

Abstract
The detection and abundance of Escherichia coli in water is used to monitor and mandate the quality of drinking and recreational water. Distinguishing commensal waterborne E. coli isolates from those that cause diarrhea or extraintestinal disease in humans is important for quantifying human health risk. A DNA microarray was used to evaluate the distribution of virulence genes in 148 E. coli environmental isolates from a watershed in eastern Ontario, Canada, and in eight clinical isolates. Their pathogenic potential was evaluated with Caenorhabditis elegans, and the concordance between the bioassay result and the pathotype deduced by genotyping was explored. Isolates identified as potentially pathogenic on the basis of their complement of virulence genes were significantly more likely to be pathogenic to C. elegans than those determined to be potentially nonpathogenic. A number of isolates that were identified as nonpathogenic on the basis of genotyping were pathogenic in the infection assay, suggesting that genotyping did not capture all potentially pathogenic types. The detection of the adhesin-encoding genes sfaD, focA, and focG, which encode adhesins; of iroN2, which encodes a siderophore receptor; of pic, which encodes an autotransporter protein; and of b1432, which encodes a putative transposase, was significantly associated with pathogenicity in the infection assay. Overall, E. coli isolates predicted to be pathogenic on the basis of genotyping were indeed so in the C. elegans infection assay. Furthermore, the detection of C. elegans-infective environmental isolates predicted to be nonpathogenic on the basis of genotyping suggests that there are hitherto-unrecognized virulence factors or combinations thereof that are important in the establishment of infection.
INTRODUCTION
The deterioration of source water quality by fecal contamination considerably increases the cost of producing potable water, as well as the risk to public health if water treatment is lacking or is insufficient (1). Monitoring water quality and tracking sources of contamination are essential to address this public health issue. The Gram-negative enteric bacterium Escherichia coli is ubiquitous in warm-blooded animals and is short lived once shed into the environment (2). These characteristics, coupled with the development of efficacious semiselective differential growth media for the enumeration of E. coli cells, have resulted in the widespread adoption of E. coli abundance as the metric of choice for evaluating and mandating the quality of water used for drinking, crop irrigation, or recreation (1, 3). The vast majority of E. coli strains are benign, but there are several virotypes (the virotype is the potential for pathogenicity as determined on the basis of genotyping results) of E. coli able to cause infections of the gastrointestinal tract, central nervous system, urinary tract, or bloodstream (4, 5). Thus, the ability to detect and quantify E. coli virotypes and distinguish these from commensal strains is a precursor to evaluating, with confidence, the human health risk from E. coli contamination of water. In this context, specific virotypes can tentatively be ascribed to environmental isolates on the basis of virulence gene profiling, the assumption being that specific complements of virulence genes are associated with the ability to cause specific diseases (6–8). The virulence gene profiling approach has been used to characterize the seasonal and spatial distribution of waterborne E. coli that are potentially pathogenic to humans or livestock and to identify associations in virotype distribution within catchments with variation in land use, climate, and the distribution of potential sources of fecal contamination (9–15). There are, however, practical problems with the tractability of the virulence gene profiling approach. The cost of obtaining data using array or PCR methods can be prohibitive, particularly when undertaking large-scale environmental surveys. Furthermore, the technology required to do so will not be available to many water quality or public health laboratories. In addition, PCR and array methods do not assess for mutations in virulence or regulatory genes that may inactivate the expression of factors required to confer a pathogenic phenotype. Finally, with a growing literature on the list of potential virulence factors and the combinations of genes required for pathogenicity, keeping microarrays up-to-date may become costly over time.
Within this context, we have sought here to explore the utility of the Caenorhabditis elegans infection assay as a high-throughput method to distinguish pathogenic from nonpathogenic environmental isolates of E. coli. The nematode C. elegans is susceptible to a wide range of medically relevant bacteria, including Pseudomonas aeruginosa (16, 17), Burkholderia (18–21), Enterococcus (22–24), Legionella pneumophila (25, 26), and E. coli (27–30). A strong correlation has been demonstrated between nematode-pathogenic extraintestinal E. coli strains and strains that are capable of killing mice (29). Furthermore, enteroaggregative E. coli (EAEC), enteropathogenic E. coli (EPEC), adherent-invasive E. coli (AIEC), and uropathogenic E. coli (UPEC) have also been shown to be pathogenic to C. elegans (29, 31–33). Pathogenic bacteria cause disease in C. elegans directly through infection or indirectly by production of toxins (34). For example, secreted toxins from enteropathogenic E. coli (27), P. aeruginosa (17), and B. cepacia (21) were found to kill C. elegans. N-Acylhomoserine lactone (AHL)-mediated quorum sensing in Yersinia pseudotuberculosis (35) and B. cepacia-complex strains (36) have also been shown to have a role in C. elegans killing.
The antibiotic resistance of environmental E. coli is also screened to assess the potential risk of antibiotic resistance being transmitted to potentially pathogenic bacteria. A survey of clinical E. coli isolates from urinary tract infections suggested that isolates expressing extended-spectrum beta-lactamases (CTX-Ms) had a low virulence potential with C. elegans (30). Overall, it is reasonable to assume that the nematode-killing mechanisms of the bacteria can be associated with the potential to cause disease in humans and animals but that relationships between virulence potential and the complement of virulence genes are highly complex.
Our objectives in the present study were to (i) screen a large number of environmental E. coli isolates obtained from within one watershed in Eastern Ontario, Canada, for pathogenic potential using C. elegans, (ii) subject the isolates to virulence gene and antibiotic resistance gene profiling, as well as phenotypic elucidation of antibiotic resistance, and (iii) evaluate associations between the pathogenic potential established with the nematode bioassay and the gene-profiling characteristics of the isolates.
MATERIALS AND METHODS
Bacterial isolates and nematode strains.
Surface water (n = 143) and fecal (n = 5; 3 dog, 1 dairy, and 1 poultry) samples were obtained in 2004 from the South Nation River drainage basin (an area of about 3,900 km2) located east of Ottawa (45°25′15″N, 75°41′24″W) in the province of Ontario, Canada. Descriptions of the experimental area, water and fecal sampling procedures, and laboratory methods for E. coli isolation, confirmation, and storage are found in Lyautey et al. (37, 38), Ruecker et al. (39), and Wilkes et al. (40). The following clinical E. coli strains were obtained from Josée Harel (University of Montreal): enteropathogenic strain E2348/69, isolated during an outbreak of infantile diarrhea in 1969 (41); enterohemorrhagic strain EDL933, isolated from meat during an EHEC outbreak in the United States in 1982 (42); uropathogenic strain CFT073, isolated from the urine and blood of a patient with acute pyelonephritis (43); enterotoxigenic strain H10407, a clinical isolate from a patient with cholera-like symptoms (44, 45); enteroaggregative 17-2, from a patient with diarrhea (46–48); sepsis- and meningitis-associated strain 536, from a patient with pyelonephritis (49); and enteroinvasive (EIEC) strain H84 (50). Strain 25922 was purchased from the ATCC and is a biosafety level 1 clinical isolate used as a negative control in antibiotic resistance assays (51).
The temperature-sensitive C. elegans strain DH26 [fer-15(b26)II] was routinely grown in the presence of E. coli OP50, a uracil auxotroph, using standard practices (52). OP50 and DH26 were purchased from the Caenorhabditis Genetics Center (CGC) from the University of Minnesota. C. elegans strains CF512 [fer-15(b26)II fem-1(hc17)IV] and BA15 [fer-15(hc15)II] were from Ann Karen Brassinga from the University of Manitoba. Burkholderia cenocepacia strain H111 was obtained from Daniel Aubert from the University of Western Ontario. E. coli strains RO8, B44, F107, PD20, AMR-472, P16M, and JG280 were obtained from Patrick Boerlin from the University of Guelph and used as positive controls for virulence genes detected by PCR (see below), along with strains E2348/69, EDL933, H10407, 17-2, 536, and H84 described above.
Genotyping environmental isolates.
All environmental and clinical isolates were evaluated for the presence of 300 virulence and antibiotic resistance genes by microarray hybridization using the methods of Hamelin et al. (53). The presence of 17 virulence-associated genes, faeG, fanA, fedA, aidAI, paa, sepA, pic, bfpB, invE, elt, escV, aggR, stx2, stx1, estIb, estIa, and ast, was determined through multiplex PCR using primers and conditions previously described (8, 54, 55). Putative intestinal and extraintestinal virotypes were identified based on the criteria elaborated by Johnson et al. (56) and Hamelin et al. (53). Briefly, isolates were classified as extraintestinal E. coli when isolates encoded two or more of the following genes: pap, sfa/foc, afa/dra, iutA, and kpsMT-II. Specific types of extraintestinal isolates were identified, including uropathogenic E. coli (UPEC; containing P fimbria-encoding genes, hlyA, S fimbria-encoding genes, chuA, fepC, cnf1, irp1, irp2, fyuA, iroN, and usp), neonatal meningitis-causing E. coli (NMEC; containing ibeA, neuA, and neuC), and septicemic E. coli [SEPEC; containing cdtB-3, f165(1)A, gafD, and F17A]. Intestinal virotypes were also identified, including enterotoxigenic E. coli (ETEC; containing heat-stable and heat-labile toxin-encoding genes and F4 and F18 fimbria-encoding genes), atypical enteropathogenic E. coli (AEPEC; containing espA, espB, tir, and eae), and enteroaggregative E. coli (EAEC; containing capU, shf, virK, and aggregative adherence fimbria-encoding genes).
The Bingen phylogenetic grouping of each isolate was determined by multiplex PCR screening for the presence of chuA, yjaA, tspE4.C2, and svg as previously described (57, 58), using cell lysates prepared by proteinase K digestion. The chuA positive control, ATCC 35381, was purchased from ATCC. Strain J96, from Patrick Boerlin, was used as the positive control for yjaA, chuA, and tspE4.C2. The criteria used to differentiate the five phylogenetic lineages were as follows: D, possesses chuA and lacks yjaA; B2, possesses chuA, yjaA, and svg; B2-1, possesses chuA and yjaA and lacks svg; B1, lacks chuA and possesses tspE4.C2; and A, lacks chuA and tspE4.C2 (57).
Antibiotic resistance profiles of environmental and clinical isolates.
The antibiotic resistance profile for each isolate was determined phenotypically. Briefly, overnight cultures were prepared in Mueller-Hinton broth in 96-well microplates. Amounts of 8 μl of the overnight cultures were transferred to 96-well plates containing 200 μl of 0.02% Tween 20 to stabilize the suspensions. Using a 96-pin floating-pin replicator (V&P Scientific, Inc.), 5-μl amounts of culture were placed on 245-mm2 square plates containing BD Bacto Mueller-Hinton agar supplemented with various antibiotics. The antibiotics and concentrations (in μg/ml) tested included amikacin (32, 64, and 128), ampicillin (16, 32, and 64) ceftiofur (4, 8, and 16), cephalothin (16, 32, and 64), ciprofloxacin (2, 4, and 8), chloramphenicol (16, 32, and 64), gentamicin (8, 16, and 32), kanamycin (32, 64, and 128), nalidixic acid (16, 32, and 64), streptomycin (32, 64, and 128), tetracycline (8, 16, and 32), trimethoprim (8, 16, and 32), and trimethoprim-sulfamethoxazole (STX) (2:38, 4:76, and 8:152). The concentrations were based on the breakpoints identified by the Canadian Integrated Program for Antimicrobial Resistance Surveillance (CIPARS) report (59).
The viability of each isolate was also determined on antibiotic-free plates. After an overnight incubation at 37°C, isolates were considered to be antibiotic resistant if they grew at the midpoint antibiotic concentration.
Determining the growth kinetics of isolates.
Overnight cultures of each E. coli isolate were diluted in 96-well microplates to an optical density at 600 nm (OD600) of 0.4 in nematode growth medium (NGMII; 0.3% [wt/vol] NaCl, 0.35% [wt/vol] Bacto peptone, 0.5% [wt/vol] cholesterol, 1 mM CaCl2, 1 mM MgSO4, and 25 mM phosphate buffer, pH 6) containing 1.22 mM tryptophan. The cultures were incubated at 26°C for 12 h, and the OD600 of each culture was monitored every 15 min using a Biotek Powerwave XS microplate spectrophotometer. The average generation time for each culture was calculated from three independent experiments.
Infection assays.
Plate-to-liquid infection assays of C. elegans with E. coli were performed as previously described (24), with some modifications. Briefly, NGMII plates (0.3% NaCl, 0.35% Bacto peptone, 5 μg/ml cholesterol, 1 mM CaCl2, 1 mM MgSO4, 25 mM phosphate buffer, 1.7% Bacto agar) containing 1.22 mM tryptophan (27) were inoculated with 20 μl of overnight bacterial cultures diluted to an OD600 of 0.8 in LB containing 1.22 mM tryptophan and incubated at 37°C overnight. Approximately 90 NGMII broth-washed, hypochlorite-synchronized young-adult nematodes were added to each lawn, and the nematodes were allowed to feed overnight at 26°C. The next day, the nematodes and bacteria were washed off NGMII agar plates using NGMII broth containing tryptophan. After the nematodes were allowed to settle to the bottom of the tube, the bacterial supernatant was diluted with NGMII broth containing tryptophan to an OD600 of between 0.8 and 1. Approximately 5 to 10 nematodes in a volume of 100 μl were transferred into each well of a 96-well plate with six wells per tested bacterial strain. Nematodes were scored as live or dead every other day. As DH26 C. elegans are unable to reproduce at 26°C, the total number of worms in the wells did not increase over time. All of the environmental and the clinical isolates (H84, H10407, 17-2, EDL933, 536, and CFT073) were tested in three independent assays, while OP50, ATCC 25922, and E2348/69 were included as controls in all assays. To test for potential differences in nematode response to the clinical isolates, the infections with the clinical isolates and OP50 were also done using C. elegans strains CF512 and BA15.
To test the pathogenicity of nonviable bacteria on C. elegans, bacterial lawns were exposed to UV light (310 nm) for 1 h prior to the addition of nematodes. The death of each bacterial strain following UV treatment was confirmed by determining viability on LB agar plates.
Nematode food preference assays.
The food preference of C. elegans for the clinical strains was determined as described previously (60). Briefly, bacterial isolates were grown overnight in LB broth containing 1.22 mM tryptophan at 37°C. The bacterial cultures were diluted to an OD600 of approximately 0.8. Assays were done on 100- by 15-mm plates containing NGMII agar containing 1.22 mM tryptophan. Thirty microliters of E. coli OP50 culture was added 1.5 cm away from the periphery of the plate, and 30 μl of the test bacterial culture was added on the other side at a similar distance away from the periphery. The plates were then incubated overnight at 37°C. The next day, approximately 50 prewashed young-adult nematodes were added to the center of the plate and incubated at 26°C. The percentage of nematodes present on each bacterial lawn was determined at 2, 5, and 23 h.
In addition, the selective grazing preference of the nematodes for all 148 environmental isolates and the clinical isolates on NGMII agar with 1.22 mM tryptophan was determined as previously described (61). Briefly, using a 96-pin floating pin replicator (V&P Scientific, Inc.), overnight cultures of the environmental isolates in LB broth containing 1.22 mM tryptophan were added to three 150- by 15-mm NGMII agar plates each. After allowing the lawns to grow overnight at 37°C, 5 to 10 prewashed young-adult nematodes were added near each of the lawns, and the plates were incubated at 26°C for 5 days. The size of the lawns was monitored daily and used as a measure of nematode feeding. The nematode-pathogenic B. cenocepacia H111 was used in both assays as a food source avoided by the worms (18–21).
Statistical analyses.
The survival of the nematodes was analyzed using the Cox proportional hazards (CPH) model in R 2.11.1 with the Survival package (62). The Efron method was used to handle censored survival data and robust jackknife estimates for estimation of 95% confidence intervals. The hazard ratio and median survival for each bacterial strain were determined in comparison to those of E. coli OP50. Due to different rates of survival of the nematodes in the presence of OP50, the data sets were stratified for the individual tests (using the function “strata”) in order to allow for different baseline hazard functions.
The multiplicative effect of individual genes and phenotypes was tested by including the binary covariate (presence/absence of gene or phenotype) as a linear predictor in a CPH analysis, separately for the data set of the environmental isolates and the clinical strains and individually for each gene. The data sets were clustered for strains encoding the genes or phenotypes of interest. Bonferroni correction for the P values obtained for each gene and phenotype within each of the two data sets was applied to correct for multiple testing. Only genes or phenotypes that were present (or absent) in at least 10 of the environmental isolates were considered in the covariate analysis of that data set (i.e., 45 genes in total). Interactions between various genes or phenotypes were not considered in the analysis, simply because most interactions could not be assessed as no strain had the respective genetic profile pattern (i.e., the data set was not fully crossed with regard to the presence and absence of each gene).
For CPH analysis, the generation time of each isolate was binarized such that 0 indicates strains that grew faster than the median generation time of all strains, including OP50, whereas 1 indicates strains growing more slowly than the median generation time. Virotypes, including UPECs, other extraintestinal pathogens, and AEPECs, were grouped into the category “potentially pathogenic” prior to CPH analysis. Isolates that could not be classified as intestinal or extraintestinal pathogens were grouped as “potentially nonpathogenic” for analysis.
RESULTS
Abundance of pathogenic environmental isolates.
The survival of C. elegans DH26 in the presence of 148 environmental E. coli isolates and 8 human clinical isolates was compared to its survival in the presence of E. coli OP50 (52, 63). The mean survival time of nematodes in the presence of OP50 was 8.3 days, consistent with what others have found (64). In contrast, the mean survival times of nematodes in the presence of the environmental and clinical isolates ranged from 3 to 14 days. A strain was considered pathogenic if its hazard ratio was significantly (Bonferroni-corrected P value of <0.05) higher than that of E. coli OP50, i.e., >1. As referenced to the survival of nematodes in the presence of OP50, 29% of the environmental isolates (n = 43) significantly decreased the survival of C. elegans, with the hazard ratios determined for these pathogenic isolates ranging from 1.3 to 6.9 (Fig. 1A). Of the eight clinical isolates, strains 17-2, CFT073, ATCC 25922, and EDL933 were not pathogenic, whereas E2348/69, 536, H10407, and H84 had hazard ratios ranging from 1.7 to 3.9 and were therefore considered to be pathogenic. Similar results were seen for the clinical isolates using C. elegans strains CF512 and BA15. Thirty-five (24%) of the environmental isolates had hazard ratios significantly smaller than 1, indicating that the survival of C. elegans was better in their presence than with E. coli OP50 (Fig. 1B).
Fig 1.

Hazard ratios of E. coli isolates in the C. elegans assay. A bacterial isolate was considered pathogenic when its hazard ratio was statistically higher than 1. Stars represent clinical strains and OP50, whereas diamonds represents environmental isolates. Red symbols indicate isolates characterized as potentially pathogenic on the basis of virotyping. Filled symbols indicate hazard ratios that are significantly different (Bonferroni-corrected P value of <0.05) than 1 (vertical dashed line). The error bars represent the 95% confidence limits. Cox proportional hazards ratios greater (A) or less (B) than 1 with respect to that of OP50 are plotted.
Associations between pathogenicity and virotype and other attributes.
Statistical associations between pathogenicity and the presence of various genes and virotypes were explored using CPH analysis. Likewise, the association of pathogenicity with the resistance to specific antibiotics and the generation time for each bacterium in NGMII broth were determined. Of the 317 genes and probes screened by PCR or microarray, 143 genes were found in at least one environmental isolate and 7 genes were found in all isolates. In order to be statistically robust, the analysis was performed only with virulence genes or antimicrobial resistance phenotypes present (or absent) in at least 10 of the 148 environmental isolates. Based on this criterion, 45 virulence genes and six antibiotic resistance phenotypes were sufficiently frequent that associations with pathogenicity could be explored. As some genes or gene probes (the latter denoted by subscripts) [ompT, ompT2, iucD, iutA2, iutA(UPEC), fyuA, irp1, irp2, chuA, fepC, focA, and focG] were always present together in the environmental isolates, the impact of these genes or phenotypes could only be determined as a group, not individually.
Within the collection of environmental isolates, 6 genes of the 45 genes analyzed were found to be very highly associated with phenotypic pathogenicity (Fig. 2; see also Table S1 in the supplemental material). The adhesin-encoding genes sfaD, focA, and focG; iroN2, a gene probe for iroN, which encodes a siderophore receptor; pic, which encodes an autotransporter protein; and b1432, which encodes a putative transposase, were all detected at high frequency in pathogenically potent isolates. On the other hand, flmA54, encoding a flagellin subunit, was associated with decreased pathogenicity in environmental isolates (Fig. 2). Alternatively, the remaining 39 genes analyzed were not found to be associated with phenotypic pathogenicity. Those genes encoded capsular and somatic antigens (kpsM-II), adhesins [lpfA, lpfA(O157), lpfA(O113), and lpfA(EHEC)], colicins and microcins (cba, cia, cma, and mchB), toxins (astA and astA2, a variant of astA with an 8-amino-acid deletion), iron acquisition or transport systems [chuA, fepC, iroN, irp1, irp2, fyuA, iucD, iutA2, and iutA(UPEC)], hemolysins or agglutinins (hylA, hra1, and tsh), new or putative EC virulence genes (b1121, ECs1282, rtx, tspE4.C2, usp, and yjaA), and genes encoding proteins or enzymes with various functions (agn43, ccdB, fliC, ibeA, iss, malX, ompT, ompT2, tia, traT, and sepA).
Fig 2.
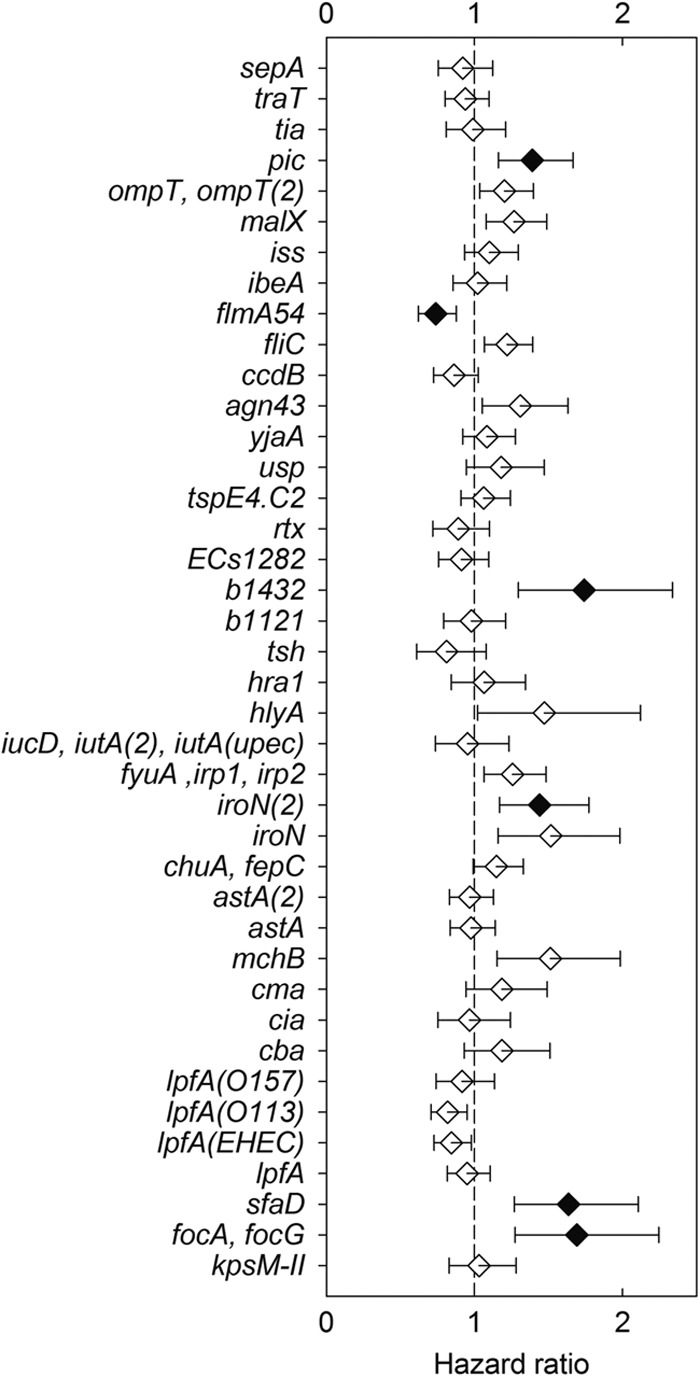
Relationships between the carriage of specific virulence genes and the pathogenic potential of E. coli environmental isolates in the C. elegans assay. CPH analysis of genes present in environmental isolates. Filled symbols indicate hazard ratios that are significantly different (Bonferroni-corrected P value of ≤0.05) than 1 (vertical dashed line). The error bars indicate the 95% confidence limits.
Pathogenicity was not associated with antibiotic resistance phenotypes, generation time, or specific Bingen phylotypes (Fig. 3 and Table 1). However, isolates ascribed to various virotypes (including other extraintestinal pathogenic E. coli [ExPEC], AEPEC, and UPEC) on the basis of genotyping and thus considered to be potentially pathogenic were significantly more likely to be pathogenic in the C. elegans infection assay than those determined to be potentially nonpathogenic on the basis of virotyping (Fig. 3 and Table 2). Of the 16 environmental isolates characterized as potentially pathogenic based on virotyping, 11 (69%) were found to be pathogenic to the nematodes. In contrast, 32 (24%) of the 132 isolates not ascribed to a specific virotype were found to be pathogenic.
Fig 3.

Relationships between characteristics of the environmental E. coli isolates, such as Bingen phylotype, virotype, generation time, and antibiotic resistance phenotypes, and pathogenicity in the C. elegans assay. Bingen phylotypes include A, D, B1, and B2. Potentially pathogenic isolates were identified via virotyping. Filled symbols indicate hazard ratios that are significantly different (Bonferroni-corrected P value of ≤0.05) than 1 (vertical dashed line). The error bars indicate the 95% confidence limits.
Table 1.
Virotypes and phylotypes of E. coli environmental isolates and clinical strains used in C. elegans bioassays
Table 2.
Genotypes of the virotyped environmental isolates and their associated pathogenicity
| Virotype (no. of isolates with virotype [% pathogenic]) and genotypea | Isolate(s) with indicated genotype |
|||||
|---|---|---|---|---|---|---|
| Total no. of isolates | % pathogenicb | Nonpathogenic (n = 105) |
Pathogenic (n = 43) |
|||
| No. of isolates | Name(s) | No. of isolates | Name(s) | |||
| AEPEC (1 [100]) | ||||||
| eae genes, espA tir chuA fepC fyuA irp1 irp2 | 1 | 100 | 0 | 1 | NRC225 | |
| Other ExPEC (6 [50]) | ||||||
| kpsM-II, sfa/foc genes, S fimbria genes, chuA fepC iroN usp ibeA | 2 | 50 | 1 | NRC59 | 1 | NRC34 |
| kpsM-II, chuA fepC fyuA irp1 iutA irp2 usp ibeA | 2 | 0 | 2 | NRC10, NRC181 | 0 | |
| kpsM-II, sfa/foc genes, S fimbria genes, cnf1 chuA fepC iroN fyuA irp1 irp2 hlyA usp | 2 | 100 | 0 | 2 | NRC1, NRC423 | |
| UPEC (9 [78]) | ||||||
| kpsM-II, sfa/foc genes, S fimbria genes, pap genes, P fimbria genes, cnf1 chuA fepC iroN fyuA irp1 irp2 hlyA usp | 2 | 50 | 1 | NRC145 | 1 | NRC410 |
| kpsM-II, aggregative adherence fimbria genes, pap genes, P fimbria genes, heat-stable toxin genes, fyuA irp1 iutA irp2 hlyA | 1 | 0 | 1 | NRC175 | 0 | |
| kpsM-II, pap genes, chuA fepC | 1 | 100 | 0 | 1 | NRC35 | |
| kpsM-II, sfa/foc genes, S fimbria genes, pap genes, P fimbria genes, cnf1 chuA fepC iroN fyuA irp1 irp2 hlyA usp ibeA | 1 | 100 | 0 | 1 | NRC517 | |
| f165(1)A, sfa/foc genes, S fimbria genes, pap genes, P fimbria genes, eae genes, heat-stable toxin genes, cnf1 chuA fepC iroN fyuA irp1 irp2 hlyA | 1 | 100 | 0 | 1 | NRC149 | |
| kpsM-II, afa/dra genes, sfa/foc genes, S fimbria genes, pap genes, P fimbria genes, cnf1 chuA fepC iroN fyuA irp1 irp2 hlyA usp | 1 | 100 | 0 | 1 | NRC2 | |
| pap genes, heat-stable toxin genes, chuA fepC iroN iutA | 1 | 100 | 0 | 1 | NRC364 | |
| kpsM-II, sfa/foc genes, S fimbria genes, pap genes, P fimbria genes, chuA fepC iroN fyuA irp1 irp2 usp | 1 | 100 | 0 | 1 | NRC162 | |
| Potentially nonpathogenic (132 [24]) | ||||||
| Heat-stable toxin genes, heat-labile toxin genes, chuA fepC iroN fyuA irp1 irp2 ibeA | 2 | 0 | 2 | NRC160, NRC74 | 0 | |
| shf virK capU | 2 | 0 | 2 | NRC237, NRC363 | 0 | |
| chuA fepC iroN fyuA irp1 irp2 usp ibeA | 2 | 0 | 2 | NRC172, NRC178 | 0 | |
| pap genes, chuA fepC fyuA irp1 irp2 | 2 | 0 | 2 | NRC150, NRC189 | 0 | |
| chuA fepC iroN fyuA irp1 irp2 | 1 | 0 | 1 | NRC154 | 0 | |
| chuA fepC usp ibeA | 1 | 0 | 1 | NRC56 | 0 | |
| Heat-stable toxin genes, heat-labile toxin genes, chuA fepC | 2 | 50 | 1 | NRC201 | 1 | NRC203 |
| Heat-stable toxin genes, heat-labile toxin genes, chuA fepC usp | 1 | 0 | 1 | NRC239 | 0 | |
| cdtB-3 chuA fepC fyuA irp1 iutA irp2 hlyA | 2 | 50 | 1 | NRC210 | 1 | NRC214 |
| Heat-stable toxin genes | 3 | 0 | 3 | NRC230, NRC232, NRC241 | 0 | |
| fyuA irp1 irp2 | 3 | 0 | 3 | NRC143, NRC168, NRC219 | 0 | |
| kpsM-II, heat-stable toxin genes, chuA fepC fyuA irp1 irp2 usp ibeA | 1 | 0 | 1 | NRC151 | 0 | |
| sfa/foc genes, S fimbria genes, heat-stable toxin genes, chuA fepC iroN fyuA irp1 irp2 | 7 | 57 | 3 | NRC157, NRC20, NRC29 | 4 | NRC275, NRC281, NRC353, NRC354 |
| Heat-stable toxin genes, fyuA irp1 irp2 | 2 | 0 | 2 | NRC231, NRC28 | 0 | |
| chuA fepC | 29 | 34 | 19 | NRC161, NRC165, NRC169, NRC182, NRC190, NRC193, NRC196, NRC206, NRC209, NRC216, NRC242, NRC31, NRC40, NRC44, NRC54, NRC58, NRC62, NRC68, NRC73 | 10 | NRC14, NRC153, NRC186, NRC187, NRC191, NRC197, NRC221, NRC7, NRC8, NRC82 |
| chuA fepC usp | 2 | 0 | 2 | NRC11, NRC148 | 0 | |
| Heat-stable toxin genes, shf virK capU | 2 | 0 | 2 | NRC158, NRC184 | 0 | |
| kpsM-II, chuA fepC | 4 | 0 | 4 | NRC218, NRC240, NRC243, NRC46 | 0 | |
| gafD F17A | 3 | 0 | 3 | NRC228, NRC229, NRC65 | 0 | |
| None of the genes | 21 | 19 | 17 | NRC13, NRC144 NRC16, NRC173, NRC174, NRC177, NRC198, NRC22, NRC226, NRC227, NRC238, NRC24, NRC244, NRC245, NRC246, NRC38, NRC78 | 4 | NRC199, NRC212, NRC83 |
| eae genes, heat-stable toxin genes, cdtB-3 chuA fepC fyuA irp1 irp2 usp | 2 | 0 | 2 | NRC23, NRC236 | 0 | |
| kpsM-II, chuA fepC fyuA irp1 irp2 | 4 | 0 | 4 | NRC15, NRC207, NRC220, NRC27 | 0 | |
| gafD F17A chuA fepC | 1 | 0 | 1 | NRC64 | 0 | |
| kpsM-II, chuA fepC fyuA irp1 irp2 usp ibeA | 4 | 0 | 4 | NRC155, NRC166, NRC49, NRC60 | 0 | |
| chuA fepC iutA | 1 | 0 | 1 | NRC52 | 0 | |
| kpsM-II, heat-stable toxin genes, heat-labile toxin genes, chuA fepC | 1 | 0 | 1 | NRC72 | 0 | |
| Heat-stable toxin genes, iroN | 1 | 0 | 1 | NRC9 | 0 | |
| kpsM-II, heat-stable toxin genes, chuA fepC | 2 | 0 | 2 | NRC167, NRC215 | 0 | |
| kpsM-II, chuA fepC iroN usp ibeA | 3 | 67 | 1 | NRC42 | 2 | NRC176, NRC37 |
| Heat-stable toxin genes, chuA fepC | 12 | 33 | 8 | NRC156, NRC163, NRC234, NRC25, NRC30, NRC66, NRC67, NRC88 | 4 | NRC194, NRC202, NRC41, NRC77 |
| kpsM-II, chuA fepC iroN | 1 | 0 | 1 | NRC63 | 0 | |
| gafD cdtB-3 iutA hlyA | 2 | 0 | 2 | NRC235, NRC70 | 0 | |
| gafD F17A, heat-stable toxin genes, chuA fepC | 1 | 100 | 0 | 1 | NRC69 | |
| chuA fepC iroN | 1 | 100 | 0 | 1 | NRC50 | |
| chuA fepC fyuA irp1 irp2 usp ibeA | 1 | 100 | 0 | 1 | NRC205 | |
| Heat-stable toxin genes, chuA fepC iroN fyuA irp1 irp2 usp ibeA | 1 | 100 | 0 | 1 | NRC179 | |
| chuA fepC iroN fyuA irp1 irp2 ibeA | 1 | 100 | 0 | 1 | NRC164 | |
| iutA | 1 | 100 | 0 | 1 | NRC43 | |
Putative intestinal and extraintestinal virotypes (UPEC, AEPEC, and ExPEC), as well as potentially nonpathogenic isolates, were assigned based on the criteria of Johnson et al. (56) and Hamelin et al. (53). Genotypes include only genes used to determine putative intestinal and extraintestinal virotypes. S fimbria genes, heat-stable toxin genes, heat-labile toxin genes, P fimbria genes, and aggregative adherence fimbria genes encompass several genes encoding components of these structures.
Percentage of isolates carrying this genotype that were found to be pathogenic in the C. elegans infection assay.
Pathogenicity requires viability, and nonpathogenic types are palatable.
Viable clinical E. coli isolates that had a high hazard ratio were benign in the assay when killed by UV irradiation prior to exposure to C. elegans, indicating that viability was required for pathogenicity (Fig. 4). The relative palatability of all isolates to C. elegans was determined and referenced to that of the food source E. coli OP50 (60, 61). The nematode-pathogenic B. cenocepacia H111 was used as a negative control to which food avoidance could be expected (18–21). There was no relationship between an isolate's hazard ratio and its preferability as a food source (Fig. 5). There was a clear preference for OP50 when the choice was between the organism and no food. Furthermore, C. elegans fed upon all environmental isolates equally after 5 days but avoided the H111 lawn (data not shown). Taken together, these results indicate that viable bacteria were required to kill C. elegans and that palatability did not play a role in determining why some environmental isolates were nonpathogenic in the bioassay.
Fig 4.
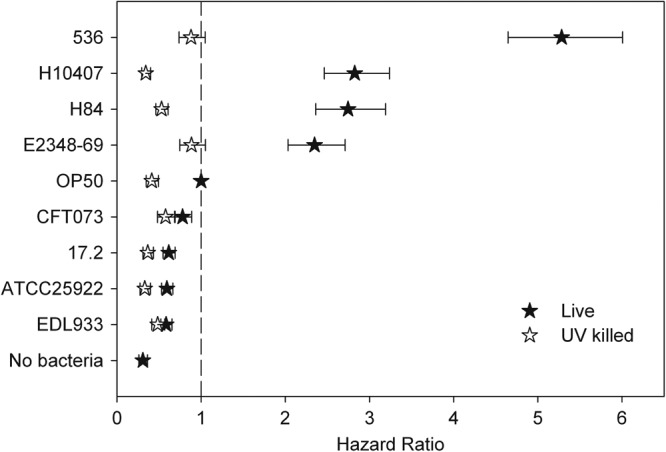
Pathogenicity of E. coli is destroyed by UV irradiation. The hazard ratio for each live and UV-killed strain was calculated with the CPH model relative to the CPH of live OP50. A bacterial isolate was considered pathogenic when its hazard ratio was statistically higher than 1 (vertical dashed line). The error bars represent the 95% confidence limits.
Fig 5.
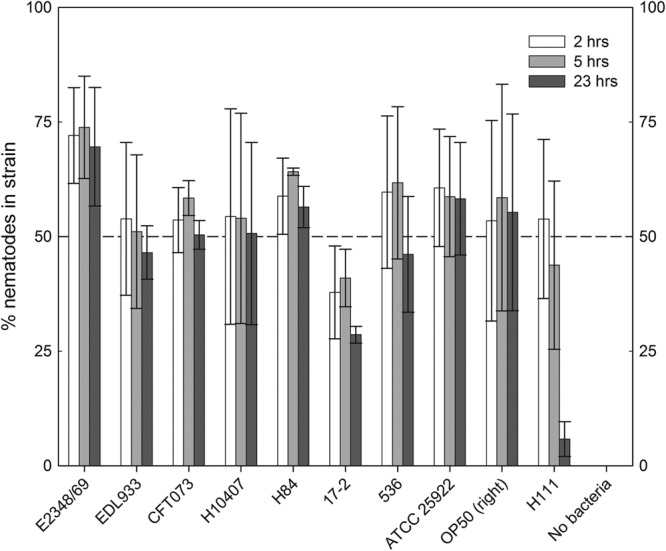
Pathogenicity of E. coli strains is not dependent on their palatability to C. elegans. Nematodes were given the choice between an OP50 lawn and clinical E. coli strains. Burkholderia cenocepacia H111 was used as a repellant. The number of nematodes present in each lawn was counted at 2, 5, and 23 h following the addition of the nematodes to the agar plate. The error bars represent the standard deviations of the results from triplicate plates.
DISCUSSION
Ascribing pathogenic potential to environmental isolates on the basis of virulence gene complement is an important facet of understanding risk from waterborne E. coli (12, 53). However, conclusions regarding the potential risk to humans or animals from isolates with defined genotypes are best confirmed by validating presumptive pathogenicity using an infection model. In the present study, we investigated the pathogenicity to C. elegans of environmental E. coli isolates and medically relevant pathogens to explore possible associations between the presence of specific virulence genes or antibiotic resistance attributes and pathogenic potential. The C. elegans model has several advantages that make it tractable and powerful for this purpose; the killing assay is relatively facile, rapid, and inexpensive, and the knowledge base regarding C. elegans-bacterial pathogen interactions is large (29, 64–68).
In the present study, 29% of the 148 environmental isolates were pathogenic to the nematode. To investigate which specific genes and phenotypes were associated with increased or decreased pathogenicity, we analyzed their contributions to pathogenicity with CPH analysis. No associations between antibiotic resistance phenotype and pathogenicity were found; environmental isolates that were resistant to ampicillin, cephalothin, streptomycin, tetracycline, trimethoprim, and STX were generally not pathogenic. Associations between pathogenicity and the carriage of specific antimicrobial genes could not be evaluated systematically as too few isolates possessed these genes. However, seven of the nine environmental isolates that possessed these genes were nonpathogenic. These results, along with similar ones obtained by Lavigne et al. (30), support the conclusion that specific virulence genes, not simply antibiotic resistance in itself, are important in pathogenicity.
A number of the virulence genes evaluated in the present study were noteworthy in that they were detected at much higher frequency in pathogenically potent isolates (Fig. 2), namely, the adhesin-encoding genes sfaD, focA, and focG, iroN2 that encodes a siderophore receptor, and pic that encodes an autotransporter protein. These genes are typically associated with strains that cause extraintestinal infections. The genes sfaD, focA, and focG are all required for synthesis of the F1C fimbriae and are upregulated during urinary tract infection by the prototypic uropathogenic E. coli strain CFT073 (13). Likewise, pic is associated both with strains that cause pyelonephritis and with fecal strains and is expressed during urinary tract infection (UTI) (69). The gene product, Pic, is a serine protease autotransporter that has mucinase activity and is associated with UTI isolates (69, 70). The siderophore receptor IroN is detected at high frequency in ExPEC strains and is an important virulence factor in UTI and neonatal meningitis (71, 72). Similar results for the adhesins and siderophore receptor were seen by Diard et al. (29), but this is the first time that an autotransporter and transposase have been shown to be associated with C. elegans pathogenicity. Alternatively, the gene encoding flagellin (flmA54) that is associated with repressed expression of the flagellin gene fliC was found to be inversely associated with pathogenicity (73). It is important to note that the genes themselves may not be directly involved in pathogenicity but instead could represent the effect of a combination of genes linked with the specific genes on pathogenicity islands that were not screened for in the present microarray or PCR analysis. Furthermore, regulatory genes required for the expression of virulence genes might be absent or nonfunctional, creating discordance between the virotype and pathogenicity in the C. elegans assay.
There was a strong relationship between the ability to kill C. elegans in the bioassay and an isolate's potential for pathogenicity as determined on the basis of genotyping results, i.e., virotype (Fig. 3). Microarray analysis of the environmental isolates identified 15 putative extraintestinal virotypes and one putative enteric virotype. C. elegans infection assays revealed that 11 (69%; the enteric AEPEC isolate and 10 of the extraintestinal isolates) of the 16 potentially pathogenic isolates were pathogenic to nematodes. These results, as well as the pathogenicity of the extraintestinal and enteric clinical isolates, indicate that the C. elegans model can be used to detect both extraintestinal and intestinal virotypes. The nematode bioassay is therefore clearly a useful tool for identifying environmental isolates that would be expected to pose some public health risk. Nonetheless, a few of the clinical isolates used as positive controls failed to kill the nematodes (Table 2). It is not expected that the nematode model will provide a 100% correlation due to limits of the model (67, 74). For example, others have shown that CFT073 is only a weak worm killer of C. elegans (29), while other clinical isolates have been found to have a level of killing of C. elegans that is not significant versus that of the negative control (75). It has been shown that the conditions of infection can play a role in the ability of human pathogens to cause disease in C. elegans (22, 76); as such, it is quite possible that the clinical isolates that were nonpathogenic to C. elegans in our assay did not express the appropriate virulence factors. Consequently, they may have been rendered nonpathogenic in C. elegans under the infection conditions described. Therefore, the observed differences in pathogenicity for some of the clinical isolates in this study versus their pathogenicity in other studies may be due to differences in the infection conditions. This observation raises interesting questions concerning how the expression of various genes differs depending on the infection conditions and how this can affect the pathogenicity of the strains in C. elegans.
Importantly, 32 (24%) of the 132 isolates that were classified as potentially nonpathogenic based on virotype killed the nematodes (Table 2). This included the environmental water isolate NRC69, which was even more pathogenic to C. elegans than the clinical UPEC isolate 536 (Fig. 1A). This may indicate that, when utilizing virotyping alone based on currently known virulence genes, pathogenic E. coli strains can be missed, i.e., erroneously not be identified as a health concern. It is reassuring, however, that all 35 isolates that were found to significantly enhance nematode survival (Fig. 1B; see also Table S1 in the supplemental material) were ascribed as potentially nonpathogenic (26%). Together, the results demonstrate that the relationships between virulence gene complement and the ability to kill C. elegans are highly complex. Fully understanding these relationships in the context of the data in the present study will require whole-genome sequencing of the E. coli isolates described here.
As C. elegans worms have been shown to be capable of recognizing and avoiding potentially pathogenic bacteria (77), it was important to determine that pathogenic strains would not be missed due to the nematodes simply not feeding on the bacteria. No specific strain avoidance was observed in the food preference assays with both the clinical isolates and environmental isolates (Fig. 5). Although it is expected that the liquid aspect of the assay prevents the nematodes from fully avoiding feeding on the bacteria, the lack of avoidance on plates also indicates that the nematodes feed on a similar dose of bacteria when initially on the agar plates. As such, the difference in pathogenic potential between isolates was dependent on the presence of specific virulence genes carried by these isolates and not simply on palatability.
The South Nation River watershed sampled in the present study is a mixed-activity watershed with livestock, human, and wildlife fecal inputs and, consequently, a high diversity of E. coli strains (38, 40). The results in the present study are in agreement with those of Hamelin et al. (12), who found a high abundance of ExPEC strains as opposed to intestinal strains in a survey of riverine, estuarine, and offshore lake water (12). Here, we demonstrate that a high percentage of these E. coli strains are pathogenic in a nematode killing assay and, therefore, represent a potential human health problem.
In conclusion, the results with the C. elegans model suggest that microarray studies and surveys of the presence of single virulence genes in the environment are not sufficient to determine the potential for organisms in this environment to be a health risk to the public. Instead, the combination of genes present is more important for pathogenicity. As more virotypes are characterized, the rules for identifying specific virotypes based on gene combinations will be enhanced to decrease the number of false negatives via microarray analysis alone. Furthermore, we identified specific genes that were associated with an increased risk for pathogenicity and demonstrated that virotype determination through microarray analysis was efficient in predicting pathogenic risk. Overall, the C. elegans infection assay is a reasonably tractable assay for helping distinguish environmental isolates of E. coli that have pathogenic potential.
Supplementary Material
ACKNOWLEDGMENTS
We thank Belinda Yung, Danielle DeBlock, Chelsea Hicks, Magda Konopka, Justine Denhomme, David Martin, Estefania Gonzalez, Andrew Scott, Yun Zhang, and Lyne Sabourin for technical assistance. Thanks to Stephanie Dixon, Daniel Aubert, and Ann Brassinga for useful discussions and to Erik Vasaasen for writing the Visual Basic script to organize the survival data for analysis.
A. Merkx-Jacques was supported by an NSERC Visiting Fellowship in Government Laboratories funded through the AAFC SAGES program.
Footnotes
Published ahead of print 1 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03501-12.
REFERENCES
- 1.WHO 2009. Global health risks: mortality and burden of disease attributable to selected major risks. WHO Press, Geneva, Switzerland [Google Scholar]
- 2.Yost CK, Diarra MS, Topp E. 2011. Animals and humans as sources of fecal indicator bacteria. In Sadowsky MJ, Whitman RL. (ed), The fecal bacteria. ASM Press, Washington, DC [Google Scholar]
- 3.National Research Council of the National Academies 2004. Indicators for waterborne pathogens. The National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 4.Croxen MA, Finlay BB. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8:26–38 [DOI] [PubMed] [Google Scholar]
- 5.Kaper JB, Nataro JP, Mobley HLT. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 6.Bruant G, Maynard C, Bekal S, Gaucher I, Masson L, Brousseau R, Harel J. 2006. Development and validation of an oligonucleotide microarray for detection of multiple virulence and antimicrobial resistance genes in Escherichia coli. Appl. Environ. Microbiol. 72:3780–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coombes BK, Wickham ME, Mascarenhas M, Gruenheid S, Finlay BB, Karmali MA. 2008. Molecular analysis as an aid to assess the public health risk of non-O157 Shiga toxin-producing Escherichia coli strains. Appl. Environ. Microbiol. 74:2153–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller D, Greune L, Heusipp G, Karch H, Fruth A, Tschape H, Schmidt MA. 2007. Identification of unconventional intestinal pathogenic Escherichia coli isolates expressing intermediate virulence factor profiles by using a novel single-step multiplex PCR. Appl. Environ. Microbiol. 73:3380–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chern EC, Tsai YL, Olson BH. 2004. Occurrence of genes associated with enterotoxigenic and enterohemorrhagic Escherichia coli in agricultural waste lagoons. Appl. Environ. Microbiol. 70:356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duris JW, Haack SK, Fogarty LR. 2009. Gene and antigen markers of Shiga-toxin producing E. coli from Michigan and Indiana river water: occurrence and relation to recreational water quality criteria. J. Environ. Qual. 38:1878–1886 [DOI] [PubMed] [Google Scholar]
- 11.Fremaux B, Prigent-Combaret C, Vernozy-Rozand C. 2008. Long-term survival of Shiga toxin-producing Escherichia coli in cattle effluents and environment: an updated review. Vet. Microbiol. 132:1–18 [DOI] [PubMed] [Google Scholar]
- 12.Hamelin K, Bruant G, El-Shaarawi A, Hill S, Edge TA, Fairbrother J, Harel J, Maynard C, Masson L, Brousseau R. 2007. Occurrence of virulence and antimicrobial resistance genes in Escherichia coli isolates from different aquatic ecosystems within the St. Clair River and Detroit River areas. Appl. Environ. Microbiol. 73:477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haugen BJ, Pellett S, Redford P, Hamilton HL, Roesch PL, Welch RA. 2007. In vivo gene expression analysis identifies genes required for enhanced colonization of the mouse urinary tract by uropathogenic Escherichia coli strain CFT073 dsdA. Infect. Immun. 75:278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabate M, Prats G, Moreno E, Balleste E, Blanch AR, Andreu A. 2008. Virulence and antimicrobial resistance profiles among Escherichia coli strains isolated from human and animal wastewater. Res. Microbiol. 159:288–293 [DOI] [PubMed] [Google Scholar]
- 15.Shelton DR, Karns JS, Coppock C, Patel J, Sharma M, Pachepsky YA. 2011. Relationship between eae and stx virulence genes and Escherichia coli in an agricultural watershed: implications for irrigation water standards and leafy green commodities. J. Food Prot. 74:18–23 [DOI] [PubMed] [Google Scholar]
- 16.Evans EA, Kawli T, Tan MW. 2008. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 4:e1000175 doi:10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan MW, Mahajan-Miklos S, Ausubel FM. 1999. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 96:715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardona ST, Wopperer J, Eberl L, Valvano MA. 2005. Diverse pathogenicity of Burkholderia cepacia complex strains in the Caenorhabditis elegans host model. FEMS Microbiol. Lett. 250:97–104 [DOI] [PubMed] [Google Scholar]
- 19.Cooper VS, Carlson WA, Lipuma JJ. 2009. Susceptibility of Caenorhabditis elegans to Burkholderia infection depends on prior diet and secreted bacterial attractants. PLoS One 4:e7961 doi:10.1371/journal.pone.0007961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huber B, Feldmann F, Kothe M, Vandamme P, Wopperer J, Riedel K, Eberl L. 2004. Identification of a novel virulence factor in Burkholderia cenocepacia H111 required for efficient slow killing of Caenorhabditis elegans. Infect. Immun. 72:7220–7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kothe M, Antl M, Huber B, Stoecker K, Ebrecht D, Steinmetz I, Eberl L. 2003. Killing of Caenorhabditis elegans by Burkholderia cepacia is controlled by the cep quorum-sensing system. Cell. Microbiol. 5:343–351 [DOI] [PubMed] [Google Scholar]
- 22.Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, Calderwood SB, Ausubel FM. 2001. A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. U. S. A. 98:10892–10897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavigne JP, Nicolas-Chanoine MH, Bourg G, Moreau J, Sotto A. 2008. Virulent synergistic effect between Enterococcus faecalis and Escherichia coli assayed by using the Caenorhabditis elegans model. PLoS One 3:e3370 doi:10.1371/journal.pone.0003370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. 2006. Identification of novel antimicrobials using a live-animal infection model. Proc. Natl. Acad. Sci. U. S. A. 103:10414–10419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brassinga AK, Kinchen JM, Cupp ME, Day SR, Hoffman PS, Sifri CD. 2010. Caenorhabditis is a metazoan host for Legionella. Cell. Microbiol. 12:343–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komura T, Yasui C, Miyamoto H, Nishikawa Y. 2010. Caenorhabditis elegans as an alternative model host for Legionella pneumophila, and protective effects of Bifidobacterium infantis. Appl. Environ. Microbiol. 76:4105–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anyanful A, Dolan-Livengood JM, Lewis T, Sheth S, Dezalia MN, Sherman MA, Kalman LV, Benian GM, Kalman D. 2005. Paralysis and killing of Caenorhabditis elegans by enteropathogenic Escherichia coli requires the bacterial tryptophanase gene. Mol. Microbiol. 57:988–1007 [DOI] [PubMed] [Google Scholar]
- 28.Anyanful A, Easley KA, Benian GM, Kalman D. 2009. Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe 5:450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diard M, Baeriswyl S, Clermont O, Gouriou S, Picard B, Taddei F, Denamur E, Matic I. 2007. Caenorhabditis elegans as a simple model to study phenotypic and genetic virulence determinants of extraintestinal pathogenic Escherichia coli. Microbes Infect. 9:214–223 [DOI] [PubMed] [Google Scholar]
- 30.Lavigne JP, Blanc-Potard AB, Bourg G, Moreau J, Chanal C, Bouziges N, O'Callaghan D, Sotto A. 2006. Virulence genotype and nematode-killing properties of extra-intestinal Escherichia coli producing CTX-M beta-lactamases. Clin. Microbiol. Infect. 12:1199–1206 [DOI] [PubMed] [Google Scholar]
- 31.Bhatt S, Anyanful A, Kalman D. 2011. CsrA and TnaB coregulate tryptophanase activity to promote exotoxin-induced killing of Caenorhabditis elegans by enteropathogenic Escherichia coli. J. Bacteriol. 193:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang J, Mattei LM, VanArendonk LG, Meneely PM, Okeke IN. 2010. A pathoadaptive deletion in an enteroaggregative Escherichia coli outbreak strain enhances virulence in a Caenorhabditis elegans model. Infect. Immun. 78:4068–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simonsen KT, Nielsen G, Bjerrum JV, Kruse T, Kallipolitis BH, Moller-Jensen J. 2011. A role for the RNA chaperone Hfq in controlling adherent-invasive Escherichia coli colonization and virulence. PLoS One 6:e16387 doi:10.1371/journal.pone.0016387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darby C. 2005. Interactions with microbial pathogens. WormBook 6 September 2005:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atkinson S, Goldstone RJ, Joshua GW, Chang CY, Patrick HL, Camara M, Wren BW, Williams P. 2011. Biofilm development on Caenorhabditis elegans by Yersinia is facilitated by quorum sensing-dependent repression of type III secretion. PLoS Pathog. 7:e1001250 doi:10.1371/journal.ppat.1001250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wopperer J, Cardona ST, Huber B, Jacobi CA, Valvano MA, Eberl L. 2006. A quorum-quenching approach to investigate the conservation of quorum-sensing-regulated functions within the Burkholderia cepacia complex. Appl. Environ. Microbiol. 72:1579–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyautey E, Lapen DR, Wilkes G, McCleary K, Pagotto F, Tyler K, Hartmann A, Piveteau P, Rieu A, Robertson WJ, Medeiros DT, Edge TA, Gannon V, Topp E. 2007. Distribution and characteristics of Listeria monocytogenes isolates from surface waters of the South Nation River watershed, Ontario, Canada. Appl. Environ. Microbiol. 73:5401–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyautey E, Lu Z, Lapen DR, Wilkes G, Scott A, Berkers T, Edge TA, Topp E. 2010. Distribution and diversity of Escherichia coli populations in the South Nation River Drainage Basin, Eastern Ontario, Canada. Appl. Environ. Microbiol. 76:1486–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruecker NJ, Braithwaite SL, Topp E, Edge T, Lapen DR, Wilkes G, Robertson W, Medeiros D, Sensen CW, Neumann NF. 2007. Tracking host sources of Cryptosporidium spp. in raw water for improved health risk assessment. Appl. Environ. Microbiol. 73:3945–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkes G, Edge T, Gannon V, Jokinen C, Lyautey E, Medeiros D, Neumann N, Ruecker N, Topp E, Lapen DR. 2009. Seasonal relationships among indicator bacteria, pathogenic bacteria, Cryptosporidium oocysts, Giardia cysts, and hydrological indices for surface waters within an agricultural landscape. Water Res. 43:2209–2223 [DOI] [PubMed] [Google Scholar]
- 41.Iguchi A, Thomson NR, Ogura Y, Saunders D, Ooka T, Henderson IR, Harris D, Asadulghani M, Kurokawa K, Dean P, Kenny B, Quail MA, Thurston S, Dougan G, Hayashi T, Parkhill J, Frankel G. 2009. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191:347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308:681–685 [DOI] [PubMed] [Google Scholar]
- 43.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect. Immun. 58:1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crossman LC, Chaudhuri RR, Beatson SA, Wells TJ, Desvaux M, Cunningham AF, Petty NK, Mahon V, Brinkley C, Hobman JL, Savarino SJ, Turner SM, Pallen MJ, Penn CW, Parkhill J, Turner AK, Johnson TJ, Thomson NR, Smith SG, Henderson IR. 2010. A commensal gone bad: complete genome sequence of the prototypical enterotoxigenic Escherichia coli strain H10407. J. Bacteriol. 192:5822–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans DJ, Jr, Evans DG. 1973. Three characteristics associated with enterotoxigenic Escherichia coli isolated from man. Infect. Immun. 8:322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine MM, Prado V, Robins-Browne R, Lior H, Kaper JB, Moseley SL, Gicquelais K, Nataro JP, Vial P, Tall B. 1988. Use of DNA probes and HEp-2 cell adherence assay to detect diarrheagenic Escherichia coli. J. Infect. Dis. 158:224–228 [DOI] [PubMed] [Google Scholar]
- 47.Nataro JP, Kaper JB, Robins-Browne R, Prado V, Vial P, Levine MM. 1987. Patterns of adherence of diarrheagenic Escherichia coli to HEp-2 cells. Pediatr. Infect. Dis. J. 6:829–831 [DOI] [PubMed] [Google Scholar]
- 48.Vial PA, Robins-Browne R, Lior H, Prado V, Kaper JB, Nataro JP, Maneval D, Elsayed A, Levine MM. 1988. Characterization of enteroadherent-aggregative Escherichia coli, a putative agent of diarrheal disease. J. Infect. Dis. 158:70–79 [DOI] [PubMed] [Google Scholar]
- 49.Berger H, Hacker J, Juarez A, Hughes C, Goebel W. 1982. Cloning of the chromosomal determinants encoding hemolysin production and mannose-resistant hemagglutination in Escherichia coli. J. Bacteriol. 152:1241–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bekal S, Brousseau R, Masson L, Prefontaine G, Fairbrother J, Harel J. 2003. Rapid identification of Escherichia coli pathotypes by virulence gene detection with DNA microarrays. J. Clin. Microbiol. 41:2113–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyle VJ, Fancher ME, Ross RW., Jr 1973. Rapid, modified Kirby-Bauer susceptibility test with single, high-concentration antimicrobial disks. Antimicrob. Agents Chemother. 3:418–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stiernagle T. 2006. Maintenance of C. elegans. WormBook 11 February 2006:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamelin K, Bruant G, El-Shaarawi A, Hill S, Edge TA, Bekal S, Fairbrother JM, Harel J, Maynard C, Masson L, Brousseau R. 2006. A virulence and antimicrobial resistance DNA microarray detects a high frequency of virulence genes in Escherichia coli isolates from Great Lakes recreational waters. Appl. Environ. Microbiol. 72:4200–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boerlin P, Travis R, Gyles CL, Reid-Smith R, Janecko N, Lim H, Nicholson V, McEwen SA, Friendship R, Archambault M. 2005. Antimicrobial resistance and virulence genes of Escherichia coli isolates from swine in Ontario. Appl. Environ. Microbiol. 71:6753–6761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duriez P, Zhang Y, Lu Z, Scott A, Topp E. 2008. Loss of virulence genes in Escherichia coli populations during manure storage on a commercial swine farm. Appl. Environ. Microbiol. 74:3935–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson JR, Murray AC, Gajewski A, Sullivan M, Snippes P, Kuskowski MA, Smith KE. 2003. Isolation and molecular characterization of nalidixic acid-resistant extraintestinal pathogenic Escherichia coli from retail chicken products. Antimicrob. Agents Chemother. 47:2161–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bidet P, Metais A, Mahjoub-Messai F, Durand L, Dehem M, Aujard Y, Bingen E, Nassif X, Bonacorsi S. 2007. Detection and identification by PCR of a highly virulent phylogenetic subgroup among extraintestinal pathogenic Escherichia coli B2 strains. Appl. Environ. Microbiol. 73:2373–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clermont O, Bonacorsi S, Bingen E. 2000. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 66:4555–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Public Health Agency of Canada 2006. Canadian Integrated Program for Antimicrobial Resistance Surveillance (CIPARS), 2004. Public Health Agency, Guelph, Ontario, Canada [Google Scholar]
- 60.Abada EA, Sung H, Dwivedi M, Park BJ, Lee SK, Ahnn J. 2009. C. elegans behavior of preference choice on bacterial food. Mol. Cells 28:209–213 [DOI] [PubMed] [Google Scholar]
- 61.Ballestriero F, Thomas T, Burke C, Egan S, Kjelleberg S. 2010. Identification of compounds with bioactivity against the nematode Caenorhabditis elegans by a screen based on the functional genomics of the marine bacterium Pseudoalteromonas tunicata D2. Appl. Environ. Microbiol. 76:5710–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Therneau T. 2010. Survival: survival analysis, including penalised likelihood, R version 2.36-2. http://cran.r-project.org/web/packages/survival/index.html
- 63.Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powell JR, Ausubel FM. 2008. Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol. Biol. 415:403–427 [DOI] [PubMed] [Google Scholar]
- 65.Alegado RA, Campbell MC, Chen WC, Slutz SS, Tan MW. 2003. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell. Microbiol. 5:435–444 [DOI] [PubMed] [Google Scholar]
- 66.Gravato-Nobre MJ, Hodgkin J. 2005. Caenorhabditis elegans as a model for innate immunity to pathogens. Cell. Microbiol. 7:741–751 [DOI] [PubMed] [Google Scholar]
- 67.Marsh EK, May RC. 2012. Caenorhabditis elegans, a model organism for investigating immunity. Appl. Environ. Microbiol. 78:2075–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Callaghan D, Vergunst A. 2010. Non-mammalian animal models to study infectious disease: worms or fly fishing? Curr. Opin. Microbiol. 13:79–85 [DOI] [PubMed] [Google Scholar]
- 69.Heimer SR, Rasko DA, Lockatell CV, Johnson DE, Mobley HLT. 2004. Autotransporter genes pic and tsh are associated with Escherichia coli strains that cause acute pyelonephritis and are expressed during urinary tract infection. Infect. Immun. 72:593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Restieri C, Garriss G, Locas M- C, Dozois CM. 2007. Autotransporter-encoding sequences are phylogenetically distributed among Escherichia coli clinical isolates and reference strains. Appl. Environ. Microbiol. 73:1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldmann F, Sorsa LJ, Hildinger K, Schubert S. 2007. The salmochelin siderophore receptor IroN contributes to invasion of urothelial cells by extraintestinal pathogenic Escherichia coli in vitro. Infect. Immun. 75:3183–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nègre VL, Bonacorsi S, Schubert S, Bidet P, Nassif X, Bingen E. 2004. The siderophore receptor IroN, but not the high-pathogenicity island or the hemin receptor ChuA, contributes to the bacteremic step of Escherichia coli neonatal meningitis. Infect. Immun. 72:1216–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ratiner YA. 1998. New flagellin-specifying genes in some Escherichia coli strains. J. Bacteriol. 180:979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pukkila-Worley R, Ausubel FM. 2012. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 24:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mellies JL, Barron AM, Haack KR, Korson AS, Oldridge DA. 2006. The global regulator Ler is necessary for enteropathogenic Escherichia coli colonization of Caenorhabditis elegans. Infect. Immun. 74:64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Couillault C, Ewbank JJ. 2002. Diverse bacteria are pathogens of Caenorhabditis elegans. Infect. Immun. 70:4705–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schulenburg H, Ewbank JJ. 2007. The genetics of pathogen avoidance in Caenorhabditis elegans. Mol. Microbiol. 66:563–570 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.