

Abstract
Background and Purpose
We have previously shown that isoprenaline-induced cardiac hypertrophy causes significant changes in the expression of cytochromes P450 (CYP) and soluble epoxide hydrolase (sEH) genes. Therefore, it is important to examine whether the inhibition of sEH by 1-(1-methanesulfonyl-piperidin-4-yl)-3-(4-trifluoromethoxy-phenyl)-urea (TUPS) will protect against isoprenaline-induced cardiac hypertrophy.
Experimental Approach
Male Sprague–Dawley rats were treated with TUPS (0.65 mg kg−1 day−1, p.o.), isoprenaline (5 mg kg−1 day−1, i.p.) or the combination of both. In vitro H9c2 cells were treated with isoprenaline (100 μM) in the presence and absence of either TUPS (1 μM) or 11,12 EET (1 μM). The expression of hypertrophic, fibrotic markers and different CYP genes were determined by real-time PCR.
Key Results
Isoprenaline significantly induced the hypertrophic, fibrotic markers as well as the heart to body weight ratio, which was significantly reversed by TUPS. Isoprenaline also caused an induction of CYP1A1, CYP1B1, CYP2B1, CYP2B2, CYP4A3 and CYP4F4 gene expression and TUPS significantly inhibited this isoprenaline-mediated effect. Moreover, isoprenaline significantly reduced 5,6-, 8,9-, 11,12- and 14,15-EET and increased their corresponding 8,9-, 11,12- and 14,15-dihydroxyeicosatrienoic acid (DHET) and the 20-HETE metabolites. TUPS abolished these isoprenaline-mediated changes in arachidonic acid (AA) metabolites. In H9c2 cells, isoprenaline caused a significant induction of ANP, BNP and EPHX2 mRNA levels. Both TUPS and 11,12-EET significantly decreased this isoprenaline-mediated induction of ANP, BNP and EPHX2.
Conclusions and Implications
TUPS partially protects against isoprenaline-induced cardiac hypertrophy, which confirms the role of sEH and CYP enzymes in the development of cardiac hypertrophy.
Keywords: Isoprenaline (isoproterenol), cardiac hypertrophy, cytochrome P450, soluble epoxide hydrolase inhibitor, TUPS
Introduction
Cardiac hypertrophy can be broadly defined as an increase in heart mass in response to an increase in biomechanical stress. The growth of heart is characterized by the growth of individual cardiomyocytes rather than an increase in cell number (Vakili et al., 2001). Cardiac hypertrophy is traditionally considered as an adaptive response that balances the stress and optimizes the cardiac pump function (Carreno et al., 2006). However, prolonged hypertrophy has been found to be a well-established risk factor for cardiovascular mortality (Muiesan et al., 1995; Verdecchia et al., 1998). Moreover, cardiac hypertrophy can lead to systolic and diastolic cardiac dysfunction and ultimately heart failure (Levy et al., 1994). Heart failure is a deadly cardiovascular disease that affects more than 23 million worldwide and more than 5 million people in North America (Bui et al., 2011). In Canada, heart failure affects more than 400 000 Canadians and costs over $1 billion annually for inpatient care alone (O'Connell, 2000). Despite advances made in heart research over the past two decades, heart disease remains the leading cause of death in North America and accounts for about 45% of all deaths (Levy et al., 2002).
The role of cytochrome P450 (P450) enzymes in cardiovascular health and disease is well established (Elbekai and El-Kadi, 2006; Zordoky and El-Kadi, 2008). P450 is a superfamily of mixed function mono-oxygenases that is involved in the oxidative metabolism of a wide range of xenobiotics and endogenous substances (Elbekai and El-Kadi, 2006). Many studies have examined the expression of P450 enzymes in the heart (Elbekai and El-Kadi, 2006). In vivo, the presence of P450 enzymes has been reported in human hearts (Thum and Borlak, 2000b; Delozier et al., 2007) and in the left ventricle of Sprague–Dawley (SD) rats and spontaneously hypertensive rats (SHRs) (Thum and Borlak, 2002; Zordoky et al., 2008). In vitro, the gene expression and protein activity of many P450 enzymes have been reported in cultured primary cardiomyocytes (Thum and Borlak, 2000a) and in the rat cardiomyoblast, H9c2 cell line (Zordoky and El-Kadi, 2007).
In the presence of NADPH and oxygen, P540 ω-hydroxylases metabolize arachidonic acid (AA) to 20-hydroxyeicosatetraenoic acid (20-HETE), whereas P450 epoxygenases metabolize AA to four regioisomers of epoxyeicosatrienoic acids (EETs), 5,6-, 8,9-, 11,12- and 14,15-EET metabolites (Roman, 2002).
EETs are the major cardioprotective products of AA metabolism by P450 enzymes. Once produced, EETs are either incorporated into membrane phospholipid pools, secreted into the extracellular space or efficiently hydrolysed by soluble epoxide hydrolase (sEH) to biologically less active dihydroxyeicosatrienoic acids (DHETs), thus reducing their beneficial cardiovascular effect (Zeldin et al., 1995; Imig et al., 2002; Spector et al., 2004). The gene encoding sEH enzyme, EPHX2, was found to be significantly induced in different models of cardiac hypertrophy such as, 3-methylcholanthrene (3-MC) and benzo(a)pyrene (BaP)-induced cardiac hypertrophy (Aboutabl et al., 2009), isoprenaline-induced cardiac hypertrophy (Zordoky et al., 2008), SHRs with heart failure (Monti et al., 2008) and angiotensin II–induced hypertrophy (Ai et al., 2009). The association between the up-regulation of EPHX2 with the development of cardiac hypertrophy in different animal models suggested its involvement in the development of cardiac hypertrophy. Therefore, sEH inhibition is considered a new potential therapeutic target for the prevention and/or treatment of cardiac hypertrophy. The cardioprotective mechanisms of sEH inhibitors involve inhibiting the degradation of EETs and hence blocking the activation of of NF-κB (Xu et al., 2006; Imig and Hammock, 2009). Interestingly, inhibition of sEH has been reported to prevent and reverse cardiac hypertrophy in a murine model of chronic pressure overload–induced cardiac hypertrophy (Xu et al., 2006), prevent angiotensin II–induced cardiac hypertrophy in rats (Ai et al., 2009) and attenuate BaP-induced cardiac hypertrophy (Aboutabl et al., 2011).
We have previously demonstrated that isoprenaline-induced cardiac hypertrophy causes significant changes in several P450 along with the changes in EPHX2 gene expression. The overall balance of these changes leads to a higher production of 20-HETE and lower production of EETs in the hypertrophied hearts (Zordoky et al., 2008). Several studies have demonstrated the association and the potential contribution of higher levels of the cardiotoxic metabolite 20-HETE and lower levels of the cardioprotective EETs in the development of cardiac hypertrophy (Chabova et al., 2007; Aboutabl et al., 2009). In the present study, we investigated whether inhibition of sEH by 1-(1-methanesulfonyl-piperidin-4-yl)-3-(4-trifluoromethoxy-phenyl)-urea (TUPS) protects against isoprenaline-induced cardiac hypertrophy. Furthermore, we examined the effect of TUPS treatment on P450 enzymes and the formation of P450-mediated AA lipid mediators. Our findings provide the first evidence for cardiac-specific changes in P450, sEH enzymes and AA metabolism during sEH inhibition in this isoprenaline-induced cardiac hypertrophy model.
Methods
Animals
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., ; McGrath et al., 2010). The investigation follows the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (Publication no. 85-23, revised 1996). All experimental animal procedures were approved by the University of Alberta Health Sciences Animal Policy and Welfare Committee. Male Sprague–Dawley rats weighing 200–250 g were obtained from Charles River Canada (St. Constant, QC, Canada). All animals were maintained on a 12 h light/dark cycle with food and water available ad libitum.
Cell culture and treatments
H9c2 cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM, without phenol red, supplemented with 0.45% glucose, 0.15% sodium bicarbonate, 0.11% sodium pyruvate, 10% FBS, 20 μM l-glutamine, 100 IU mL−1 penicillin, 10 μg mL−1 streptomycin and 25 ng mL−1 amphotericin B. Cells were grown in 75 cm2 tissue culture flasks at 37°C in a 5% CO2 humidified incubator. For analysis of mRNA, cells were grown at a density of 1–1.5 × 106 cells per well in a six-well tissue culture plate. On 60–80% confluence (2–3 days), an appropriate stock solution of isoprenaline at a concentration of 100 μM was added to the culture medium in the presence or absence of 11,12-EET, which was added every 8 h for 24 h at a final concentration of 1 μM. An appropriate stock solution of TUPS was added to the culture medium to reach a final concentration of 1 μM for 24 h.
Chemicals and reagents
AA, isoprenaline, 4-hydroxybenzophenone, DMEM base and anti-goat IgG with HRP secondary antibody were purchased from Sigma-Aldrich Chemical Co (St Louis, MO). Amphotericin B was purchased from ICN Biomedicals Canada (Montreal, QC, Canada). Penicillin–streptomycin, l-glutamine, FBS and TRIzol reagent was purchased from Invitrogen (Carlsbad, CA). High-capacity cDNA Reverse Transcription Kit, SYBR Green SuperMix and 96-well optical reaction plates with optical adhesive films were purchased from Applied Biosystems (Foster City, CA). Real-time PCR primers were synthesized by Integrated DNA Technologies Incorporation (San Diego, CA) according to previously published sequences. AA metabolite standards 5,6-EET, 8,9-EET, 11,12-EET, 14,15-EET, 5,6-DHET, 8,9-DHET, 11,12-DHET, 14,15-DHET and 20-HETE were obtained from Cayman Chemical (Ann Arbor, MI). TUPS was synthesized by Dr Paul Jones (University of California, Davis) as described previously (Tsai et al., 2010). Acrylamide, N′N′-bis-methylene-acrylamide, β-mercaptoethanol, ammonium persulfate, glycine, pure nitrocellulose membrane (0.45 mm) and N,N,N′,N′-tetramethylethylenediamine (TEMED) were purchased from Bio-Rad Laboratories (Hercules, CA). Chemiluminescent Western blotting detection reagents were purchased from GE Healthcare Life Sciences (Piscataway, NJ). CYP1B1 rabbit anti-rat polyclonal primary antibody was purchased from BD Gentest (Woburn, MA). sEH rabbit anti-mouse primary antibodies were obtained as generous gifts from Dr Darryl Zeldin (National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC). CYP1A1 gout anti-rat, mouse anti-rat CYP2B1/2, mouse anti-rat CYP4A and rabbit anti-rat actin polyclonal primary antibodies with secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All other chemicals were purchased from Fisher Scientific Co (Toronto, ON, Canada).
Experimental design
Male Sprague–Dawley rats (200–250 g) were injected i.p. with isoprenaline (dissolved in saline; final volume = 0.25 mL per rat) at 5 mg kg−1 day−1 for 7 days, with or without 0.65 mg kg−1 day−1 TUPS (TUPS was dissolved in PEG400, followed by further dilution in saline to 1:3 ratio; final volume = 1 mL per rat). The rats were randomly segregated into four groups. The first group (n = 6) consisted of control rats that received saline (i.p.) plus 25% PEG400 (oral gavage). The second group (n = 6) consisted of TUPS-treated rats that received TUPS dissolved in 25% PEG (oral gavage) and saline (i.p.). The third group (n = 6) consisted of isoprenaline-treated rats that received isoprenaline dissolved in saline (i.p.) plus 25% PEG400 (oral gavage). The fourth group (n = 6) consisted of isoprenaline and TUPS-treated rats that received isoprenaline dissolved in saline (i.p.) plus TUPS dissolved in 25%PEG (oral gavage). Thereafter, animals were killed by an overdose of isoflurane anaesthesia 24 h after their last injection. Heart, kidney and liver were excised, immediately frozen in liquid nitrogen and stored at −80°C until analysis.
RNA extraction and relative gene expression analysis by real-time PCR
Total RNA from the frozen heart, kidney and liver tissues was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol and quantified by measuring the absorbance at 260 nm.
The relative gene expression was determined by real-time PCR using the ABI Prism 7500 System (Applied Biosystems) according to the manufacturer's protocol. The primers used in the current study were as previously published (Bleicher et al., 2001; Kalsotra et al., 2002; Kuwahara et al., 2002; Grygielko et al., 2005; Hirasawa et al., 2005; Rollin et al., 2005; Sellers et al., 2005; Baldwin et al., 2006; Soppa et al., 2008; Zordoky et al., 2011) and are listed in Table 1. A melting curve was determined at the end of each cycle to confirm the specificity of the primers and the purity of the PCR product. Thereafter, real-time PCR data were analysed using the relative gene expression method as described previously (Livak and Schmittgen, 2001). β-actin was used as the endogenous control, and the untreated control was used as the calibrator when any changes in gene expression induced by TUPS, isoprenaline and isoprenaline + TUPS were being studied.
Table 1.
Primers sequences used for real-time PCR reactions
| Gene | Forward primer | Reverse primer |
|---|---|---|
| ANP | GGAGCCTGCGAAGGTCAA | TATCTTCGGTACCGGAAGCTGT |
| BNP | CAGAAGCTGCTGGAGCTGATAAG | TGTAGGGCCTTGGTCCTTTG |
| B-MHC | AGC TCC TAA GTA ATC TGT TTG CCAA | AAA GGATGAGCCTTTCTTTGCT |
| PROCOLLAGEN I | TATGCTTGATCTGTATCTGCCACAAT | TCGCCCTCCCGTTTTTG |
| PROCOLLAGEN III | CAGCTGGCCTTCCTCAGACT | TGCTGTTTTTGCAGTGGTATGTAA |
| TGF-1 | ACCTGCAAGACCATCGACATG | CGAGCCTTAGTTTGGACAGGAT |
| CYP1A1 | CCAAACGAGTTCCGGCCT | TGCCCAAACCAAAGAGAATGA |
| CYP1B1 | GCTTTACTGTGCAAGGGAGACA | GGAAGGAGGATTCAAGTCAGGA |
| CYP2B1 | AACCCTTGATGACCGCAGTAAA | TGTGGTACTCCAATAGGGACAAGATC |
| CYP2B2 | CCATCCCTTGATGATCGTACCA | AATTGGGGCAAGATCTGCAAA |
| CYP4A3 | CTCGCCATAGCCATGCTTATC | CCTTCAGCTCATTCATGGCAATC |
| CYP4F4 | CAGGTCTGAAGCAGGTAACTAAGC | CCGTCAGGGTGGCACAGAGT |
| EPHX2 | GATTCTCATCAAGTGGCTGAAGAC | GGACACGCCACTGGCTAAAT |
| β-actin | CCAGATCATGTTTGAGACCTTCAA | GTGGTACGACCAGAGGCATACA |
Microsomal preparation and western blot analysis
Preparation of heart microsomal protein was performed as described previously (Barakat et al., 2001). Briefly, all tissues were washed in ice-cold KCl (1.15%, KCl w v−1), cut into pieces and homogenized separately in ice-cold sucrose solution (1 g of tissue in 25 mL of 0.25 M sucrose). Tissue homogenates were centrifuged at 600× g for 8 min. The supernatant was then centrifuged at 12 000× g for 10 min. Thereafter, supernatants resulting from the previous step were mixed with 8 mM CaCl2 and centrifuged at 27 000× g for 15 min. The resulting pellets were suspended in 0.15 M KCl and re-centrifuged at 27 000× g for 15 min. Final pellets were re-suspended in cold sucrose and supernatant, cytosol, were stored at −80°C. Thereafter, the Lowry method was used for measuring heart microsomal protein concentration using BSA as a standard (Lowry et al., 1951). Western blot analysis was carried out using a previously described method (Gharavi and El-Kadi, 2005).
Microsomal incubation and separation of different arachidonic acid metabolites by LC-ESI-MS
Heart microsomes (1 mg protein mL−1) were incubated in the incubation buffer (5 mM magnesium chloride hexahydrate dissolved in 0.5 M potassium phosphate buffer, pH 7.4) at 37°C in a shaking water bath (50 r.p.m.). A pre-equilibration period of 5 min was performed. The reaction was initiated by the addition of 1 mM NADPH. AA was added to a final concentration of 50 μM and incubated for 30 min. The reaction was terminated by the addition of 600 μL of ice-cold acetonitrile followed by the internal standard, 4-hydroxybenzophenone. AA metabolites were extracted twice by 1 mL of ethyl acetate and dried using a speed vacuum (Thermo Fisher Scientific, Ottawa, ON, Canada). The concentrations of these eicosanoids in the samples were calculated by comparing the ratios of peak heights with their corresponding standards. Extracted AA metabolites were analysed using the LC-ESI-MS (Waters Micromass ZQ 4000 spectrometer; Waters, Milford, MA) method as described previously (Nithipatikom et al., 2001). The mass spectrometer was operated in negative ionization mode with single-ion recorder acquisition. The nebulizer gas was obtained from an in-house high-purity nitrogen source. The temperature of the source was set at 150°C, and the voltages of the capillary and the cone were 3.51 kV and 25 V respectively. The samples (10 μL) were separated on a reverse-phase C18 column (Kromasil, 250 × 3.2 mm) using a linear gradient mobile phase system water/acetonitrile with 0.005% acetic acid as mobile phase at a flow rate of 0.2 mL min−1. The mobile phase system was started at 60% acetonitrile, linearly increased to 80% acetonitrile in 30 min, increased to 100% acetonitrile in 5 min and held for 5 min.
sEH activity assay
sEH activity was measured using the method of Morisseau and Hammock with modifications; 14,15-EET was used as the natural substrate (Morisseau and Hammock, 2007). Briefly, the cytosolic fraction was diluted with sodium phosphate buffer (0.076 M, pH 7.4) supplemented with BSA (2.5 mg mL−1) to 0.4 mg mL−1. The assay was initiated by the addition of 14,15-EET (final concentration of 14,15-EET is 2 μg mL−1) final volume of incubates was 200 μL. The mixture was incubated at 37°C for 10 min. The reaction was terminated by the addition of 600 μL ice-cold acetonitrile followed by the internal standard, 4-hydroxybenzophenone. 14,15-EET and its corresponding 14,15-DHET were extracted by 1 mL ethyl acetate twice and dried using a speed vacuum (Savant, Farmingdale, NY). Extracted 14,15-EET and its metabolite were analysed using the LC-ESI-MS (Waters Micromass ZQ 4000 spectrometer) method, as described previously (Anwar-Mohamed et al., 2012).
Statistical analysis
Data are presented as mean ± SEM. The expression of genes was compared among tissues by using Kruskal–Wallis one-way anova on ranks. A result was considered statistically significant when P < 0.05.
Results
Effect of the sEH Inhibitor, TUPS, on the cardiac hypertrophy induced by isoprenaline
To investigate whether the inhibition of sEH confers cardioprotection in isoprenaline-treated rats, we measured the cardiac gene expression of the hypertrophic markers, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) relative to those in isoprenaline-treated rats. Isoprenaline caused a significant induction in the hypertrophic markers ANP, BNP and β-MHC by 500%, 50% and 265%, respectively (Figure 1A). On the other hand, TUPS treatment significantly decreased the isoprenaline-mediated induction of ANP, BNP and β-MHC by 47%, 71%, and 72% respectively (Figure 1A). In addition, TUPS treatment alone did not alter the gene expression of ANP, BNP and β-MHC. Moreover, isoprenaline significantly increased the heart weight to body weight ratio by 22%, whereas treatment with TUPS significantly decreased this isoprenaline-mediated increase in the heart weight to body weight ratio by 35%, compared with isoprenaline alone. Furthermore, no significant difference was observed between the control and the TUPS treatment alone (Figure 1B).
Figure 1.
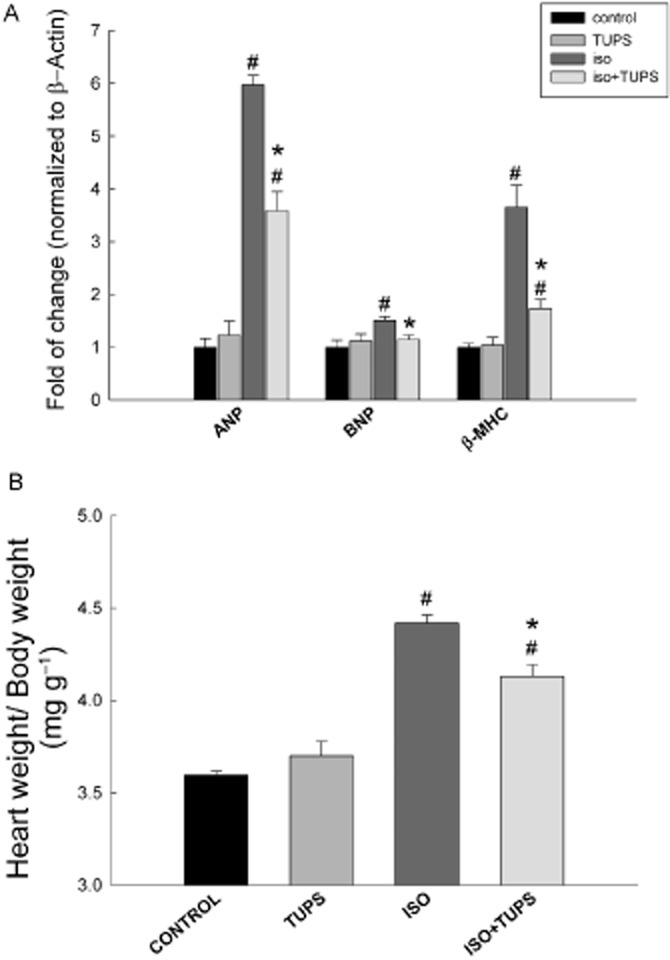
Effect of the sEH inhibitor, TUPS, on the cardiac hypertrophy induced by isoprenaline. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, oral) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. (A) The expression of the hypertrophic genes, ANP, BNP and β-MHC were determined in the heart. (B) Heart to body weight ratio (in mg g−1) was determined for each animal after 7 days of treatment with vehicles, TUPS, isoprenaline or the combination of these reagents. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline-treated rats.
Effect of the sEH inhibitor, TUPS, on fibrotic markers associated with cardiac hypertrophy
To investigate whether the inhibition of sEH has an effect on the fibrosis associated with isoprenaline-induced cardiac hypertrophy, we measured the cardiac gene expression of the fibrotic markers, procollagen I, procollagen III and TGF-1 relative to isoprenaline-treated rats. Isoprenaline treatment caused a significant induction in the fibrotic markers, procollagen I, procollagen III and TGF-1 by 456%, 443% and 123% respectively (Figure 2). On the other hand, TUPS treatment significantly decreased the isoprenaline-mediated induction of procollagen I, procollagen III and TGF-1 by 40%, 45% and 40%, respectively, compared with isoprenaline alone (Figure 2). Furthermore, no significant difference was observed between the control and the TUPS treatment alone (Figure 2).
Figure 2.
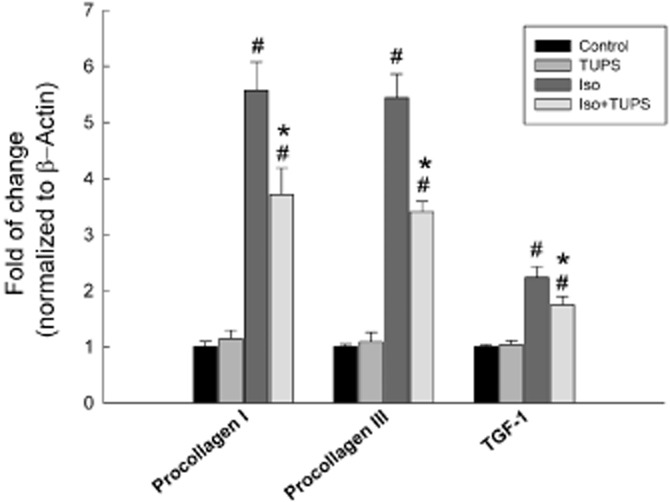
Effect of the sEH inhibitor, TUPS, on fibrotic markers associated with cardiac hypertrophy. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Total RNA was isolated from heart of control, TUPS, isoprenaline and isoprenaline+TUPS-treated rats. Gene expressions were determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline.
Effect of the sEH inhibitor, TUPS, on the changes in CYP450 gene expression mediated by isoprenaline
To examine the effect of TUPS on isoprenaline-mediated alterations in P450 gene expressions, total RNA was extracted from the heart, kidney and liver of control, TUPS, isoprenaline and isoprenaline + TUPS-treated rats. Thereafter, the expression of the different P450 genes was measured using reverse transcription followed by real-time PCR.
Figure 3A shows the effect of TUPS treatment on cardiac hypertrophy-induced CYP1A1 gene expression; isoprenaline treatment caused a significant increase in CYP1A1 gene expression in heart and kidney by about 110% and 94% respectively (Figure 3A). On the other hand, TUPS treatment caused significant inhibition of isoprenaline-induced CYP1A1 gene expression by 83% in the heart compared with isoprenaline alone treatment. In kidney, TUPS alone caused a significant induction of CYP1A1 gene expression by 260% and isoprenaline+TUPS treatment was similar to TUPS alone. In liver, CYP1A1 mRNA levels were not altered in response to either TUPS or isoprenaline treatment when compared with the control group (Figure 3A). Isoprenaline treatment also significantly induced CYP1B1 gene expression in the heart by 320%. In contrast, the CYP1B1 mRNA level was significantly decreased in hearts after treatment with TUPS, by 88% compared with the isoprenaline alone treated group. In the kidney, CYP1B1 mRNA levels were significantly induced in the TUPS- and isoprenaline+TUPS- but not in isoprenaline alone-treated groups by 151% and 82%, respectively, compared with the control group. However, no significant changes were observed in the liver (Figure 3B).
Figure 3.
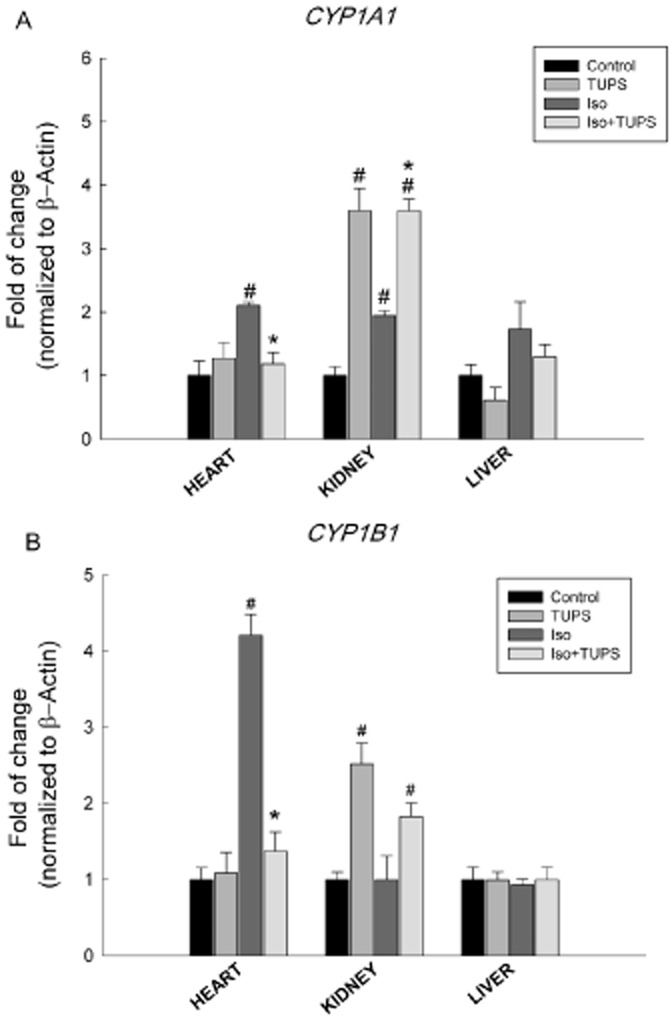
Effect of the sEH inhibitor, TUPS, on the changes in the CYP1 family gene expression mediated by isoprenaline. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Total RNA was isolated from heart, kidney and liver of control, TUPS, isoprenaline and isoprenaline+TUPS-treated rats; and the relative gene expression of (A) CYP1A1 and (B) CYP1B1 was determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline.
With respect to the CYP2 family, isoprenaline treatment caused a significant induction in CYP2B1 and CYP2B2 mRNA levels in the heart by 324% and 407% respectively. In contrast, TUPS treatment significantly inhibited the isoprenaline-mediated induction of CYP2B1 and CYP2B2 mRNA by 48% and 64%, respectively, compared with isoprenaline alone. However, no significant changes were observed in either the kidney or the liver (Figure 4A and B).
Figure 4.
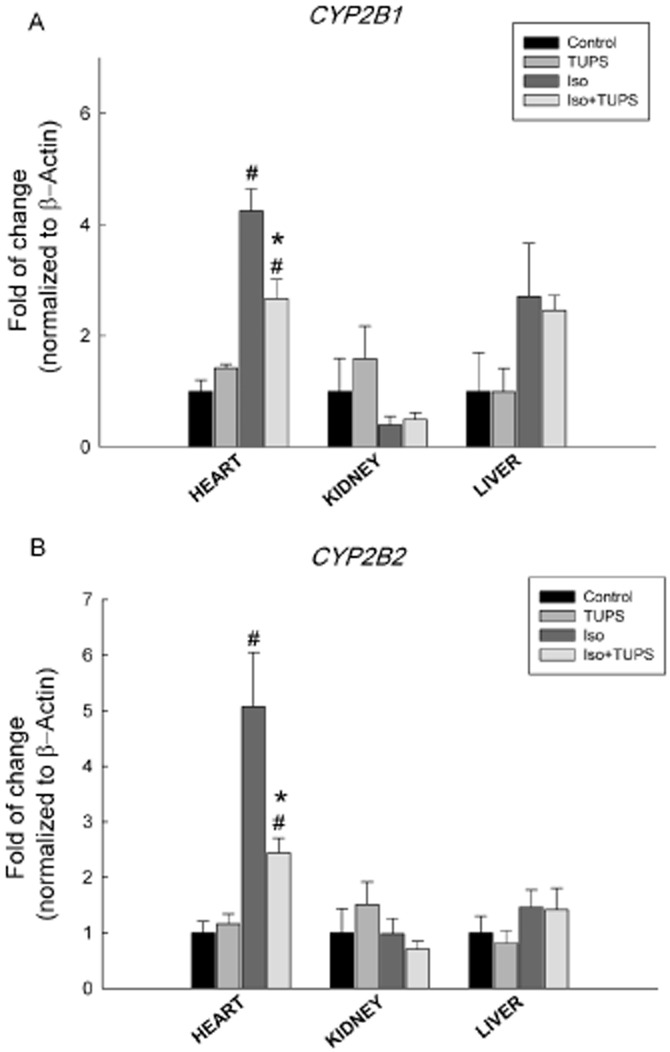
Effect of the sEH inhibitor, TUPS, on the changes in the CYP2 family gene expression mediated by isoprenaline. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Total RNA was isolated from the heart, kidney and liver of control, TUPS, isoprenaline and isoprenaline+TUPS-treated rats; and the relative gene expression of (A) CYP2B1 and (B) CYP2B2 was determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline.
With regard to the CYP4 family, CYP4A3 gene expression was significantly increased by 1169%, 46% and 103% in the heart, kidney and liver of isoprenaline-treated rats. On the other hand, the gene expression of CYP4A3 was significantly reduced by 82%, 100% and 89% in the heart, kidney and liver of rats treated with isoprenaline+TUPS, respectively, compared with the isoprenaline alone group (Figure 5A). In addition, CYP4F4 gene expression was induced in the heart by 343% but not in the kidney or the liver of isoprenaline-treated rats. Moreover, TUPS treatment significantly reduced the isoprenaline-mediated induction of CYP4F4 mRNA in the heart by 46% (Figure 5B).
Figure 5.
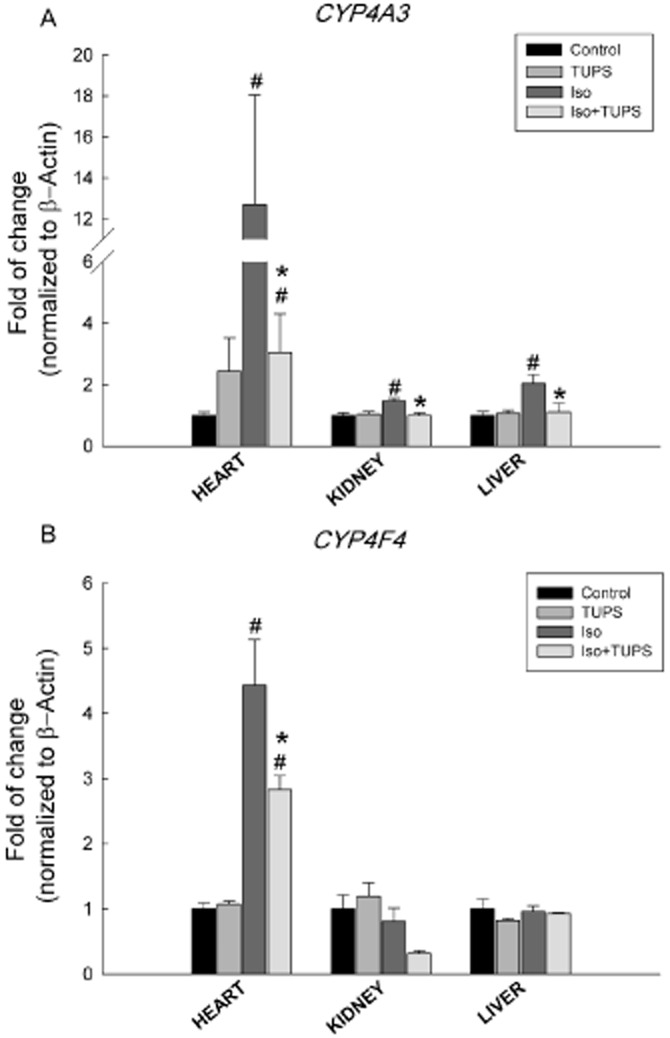
Effect of the sEH inhibitor, TUPS, on the changes in the CYP4 family gene expression mediated by isoprenaline. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Total RNA was isolated from heart, kidney and liver of control, TUPS, isoprenaline and isoprenaline+TUPS-treated rats; and the relative gene expression of (A) CYP4A3 and (B) CYP4F4 was determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline.
Effect of the sEH inhibitor, TUPS on P450 protein expression
To investigate whether the changes in P450 were further translated into protein, microsomal protein was prepared from the hearts of control, isoprenaline and isoprenaline+TUPS-treated rats. Similar to mRNA levels, isoprenaline treatment significantly increased the protein levels of CYP1A1, CYP1B1, CYP2B1/2 and CYP4A by 480%, 272%, 70% and 96% respectively (Figure 6). In contrast, TUPS treatment caused a significant inhibition of isoprenaline-mediated induction of CYP1A1, CYP1B1, CYP2B1/2 and CYP4A protein expression by 76%, 60%, 150% and 160% relative to the isoprenaline alone group, respectively (Figure 6). Unfortunately, we were unable to measure the protein level of the CYP4F enzyme because the antibodies for rat CYP4F were not commercially available.
Figure 6.
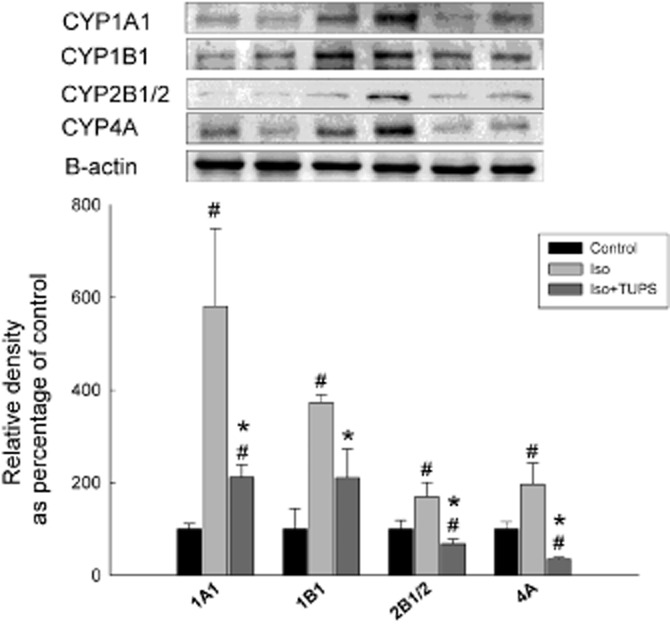
Effect of the sEH inhibitor, TUPS on the on P450 protein expression. Sprague–Dawley rats received daily injections of vehicles, isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Microsomal protein was isolated from the hearts of control, isoprenaline-treated, isoprenaline+TUPS-treated rats. Thereafter, 25 mg of microsomal protein was separated on a 10% SDS-PAGE. CYP1A1, CYP1B1, CYP2B1/2 and CYP4A proteins were detected using the enhanced chemiluminescence method. The graph represents the relative amount of protein normalized to β-actin signals (mean ± SEM, n = 4), and the results are expressed as percentage of the control values taken as 100%. #P < 0.05 compared with control, *P < 0.05 compared with isoprenaline-treated rats.
Effect of the sEH inhibitor, TUPS, on sEH at the gene expression, protein and activity levels
The enzyme sEH is a major determinant of the levels of EETs; therefore, we determined the effect of TUPS treatment on the expression of EPHX2 gene, which encodes for the sEH enzyme. Isoprenaline treatment did not cause any changes in EPHX2 gene expression in the heart, kidney or liver. Likewise, treatment with TUPS did not alter EPHX2 gene expression in any of the tissues tested (Figure 7A). To investigate whether changes in EPHX2 gene expression were translated to protein level, we determined sEH protein levels. Isoprenaline treatment significantly increased the protein level of sEH by 1300% relative to the control group. On the other hand, TUPS treatment caused a significant inhibition of isoprenaline-mediated induction of sEH protein expression by 90% (Figure 7B). In accordance with this induction in sEH protein expression, isoprenaline treatment caused a significant induction of sEH activity by 86% (Figure 7C). On the other hand, TUPS significantly decreased sEH activity by 61% compared with isoprenaline only-treated rats (Figure 7C).
Figure 7.
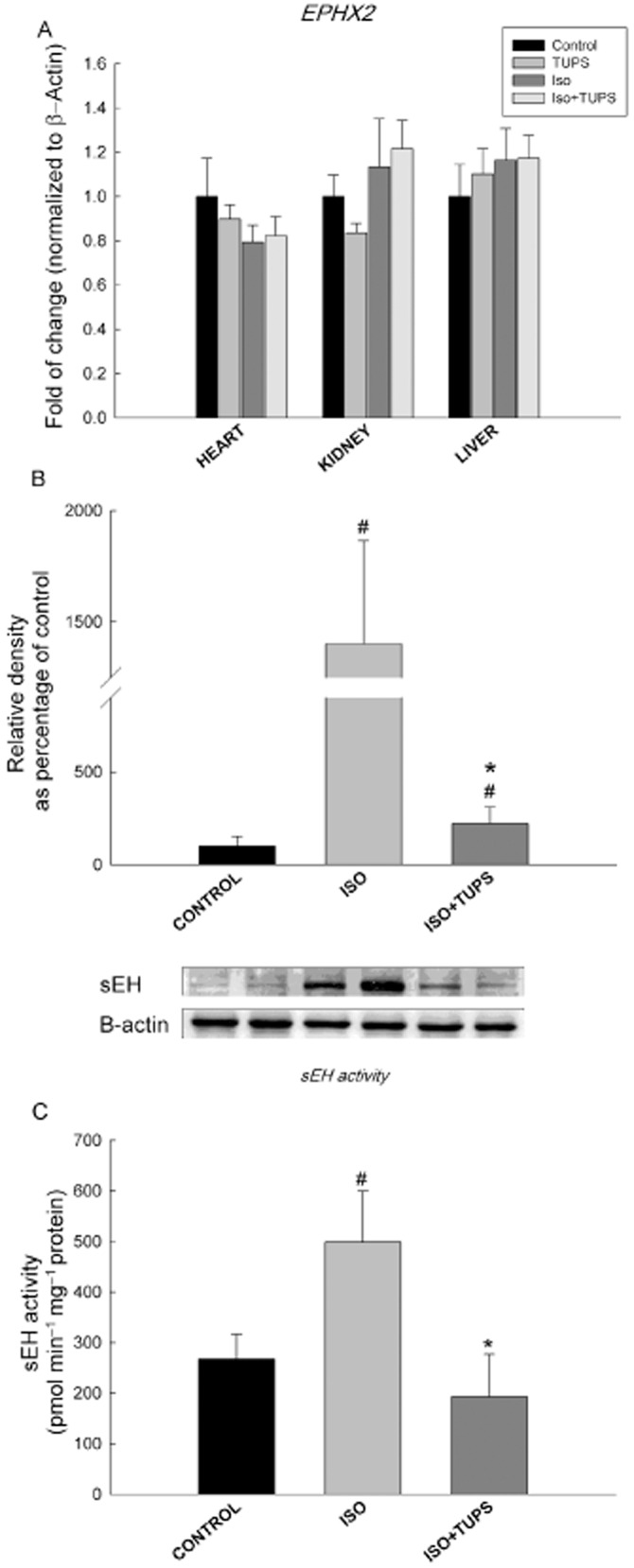
Effect of the sEH inhibitor, TUPS, on sEH at gene expression, protein and activity levels. Sprague–Dawley rats received daily injections of vehicles, TUPS (0.65 mg kg−1, p.o.), isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Total RNA was isolated from heart, kidney and liver of control, TUPS, isoprenaline and isoprenaline+TUPS-treated rats; and the relative gene expression of EPHX2 was determined by real-time PCR (A). sEH protein level was determined by Western blot analysis (B). sEH activity was calculated using sEH assay (C). Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). *P < 0.05 compared with isoprenaline, #P < 0.05 compared with control.
Effect of the sEH inhibitor, TUPS, on P450-mediated AA metabolism
To investigate the effect of TUPS treatment on P450-derived AA metabolites, heart microsomes of control, isoprenaline and isoprenaline+TUPS-treated rats were incubated with 50 μM AA for 30 min. Thereafter, AA metabolites were determined using LC-ESI-MS. The net rate of 5,6-, 8,9-, 11,12- and 14,15-EET formation were significantly lower by 16%, 34%, 50% and 35%%, respectively, in hypertrophied heart microsomes compared with control microsomes (Figure 8A). In contrast, the net formation rates of 8,9- and 14,15-DHET were significantly higher by 120% and 75%, respectively, in hypertrophied hearts microsomes compared with control microsomes. However, the net formation rates of 5,6-and 11,12-DHET were not significantly altered (Figure 8B). TUPS treatment reversed significantly the isoprenaline-induced inhibitory effects on 8,9-, 11,12- and 14,15-EET by 53%, 154% and 151%, respectively, but not that on 5,6-EET levels (Figure 8A). Furthermore, TUPS treatment caused a significant reduction in the net formation rate of 8,9-, 11,12- and 14,15-DHET by 87%, 70% and 61%, respectively, but not of 5,6-DHET levels (Figure 8B).
Figure 8.
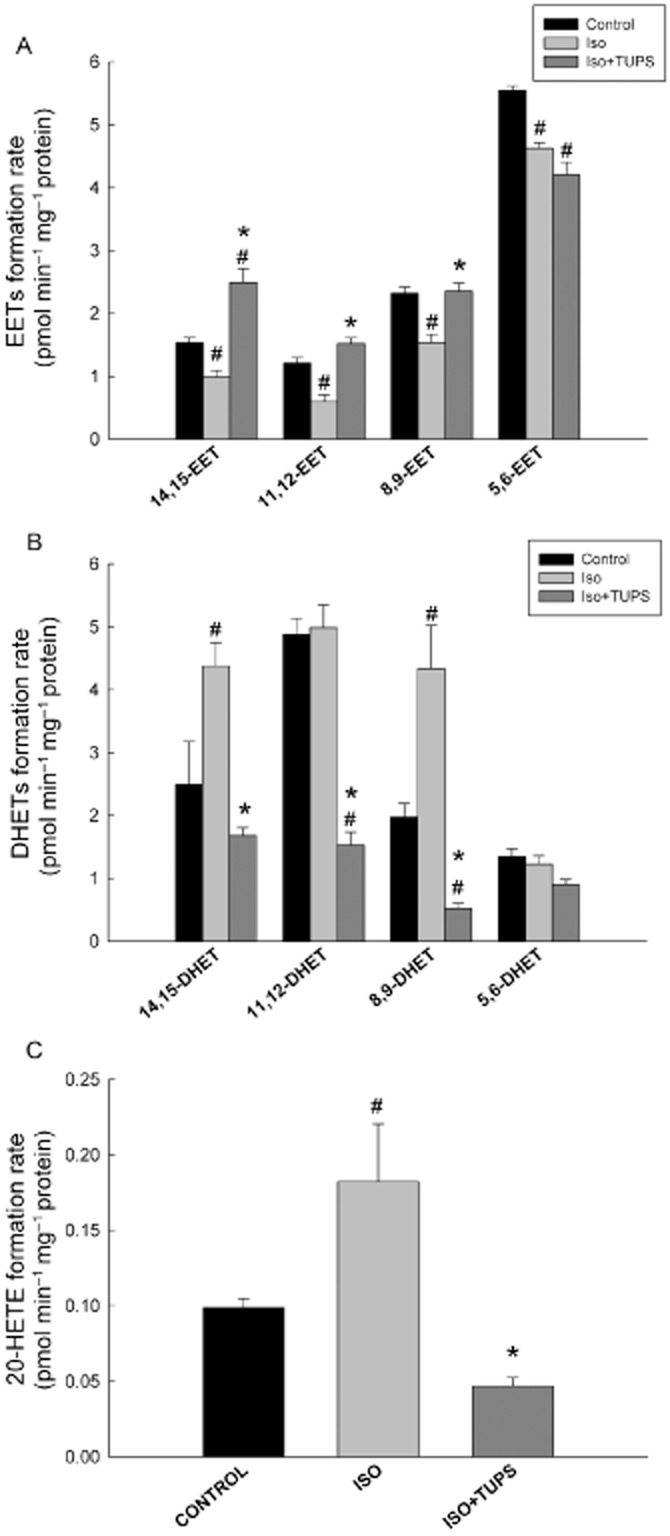
Effect of the sEH inhibitor, TUPS, on P450-mediated arachidonic acid metabolism. Sprague–Dawley rats received daily injections of vehicles, isoprenaline (5 mg kg−1, i.p.) or isoprenaline (5 mg kg−1., i.p.) plus TUPS (0.65 mg kg−1, p.o.) for 7 days; while weight-matched controls received the same volume of 25% PEG400 and saline. Heart microsomes of control, isoprenaline or isoprenaline +TUPS-treated rats were incubated with 50 μM arachidonic acid. The reaction was started by the addition of 1 mM NADPH and lasted for 30 min. The reaction was terminated by the addition of ice-cold acetonitrile. (A) EETs, (B) DHETs and (C) 20-HETE were extracted twice by 1 mL of ethyl acetate and dried using speed vacuum. Reconstituted metabolites were injected into LC-ESI-MS for metabolite determination. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). *P < 0.05 compared with isoprenaline, #P < 0.05 compared with control.
To determine the effect of TUPS treatment on P450 ω-hydroxylase activity, we measured the net formation rate of 20-HETE in microsomes from control, isoprenaline and isoprenaline+TUPS-treated rats. Isoprenaline treatment significantly increased the 20-HETE net formation by 84% in comparison with the control microsomes. On the other hand, TUPS treatment caused a significant reduction in 20-HETE net formation by 74% compared with the isoprenaline alone group (Figure 8C).
Effect of 11,12 EET and the sEH inhibitor, TUPS, on the hypertrophic markers and EPHX2 mRNA in H9c2 cells
To confirm the role of EETs in the cardioprotective effect against isoprenaline-induced cardiac hypertrophy, the cardiac-derived H9c2 cells were treated with isoprenaline (100 μM) in the presence and absence of either TUPS (1 μM) or 11,12 EET (1 μM). Thereafter, the expression of ANP, BNP and EPHX2 was measured using real-time PCR. Isoprenaline treatment caused a significant induction of hypertrophic markers, ANP and BNP, by 169% and 65%, respectively, compared with control (Figure 9A). Moreover, isoprenaline caused a significant induction in EPHX2 mRNA level by 62% compared with control (Figure 9B). On the other hand, TUPS treatment significantly decreased the isoprenaline-mediated induction of ANP, BNP and EPHX2 by 46%, 57% and 56% respectively (Figure 9). Interestingly, 11,12 EET was able to replicate the protective effects of TUPS and caused a significant decrease in ANP, BNP and EPHX2 mRNA levels by 58%, 43% and 36%, respectively, compared with the isoprenaline alone group (Figure 10).
Figure 9.
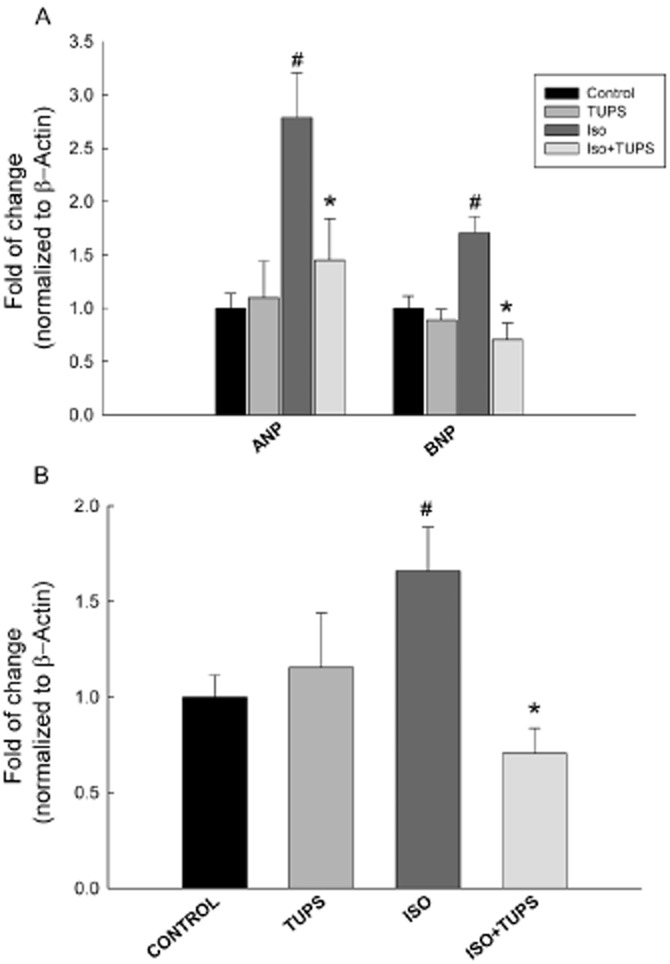
Effect of the sEH inhibitor, TUPS, on the cardiac hypertrophic markers and EPHX2 induced by isoprenaline in H9c2 cells. Total RNA was isolated from H9c2 treated with vehicle, TUPS (1 μM), isoprenaline (100 μM) or isoprenaline (100 μM) + TUPS (1 μM). Gene expressions of (A) hypertrophic markers, ANP and BNP, and (B) EPHX2 were determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). *P < 0.05 compared with isoprenaline, #P < 0.05 compared with control.
Figure 10.
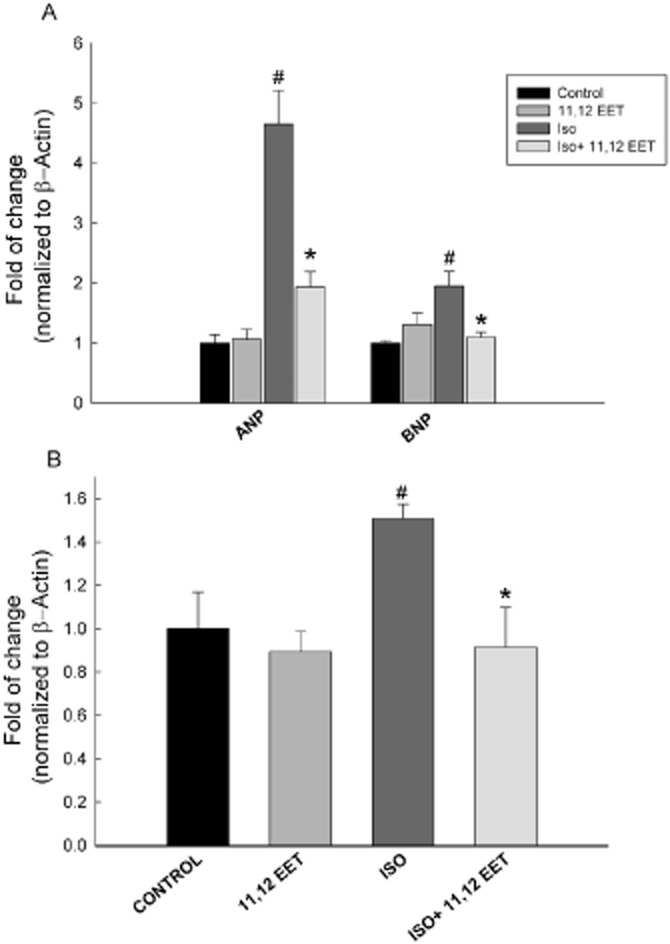
Effect of 11,12 EET on the cardiac hypertrophic markers and EPHX2 induced by isoprenaline in H9c2 cells. Total RNA was isolated from H9c2 treated with vehicle, 11,12 EET (1 μM), isoprenaline (100 μM) or isoprenaline (100 μM) +11,12 EET (1 μM). Gene expressions of (A) hypertrophic markers, ANP and BNP, and (B) EPHX2 were determined by real-time PCR. Duplicate reactions were performed for each experiment, and the results are presented as the means of six independent experiments ± SEM (n = 6). *P < 0.05 compared with isoprenaline, #P < 0.05 compared with control.
Discussion
The cardioprotective effect of sEH inhibitors appears to be due to their ability to inhibit the degradation of EETs and epoxy fatty acids and hence enhance the cardioprotective effect of EETs. In this context, several sEH inhibitors have been synthesized, among which the newly discovered, 1,3-disubstituted ureas, carbamates and amides are considered cutting-edge sEH inhibitors. They are potent and stable transition-state inhibitors of sEH that act through inhibition of the carboxy-terminal domain that possess the epoxide hydrolase activity of the sEH enzyme (Morisseau and Hammock, 2005). Among the different sEH pharmacophores, the urea pharmacophore seems to produce the most potent, competitive and tight-binding inhibitors of sEH (Morisseau et al., 1999). The sEH inhibitor of choice in this study is TUPS, which comprises a highly potent urea pharmacophore (Chiamvimonvat et al., 2007).
In the current study, we investigated the cardioprotective effect of TUPS treatment on isoprenaline-induced cardiac hypertrophy. Our results demonstrated that TUPS significantly decreased the isoprenaline-mediated induction of the hypertrophic markers ANP, BNP and β-MHC and the increase in the heart to body weight ratio. Likewise, TUPS significantly decreased the isoprenaline-mediated induction of ANP and BNP mRNA level in H9c2 cells, which further confirms the direct effect of both isoprenaline and TUPS at the cardiomyocyte level. In addition to sEH inhibition, we examined other means of increasing the level of EETs by direct addition of EET and investigating whether such treatment would replicate the sEH inhibitor-mediated cardioprotective effect. Interestingly, we showed that direct addition of 11,12-EET replicated the cardioprotective effect of sEH inhibitor. In agreement with our results, it has been previously demonstrated that TUPS decreases the left ventricular hypertrophy, heart to body weight ratio and the hypertrophic markers in angiotensin II–induced cardiac hypertrophy and BaP-induced cardiac hypertrophy, reflecting its cardioprotective effect (Ai et al., 2009; Aboutabl et al., 2011). Furthermore, sEH inhibitors were reported to prevent the development of cardiac hypertrophy in a murine model of thoracic aortic constriction (Xu et al., 2006). In addition to its effect on cardiac hypertrophy, TUPS treatment significantly decreased the isoprenaline-mediated increase in fibrotic markers procollagen I, procollagen III and TGF-1, which interestingly seems to be another protective effect of TUPS that extends beyond cardiac hypertrophy. Therefore, our study provides a new model in which TUPS is shown to protect against cardiac hypertrophy and fibrosis.
We have previously demonstrated that the induction of cardiac hypertrophy in isoprenaline-treated rats causes significant changes in the expression of several P450 and sEH genes, which is mostly specific to the heart. The overall balance of these changes has led to a higher production of the cardiotoxic metabolite, 20-HETE and a lower production of cardioprotective metabolites, EETs in the hypertrophied hearts (Zordoky et al., 2008). 20-HETE is known to be involved in many cardiovascular diseases. Interestingly, we have recently demonstrated that the CYP ω-hydroxylase inhibitor, HET0016, partially reverses the BaP-induced cardiac hypertrophy through inhibition of 20-HETE formation, suggesting the involvement of CYP ω-hydroxylases and 20-HETE in the development of cardiac hypertrophy (Aboutabl et al., 2009). On the other hand, EETs play an important role in the inhibition of NF-κB, which is a downstream target of several signalling pathways implicated in cardiac hypertrophy such as angiotensin II, α-adrenoceptor stimulation, PI3K/Akt, ras, P38, MEKK1/4, PKC and gp130 pathways. In addition to NF-κB inhibition, EETs have several other downstream targets that may explain their cardioprotective effect; EETs activate the p42/p44 MAPK pathway, ATP-sensitive potassium channels and PKA-dependent signalling pathway (Seubert et al., 2004; Lu et al., 2006; Batchu et al., 2009; Zordoky and El-Kadi, 2010).
Due to the importance of CYP450 in the pathogenesis of cardiac hypertrophy, in the current study we investigated the effect of sEH inhibition on the expression of different CYP genes involved in isoprenaline-induced cardiac hypertrophy. Our results demonstrated that isoprenaline significantly induces the gene expression of CYP1A1 in the heart and kidney but not in the liver, whereas CYP1B1 gene expression was induced only in the hypertrophied heart. We previously demonstrated the induction of these enzymes in the heart tissue in an isoprenaline-induced cardiac hypertrophy model (Zordoky et al., 2008). In agreement with our results, the expression of CYP1A1 and CYP1B1 were significantly increased in left ventricular tissues of SHRs as compared with normotensive SD rats (Thum and Borlak, 2002). CYP1A1 has been shown to be involved in ω-terminal HETE synthesis, whereas CYP1B1 can metabolize AA to both mid-chain HETEs and EETs (Choudhary et al., 2004). Moreover, it has been recently demonstrated that CYP1B1 contributes to angiotensin II–induced hypertension and cardiac hypertrophy (Jennings et al., 2010). In the present study, treatment with TUPS significantly reduced the isoprenaline-induced CYP ω-hydroxylase enzymes CYP1A1, CYP1B1 in the heart tissue. However, treatment with TUPS alone caused significant induction of CYP1A1 and CYP1B1 in the kidney and further potentiated the isoprenaline induction of CYP1A1. In contrast to this finding, it has been shown that TUPS alone does not induce any significant changes in the gene expression of CYP1A1 or CYP1B1 in the kidney (Aboutabl et al., 2011). This discrepancy is probably due to differences in the route of administration and sample size.
Previously, we found that treatment with isoprenaline did not cause any significant changes in the expression of CYP2B1 or CYP2B2 genes. However, in the present study, isoprenaline significantly induced the gene expression of both CYP2B1 and CYP2B2 in the heart tissue but not in the kidney or liver. This discrepancy might be attributed to differences in the age and weight of animals used in this study as it has been reported that the effect of isoprenaline differs in older rats than in younger ones, so the degree of cardiac hypertrophy achieved in this study is not the same as before (Kunos et al., 1978). Moreover, P450 gene expressions have been found to vary with age at both the transcriptional and post-translational level, which provides another possible reason for this discrepancy (Wauthier et al., 2007). In line with our current findings, it has been demonstrated that the gene expression of CYP2B1 and CYP2B2 are increased in SHR as compared with normotensive SD rats (Thum and Borlak, 2002). Interestingly, treatment with TUPS significantly reduced the isoprenaline-mediated induction of both CYP2B1 and CYP2B1.
With regard to the CYP4 family, our results demonstrated that CYP4A3 gene expression was significantly induced in the heart, liver and kidney of isoprenaline-treated rats as compared with the control. In addition, isoprenaline caused a significant induction of CYP4F4 in the heart alone. Interestingly, treatment with TUPS reduced the increased expression of the CYP4A3 gene in all the tissues examined. Moreover, TUPS significantly reduced the isoprenaline-mediated induction of CYP4F4 gene expression in the heart. The premise of this observation emerges from the fact that the CYP4A and 4F subfamilies are important enzymes involved in AA metabolism to 20-HETE (Wang et al., 1996; 1999; Roman, 2002).
To investigate whether the alterations in CYP450 gene expression are further translated into protein levels, we determined the protein expression of CYP1A1, CYP1B1, CYP2B1/2 and CYP4A. Our results show that the protein levels of CYP1A1, CYP1B1, CYP2B1/2 and CYP4A were significantly increased in the heart of isoprenaline-treated rats; whereas TUPS treatment significantly decreased the isoprenaline-mediated induction of protein expressions of these enzymes.
The sEH enzyme is a crucial determinant of EETs levels because it catalyses the conversion of EETs to DHETs, thus abolishing their biological activity (Imig et al., 2002). Therefore, any change in AA metabolism caused by modification of P450 can be augmented or opposed by an altered level of sEH. Moreover, the gene encoding sEH was found to be a vulnerability factor for heart failure in SHHF rats (Monti et al., 2008). Therefore, it seems essential to investigate the effect of cardiac hypertrophy on EPHX2 expression. Although isoprenaline treatment did not induce any changes in EPHX2 gene expression in heart tissue, as reported previously, it caused a significant induction of EPHX2 in H9c2 cells, which confirms that isoprenaline causes an induction of EPHX2 gene expression in the heart. It is probable that this effect was not apparent in the present study because of the time that the heart was harvested occurred beyond the time of its induction. Furthermore, isoprenaline caused a significant induction of sEH at both the protein and activity level in the heart. Our results accord with those obtained previously where it was shown that sEH activity is higher in SHHF rats (Monti et al., 2008).
To investigate the effect TUPS treatment on P450 gene expression and AA metabolism, we incubated heart microsomes with AA in vitro. We confirmed our previous findings as we showed that the formations of 5,6-, 8,9-, 11,12- and 14,15-EET were significantly decreased in microsomes of hypertrophied hearts in comparison with those from control. The decrease in the formation of EETs was accompanied by a significant increase in 8,9- and 14,15-DHET formation, whereas 5,6- and 11,12-DHET were not significantly altered. The decreased formation of EETs during isoprenaline-induced cardiac hypertrophy may be attributed to a higher activity of sEH. The significantly higher formation of 8,9- and 14,15-DHET is consistent with the higher expression of sEH because 14,15-EET is the optimum substrate for sEH followed by 8,9-EET (Karara et al., 1991). On the other hand, treatment with TUPS abolished the isoprenaline-mediated reduction in 8,9- and 11,12-EETs and further significantly increased the formation of 14,15-EET as compared with the control group. However, TUPS treatment did not affect the isoprenaline reduction of 5,6-EETs. Moreover, TUPS treatment significantly reduced the increase in 8,9- and 14,15-DHET formation caused by isoprenaline and further reduced the formation of 11,12-DHET significantly in comparison with the control microsomes. In the present study we demonstrated that 20-HETE formation is significantly higher in hypertrophied hearts. The increased formation of 20-HETE is suggestive of its role in cardiac hypertrophy. 20-HETE formation is mainly catalysed by P450 ω-hydroxylases (Kroetz and Xu, 2005). The P450 ω-hydroxylases involved in the formation of HETEs are CYP1A, CYP1B1, CYP4A and CYP4F (Wang et al., 1996; Wang et al., 1999; Roman, 2002; Choudhary et al., 2004; Elbekai and El-Kadi, 2006). Therefore, the increase in 20-HETE formation in the present work could be attributed to the increased expression of CYP1A1, CYP1B1, CYP4A3 and CYP4F4. Interestingly, in heart microsomes treated with TUPS, the isoprenaline mediated increase in 20-HETE formation was abolished.
In conclusion, in the present study, we demonstrated for the first time that treatment with the sEH inhibitor TUPS significantly attenuated the isoprenaline-induced cardiac hypertrophy both in vivo and in vitro. In addition, the effects of TUPS on isoprenaline-induced CYP450 enzymes showed some degree of cardioselectivity, which is thought to be secondary to its cardioprotective effect. From accumulating evidence that sEH has a role in the pathogenesis of cardiac hypertrophy, sEH inhibition will provide a new therapeutic tool to protect against cardiac hypertrophy. However, more studies are needed to explore the mechanisms by which inhibition of sEH protects against cardiac hypertrophy.
Acknowledgments
This work was supported by Canadian Institute of Health Research Grant (CIHR) MOP106665 to A.O.S. The work was supported in part by NIEHS Ro1 ES002710 (BDH) and NIH/NIHLBI R01 HL059699 (BDH). BDH is a George and Judy Marcus Senior Fellow of the American Asthma Society. HNA is the recipient of Salman Bin Abdulaziz University Scholarship, Saudi Arabia.
Glossary
- AA
arachidonic acid
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- DHET
dihydroxyeicosatrienoic acid
- EET
epoxyeicosatrienoic acid
- HETE
hydroxyeicosatetraenoic acid
- sEH
soluble epoxide hydrolase
- P450
cytochrome P450
- TUPS
1-(1-methanesulfonyl-piperidin-4-yl)-3-(4-trifluoromethoxy-phenyl)-urea
- β-MHC
β-myosin heavy chain
Conflict of interests
There are no conflicts of interests for HNA, MMYT, GA, BNMZ. BDH is an author of several University of California patents in the area of sEH inhibitors.
References
- Aboutabl ME, Zordoky BN, El-Kadi AO. 3-methylcholanthrene and benzo(a)pyrene modulate cardiac cytochrome P450 gene expression and arachidonic acid metabolism in male Sprague Dawley rats. Br J Pharmacol. 2009;158:1808–1819. doi: 10.1111/j.1476-5381.2009.00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboutabl ME, Zordoky BN, Hammock BD, El-Kadi AO. Inhibition of soluble epoxide hydrolase confers cardioprotection and prevents cardiac cytochrome P450 induction by benzo(a)pyrene. J Cardiovasc Pharmacol. 2011;57:273–281. doi: 10.1097/FJC.0b013e3182055baf. [DOI] [PubMed] [Google Scholar]
- Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:564–569. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar-Mohamed A, El-Sherbeni AA, Kim SH, Althurwi HN, Zordoky BN, El-Kadi AO. Acute arsenic toxicity alters cytochrome P450 and soluble epoxide hydrolase and their associated arachidonic acid metabolism in C57Bl/6 mouse heart. Xenobiotica. 2012;42:1235–1247. doi: 10.3109/00498254.2012.693971. [DOI] [PubMed] [Google Scholar]
- Baldwin SJ, Bramhall JL, Ashby CA, Yue L, Murdock PR, Hood SR, et al. Cytochrome P450 gene induction in rats ex vivo assessed by quantitative real-time reverse transcriptase-polymerase chain reaction (TaqMan) Drug Metab Dispos. 2006;34:1063–1069. doi: 10.1124/dmd.105.008185. [DOI] [PubMed] [Google Scholar]
- Barakat MM, El-Kadi AO, du Souich P. L-NAME prevents in vivo the inactivation but not the down-regulation of hepatic cytochrome P450 caused by an acute inflammatory reaction. Life Sci. 2001;69:1559–1571. doi: 10.1016/s0024-3205(01)01241-3. [DOI] [PubMed] [Google Scholar]
- Batchu SN, Law E, Brocks DR, Falck JR, Seubert JM. Epoxyeicosatrienoic acid prevents postischemic electrocardiogram abnormalities in an isolated heart model. J Mol Cell Cardiol. 2009;46:67–74. doi: 10.1016/j.yjmcc.2008.09.711. [DOI] [PubMed] [Google Scholar]
- Bleicher KB, Pippert TR, Glaab WE, Skopek TR, Sina JF, Umbenhauer DR. Use of real-time gene-specific polymerase chain reaction to measure RNA expression of three family members of rat cytochrome P450 4A. J Biochem Mol Toxicol. 2001;15:133–142. doi: 10.1002/jbt.10. [DOI] [PubMed] [Google Scholar]
- Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno JE, Apablaza F, Ocaranza MP, Jalil JE. [Cardiac hypertrophy: molecular and cellular events] Rev Esp Cardiol. 2006;59:473–486. [PubMed] [Google Scholar]
- Chabova VC, Kramer HJ, Vaneckova I, Vernerova Z, Eis V, Tesar V, et al. Effects of chronic cytochrome P-450 inhibition on the course of hypertension and end-organ damage in Ren-2 transgenic rats. Vascul Pharmacol. 2007;47:145–159. doi: 10.1016/j.vph.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225–237. doi: 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos. 2004;32:840–847. doi: 10.1124/dmd.32.8.840. [DOI] [PubMed] [Google Scholar]
- Delozier TC, Kissling GE, Coulter SJ, Dai D, Foley JF, Bradbury JA, et al. Detection of human CYP2C8, CYP2C9, and CYP2J2 in cardiovascular tissues. Drug Metab Dispos. 2007;35:682–688. doi: 10.1124/dmd.106.012823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbekai RH, El-Kadi AO. Cytochrome P450 enzymes: central players in cardiovascular health and disease. Pharmacol Ther. 2006;112:564–587. doi: 10.1016/j.pharmthera.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gharavi N, El-Kadi AO. tert-Butylhydroquinone is a novel aryl hydrocarbon receptor ligand. Drug Metab Dispos. 2005;33:365–372. doi: 10.1124/dmd.104.002253. [DOI] [PubMed] [Google Scholar]
- Grygielko ET, Martin WM, Tweed C, Thornton P, Harling J, Brooks DP, et al. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J Pharmacol Exp Ther. 2005;313:943–951. doi: 10.1124/jpet.104.082099. [DOI] [PubMed] [Google Scholar]
- Hirasawa F, Kawagoe M, Arany S, Koizumi Y, Ueno Y, Sugiyama T. Styrene monomer primarily induces CYP2B1 mRNA in rat liver. Xenobiotica. 2005;35:1089–1099. doi: 10.1080/00498250500356373. [DOI] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39(2 Pt 2):690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- Jennings BL, Sahan-Firat S, Estes AM, Das K, Farjana N, Fang XR, et al. Cytochrome P450 1B1 contributes to angiotensin II-induced hypertension and associated pathophysiology. Hypertension. 2010;56:667–674. doi: 10.1161/HYPERTENSIONAHA.110.154518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Anakk S, Boehme CL, Strobel HW. Sexual dimorphism and tissue specificity in the expression of CYP4F forms in Sprague Dawley rats. Drug Metab Dispos. 2002;30:1022–1028. doi: 10.1124/dmd.30.9.1022. [DOI] [PubMed] [Google Scholar]
- Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem. 1991;266:7561–7569. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol. 2005;45:413–438. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- Kunos G, Robertson B, Kan WH, Preiksaitis H, Mucci L. Adrenergic reactivity of the myocardium in hypertension. Life Sci. 1978;22:847–854. doi: 10.1016/0024-3205(78)90608-2. [DOI] [PubMed] [Google Scholar]
- Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- Levy D, Salomon M, D'Agostino RB, Belanger AJ, Kannel WB. Prognostic implications of baseline electrocardiographic features and their serial changes in subjects with left ventricular hypertrophy. Circulation. 1994;90:1786–1793. doi: 10.1161/01.cir.90.4.1786. [DOI] [PubMed] [Google Scholar]
- Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lu T, Ye D, Wang X, Seubert JM, Graves JP, Bradbury JA, et al. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J Physiol. 2006;575(Pt 2):627–644. doi: 10.1113/jphysiol.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, et al. Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet. 2008;40:529–537. doi: 10.1038/ng.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Measurement of soluble epoxide hydrolase (sEH) activity. Curr Protocol Toxico. 2007;33:1–18. doi: 10.1002/0471140856.tx0423s33. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, et al. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muiesan ML, Salvetti M, Rizzoni D, Castellano M, Donato F, Agabiti-Rosei E. Association of change in left ventricular mass with prognosis during long-term antihypertensive treatment. J Hypertens. 1995;13:1091–1095. doi: 10.1097/00004872-199510000-00003. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Grall AJ, Holmes BB, Harder DR, Falck JR, Campbell WB. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome P450 metabolites of arachidonic acid. Anal Biochem. 2001;298:327–336. doi: 10.1006/abio.2001.5395. [DOI] [PubMed] [Google Scholar]
- O'Connell JB. The economic burden of heart failure. Clin Cardiol. 2000;23(3 Suppl):III6–II10. doi: 10.1002/clc.4960231503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollin R, Mediero A, Fernandez-Cruz A, Fernandez-Durango R. Downregulation of the atrial natriuretic peptide/natriuretic peptide receptor-C system in the early stages of diabetic retinopathy in the rat. Mol Vis. 2005;11:216–224. [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Sellers KW, Sun C, Diez-Freire C, Waki H, Morisseau C, Falck JR, et al. Novel mechanism of brain soluble epoxide hydrolase-mediated blood pressure regulation in the spontaneously hypertensive rat. FASEB J. 2005;19:626–628. doi: 10.1096/fj.04-3128fje. [DOI] [PubMed] [Google Scholar]
- Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, et al. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- Soppa GK, Lee J, Stagg MA, Felkin LE, Barton PJ, Siedlecka U, et al. Role and possible mechanisms of clenbuterol in enhancing reverse remodelling during mechanical unloading in murine heart failure. Cardiovasc Res. 2008;77:695–706. doi: 10.1093/cvr/cvm106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Cytochrome P450 mono-oxygenase gene expression and protein activity in cultures of adult cardiomyocytes of the rat. Br J Pharmacol. 2000a;130:1745–1752. doi: 10.1038/sj.bjp.0703465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T, Borlak J. Gene expression in distinct regions of the heart. Lancet. 2000b;355:979–983. doi: 10.1016/S0140-6736(00)99016-0. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Testosterone, cytochrome P450, and cardiac hypertrophy. FASEB J. 2002;16:1537–1549. doi: 10.1096/fj.02-0138com. [DOI] [PubMed] [Google Scholar]
- Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, et al. Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci. 2010;40:222–238. doi: 10.1016/j.ejps.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation. 1998;97:48–54. doi: 10.1161/01.cir.97.1.48. [DOI] [PubMed] [Google Scholar]
- Wang MH, Stec DE, Balazy M, Mastyugin V, Yang CS, Roman RJ, et al. Cloning, sequencing, and cDNA-directed expression of the rat renal CYP4A2: arachidonic acid omega-hydroxylation and 11,12-epoxidation by CYP4A2 protein. Arch Biochem Biophys. 1996;336:240–250. doi: 10.1006/abbi.1996.0554. [DOI] [PubMed] [Google Scholar]
- Wang MH, Guan H, Nguyen X, Zand BA, Nasjletti A, Laniado-Schwartzman M. Contribution of cytochrome P-450 4A1 and 4A2 to vascular 20-hydroxyeicosatetraenoic acid synthesis in rat kidneys. Am J Physiol. 1999;276(2 Pt 2):F246–F253. doi: 10.1152/ajprenal.1999.276.2.F246. [DOI] [PubMed] [Google Scholar]
- Wauthier V, Verbeeck RK, Calderon PB. The effect of ageing on cytochrome p450 enzymes: consequences for drug biotransformation in the elderly. Curr Med Chem. 2007;14:745–757. doi: 10.2174/092986707780090981. [DOI] [PubMed] [Google Scholar]
- Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC, DuBois RN, Falck JR, Capdevila JH. Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch Biochem Biophys. 1995;322:76–86. doi: 10.1006/abbi.1995.1438. [DOI] [PubMed] [Google Scholar]
- Zordoky BN, El-Kadi AO. H9c2 cell line is a valuable in vitro model to study the drug metabolizing enzymes in the heart. J Pharmacol Toxicol Methods. 2007;56:317–322. doi: 10.1016/j.vascn.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Zordoky BN, El-Kadi AO. Modulation of cardiac and hepatic cytochrome P450 enzymes during heart failure. Curr Drug Metab. 2008;9:122–128. doi: 10.2174/138920008783571792. [DOI] [PubMed] [Google Scholar]
- Zordoky BN, El-Kadi AO. Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacol Ther. 2010;125:446–463. doi: 10.1016/j.pharmthera.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Zordoky BN, Aboutabl ME, El-Kadi AO. Modulation of cytochrome P450 gene expression and arachidonic acid metabolism during isoproterenol-induced cardiac hypertrophy in rats. Drug Metab Dispos. 2008;36:2277–2286. doi: 10.1124/dmd.108.023077. [DOI] [PubMed] [Google Scholar]
- Zordoky BN, Anwar-Mohamed A, Aboutabl ME, El-Kadi AO. Acute doxorubicin toxicity differentially alters cytochrome P450 expression and arachidonic acid metabolism in rat kidney and liver. Drug Metab Dispos. 2011;39:1440–1450. doi: 10.1124/dmd.111.039123. [DOI] [PubMed] [Google Scholar]