions of these agents on mitochondrial content and gene expression is robustly impaired in models with defective AMPK activity [61, 63–65], implying that AMPK is a master regulator of the transcriptional mechanisms controlling mitochondrial biogenesis. This notion was further confirmed by a number of different gain-of function and loss-of function transgenic approaches. For example, mice overexpressing a kinase dead (KD) AMPKα2 subunit in muscle displayed less voluntary running activity and reduced endurance perfomance than wild-type littermates [31], indicating impaired mitochondrial function. Similarly, muscle-specific expression of an inactive form of AMPKα2, in which Asp157 is mutated to Ala, promoted a marked decrease in mitochondrial gene expression and rendered the mice exercise intolerant [66, 67]. These defects in mitochondrial gene expression were also prominent in resting muscles from global AMPKα2 knock-out mice [63, 68]. Conversely, different genetic manipulations aimed to promote AMPK activation clearly illustrate the positive effects of AMPK activation on mitochondrial activity. Genetic AMPK activation in mice is achieved through different mutations in the γ subunits. Muscle-specific overexpression of a mutated form of AMPK, in which Arg70 from the γ1 subunit is mutated to to Gln, promoted a three-fold higher basal AMPK activity [69], which translated into an increase in mitochondrial markers' gene expression [67]. A different gain of function model, in which a mutated form of the γ3 subunit (Arg225Gln) is overexpressed, also displayed a prominent increase in mitochondrial gene expression and muscle oxidative profile [70]. Altogether, both pharmacological and transgenic manipulations clearly indicate that AMPK acts as a master transcriptional regulator of mitochondrial genes.
The effects of AMPK activation on mitochondrial genes can be achieved through the regulation of a number of transcriptional factors and cofactors (Fig. 1). For example, AMPK is a master controller of PGC-1α [60, 65, 71, 72], a transcriptional coactivator that orchestrates a constellation of transcription factors, such as ERRα, NRF1, NRF2 or PPARs, to induce mitochondrial gene expression [73–75]. The link between AMPK and PGC-1α is further reinforced by the phenotypic similarities of mice with muscle-specific deletions of PGC-1α or AMPK, both of which have a general reduction in mitochondrial gene expression and exercise intolerance [31, 66–68, 76]. Conversely, a number of pharmacological or transgenic strategies that increase AMPK or PGC-1α activity in muscle have all consistently potentiated the endurance capabilities of mice and led to a higher oxidative profile of muscle fibers [60, 62, 70, 77, 78]. Firm proof for this link was provided by the fact that AICAR was unable to increase mitochondrial gene expression in muscles of mice lacking PGC-1α [71]. As such, PGC-1α seems the key downstream mediator of the effects of AMPK on mitochondrial biogenesis. Several mechanisms explain how AMPK impacts PGC-1α. AMPK can directly phosphorylate PGC-1α at Thr177 and Ser538 in in vitro assays [71]. PGC-1α phosphorylation might not directly affect its intrinsic coactivation activity, but, rather, release it from its repressor protein p160myb [79] and/or allow deacetylation and subsequent activation by SIRT1 [65, 72]. Additionally, AMPK activation increases PGC-1α expression in muscle [60, 80], an effect that is likely to be achieved though PGC-1α autoregulation on its own promoter [72, 81–83]. Trimers containing the γ3 subunit are responsible for the majority of the effect of AMPK on PGC-1α deacetylation and activation upon exercise or fasting [65]. This is an interesting finding with long-reaching consequences, as the γ3 subunit is enriched in fast glycolytic muscle, while it is almost absent in oxidative muscle [42]. This helps explain why PGC-1α is not deacetylated in the oxidative soleus muscle or in the heart upon AMPK activation, but only in glycolytic skeletal muscle [62, 72]. Similarly, trimers containing the γ3 subunit are the ones more sensitive to exercise-induced energy stress in mouse muscle [28], making them the more apt to fine-tune exercise intensity/duration to transcriptional outputs.
Fig. 1.

AMPK regulates muscle transcriptional events through distinct mechanisms. Activation of AMPK upon energy stress increases mitochondrial and oxidative metabolism gene expression through direct and indirect events. SIRT1 is an example of a transcriptional regulator whose activity is increased by AMPK through an indirect mechanism (i.e., by promoting an increase in NAD+). Direct phosphorylation of AMPK occurs, for example, on the coactivator PGC-1α and the FOXO family of transcription factors, whose subsequent deacetylation by SIRT1 increases their activity. The activation of PGC-1α leads to the coactivation of a myriad of transcription factors, such as PPARα, PPARβ/δ and CREB, which is also phosphorylated and activated by AMPK. Phosphorylation of GEF promotes co-translocation with MEF2 to the nucleus. Furthermore, phosphorylation of HDAC5 by AMPK relieves the inhibition on the MEF2/GEF complex and allows transcriptional activation. These examples illustrate the mechanisms involved when AMPK directly and indirectly regulates transcriptional events
However, PGC-1α is a coactivator, and its transcriptional effects depend on the transcription factors it coactivates. Therefore, it is also likely that AMPK can somehow target PGC-1α towards the transcription factors of interest. This is important, as discussed below for liver metabolism, and helps to understand how AMPK activation does not activate all possible PGC-1α-regulated gene programs. A key transcriptional factor coactivated by PGC-1α in muscle to promote oxidative metabolism is MEF2 [78], which in turn also regulates PGC-1α expression through directly binding the PGC-1α promoter [84]. Interestingly, MEF2 activity is also crucially regulated by AMPK [85, 86], as demonstrated by studies on the GLUT4 promoter [86]. Activation of AMPK leads to the translocation of MEF2 to the nucleus and its binding to its target promoters in vivo in a time frame concordant with the increased expression of GLUT4 and PGC-1α in exercised or AICAR-treated mice [84, 86, 87]. The mechanism by which AMPK impacts on MEF2 is likely to be indirect, as AMPK does not phosphorylate MEF2 [86] and no interaction has been reported to date. One suggested hypothesis was that MEF translocation could be aided by its interacting partner GEF (GLUT4 Enhancer Factor) [86, 88]. Interestingly, AMPK phosphorylates GEF and promotes its import into the nucleus and DNA binding [86], strengthening the possibility that both transcription factors are regulated in coordination by AMPK as a unit.
The CREB family of transcription factors has also been implicated in muscle metabolism through the regulation of hexokinase II or PGC-1α, among others [84, 89]. Recent data indicate that AMPK can phosphorylate the CREB family of transcription factors, including CREB1, ATF1 and CREM [90]. AMPK phosphorylates CREB at the same residue as PKA, Ser133, and enhances CREB-dependent transcription [90]. As discussed in the next chapter this coordination between AMPK and CREB might be conditioned by a number of circumstances and display some tissue/time specificity, as AMPK is also known to block the action of some CREB coactivators [91]. While phosphorylation of CREB is not essential for the binding of CREB to CRE sites, it promotes the recruitment of essential coactivators like CBP/p300 [92]. Interestingly, AMPK has also been shown to directly phosphorylate CBP/p300 at Ser89 [93]. This phosphorylation presumably alters the structure of the N-terminal region of the protein, impeding its interaction with nuclear receptors, such as PPARs, but not with other families of transcription factors, such as CREB [93]. While this constitutes a beautiful model to explain a “channelled” activation of gene expression, it potentially contradicts the notion that AMPK exerts a number of its biological effects on lipid oxidative genes through the activation of PPARα [52, 94]. Indeed, PPARα and PPARβ/δ constitute attractive mediators for the transcriptional actions of AMPK, as the metabolic profile achieved by AMPK activation shares many common features with that obtained through PPARα and PPARβ/δ activation, i.e., stimulation of mitochondrial biogenesis, of endurance performance and of lipid oxidation metabolism [95–99]. Some results already support that PPARα mediates the transcriptional actions of AMPK on oxidative metabolism [94], and recent data suggest that simultaneous AMPK and PPARα or PPARβ/δ activation may act synergistically in the induction of such genes [77, 100]. It has also been proposed that the AMPK can interact with PPARα or PPARβ/δ through the α subunit, leading to a synergistic effect with the ligand-dependent activation of the nuclear receptor [77, 100, 101]. Interesting in this context, despite many efforts, no consistent evidence exists for the requirement of a direct phosphorylation event to link AMPK with PPARα or PPARβ/δ activity [77, 101]. Another plausible explanation for the synergism between AMPK and PPAR activation could be the fact that the activation of PGC-1α by AMPK would further increase transcriptional co-activation of the ligand-bound PPARα or PPARβ/δ. The ability of AMPK to acutely promote lipid oxidation could provide endogenous ligands for PPARs, hence contributing as such to the synergism between the kinase and the PPARs. Unravelling these links between AMPK and PPARs will constitute a promising ground for investigation in the years to come. Expanding on this field, it will be interesting to test the possible relationship and synergistic effects that AMPK could have with other nuclear receptors that strongly influence mitochondrial biogenesis, such as the estrogen-related receptors (ERRs) [102].
The FOXO family of transcription factors is another seducing target for AMPK. The actions of FOXO have been linked to lifespan extension [103], and in muscle they are commonly associated with protection against oxidative stress, enhancement of lipid metabolism and induction of autophagy [104]. The relation of AMPK with FOXOs was brought to light when FOXOs were reported as possible mediators of the effects of AMPK on autophagy [105]. Furthermore, AMPK can directly phosphorylate different members of the FOXO family of transcription factors [106]. Among them, FOXO3 is phosphorylated by AMPK in up to six residues [106]. Mutation of these residues impaired the ability of AMPK to promote key transcriptional responses during glucose deprivation, including the transcriptional activation of oxidative protection genes [106]. FOXO phosphorylation by AMPK does not influence FOXO subcellular localization, but rather its activity [106]. However, it must be noted that, as with PGC-1α, FOXO activity is also critically controlled through acetylation/deacetylation, which is altered by SIRT1 [107–109]. It is tempting to speculate that AMPK phosphorylation of FOXO could also serve as a signal for the deacetylation by SIRT1, which, in turn, seems to provide FOXO with specificity towards the regulation of oxidative stress genes [107], suggesting that the modifications of FOXO by AMPK and SIRT1 might be interconnected.
The transcriptional actions of AMPK in muscle not only take place through the activation of transcriptional factors, but also through the modulation of corepressors and histone deacetylase activities. For example, SIRT1 has already been mentioned as an enzyme whose activity is highly linked to AMPK [72, 110]. SIRT1 is an evolutionarily conserved NAD+-dependent deacetylase, whose action impacts on a number of transcriptional regulators [111]. Activation of SIRT1 has generally been linked to the induction of lipid oxidation and mitochondrial metabolism in muscle [112]. The similar phenotypic outputs from AMPK and SIRT1 activation suggest that there might be a functional link between both activities. Direct interaction or phosphorylation events, however, do not seem to take place between these enzymes [72, 106]. Rather, AMPK seems to influence SIRT1 activity through an AMPK-induced modulation of NAD+ metabolites [72, 110], which are critical determinants of SIRT1 activity [113, 114]. For example, pharmacological or physiological activation of AMPK is followed by a robust increase in NAD+ within hours, which derives from the metabolic rearrangements promoted by an increase in fatty acid oxidation rates [72]. This metabolic and fast increase in NAD+ levels induced by AMPK is sustained by the induction of Nampt expression, a gene that resynthesizes NAD+ from its metabolic breakdown product, nicotinamide [110]. This constitutes a two-way impact of AMPK on SIRT1 activity as it generates the SIRT1 activator NAD+, while reducing the levels of nicotinamide, a physiological inhibitor of SIRT1 activity [114]. The intimate link between SIRT1 and AMPK is further reinforced by studies using resveratrol, a polyphenol compound that has long been used as a SIRT1 agonist. Resveratrol increases lifespan in a number of lower eukaryotes [115]. In higher eukaryotes, resveratrol increases muscle mitochondrial content and enhances endurance perfomance [62]. This increased ability to oxidize lipids confers the mice with protection against metabolic disease upon high-fat feeding [62, 116]. While it is true that an important number of biological actions of resveratrol depend on SIRT1 [115], the initial belief that resveratrol could act as a direct SIRT1 agonist [117] is long gone now, as in vivo evidence suggests that resveratrol rather acts primordially through AMPK, and any effect on SIRT1 activity is a downstream consequence of AMPK activation [64, 65]. These observations stress the relevance of AMPK/SIRT1 as a conserved signaling axis that is activated upon energy stress. Resveratrol effects on AMPK probably derive from the overlooked fact that resveratrol can act as a mild mitochondrial “poison” by inhibiting complex III and V of the mitochondrial respiratory chain [118, 119]. Therefore, resveratrol's actions, like those of metformin [20, 120], likely derive from a mild impairment in ATP synthesis.
Another enzyme, HDAC5, is the predominant type II histone deacetylase in adult skeletal muscle. In general, HDAC5 acts as a transcriptional repressor by direcly deacetylating histone lysine residues within the nucleosome, forming a compact structure that limits the accessibility of transcriptional regulators to DNA [121]. The specificity of genes repressed by HDAC5 is provided by the ability of this deacetylase to bind only certain transcription factors, such as MEF2 [122, 123]. This way, HDAC5 controls a myriad of processes in skeletal muscle, from glucose and oxidative metabolism [124, 125] to myocyte differentiation [126]. GLUT4 expression is controlled by interactions among AMPK, HDAC5 and MEF2. This involves an interesting cascade of events in which translocation of certain AMPK trimers to the nucleus upon activation allows the direct phosphorylation of HDAC5 in two residues, Ser259 and Ser498 [124]. This AMPK-dependent phosphorylation of HDAC5 triggers its dissociation from MEF2 and provides binding sites for 14-3-3 proteins, which export HDAC5 out from the nucleus [124]. The release of HDAC5 will increase histone acetylation and enable the recruitment of MEF2 coactivators, such as PGC-1α [127], and the basic transcriptional machinery to the GLUT4 promoter. The mutation of these HDAC5 phosphorylable residues is enough to prevent AMPK-dependent induction of the GLUT4 gene [124], clearly illustrating the relevance of this mechanism of action.
Transcriptional regulation by AMPK in liver
The liver is key to maintain the whole body's nutrient homeostasis, as it adapts its ability to store and release carbohydrates to the metabolic needs of the organism. Deficiencies in this regulatory mechanism are manifested in type 2 diabetic patients, where elevated hepatic glucose production leads to hyperglycemia. Consequent to the fact that energy stress triggers its activity, AMPK activation in liver shuts down glucose, cholesterol and triglyceride biosynthetic pathways in liver while promoting fatty acid oxidation [41]. Most manipulations of AMPK activity in liver confirm this paradigm. Deletion of the α2 subunit of AMPK in the liver promotes hyperglycemia and glucose intolerance because of increased hepatic glucose production [128]. Similarly, defective AMPK activity compromises fatty acid metabolism as a consequence of decreased mitochondrial gene expression [129], leading to increased plasma free fatty acids and decreased production of ketone bodies. Conversely, overexpression of an active form of AMPKα in liver is enough to improve glucose profiles in diabetic mice [130]. Similarly, overexpression of the α2 subunit in the liver decreases plasma triglycerides and increases production of ketone bodies, reflecting an increase in lipid oxidation versus synthesis [130].
Some of the above-mentioned actions of AMPK happen through direct phosphorylation of key enzymes. This is the case with, for example, the regulation of cholesterol biosynthesis, which is rapidly decreased by AMPK through direct phosphorylation and inhibition of the rate-limiting enzyme hydroxy-3-methylgltaryl-coenzyme A (HMG-CoA) reductase [131]. Another example is ACC, whose phosphorylation by AMPK prevents lipid synthesis and favors fatty acid import into the mitochondria for oxidation [36]. However, processes like gluconeogenesis and lipid biosynthesis are also highly regulated by transcriptional changes. Most of them are crucially affected by AMPK, as described below.
Gluconeogenesis, the de novo synthesis of glucose, takes place in liver through the fast induction of genes encoding rate-limiting enzymes of this process, such as phospho-enol pyruvate carboxykinase (PEPCK) or glucose-6-phosphatase (G6P). Gluconeogenesis is triggered by an increase in intracellular cAMP, as a consequence of low insulin and increasing glucagon blood levels. Through a cascade of events, increased cAMP levels will activate the transcription factor CREB, which binds to and activates the promoters of the above-mentioned genes [132]. Furthermore, binding of the CREB coactivator CRTC2 to CREB allows the recruitment of the transcriptional machinery [133]. AMPK regulates CRTC2 in a similar fashion to that described above for HDAC5 [91]. AMPK can directly phosphorylate CRTC2 on Ser171 [91]. Interestingly, the ability to phosphorylate this residue is shared by other members of the AMPK-related kinases subfamily, such as SIK2 [133]. This phosphorylation event promotes the binding of 14-3-3 to CRTC2 and induces its export to the cytosol [133]. The immediate consequence of this is that CREB loses the interaction with its coactivator and, consequently, CREB-dependent gluconeogenic gene expression is reduced. It is important to note that activation of AMPK led to increased CRTC2 cytoplasmic localization even in the presence of cAMP agonists [91], indicating that cellular energy stress overrides the systemic needs for glucose synthesis. Importantly, this characteristic is unique to AMPK, as phorphorylation of CRTC2 by SIK2 is prenvented by cAMP agonists [133]. This model also raises a number of questions. For example, there are situations in which agents that increase cAMP, such as forskolin, isoproterenol or glucagon, lead to AMPK activation [134, 135] which, in liver, would be antagonistic with the induction of gluconeogenic genes. Recent results indicate that PKA can phosphorylate and negatively regulate certain AMPK trimers containing the α1 subunit [136], which could keep AMPK activity low during gluconeogenic periods. Another complexity relies in the fact that, at least in muscle, AMPK can phosphorylate and activate CREB [90]. If this happened in liver, then there should be additional mechanisms targeting CREB to non-gluconeogenic gene sets.
An additional critical transcription factor regulating glucose metabolism in liver is HNF4α, which controls the expression of GLUT2, pyruvate kinase (L-PK) and aldolase B, among others [137]. Initial findings showed how pharmacological activation of AMPK by AICAR led to a downregulation of HNF4α target genes [138]. This phenomenon was linked to a robust reduction in HNF4α protein levels, apparently caused by a decrease in HNF4α protein stability [139]. Furthermore, HNF4α was identified as a direct target for AMPK. Specifically, AMPK phosphorylated Ser303 (Ser313 in humans) [139], a residue located in the ligand-binding domain that directly participates in homodimerization, the functional form of these transcription factors. Consequently, it was reported that mutation of Ser303 to Asp, mimicking constant phosphorylation, impeded HNF4α homodimerization and DNA binding [139]. Of note, the implications of these findings might not be limited to the liver, as HNF4α is a critical regulator of glucose metabolism through actions in the pancreas, kidney and intestine [137].
AMPK might also participate in the modulation of a third transcription factor involved in the sensing and regulation of liver glucose metabolism. The carbohydrate response element binding protein (ChREBP) is a liver-specific transcription factor that promotes the expression of glycolytic and fatty acid synthesis genes in situations of high glucose availability [140]. Like HNF4α, ChREBP induces L-PK expression by binding to its promoter [141]. It has been reported that AMPK phosphorylates ChREBP on Ser568, thereby compromising its DNA binding and transcriptional activities [142]. By inhibiting ChREBP, AMPK promotes the use of fatty acids as the main energy source. These findings, however, have been challenged by a report showing that ChREBP nuclear translocation is normal in AMPK-deficient animal models [143]. It must be remembered, however, that as with CREB and CRTC2, AMPK may not be the only kinase acting on ChREBP, and compensatory mechanisms could explain the unaltered phenotype in AMPK-deficient models.
Some conflicting points arise from the extrapolation of AMPK’s effects on certain transcriptional regulators in muscle, such as FOXO, SIRT1 and PGC-1α. AMPK activation in liver promotes an increase in the ratio between β-oxidation and lipogenesis, in part through the induction of mitochondrial content and function ([116], Cantó C and Auwerx J, unpublished observations). Conversely, ablation of AMPK in liver reduces mitochondrial content and activity [129, 144], probably as a consequence of decreased PGC-1α expression and activity [129]. However, in contrast to the role of AMPK, most reports to date indicate that PGC-1α induces gluconeogenesis [145, 146]. Activation of PGC-1α through SIRT1-mediated deacetylation seems to be a key step in the induction of the gluconeogenic program [147, 148]. Intriguingly, AMPK-induced PGC-1α expression and deacetylation can also be observed in liver, indicating that AMPK increases SIRT1 and PGC-1α activity (Cantó C., Auwerx J., unpulished observation). This being so, why does AMPK activation not promote gluconeogenesis? A very likely explanation lies in the fact that PGC-1α is a coactivator, and consequently, its action depends on the transcription factors it binds to. As AMPK inactivates CRTC2 and HNF4 actions, it is possible that PGC-1α cannot properly bind CRTC2/CREB and HNF4 transcriptional complexes, therefore redirecting its coactivating activities to other transcription factors linked to mitochondrial biogenesis. While such an explanation might be valid in the case of PGC-1α, it is more difficult to apply to the case of the FOXO family of transcription factors, which are activated by AMPK and mediate a significant part of AMPK’s effects in a number of tissues [106, 149, 150]. Most results to date make it unlikely that this also should be the case in liver, as the FOXO transcription factors are critical positive gluconeogenic regulators [104]. Furthermore, deacetylation by SIRT1 seems to promote nuclear trapping of FOXOs and transcription of gluconeogenic genes [109], which is diametrically opposite to what would be expected for AMPK activation. Strikingly, a great deal of evidence show that resveratrol, which activates AMPK in liver and cultured hepatocytes [116, 151], leads to FOXO deacetylation [109]. Therefore, the paradigm that SIRT1 is pro-gluconeogenic through its actions on FOXO and PGC-1α need to be revised in light of the number of conflicting observations, for example:
While SIRT1 downregulation in liver through adenoviral delivery of SIRT1 shRNAs leads to fasting hypoglycemia and decreased expression of gluconeogenic genes [148], liver-specific SIRT1 knock-out mice show normal blood parameters upon fasting and nicely adapt to calorie restriction [152].
SIRT1 activation in liver does not seem to happen in the initial phase of gluconeogenesis, which is controlled by CRTC2, but rather occurs during a later phase, leading to the deacetylation and degradation of CRTC2, which attenuates gluconeogenic rate [153].
Mice mildly overexpressing SIRT1 are largely normal when fed a standard chow [154–156], with a tendency towards lower fasting blood glucose levels [155]. SIRT1 overexpression, however, effectively protected against hyperglycemia in a number models of metabolic disease because of reduced hepatic glucose output [154, 156] and lower FOXO and PGC-1α acetylation levels [154], indicating that SIRT1 activity can actually be linked to a decrease in gluconeogenic rates.
In all murine models of metabolic disease and diabetes tested to date, resveratrol or similar compounds consistently protect against hyperglycemia, triglyceride accumulation and excessive cholesterol production [62, 116, 157, 158], very much in line with the results obtained in mice overexpressing SIRT1 [154, 156]. AMPK is robustly activated in the livers of mice fed with resveratrol [116], and the phenotypic outputs are perfectly in line with those expected for AMPK activation. Since these mice displayed higher SIRT1 and PGC-1α activity [116], physiological activation of SIRT1 or PGC-1α in liver is not per se linked to gluconeogenesis. Similar observations were made with the SIRT1 agonist described by Sirtris, SRT1720 [159, 160], even though the direct and specific effects of this compound on SIRT1 activation are controversial [161].
The observations that resveratrol deacetylates FOXO1 [109] and protects against hyperglycemia [62, 116] indicate that FOXO activation of the gluconeogenic program might be avoided or be very moderate in situations of AMPK activation, while the induction of other FOXO target genes is prioritized. This might be explained by the fact that FOXO actions sometimes require interplay with other transcription factors, such as HNF4α [162, 163], to modulate glucose metabolism genes. Therefore, AMPK might also channel FOXO activity to specific gene sets through post-translational modifications, such as phosphorylation [106] and deacetylation [107], and by preventing its interplay with certain transcription factors.
Recent evidence indicates that SIRT1 enhances AMPK action in the liver by deacetylating LKB1, altering its cellular localization and its association with STRAD, ultimately stimulating its activation of AMPK [16]. This suggests that SIRT1 and AMPK might reciprocally activate each other in liver and HepG2 cells [16, 164, 165], creating a positive feedback loop. Such observations imply that AMPK and SIRT1 activities would also go hand in hand in liver, which contradicts the notion of SIRT1 as pro-gluconeogenic factor.
Given these observations, it is clear that we are only at the beginning of our understanding about how the transcriptional effectors of AMPK are regulated, but it seems clear that different mechanisms of action might be coexisting (Fig. 2). The lack of a linear extrapolation of the way in which SIRT1, PGC-1 and FOXOs act downstream of AMPK complicates the picture. Furthermore, we are still far from grasping how AMPK quickly downregulates some key players in liver lipid metabolism, such as SREBP1c [120, 130]. Given the proven efficacy of AMPK-activating drugs, such as metformin, in type 2 diabetes, the clarification of these enigmas should be a priority for the field.
Fig. 2.
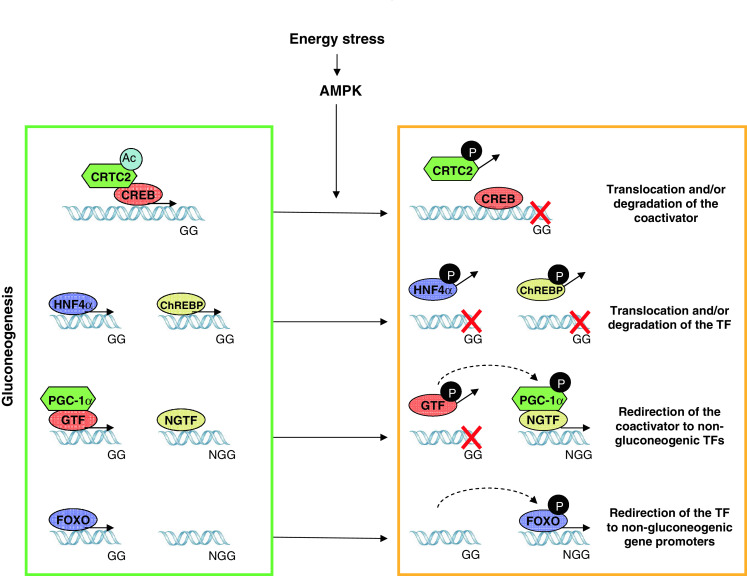
AMPK anti-gluconeogenic effects are achieved through a combination of different transcriptional mechanisms. A constellation of transcriptional regulators modulates gluconeogenesis, such as CRTC2, FOXO, ChREBP, HNF4α and PGC-1α. AMPK impacts on them all through different strategies. For example, AMPK phosphorylates CRTC2 and promotes its nuclear exclusion, disassembling the coactivator from CREB on gluconeogenic genes (GG). AMPK can also directly phosphorylate transcription factors (TFs), as happens with HNF4α and ChREBP, promoting their nuclear exclusion and/or degradation. In the case of PGC-1α, phosphorylation by AMPK might direct its coactivating actions towards non-gluconeogenic gene (NGG) regulation. Similarly, phosphorylation of FOXOs by AMPK may drive its action from gluconeogenic genes towards other gene sets, such as oxidative protection
Additional transcriptional regulators controlled by AMPK
Most of the attention on AMPK has been focused on transcriptional regulation in metabolic tissues, as those described above, or in the immediate phosphorylation of metabolic enzymes and signaling pathways. Still, AMPK may regulate additional transcriptional events, which are worthy of attention.
Cell cycle and differentiation regulators
A riveting field for future study is the regulation of p53 by AMPK, which potentially will shed light on the link among metabolism and cell cycle and division. Evidence is accumulating that AMPK could control the cell cycle by promoting G1 arrest and reduce the number of S phase cells [166, 167]. Studies showing that AMPK can directly phosphorylate p53 on Ser15 (Ser18 in mice) were key to understanding the effects of AMPK on proliferation [166]. In normal circumstances, p53 is rapidly ubiquitinated and degraded. A number of post-translational modifications, such as phosphorylation and acetylation, can stabilize the protein and promote cell cycle arrest and anti-tumorigenic effects [168]. In line with this, phosphorylation of p53 by AMPK stabilizes p53 and induces the expression of its target gene p21 [166, 167], an inhibitor of cyclin-dependent kinases, which promotes a cell cycle arrest at the level of G1 and G2 [169]. Therefore, situations of low nutrient availability and/or energy stress could translate into a natural inhibition of cell division in order to ensure cellular sustainability. These findings have serious implications for the possibility of using AMPK-activating compounds as anti-cancer drugs. Additionally, p53 has also been linked to the transcriptional regulation of mitochondrial metabolism [170], providing a new mechanism by which AMPK could impact on mitochondrial gene expression. An intriguing paradox in the link between AMPK and SIRT1 is the fact that SIRT1 is known to deacetylate and inactivate p53 [171, 172], while the role of AMPK seems to be the opposite. Elucidation of this apparent contradiction deserves investigation. To date, most of the studies on the AMPK/SIRT1 link have been done in adult normal tissue, making it possible that this signaling pathway is altered in tumors. Similarly, a recent report indicates that SIRT1 activity can somehow be oriented towards certain targets, as phosphorylation of SIRT1 by JNK leads to specific deacetylation of p53, but not of other substrates [173]. This concept is in line with our observations showing that PGC-1α needs to be primed by prior AMPK-mediated phosphorylation in order to be deacetylated [72] and makes it possible that in a similar fashion AMPK phosphorylation of p53 could be preventing or not affecting SIRT1 interaction with this substrate.
Another transcriptional regulator controlled by AMPK is the retinoblastoma protein (Rb). Rb regulates the progression, fate and differentiation of a number of cell types by binding and modulating the activity of members of the E2F family of transcription factors [174]. In neuronal precursor and stem cells, AMPK can directly phosphorylate Rb on Ser804, which then leads to its dissociation from E2F [175]. This is in line with the fact that low glucose promotes Rb/E2F dissociation [175]. The regulation of the Rb/E2F axis by AMPK has long-reaching consequences. For example, Rb phosphorylaton status determines a number of fate choices [176] and interactions with other transcriptional regulators, such as PPARγ [177]. However, as several kinases can impact on the phosphorylation of the same residue in Rb, it is difficult to extrapolate from these data the relevance of AMPK signaling on the Rb/E2F axis. In fact, a number of scenarios are potentially opposed to the hypothesis that AMPK inhibits Rb and favors E2F transcription, such as those implying that Rb is a tumor suppressor [174] or that E2F can negatively regulate mitochondrial biogenesis [178]. It is also interesting to note that, again, AMPK and SIRT1 find a convergent substrate in Rb [179], even though any possible interplay between AMPK-mediated phosphorylation and SIRT1-dependent deacetylation of Rb is yet to be explored.
Direct regulation of the epigenetic and transcriptional machinery
Other possible substrates of interest that need confirmation are those intimately related to epigenetic phenomena. The finding that AMPK trimer containing the γ3 subunit could be detected in the nucleoli [46] led to the hypothesis that it could participate in the regulation of rRNA synthesis, which is necessary for the whole ribosomal structure and mRNA translation. As AMPK is known to decrease protein translation by inhibiting the mTOR pathway [180], it would make sense that it could also shut down this process directly through an alternative mechanism. In line with this, AMPK activation decreased RNApol I activity [46, 181]. This raised the hypothesis of a possible direct regulation through phosphorylation events in the nucleoli, as recently shown by the fact that AMPK phosphorylates the RNA polymerase I (Pol I)-associated transcription factor TIF-IA at Ser635 [181]. Phosphorylation by AMPK impairs the interaction of TIF-IA with SL1, precluding the assembly of functional transcription initiation complexes [181]. Further supporting this hypothesis, mutation of Ser635 prevents down-regulation of Pol I transcription in response to low energy supply [181]. All these results provide evidence that activation of AMPK adapts rRNA synthesis to nutrient availability [181]. Another intriguing link is that between AMPK and histone phosphorylation, which derives from pioneer findings in yeast indicating that the yeast AMPK homolog, snf1, could phosphorylate histone 3 on Ser10, enabling the subsequent recruitment of the GCN5 acetyltransferase to acetylate Lys14, unfold DNA strands and initiate transcription [51]. While the possibility of AMPK directly phosphorylating histones on target genes would open doors for innumerable hypothesis, this finding has not yet been confirmed in mammalian cells. Additionally, it would also imply the requirement of a currently unknown additional specificity mechanism in order to select target genes.
Conclusions and future perspectives
The fact that AMPK activation tightly controls the transcriptional regulation of a number of gene sets has been known for years. A number of transcriptional regulators have arisen as immediate AMPK phosphorylation targets, but the implications of such findings at the gene promoter level are far from understood. We are now beginning to elucidate the way phosphorylation by AMPK influences the activity and interaction of transcriptional regulators in different tissues, which will provide clues on how AMPK determines gene set specification. Furthermore, AMPK regulates transcription not only through direct events (i.e., phosphorylation of transcriptional regulators), but also indirectly (for example, by increasing NAD+ and inducing SIRT1 activity). Further possibilities yet to be explored would involve the direct binding of AMPK to target promoters. Another challenging point for future research will be the complete understanding of how AMPK actually shuttles in and out of the nucleus and of how the nuclear functions of AMPK depend on the trimer composition. All these questions will need answers in order to fully understand AMPK action.
Acknowledgments
The work in the laboratory of the authors was supported by grants of the Ecole Polytechnique Fédérale de Lausanne, Swiss National Science Foundation, NIH (DK59820), and the European Research Council Ideas programme (Sirtuins; ERC-2008-AdG23118). CC is supported by an EMBO fellowship. The authors thank all the members of the Auwerx laboratory for inspiring discussions.
References
- 1.Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front Biosci. 2008;13:2408–2420. doi: 10.2741/2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beale EG. 5′-AMP-activated protein kinase signaling in Caenorhabditis elegans. Exp Biol Med (Maywood) 2008;233:12–20. doi: 10.3181/0705-MR-117. [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 4.Halford NG, Hey SJ. Snf1-related protein kinases (SnRKs) act within an intricate network that links metabolic and stress signalling in plants. Biochem J. 2009;419:247–259. doi: 10.1042/BJ20082408. [DOI] [PubMed] [Google Scholar]
- 5.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 6.Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 7.Hudson ER, Pan DA, James J, Lucocq JM, Hawley SA, Green KA, Baba O, Terashima T, Hardie DG. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol. 2003;13:861–866. doi: 10.1016/s0960-9822(03)00249-5. [DOI] [PubMed] [Google Scholar]
- 8.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 9.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 11.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 12.Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Gorgun CZ, Carling D, Hotamisligil GS, Febbraio MA, Kay TW, Kemp BE. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 15.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab. 2004;287:E310–E317. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- 18.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Rose AJ, Kiens B, Richter EA. Ca2+ calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol. 2006;574:889–903. doi: 10.1113/jphysiol.2006.111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol. 2008;586:1731–1741. doi: 10.1113/jphysiol.2007.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292:E1308–E1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- 27.Witczak CA, Fujii N, Hirshman MF, Goodyear LJ. Ca2 +/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes. 2007;56:1403–1409. doi: 10.2337/db06-1230. [DOI] [PubMed] [Google Scholar]
- 28.Jensen TE, Wojtaszewski JF, Richter EA. AMP-activated protein kinase in contraction regulation of skeletal muscle metabolism: necessary and/or sufficient? Acta Physiol (Oxf) 2009;196:155–174. doi: 10.1111/j.1748-1716.2009.01979.x. [DOI] [PubMed] [Google Scholar]
- 29.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418:261–275. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–1671. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 31.Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 32.Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, Bowles N, Hirshman MF, Xie J, Feener EP, Goodyear LJ. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem. 2008;283:9787–9796. doi: 10.1074/jbc.M708839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab. 2007;32:557–566. doi: 10.1139/H07-026. [DOI] [PubMed] [Google Scholar]
- 34.Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012:81–86. doi: 10.1016/0167-4889(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 35.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 36.Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30:1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 37.Mills SE, Foster DW, McGarry JD. Interaction of malonyl-CoA and related compounds with mitochondria from different rat tissues. Relationship between ligand binding and inhibition of carnitine palmitoyltransferase I. Biochem J. 1983;214:83–91. doi: 10.1042/bj2140083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 39.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209–218. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 41.Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R, Bertrand L. AMPK: lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahlapuu M, Johansson C, Lindgren K, Hjalm G, Barnes BR, Krook A, Zierath JR, Andersson L, Marklund S. Expression profiling of the gamma-subunit isoforms of AMP-activated protein kinase suggests a major role for gamma3 in white skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286:E194–E200. doi: 10.1152/ajpendo.00147.2003. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Heierhorst J, Mann RJ, Mitchelhill KI, Michell BJ, Witters LA, Lynch GS, Kemp BE, Stapleton D. Expression of the AMP-activated protein kinase beta1 and beta2 subunits in skeletal muscle. FEBS Lett. 1999;460:343–348. doi: 10.1016/s0014-5793(99)01371-x. [DOI] [PubMed] [Google Scholar]
- 44.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003;52:926–928. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- 46.Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans. 2003;31:224–227. doi: 10.1042/bst0310224. [DOI] [PubMed] [Google Scholar]
- 47.Hutber CA, Hardie DG, Winder WW. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am J Physiol. 1997;272:E262–E266. doi: 10.1152/ajpendo.1997.272.2.E262. [DOI] [PubMed] [Google Scholar]
- 48.Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, Witters LA, Ruderman NB. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 49.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 50.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo WS, Duggan L, Emre NC, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL. Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science. 2001;293:1142–1146. doi: 10.1126/science.1062322. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kodiha M, Rassi JG, Brown CM, Stochaj U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK→ERK1/2 pathway. Am J Physiol Cell Physiol. 2007;293:C1427–C1436. doi: 10.1152/ajpcell.00176.2007. [DOI] [PubMed] [Google Scholar]
- 54.DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cahill GF, Jr, Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, Reichard GA, Jr, Kipnis DM. Hormone-fuel interrelationships during fasting. J Clin Invest. 1966;45:1751–1769. doi: 10.1172/JCI105481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fink WJ, Costill DL, Pollock ML. Submaximal and maximal working capacity of elite distance runners. Part II. Muscle fiber composition and enzyme activities. Ann N Y Acad Sci. 1977;301:323–327. doi: 10.1111/j.1749-6632.1977.tb38210.x. [DOI] [PubMed] [Google Scholar]
- 57.Costill DL, Fink WJ, Pollock ML. Muscle fiber composition and enzyme activities of elite distance runners. Med Sci Sports. 1976;8:96–100. [PubMed] [Google Scholar]
- 58.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- 59.Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 60.Suwa M, Nakano H, Kumagai S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–968. doi: 10.1152/japplphysiol.00349.2003. [DOI] [PubMed] [Google Scholar]
- 61.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab. 2007;292:E331–E339. doi: 10.1152/ajpendo.00243.2006. [DOI] [PubMed] [Google Scholar]
- 64.Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH. AMP-Activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujii N, Seifert MM, Kane EM, Peter LE, Ho RC, Winstead S, Hirshman MF, Goodyear LJ. Role of AMP-activated protein kinase in exercise capacity, whole body glucose homeostasis, and glucose transport in skeletal muscle -insight from analysis of a transgenic mouse model. Diabetes Res Clin Pract. 2007;77(Suppl 1):S92–S98. doi: 10.1016/j.diabres.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 67.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–2069. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 68.Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. Faseb J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- 69.Barre L, Richardson C, Hirshman MF, Brozinick J, Fiering S, Kemp BE, Goodyear LJ, Witters LA. Genetic model for the chronic activation of skeletal muscle AMP-activated protein kinase leads to glycogen accumulation. Am J Physiol Endocrinol Metab. 2007;292:E802–E811. doi: 10.1152/ajpendo.00369.2006. [DOI] [PubMed] [Google Scholar]
- 70.Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008;283:35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]
- 71.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 74.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 75.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 76.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem. 2007;282:30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 77.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 79.Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, Spiegelman BM. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. Faseb J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 81.Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- 83.Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J Biol Chem. 2009;284:21872–21880. doi: 10.1074/jbc.M109.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. 2004;287:C790–C796. doi: 10.1152/ajpcell.00425.2003. [DOI] [PubMed] [Google Scholar]
- 85.Al-Khalili L, Chibalin AV, Yu M, Sjodin B, Nylen C, Zierath JR, Krook A. MEF2 activation in differentiated primary human skeletal muscle cultures requires coordinated involvement of parallel pathways. Am J Physiol Cell Physiol. 2004;286:C1410–C1416. doi: 10.1152/ajpcell.00444.2003. [DOI] [PubMed] [Google Scholar]
- 86.Holmes BF, Sparling DP, Olson AL, Winder WW, Dohm GL. Regulation of muscle GLUT4 enhancer factor and myocyte enhancer factor 2 by AMP-activated protein kinase. Am J Physiol Endocrinol Metab. 2005;289:E1071–E1076. doi: 10.1152/ajpendo.00606.2004. [DOI] [PubMed] [Google Scholar]
- 87.McGee SL, Sparling D, Olson AL, Hargreaves M. Exercise increases MEF2- and GEF DNA-binding activity in human skeletal muscle. Faseb J. 2006;20:348–349. doi: 10.1096/fj.05-4671fje. [DOI] [PubMed] [Google Scholar]
- 88.Knight JB, Eyster CA, Griesel BA, Olson AL. Regulation of the human GLUT4 gene promoter: interaction between a transcriptional activator and myocyte enhancer factor 2A. Proc Natl Acad Sci USA. 2003;100:14725–14730. doi: 10.1073/pnas.2432756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Osawa H, Robey RB, Printz RL, Granner DK. Identification and characterization of basal and cyclic AMP response elements in the promoter of the rat hexokinase II gene. J Biol Chem. 1996;271:17296–17303. doi: 10.1074/jbc.271.29.17296. [DOI] [PubMed] [Google Scholar]
- 90.Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, Winder WW. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–438. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- 91.Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 92.De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol. 2000;64:343–369. doi: 10.1016/s0079-6603(00)64009-6. [DOI] [PubMed] [Google Scholar]
- 93.Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- 94.Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, Lee KU, Park JY. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 95.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 96.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. Faseb J. 2003;17:2299–2301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- 98.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- 100.Houten SM, Chegary M, Te Brinke H, Wijnen WJ, Glatz JF, Luiken JJ, Wijburg FA, Wanders RJ. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci. 2009;66:1283–1294. doi: 10.1007/s00018-009-9066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bronner M, Hertz R, Bar-Tana J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem J. 2004;384:295–305. doi: 10.1042/BJ20040955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- 103.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009;9:208–214. doi: 10.1007/s11892-009-0034-5. [DOI] [PubMed] [Google Scholar]
- 105.Nakashima K, Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem. 2007;71:1650–1656. doi: 10.1271/bbb.70057. [DOI] [PubMed] [Google Scholar]
- 106.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 107.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 108.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 109.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 110.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feige JN, Auwerx J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr Opin Cell Biol. 2008;20:303–309. doi: 10.1016/j.ceb.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu J, Auwerx J. The role of sirtuins in the control of metabolic homeostasis. Ann N Y Acad Sci. 2009;1173(Suppl 1):E10–E19. doi: 10.1111/j.1749-6632.2009.04952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 114.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 115.Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 118.Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zini R, Morin C, Bertelli A, Bertelli AA, Tillement JP. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp Clin Res. 1999;25:87–97. [PubMed] [Google Scholar]
- 120.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev. 2001;11:497–504. doi: 10.1016/s0959-437x(00)00224-0. [DOI] [PubMed] [Google Scholar]
- 122.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 123.Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- 125.Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 2006;147:2432–2441. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- 129.Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem J. 2007;404:499–507. doi: 10.1042/BJ20070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54:1331–1339. doi: 10.2337/diabetes.54.5.1331. [DOI] [PubMed] [Google Scholar]
- 131.Carling D, Clarke PR, Zammit VA, Hardie DG. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem. 1989;186:129–136. doi: 10.1111/j.1432-1033.1989.tb15186.x. [DOI] [PubMed] [Google Scholar]
- 132.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 133.Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, Okamoto M, Montminy M. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 134.Yin W, Mu J, Birnbaum MJ. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3–L1 adipocytes. J Biol Chem. 2003;278:43074–43080. doi: 10.1074/jbc.M308484200. [DOI] [PubMed] [Google Scholar]
- 135.Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–54109. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- 136.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. Embo J. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gupta RK, Kaestner KH. HNF-4alpha: from MODY to late-onset type 2 diabetes. Trends Mol Med. 2004;10:521–524. doi: 10.1016/j.molmed.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 138.Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- 139.Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- 140.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 141.Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D, Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- 143.Dentin R, Benhamed F, Pegorier JP, Foufelle F, Viollet B, Vaulont S, Girard J, Postic C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest. 2005;115:2843–2854. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 145.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 146.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 147.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 148.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans . Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Williams DS, Cash A, Hamadani L, Diemer T. Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell. 2009;8:765–768. doi: 10.1111/j.1474-9726.2009.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- 152.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, 3rd, Olefsky J, Guarente L, Montminy M. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 156.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhang F, Sun C, Wu J, He C, Ge X, Huang W, Zou Y, Chen X, Qi W, Zhai Q. Combretastatin A-4 activates AMP-activated protein kinase and improves glucose metabolism in db/db mice. Pharmacol Res. 2008;57:318–323. doi: 10.1016/j.phrs.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 158.Chi TC, Chen WP, Chi TL, Kuo TF, Lee SS, Cheng JT, Su MJ. Phosphatidylinositol-3-kinase is involved in the antihyperglycemic effect induced by resveratrol in streptozotocin-induced diabetic rats. Life Sci. 2007;80:1713–1720. doi: 10.1016/j.lfs.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 159.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 161.Pacholec M, Chrunyk BA, Cunningham D, Flynn D, Griffith DA, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285(11):8340–8345. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hirota K, Sakamaki J, Ishida J, Shimamoto Y, Nishihara S, Kodama N, Ohta K, Yamamoto M, Tanimoto K, Fukamizu A. A combination of HNF-4 and Foxo1 is required for reciprocal transcriptional regulation of glucokinase and glucose-6-phosphatase genes in response to fasting and feeding. J Biol Chem. 2008;283:32432–32441. doi: 10.1074/jbc.M806179200. [DOI] [PubMed] [Google Scholar]
- 163.Ganjam GK, Dimova EY, Unterman TG, Kietzmann T. FoxO1 and HNF-4 are involved in regulation of hepatic glucokinase gene expression by resveratrol. J Biol Chem. 2009;284:30783–30797. doi: 10.1074/jbc.M109.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Suchankova G, Nelson LE, Gerhart-Hines Z, Kelly M, Gauthier MS, Saha AK, Ido Y, Puigserver P, Ruderman NB. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun. 2009;378:836–841. doi: 10.1016/j.bbrc.2008.11.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 167.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-d-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 168.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 169.Liu G, Lozano G. p21 stability: linking chaperones to a cell cycle checkpoint. Cancer Cell. 2005;7:113–114. doi: 10.1016/j.ccr.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 170.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 171.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 172.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 173.Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One. 2009;4:e8414. doi: 10.1371/journal.pone.0008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol. 2008;9:713–724. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- 175.Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16:256–270. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- 177.Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, Miard S, Auwerx J. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- 178.Goto Y, Hayashi R, Kang D, Yoshida K. Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J Cell Physiol. 2006;209:923–934. doi: 10.1002/jcp.20802. [DOI] [PubMed] [Google Scholar]
- 179.Wong S, Weber JD. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem J. 2007;407:451–460. doi: 10.1042/BJ20070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 181.Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, Voit R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA. 2009;106:17781–17786. doi: 10.1073/pnas.0909873106. [DOI] [PMC free article] [PubMed] [Google Scholar]