

Abstract
Enzymatic transamination of amino acids yields α-keto acids and is the initial step for the production of volatile compounds that contribute to the sensory perception of fermented foods such as salami. Lactobacillus sakei is one of the lactic acid bacterial strains commonly used in starter cultures. Although the genome sequence of L. sakei 23K lacks genes encoding typical branched-chain amino acid transaminases, transamination activity and the formation of amino acid-derived volatile metabolites could be demonstrated. A protein purified from L. sakei is held responsible for the transamination activity. By heterologous expression of the corresponding gene in Escherichia coli, we were able to characterize the transamination side activity of an enzyme annotated as a putative acylphosphatase (AcP). A transamination side activity of hen egg white lysozyme (HEWL) was also discovered. Both enzymes showed substrate specificity toward branched-chain and aromatic amino acids. AcP also accepted l-methionine. Activity was optimal at neutral pH for both enzymes, whereas AcP showed a significantly higher temperature optimum (55°C) than that of HEWL (37°C). Kinetic parameters revealed high affinity toward l-leucine for AcP (Km = 1.85 mM) and toward l-isoleucine for HEWL (Km = 3.79 mM). AcP seems to play a major role in the metabolism of amino acids in L. sakei.
INTRODUCTION
The flavor of dry fermented sausages derives from the ingredients (meat, spices, and smoke) and the chemical changes occurring during the fermentation and drying process. Flavor formation in dry fermented sausage occurs by meat enzymes and bacterial fermentation, which are responsible for the metabolism of fats, carbohydrates, and proteins (1, 2). The autochthonous microbiota of fermented meat products is dominated by coagulase-negative staphylococci and lactic acid bacteria (LAB), mainly Lactobacillus sakei, Lactobacillus curvatus, and Lactobacillus plantarum (3, 4). L. sakei, formerly known as L. sake, is one of the dominating LAB in dry spontaneously fermented sausages (3, 5). Therefore, it is commonly used in starter cultures for the production of dry fermented sausages (6).
Free amino acids, incorporated into LAB cells by amino acid transporters or released intracellularly by peptidase activity, are converted, among other metabolic pathways, to various volatile compounds by transamination and decarboxylation. Enzymatic transamination leading to the formation of the corresponding α-keto acids is the first step of this intracellular amino acid catabolism to volatile metabolites in LAB (Fig. 1) (2, 7). The transformation of branched-chain, aromatic, and sulfur-containing amino acids by these reactions yields volatile compounds, which contribute to the sensory perception of fermented food (8, 9). It has been calculated that 11.8% of the volatile compounds in salami probably originate from amino acid catabolism (10).
Fig 1.
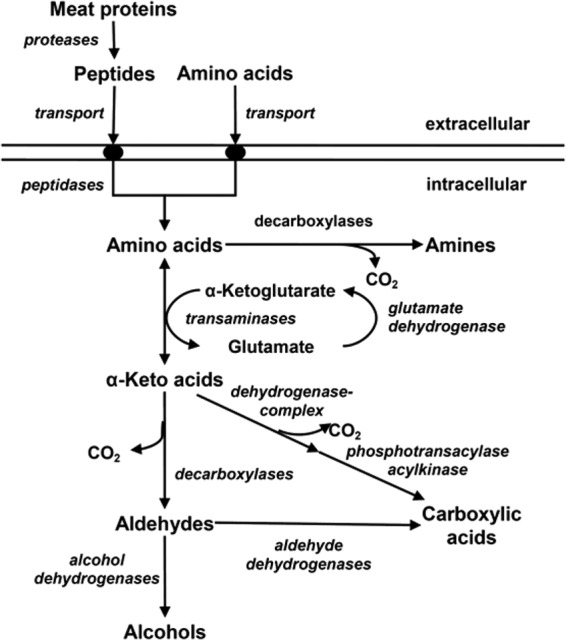
Possible intracellular pathway of amino acids in lactic acid bacteria. (Adapted from reference 9.)
Analysis of the genome sequence of L. sakei 23K delivered insights into the strain's potential concerning aroma formation from amino acid substrates. Genes encoding typical branched-chain amino acid (BCAA) transaminases, specific for leucine, isoleucine, and valine, could not be found in the genome sequence of L. sakei 23K or in more than 50 L. sakei strains (11–13). However, BCAA transamination activity and the formation of amino acid-derived volatile metabolites could be demonstrated for L. sakei 23K and other L. sakei strains (14, 15). Yet compared with other starter cultures used for the production of dry fermented sausages, the metabolic impact of L. sakei strains on the formation of volatiles has been considered low (14, 15). Two putative aspartate transaminase genes, namely, arcT and aspD, were annotated in the L. sakei 23K genome (11), and the consensus pattern for the class I transaminase pyridoxal-phosphate attachment site K could be detected in both sequences. However, the heterologously expressed and purified enzymes did not show transamination activity toward amino acids (16). In the same study, the heterologous expression of a BCAA transaminase (IlvE) in L. sakei led to significantly increased transaminase activity. These results showed that the enzyme catalyzing the transamination of BCAAs in wild-type L. sakei 23K, a reaction which is crucial for the formation of potentially aroma-relevant volatile metabolites, is still unknown.
In this study, we attempted to purify a protein with transamination activity from L. sakei TMW 1.1322, an isogenic clone of the sequenced strain L. sakei 23K (11), which would explain the production of amino acid-derived volatiles by this strain. The result is important for understanding the intracellular pathways of amino acids in L. sakei and will affect system biology studies.
In our first attempts to purify the enzyme from an L. sakei crude cell extract (CFE), cells were treated with hen egg white lysozyme (HEWL) before lysis by ultrasonication. In sequencing the protein from active fractions after different purification steps, the amino acid sequence of HEWL was obtained. This suggested a transamination side activity of lysozyme, which was investigated and characterized as well.
MATERIALS AND METHODS
Bacteria, media, and growth conditions.
The strains used in this study were L. sakei TMW 1.1322 and Escherichia coli TOP10. They were obtained from the laboratory collection of Technische Universität München, Lehrstuhl für Technische Mikrobiologie, Freising, Germany. L. sakei TMW 1.1322 is an isogenic clone of the sequenced strain L. sakei 23K (11). In this communication, the TMW clone number is used for correctness, as strains may change upon prolonged lab propagation. LAB were grown at 30°C in modified De Man-Rogosa-Sharpe (mMRS) medium (17, 18). E. coli TOP10 was cultivated in Luria-Bertani (LB) medium (19) at 37°C. Strains harboring plasmids with antibiotic resistance were propagated in media containing 100 μg/ml ampicillin. Cells were harvested by centrifugation (3,000 × g for 15 min).
Chemicals.
Peptone from casein, yeast extract, meat extract, ammonium chloride, cysteine-HCl, Tween 80, d-maltose, d-glucose, K2HPO4 · 3H2O, KH2PO4, Na2HPO4, and NaH2PO4 were obtained from Carl Roth GmbH & Co. KG (Karlsruhe, Germany). HEWL, pyridoxal-5-phosphate (PLP), α-ketoglutaric acid, 3-methyl-2-oxobutanoic acid (α-ketoisovaleric acid [KIV]), 4-methyl-2-oxovaleric acid (α-ketoisocaproic acid [KIC]), 3-methyl-2-oxovaleric acid (α-keto-3-methylvaleric acid [KMV]), 2-oxo-3-phenylpropanoic acid (phenylpyruvic acid [PPA]), 4-methylthio-2-oxobutanoic acid [α-keto-γ-(methylthio)butyric acid (KMBA)], and vitamins and metals for mMRS medium were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO).
Preparation of crude CFE for enzyme purification.
Harvested cells were washed twice with 50 mM potassium phosphate buffer (KPi), pH 7.4. Crude extract was obtained by ultrasonication (10 times, 10%, 30 s) (Sonoplus UW 2070/MS 73; Bandelin Electronic GmbH & Co. KG, Berlin, Germany) of resuspended cells in the same buffer on ice. Cell debris was removed by centrifugation (12,000 × g for 20 min at 4°C). In the beginning of this study, cells were treated with lysozyme (HEWL at 10 mg/ml, 30°C, 1 h) and washed once with KPi (50 mM, pH 7.4) before lysis by ultrasonication. After demonstration of the promiscuous transaminase activity of HEWL, treatment with HEWL was omitted in the following purification experiments, and cells were lysed only by ultrasonication. The protein concentrations of CFEs and purified enzyme (acylphosphatase [AcP]) were determined according to the method of Bradford (20), with bovine serum albumin as the standard.
Enzyme purification protocol.
All purification steps were performed at 4°C. Crude CFE was prepared shortly before purification. The system used was an Äkta purifier equipped with an Äkta P-900 pump, an Äkta UV-900 as well as Äkta UPC-900 detector, and an Äkta Frac-950 fraction collector (GE Healthcare Biosciences AB, Uppsala, Sweden). (i) The crude CFE was applied to a HiLoad 16/10 Q Sepharose High Performance (GE Healthcare Biosciences AB, Uppsala, Sweden) anion-exchange column. The column was equilibrated initially with 110 ml KPi (50 mM, pH 7.4). After applying the crude CFE to the column, it was washed with 25 ml of the same buffer, followed by a linear NaCl gradient (0 to 0.5 M) in 300 ml. The flow rate was 3 ml/min, and 2.5-ml fractions were collected. Aliquots of 100 μl of the collected fractions were immediately tested for transamination activity. (ii) Active fractions from the first purification step were pooled and concentrated to a volume of 1.5 ml, using a Vivaspin 20 3,000-molecular-weight-cutoff (MWCO) PES concentrator (Sartorius Stedim Biotech S.A., Aubagne Cedex, France) according to the manufacturer's instructions. After adjusting the NaCl concentration in the sample to 0.15 M, 700 μl of sample was applied to a preconditioned (50 mM KPi, pH 7.4, 0.5 M NaCl; 60 ml) Superdex 200 10/300 GL (GE Healthcare Biosciences AB, Uppsala, Sweden) gel permeation column. The proteins were eluted isocratically with 45 ml KPi (50 mM, pH 7.4, 0.5 M NaCl) at a flow rate of 0.4 ml/min. Fractions (0.5 ml) were collected, and 100 μl was immediately tested for transamination activity.
Desalting and lyophilization of active fractions.
To allow separation by SDS-PAGE, the pooled fractions showing transamination activity after the second purification step were desalted using 5-ml Zeba Desalt spin columns (Thermo Fisher Scientific, Bonn, Germany) according to the manufacturer's instructions. The desalted protein solution was lyophilized overnight and resolved in 45 μl KPi (50 mM, pH 7.4).
SDS-PAGE, staining, and sequencing of bands.
The purity of proteins in fractions from the different purification steps and estimations of molecular mass were determined using SDS-PAGE. Samples were mixed 4:1 with dying buffer (0.25 M Tris-HCl, pH 8.5, 25% glycerol, 12.5% 2-mercaptoethanol, 7.5% SDS, 0.025% bromophenol blue). The molecular size marker SDS7 M.W. 14000–66000 (Sigma-Aldrich Chemical Co., St. Louis, MO) was mixed 1:1 with the reducing agent dithiothreitol (DTT). Samples and the marker were boiled for 5 min for denaturation and then applied to 12% and 18% polyacrylamide gels (Anamed Elektrophorese GmbH, Gross-Bieberau, Germany), respectively. The molecular size marker PageRuler Plus (Thermo Fisher Scientific, Bonn, Germany) was used according to the manufacturer's instructions. Electrophoresis was carried out in a vertical gel chamber at 125 V. Gels were stained with Coomassie brilliant blue G250 (0.33 g in 120 ml methanol, 24 ml acetic acid, 120 ml water) for 1 h and then destained for 2 h in water-ethanol-acetic acid (70/20/10). For silver staining, a modified method (21) was used, as follows: at least 2 h in fix solution (40% ethanol, 10% acetic acid), three washes in wash solution (30% ethanol) for 20 min each, 1 min in thiosulfate solution (0.02% Na2S2O3), three water washes for 20 s each, 1 h in silver nitrate solution (0.2% AgNO3), three water washes for 20 s each, incubation in developing solution (0.0004% Na2S2O3, 0.05% H2CO, 3% Na2CO3) until bands were visible (10 to 15 min), a water wash for 1 min, 5 min in stop solution (0.5% glycine), and finally a water wash for 30 min. Protein bands of interest were cut from SDS-polyacrylamide gels and stored in fix solution. Sequencing was done by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after digestion by the Central Lab for Protein Analysis (ZfP), Ludwig Maximilian University, Munich, Germany. The program and database used for comparison of peptide fragments obtained by LC-MS/MS were both Mascot Search (Matrix Science Ltd., London, England).
Transamination activity assay.
Transamination activity assays of crude CFEs, purified enzyme (AcP), and lysozyme were performed as previously described (22), except that the concentrations varied. KPi (50 mM, pH 7.4), the substrate l-leucine (5 mM), α-ketoglutaric acid (5 mM), PLP (50 μM), and 100 μl protein solution in a total volume of 250 μl were used. All solutions except for the buffer were prepared shortly before use. The enzymatic reaction was performed for 24 h at 37°C and stopped by heating at 80°C for 15 min. The enzymatically catalyzed formation of α-keto acids from amino acids was monitored by high-pressure liquid chromatography–UV spectroscopy (HPLC-UV) or LC-electrospray ionization multidimensional mass spectrometry (LC–ESI-MSn). The formation of l-glutamic acid from α-ketoglutaric acid was monitored by an enzymatic reaction, using a colorimetric l-glutamic acid assay kit (R-Biopharm GmbH, Darmstadt, Germany). Controls were performed without the addition of enzyme (control A), without the addition of substrate (control B), and with inactivated enzyme (99°C for 15 min) (control C).
HPLC-UV.
For the detection of KIC in fractions with transamination activity, the precipitated proteins were removed by centrifugation, and 20 μl of the assay solution was applied to an HPLC-UV system. The system was a Jasco HPLC equipped with a Jasco PU-1580 quaternary pump and a Jasco UV-1575 variable-wavelength detector (Jasco GmbH, Gross-Umstadt, Germany). The column was a Luna 3μ C18(2) 100-Å by 15-cm by 2-mm column (Phenomenex, Torrance, CA). The HPLC solvents were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient went from 10% B and 90% A to 30% B and 70% A in 15 min and then increased to 100% B in 5 min, remained under these conditions for 10 min, and returned to 90% A and 10% B in 5 min, at a flow rate of 0.2 ml/min. The detection wavelength was 210 nm. Data analysis was performed using Jasco BORWIN, version 1.50, software (Jasco GmbH, Gross-Umstadt, Germany). Quantification was done by calibration with increasing concentrations of the authentic standard (0.01 to 1 mg/ml).
LC–ESI-MSn.
Levels of α-keto acids in transamination activity assays for enzyme characterizations were measured by LC–ESI-MSn. Precipitated proteins were removed by centrifugation, and 10 μl of the supernatant was injected. The system used for LC–ESI-MSn analysis was an Agilent 6340 ion trap LC-MS equipped with an Agilent 1100 HPLC system composed of an Agilent 1100 capillary pump, an Agilent 1100 microwell plate autosampler, and an Agilent 1100 variable-wavelength detector (Agilent, Waldbronn, Germany). The column was a Luna 3μ C18(2) 100-Å by 15-cm by 2-mm column (Phenomenex, Torrance, CA). The ionization parameters were as follows: the voltage of the capillary was −4,000 V, and the end plate was set to −500 V. The temperature of the dry gas (N2) was 330°C, and the flow rate was 9 liters/min. The full-scan mass spectra of the metabolites were measured from m/z 50 to 500 until the ion charge control (ICC) target reached 500,000 (positive ions) or 300,000 (negative ions) or for 200 ms, whichever was reached first. Tandem mass spectrometry was performed using helium as the collision gas, and the collision energy was set at 1.25 V. The target masses for MS/MS spectra were set to m/z 132 (KIC and KMV), m/z 163 (PPA), and m/z 147 (substrate screening, KIV, and KMBA). Mass spectra were acquired in the negative and positive ionization modes. Auto-tandem mass spectrometry was used to break down the most abundant [M + H]+ or [M − H]− ion. The LC solvents were 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B). KIC, KMV, and PPA were eluted isocratically with 50% A and 50% B at a flow rate of 0.1 ml/min for 27 min. For substrate specificity assay and the separation of KIV and KMBA, a gradient from 0% B and 100% A to 100% B and 0% A in 20 min, with 8 min under these conditions and then a return to 100% A and 0% B in 2 min at a flow rate of 0.1 ml/min, was used. Data analysis was performed using DataAnalysis for 6300 series ion trap LC/MS, version 4.0, and QuantAnalysis for 6300 series ion trap LC/MS, version 2.0, software (Bruker Daltonics GmbH, Bremen, Germany). Quantification was done by calibration with increasing concentrations of the authentic standards (0.001 to 0.1 mg/ml). The [M − H]− ion spectra were chosen for integration (for KIV, m/z 115 → 71; for KIC, m/z 129 → 85; for KMV, m/z 129 → 85; for PPA, m/z 163 → 91; for KMBA, m/z 147 → 99; and for hydroxyphenylpyruvic acid (HPPA), m/z 179 → 107). In the substrate screening process, the total ion chromatograms for negative ions were compared to the control without enzyme in searches for differences.
Colorimetric l-glutamic acid kit.
For determination of the amounts of l-glutamic acid in transamination activity assays, a colorimetric l-glutamic acid kit (R-Biopharm GmbH, Darmstadt, Germany) was used according to the manufacturer's instructions.
General molecular techniques.
Cloning, DNA manipulation, and gel electrophoresis followed standard procedures, as described previously (23). DNA of L. sakei TMW 1.1322 was isolated with an E.Z.N.A. bacterial DNA kit from Omega Bio-Tek Inc. (Norcross, GA). E. coli TOP10 plasmid DNA was isolated with a peqGOLD plasmid miniprep kit from peqlab Biotechnologie GmbH (Erlangen, Germany). Restriction endonuclease digestions and ligations were performed as recommended by the supplier (MBI Fermentas, St. Leon-Roth, Germany). DNA for cloning purposes was amplified by PCR techniques, using KOD hot-start DNA polymerase (Merck KGaA, Darmstadt, Germany). Colony PCRs were performed with a Taq Core kit (MP Biomedicals, Solon, OH). The primers used were obtained from Eurofins MWG Operon (Ebersberg, Germany). DNA was purified with an E.Z.N.A. Cycle Pure kit from Omega Bio-Tek Inc. (Norcross, GA). Standard heat shock transformation was used for plasmid transfer into E. coli (23). For the preparation of competent E. coli TOP10 cells, a 1% inoculated culture was grown in LB medium at 37°C. Cells were harvested at an optical density at 590 nm (OD590) of 0.5, washed twice with 0.1 M CaCl2, and subsequently resuspended in 2 ml CaCl2 (0.1 M, 15% glycerol). Aliquots (200 μl) were stored at −80°C. All washing solutions were ice cold.
Cloning, heterologous expression, and purification of AcP from L. sakei TMW 1.1322 in E. coli TOP10.
AcP was amplified using the primer pair AcP-for (5′-TAT ACC ATG GTT ATG CAA AAA GCA GTC CAA TTA G-3′) and AcP-rev (5′-TAT ATC TAG AAA ACC GAC AAC TGA AAA TTT ATG A-3′), with genomic DNA of L. sakei TMW 1.1322 as the template. The amplified PCR products were digested with NcoI and XbaI and subsequently ligated into similarly digested pBAD/Myc-HisB vector. The resulting constructs were introduced into competent E. coli TOP10 cells. Pure plasmid DNAs were isolated from positive clones, and the correctness of constructs was verified by sequencing (GATC Biotech GmbH, Constance, Germany). LB medium (1 liter) containing 0.3 M sorbitol and 100 μg/ml ampicillin was inoculated with 10 ml of an overnight culture of E. coli TOP10 carrying pBAD/Myc-HisB-AcP. Incubation at 37°C and 160 rpm was continued until the OD600 reached 0.6 to 0.8. After cooling down to 30°C, the culture was further incubated until the OD600 reached ∼1. The cofactor PLP (0.05 mM) was then added. Expression was subsequently induced by adding 100 μM l(+)-arabinose and incubating the culture overnight (16 to 18 h) at 30°C and 120 rpm. Cells were harvested by centrifugation (5,000 rpm for 10 min at 4°C). The supernatant was discarded, and cells were washed once with precooled binding buffer (20 mM NaPi, 0.5 M NaCl, 25 mM imidazole, pH 7.4) and resuspended in 10 ml of the same buffer. Crude extract was obtained by ultrasonication (10 times, 10%, 30 s) of cell suspension on ice (Sonoplus UW 2070/MS 73; Bandelin Electronic GmbH & Co. KG, Berlin, Germany). Cell debris was separated from the crude cell extract by centrifugation (12,000 × g for 20 min at 4°C). After filtration (0.45 μm), the supernatant was applied to a 5-ml HisTrap FF column (GE Healthcare Biosciences AB, Uppsala, Sweden) at a flow rate of 1 ml/min, using the same system as that for enzyme purification. Residual proteins were flushed out in a wash step with 10 column volumes of binding buffer at 2 ml/min. A linear gradient of 0% elution buffer (20 mM NaPi, 0.5 M NaCl, 400 mM imidazole, pH 7.4) to 100% elution buffer in 20 min at 1 ml/min eluted the target protein from the HisTrap column. Fractions (1 ml) were collected, and fractions containing the target protein, as shown by the UV trace at 280 nm, were pooled and concentrated to a volume of about 1 ml by use of a Vivaspin 20 3,000-MWCO PES concentrator (Sartorius Stedim Biotech S.A., Aubagne Cedex, France) according to the manufacturer's instructions.
Acylphosphatase activity assay.
Acylphosphatase activity assay of purified AcP was performed as previously described (24). The enzymatically catalyzed cleavage of acetyl phosphate was monitored. Triethanolamine buffer (20 mM, pH 7.6), the substrate acetyl phosphate (4 mM), NAD+ (10 mM), and 50 μl purified protein in a total volume of 200 μl were used. All solutions except for the buffer were prepared shortly before use. Controls were performed without the addition of enzyme (control A) and without the addition of substrate (control B). The concentration of acetyl phosphate remaining after 2 h of incubation at room temperature was determined spectrophotometrically at 510 nm (25).
Characterization of AcP and HEWL.
Because transamination reactions also occur chemically and to measure only the enzymatically catalyzed formation of α-keto acid, the amount formed without enzyme addition (control A) was subtracted from the amount obtained in assays with enzyme. For characterization, 50 μM purified enzymes were chosen (0.64 mg/ml AcP and 0.72 mg/ml HEWL). AcP was expressed heterologously and purified shortly before use for kinetic characterization. The effect of pH on transaminase activity against l-leucine was determined in the ranges of pH 3.0 to 10.0 (AcP) and pH 2.5 to 10.0 (HEWL) by using the following buffers: 50 mM citric acid, pH 2.0 to 6.0; 50 mM KPi, pH 5.0 to 9.0; and 50 mM Tris-HCl, pH 6.5 to 10.0. The effect of temperature was determined by testing six temperatures (25, 30, 37, 45, 55, and 65°C) at optimal pH. The maximum incubation time was deduced by determination of the longest possible incubation time before consistent levels of product were measured. The transamination activities against 19 proteinogenic amino acids were determined by the standard transamination activity assay (substrate screening). Kinetic parameters of HEWL and purified AcP were estimated for positive substrates by using concentrations ranging from 0.5 to 50 mM. Activity was measured at the optimal pH, temperature, and time as described above. Kinetic parameters were calculated from Lineweaver-Burk plots and compared with Cornish-Bowden plots for confirmation. The turnover number kcat was calculated from the velocity at saturation per active site, as follows: kcat (h−1) = Vmax (pM h−1)/c (enzyme concentration) (pM), where c is the concentration of enzyme. The catalytic efficiency was calculated as the ratio of kcat (h−1) to Km (μM).
RESULTS
Enzyme purification, purity, and molecular mass.
Although L. sakei strains degrade BCAAs via a transamination reaction, the genome of L. sakei 23K lacks the corresponding homologous genes (11–13). To clarify the metabolism of leucine, isoleucine, and valine in LAB, proteins with aminotransferase activity were isolated and characterized. Purification from crude CFE of L. sakei TMW 1.1322 was achieved by ion-exchange and gel permeation chromatography (Table 1). The enzyme eluted from a HiLoad 16/10 Q Sepharose High Performance column with 0 to 0.03 M NaCl. Specific transamination activity against l-leucine was enriched 13-fold. Fractions with the highest activities were pooled, concentrated, and applied to a Superdex 200 10/300 GL column. Gel filtration yielded four successive fractions with transamination activity. These fractions contained low-molecular-weight proteins. Protein concentrations in active fractions after gel filtration were below the detection limit of the Bradford assay. A total activity of 7.8 nkat could be determined, corresponding to 3.2% of the activity measured in the crude CFE. Transamination activity was confirmed after desalting of the pooled active fractions obtained by gel filtration (data not shown). Since the last purification step led to a substantial loss of activity, it is conceivable that additional transaminases were overlooked or that the enzyme is unstable.
Table 1.
Purification table for an enzyme with transamination activity from L. sakei TMW 1.1322
| Purification step | Total protein (mg) | Total activity (nkat) | Sp act (pkat/mg) | Purification factor | Yield (%) |
|---|---|---|---|---|---|
| Crude CFE | 426.6 | 245.6 | 575.6 | 1.0 | 100 |
| Q Sepharose chromatography | 29.3 | 219.7 | 7,505.9 | 13.0 | 89.5 |
| Superdex chromatography | NDa | 7.8 | 3.2 |
ND, below the detection threshold of the Bradford assay.
The purity of the enzyme was confirmed by SDS-PAGE with silver staining for visualization. A single protein band was detected on the gel, corresponding to approximately 10 to 12 kDa (Fig. 2). The band was cut from the gel and sequenced.
Fig 2.

SDS-PAGE analysis of active fractions from purification of an enzyme with transamination activity from L. sakei TMW 1.1322. Cells were broken down by ultrasonication. HEWL was not added. Lanes: M, molecular size marker; A, crude CFE from L. sakei TMW 1.1322; B, active fractions after anion-exchange chromatography; C, purified enzyme after gel filtration. Protein bands were visualized by silver staining. The catalytically active protein at 12 kDa in lane C was identified as AcP.
In the beginning of this study, cells were treated with an excessive amount of lysozyme (HEWL) to improve lysis by ultrasonication for the preparation of the crude CFE. Similar to the purification process described above (without addition of HEWL), activity could be measured in fractions after anion-exchange chromatography followed by gel filtration (data not shown). The purity of the protein was confirmed by SDS-PAGE. A single protein band for one of the active fractions could be visualized by Coomassie blue staining (see Fig. S1 in the supplemental material), corresponding to approximately 14 kDa. The band was cut from the gel and sequenced.
Protein sequencing.
LC-MS/MS analysis of the digested protein with transamination activity from L. sakei TMW 1.1322 (Fig. 2) yielded sequence information on peptide fragments that could be assigned to several proteins annotated from the L. sakei 23K genome. The highest score calculated by the program Mascot Search, using the Mowse scoring algorithm (26), was obtained for a putative acylphosphatase, followed by a number of other proteins with much lower probabilities (Table 2).
Table 2.
Protein hits obtained by sequencing of the protein with transamination activity from L. sakei TMW 1.1322
| Protein | NCBI reference sequence (GenBank accession no.) | % Similaritya | Protein scoreb | Calculated mol mass (kDa)c |
|---|---|---|---|---|
| Putative acylphosphatase | YP_395991.1 | 69.9 | 18,743 | 9.6 |
| 50S ribosomal protein L33 | YP_395944.1 | 89.8 | 4,110 | 5.9 |
| Thioredoxin | YP_394828.1 | 82.9 | 2,806 | 11.4 |
| Phospho-carrier protein HPr (histidine-containing protein) | YP_396072.1 | 38.6 | 2,233 | 9.2 |
| Histone-like DNA-binding protein HU | YP_395626.1 | 71.4 | 2,019 | 9.5 |
| Cold shock protein, CspA family | YP_396175.1 | 65.2 | 295 | 7.2 |
Similarity of peptide fragments obtained by sequencing to NCBI reference sequence of L. sakei 23K.
Calculated by Mascot Search, using the Mowse scoring algorithm (26).
According to the NCBI reference sequence.
LC-MS/MS analysis of the digested protein with a molecular mass of approximately 14 kDa (data not shown) revealed the amino acid sequence of HEWL, with an amino acid similarity of 63.6%. This led to the assumption that HEWL, which was added to improve cell lysis, was purified from the crude CFE of L. sakei TMW 1.1322. Because transamination activity was detected throughout the purification protocol, HEWL was tested in a transamination activity assay with various concentrations of the protein. The results clearly showed a transaminase side activity of HEWL (see Fig. S2 in the supplemental material).
Heterologous expression and purification of a putative acylphosphatase from L. sakei TMW 1.1322 in E. coli TOP10.
The putative AcP was chosen for heterologous expression in E. coli followed by biochemical examination of the recombinant protein. Isolated plasmid DNAs from positive clones showed the correct nucleotide sequence, and significant amounts of pure target protein could be purified by affinity chromatography. Recombinant AcP showed the expected molecular mass (12.7 kDa) on SDS-PAGE (see Fig. S3 in the supplemental material).
Acylphosphatase activity of heterologously expressed and purified AcP.
To test if the putative acylphosphatase was heterologously expressed as a functional enzyme and showed the postulated activity, the hydrolysis of acetyl phosphate was monitored with an enzyme assay. The concentration of residual acetyl phosphate remaining after incubation was determined spectrophotometrically. The enzyme catalyzed the cleavage of acetyl phosphate nearly quantitatively. Only 0.01 mM acetyl phosphate from the 4 mM added acetyl phosphate was detected after incubation. In the assay without addition of AcP (control A), the initial amount of substrate (4 mM) was found, whereas in the assay without substrate (control B), negligible traces of acetyl phosphate could be measured.
Transamination activity of heterologously expressed and purified AcP.
Transamination activity of AcP was shown by the formation of α-keto acids, which were detected by HPLC-UV and confirmed by LC–ESI-MSn analysis. Furthermore, formation of l-glutamic acid from α-ketoglutaric acid was determined by an enzymatic reaction, using a colorimetric l-glutamic acid assay kit. PLP and α-ketoglutaric acid are essential for the transamination activity of AcP, as an assay without addition of the coenzymes resulted in a loss of activity (data not shown).
Effects of pH, temperature, and time on transamination activity of AcP and HEWL.
The purified AcP and HEWL showed transaminase activity, expressed as relative product amounts, from pH 5.0 to 9.0, with an optimum at pH 7.4, and from pH 5.5 to 8.0, with an optimum at pH 7.0, respectively (see Fig. S4 in the supplemental material). AcP retained activity (59 to 96%) at pH values between 6.0 and 9.0 in KPi. In citric acid and Tris-HCl, AcP produced less product than that in KPi. HEWL showed a narrow pH optimum range but still retained about 40% of the maximum activity at pH values of 6 to 7.5. The activity at pHs beyond the mentioned values was negligible. The optimum temperature was 55°C for AcP and 37°C for HEWL. The maximum amount of product was detected after 16 h of incubation with AcP and 27 h of incubation with HEWL.
Substrate specificity.
Of the 19 proteinogenic amino acids tested, AcP accepted l-valine, l-leucine, l-isoleucine, l-phenylalanine, l-methionine, and l-tyrosine as substrates for transamination reactions (Table 3). HEWL showed transamination activity toward l-leucine, l-isoleucine, and l-phenylalanine. l-Leucine and l-phenylalanine were the preferred substrates for both enzymes.
Table 3.
Substrate specificities of AcP from L. sakei TMW 1.1322 and of HEWL
| Substrate | Relative activity (%)a |
|
|---|---|---|
| AcP | HEWL | |
| l-Leucine | 100 ± 1.1 | 100 ± 5.3 |
| l- Phenylalanine | 85.9 ± 2.6 | 83.2 ± 1.5 |
| l-Isoleucine | 53.5 ± 4.0 | 5.1 ± 0.2 |
| l-Tyrosine | 30.7 ± 0.8 | |
| l-Valine | 16.4 ± 0.2 | |
| l-Methionine | 9.3 ± 0.01 | |
Expressed as a percentage of the activity against l-leucine, which was given a value of 100%. Results are presented as means ± standard deviations (SD).
Kinetic parameters.
The kinetic parameters Km, Vmax, kcat, and catalytic efficiency for the different substrates and enzymes were determined from Lineweaver-Burk plots (Table 4; see Fig. S5 and S6 in the supplemental material). The kinetic parameters for l-tyrosine could not be obtained due to the poor solubility of this substrate. The enzymatic reaction became saturated at higher substrate concentrations, whereas the amount of chemically formed product increased. Subtraction of control A values (chemically formed amount of α-keto acids) from levels detected in the enzyme assay therefore led to a decrease in α-keto acid concentrations. For the calculation of enzymatic parameters, only the linearly increasing range of enzymatically catalyzed α-keto acid concentrations was used. The Km values for AcP indicated the highest affinity for l-leucine (1.85 mM) and the lowest affinity for l-phenylalanine (5.34 mM) with regard to transamination activity. HEWL generally showed lower affinities for the substrates, with affinities ranging from 3.79 mM (l-isoleucine) to 17.9 mM (l-leucine). The values for the catalytic efficiency confirmed the lower transamination activity of HEWL, with turnover rates of <1 μM−1 h−1 for all substrates. AcP, on the other hand, showed catalytic efficiencies ranging from 3 μM−1 h−1 (l-methionine) to 15.44 μM−1 h−1 (l-isoleucine).
Table 4.
Kinetic parameters for transamination side activity of recombinant AcP from L. sakei TMW 1.1322 and of HEWL
| Substrate |
Km (mM) |
Vmax (pkat/mg) |
kcat (h−1) |
Catalytic efficiency (μM−1 h−1) |
||||
|---|---|---|---|---|---|---|---|---|
| AcP | HEWL | AcP | HEWL | AcP | HEWL | AcP | HEWL | |
| l-Leucine | 1.85 | 17.9 | 997 | 506 | 0.011 | 0.0066 | 6.2 | 0.37 |
| l-Phenylalanine | 5.34 | 5.94 | 2,181 | 60 | 0.025 | 0.0008 | 4.7 | 0.13 |
| l-Isoleucine | 2.22 | 3.79 | 2,974 | 115 | 0.034 | 0.0015 | 15.4 | 0.39 |
| l-Methionine | 2.25 | 585 | 0.007 | 3.0 | ||||
| l-Valine | 4.87 | 1,424 | 0.016 | 3.3 | ||||
DISCUSSION
Isolation and identification of acylphosphatase.
In this study, a protein with transaminase activity for BCAAs was purified from L. sakei TMW 1.1322, an isogenic clone of the sequenced strain L. sakei 23K (11), despite the fact that the genome sequence of L. sakei 23K lacks BCAA transaminase genes. Sequencing of the isolated protein, with a molecular mass of about 10 to 12 kDa, yielded peptide fragments which could be assigned to several proteins whose corresponding genes are annotated in the L. sakei 23K genome (Table 2). The highest probability was calculated for a putative acylphosphatase, followed by 50S ribosomal protein L33, thioredoxin, the phospho-carrier protein HPr (histidine-containing protein), the histone-like DNA-binding protein HU, and a cold shock protein (CSP) (CspA family).
The 50S ribosomal protein L33 has a calculated molecular mass of 5.9 kDa and is therefore much smaller than the protein band on the gel. Ribosomal proteins in L. sakei (L. sakei subsp. sakei 2a) have been identified as bacteriocins against Listeria monocytogenes and Enterococcus faecalis (27). In the same study, the histone-like DNA-binding protein HU (9.5 kDa) was also identified as an antimicrobial compound. Both proteins exert similar functions and participate in DNA-dependent processes, e.g., DNA binding. They participate less in catalytic reactions. This fact and the low molecular mass of the ribosomal protein made it unlikely that one of these proteins is the enzyme with transamination activity.
Thioredoxin (11.4 kDa) and the phospho-carrier protein HPr (9.2 kDa) exhibit molecular masses similar to that of the isolated protein. Thioredoxin activates various biosynthesis enzymes by reducing disulfide bonds, while several other enzymes are inhibited in the same way. Furthermore, it participates in redox reactions through the reversible oxidation of its active center dithiol to a disulfide and catalyzes dithiol-disulfide exchange reactions (28). The phospho-carrier protein HPr is of major importance for the uptake of carbohydrates into the cell by the phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS) and for the regulation of carbohydrate balance by inhibiting the expression of the enzymes involved (29). A transamination activity of one of these proteins has not been reported. Since LC-MS analysis yielded low protein scores for these two candidates (Table 2), they were not held responsible for the enzymatic activity detected in the purified extract.
The cold shock protein (CspA family) has a calculated molecular mass of 7.2 kDa. CSPs are synthesized as a response to cold stress in LAB and other organisms (30). The identification of several CSPs from Lactococcus lactis suggested their involvement in various cellular processes, such as translation, sugar metabolism, chromosome structuring, and signal transduction (31). Hence, cold shock proteins participate primarily in regulatory and less in catalytic processes. This and the low molecular weight and amino acid similarity argued against the CSP as the enzyme with transamination activity.
AcPs catalyze the hydrolysis of acylphosphates, which are compounds containing a carboxyl phosphate group, e.g., 1,3-bisphosphoglycerate, carbamoyl phosphate, succinyl phosphate, acetyl phosphate, and aspartyl phosphate. These molecules play an important metabolic and physiological role as intermediates in glycolysis, the citric acid cycle, and pyrimidine synthesis. Thus, acylphosphatase has been implicated in the control of the ionic conditions of the cell and the control of the glycolytic pathway, although its biological roles are not yet fully understood (32). Furthermore, the AcP from the hyperthermophilic archaeon Sulfolobus solfataricus (Sso-AcP) has been well elucidated for understanding native-like aggregation. By comparing the amino acid sequences of AcPs, a universally conserved signature (Val-Gln-Gly-Val-X-X-Arg; residues 17 to 23 in mitochondrial AcP (mt-AcP) and cytosolic AcP (ct-AcP) and residues 24 to 30 in Sso-AcP) defining a cradle-like conformation of the polypeptide backbone was identified. The arginine residue at the end of the signature (Arg-30) and Asn-46 describe the substrate binding site in Sso-AcP (33). In our study, the most frequently detected peptide (Table 2) had the amino acid sequence Val-Gln-Gly-Val-Gly-Phe-Arg, which corresponds to the universally conserved signature identified for AcPs (33). The whole sequence of the putative AcP obtained by a database search also exhibited an asparagine residue (Asn-37) in an interval of 18 amino acids to the Arg-19 at the end of the signature. An acylphosphatase activity of the putative AcP from L. sakei 23K was therefore supposed. Transaminase activity of AcPs was not yet known; since we were able to demonstrate transamination activity for another hydrolase (HEWL) in the beginning of the study, and due to the high probability calculated by the program Mascot Search, we chose the putative AcP gene from L. sakei for cloning and heterologous expression in E. coli.
Acylphosphatase assays showed that the putative AcP exhibited the postulated catalytic activity and was heterologously expressed in an active form. AcPs show catalytic efficiencies ranging from 550 s−1 mM−1 (Sso-AcP) to 9,500 s−1 mM−1 (ct-AcP) (33). The recombinant protein hydrolyzed the substrate acetyl phosphate (4 mM) nearly quantitatively within 2 h at pH 7.6 and room temperature. Kinetic data for the acylphosphatase activity were not determined in this study, as the focus was on the promiscuous activity of AcP. Little is known about AcPs in LAB. AcP probably participates in pyruvate degradation in Lactobacillus delbrueckii subsp. lactis, by hydrolyzing acetyl phosphate to acetic acid (34). Acetic acid has been identified as the compound with the highest aroma impact in Hungarian salami (35).
Transaminase activity.
A transamination activity has not been reported for either native HEWL or AcPs. HEWL catalyzed an amination reaction after chemical modification, introducing pyridoxamine (36). However, promiscuous transamination activity has been shown for various PLP-dependent enzymes, e.g., aspartate β-decarboxylase (37), methionine α-decarboxylase (38), arginine racemase (39), α-amino ε-caprolactam racemase (40), and alanine racemase (41). Artificial transaminases have been obtained by linking a pyridoxamine derivative within a fatty acid binding protein (42). PLP-dependent kynureninase and phosphonoacetaldehyde hydrolase (2-aminoethylphosphonate pyruvate transaminase) are examples of hydrolases showing a transamination side activity (43, 44).
We were able to characterize the as yet unknown transamination side activity of two largely unrelated (<15% similarity) hydrolases, AcP from L. sakei TMW 1.1322 and HEWL. Both proteins were PLP independent with regard to their hydrolytic activity but showed transaminase activity when the prosthetic group and an amino group acceptor were available. Chemical formation of transamination products was also detected under these conditions, but the levels were significantly lower than those in assays with enzymes added. This result confirmed the findings of a study in which transamination products of radioactively labeled leucine were formed by L. sakei 23K only when α-ketoglutaric acid and PLP were available (14). In summary, the detection of α-keto acids and glutamic acid and the dependency of catalysis on PLP support a true aminotransferase mechanism.
L. sakei is commonly used in starter cultures for the production of dry fermented sausages. The only meat-relevant Lactobacillus species for which a transaminase has been characterized until now is Lactobacillus paracasei. The enzyme showed temperature and pH optima at 43°C and pH 7.3, respectively (22). Comparatively, high temperatures seem to be essential for transamination in lactic acid bacteria. pH values of 7 to 8.5 and temperatures between 45 and 50°C were reported as optimal for transamination of methionine by a partially purified BCAA transaminase from Lactococcus lactis (45). The activity of an aromatic amino acid transaminase from L. lactis was optimal between pH 6.5 and 8 and between 35 and 45°C (46). The optimum conditions for transaminase activity of AcP from L. sakei TMW 1.1322 were pH 7.4 and 55°C. For HEWL, the optima were pH 7.0 and 37°C. Values of pH 7.0 and 40°C were chosen for amination of PPA to phenylalanine by employing pyridoxamine derivatives of lysozyme (36). The transamination side activity of kynureninase was highest at pH 8.0 for incubation at 30°C (43).
The substrate specificity of AcP for branched-chain (l-valine, l-leucine, and l-isoleucine) and aromatic (l-phenylalanine and l-tyrosine) amino acids as well as l-methionine is consistent with our earlier study (15). The corresponding α-keto acids are precursors for the production of aroma-relevant compounds such as 3-methylbutanal, 2-methylpropanoic acid, phenylacetaldehyde, and 3-(methylthio)-propanal. The formation of these metabolites from amino acid substrates by L. sakei TMW 1.1322 has been shown. Transaminases can be classified, among other methods, by specificity for branched-chain or aromatic amino acids. A transaminase from L. paracasei showed specificity only for BCAAs (22). Aromatic amino acid transaminases act on the three aromatic amino acids, and some of them, like transaminase D from E. coli (47) and a transaminase from L. lactis (46), also accept l-leucine and l-methionine as substrates. Thus, the promiscuous transaminase activity of AcP from L. sakei TMW 1.1322 resembles that of aromatic amino acid transaminases that also accept hydrophobic BCAAs. The substrate specificity of HEWL toward l-leucine, l-isoleucine, and l-phenylalanine is unprecedented, as neither l-valine nor l-tyrosine and l-tryptophan were accepted.
AcP and HEWL showed low transamination side activities, as confirmed by kinetic parameters, in comparison with values for classical transaminases (http://www.brenda-enzymes.info). Although the Km values were in the range for classical aminotransferases, the kcat values were significantly lower. Thus, the catalytic efficiencies were only about 1/1,000 the values obtained for BCAA transaminase from Homo sapiens (http://www.brenda-enzymes.info). Generally, HEWL showed lower substrate affinities, lower turnover rates, and therefore also lower catalytic efficiencies (Table 4). Catalytic efficiencies of 8.7 and 37 M−1 h−1 were achieved with PPA as the substrate when lysozyme was chemically modified with pyridoxamine (36). Since this reaction represents an amination rather than a transaminase reaction, the values cannot be compared directly with those of true aminotransferase catalysis. Catalytic efficiencies of 0.034 to 1.83 mM−1 h−1 (l-phenylalanine) for different pyridoxamine derivatives of a fatty acid binding protein have been reported (42). The highest catalytic efficiency for the enzymes characterized in our study (15.4 μM−1 h−1 for AcP and l-isoleucine) is comparable with the lowest catalytic efficiency obtained for the artificial transaminase. Kinetic data have not been obtained for transamination side reactions of the PLP-dependent enzymes mentioned above.
The promiscuous transaminase activities of the enzymes characterized in this study are low, but AcP is the only enzyme characterized so far to show transamination activity in L. sakei TMW 1.1322. Although genes encoding typical BCAA transaminases could not be found in more than 50 strains of L. sakei and the genome of L. sakei 23K lacks BCAA transaminase genes (13), transamination activity and the formation of amino acid-derived volatile metabolites could be demonstrated for L. sakei 23K and other L. sakei strains. Since the formation of volatile metabolites can be enhanced by supplying α-keto acids instead of amino acids, amino acid transport and transamination seem to create a bottleneck in the formation of potentially aroma-relevant compounds (11–16). This is consistent with the low transamination activity of the AcP found in this study. The result implies that the promiscuous activity of AcP is of biological relevance and is important for understanding the intracellular metabolism of amino acids in this strain.
When we assume that LAB share an ancestor containing the full set of aminotransferase genes, it seems that the species L. sakei lost its transaminase genes involved in the degradation of BCAAs during evolution (13). Similarly, Lactobacillus johnsonii and Lactobacillus reuteri lack BCAA transaminase genes, although these genes are present in lactococcal and streptococcal strains representing two other clades of the phylogenetic tree of LAB (13). The loss did not affect the viability of the strains, probably because degradation of branched amino acids can be compensated by nontransaminating reactions (48), or the promiscuous activity of AcP and PLP-dependent enzymes was sufficient to account for the lack of the typical transaminases. In all known PLP-utilizing enzymes, the aldehyde form of the cofactor is bound covalently to the ε-amino group of a lysine residue via a Schiff base linkage (Fig. 3) (49, 50). Upon nucleophilic attack of this internal imine by the amino group of an incoming substrate, the lysine residue is released and an internal ketimine complex between the substrate and the aldehyde is formed. The lysine residue also serves as a general base in the subsequent conversion (40). In addition to lysine, aspartate or glutamate and arginine residues interact with the cofactor and are highly conserved throughout native aminotransferases (50, 51). In principle, all proteins that can bind PLP to a lysine residue and correctly position the functional group in a three-dimensional fashion can act as transaminase catalyst. A crystal structure of AcP containing pyridoxal-5-phosphate and amino acids is not available. Therefore, the mechanism of the aminotransferase activity of AcP remains unknown. An AcP homology model built by SWISS-MODEL workspace (52; http://swissmodel.expasy.org/) showed an α/β sandwich domain composed of four antiparallel and one parallel β-strand assembled in a five-stranded β-sheet facing two antiparallel α-helices, in accordance with published AcP structures (Fig. 4) (53). The amino acid sequence of AcP from L. sakei contains five lysine, six aspartate, one glutamate, and five arginine residues. The side chains of these amino acids that might be involved in the interaction with the cofactor point to the outside of the α/β sandwich domain (Fig. 4). Therefore, it is assumed that the catalysis takes place on the outer surface of the protein. Our results confirm that an organism can have more PLP-dependent activities than it has genes for PLP-dependent enzymes (54).
Fig 3.

Proposed reaction mechanism for the half-reaction catalyzed by aminotransferases. AcP-Lys-NH2 was proposed as the catalytically active lysine residue in AcP. (Adapted from reference 50.)
Fig 4.
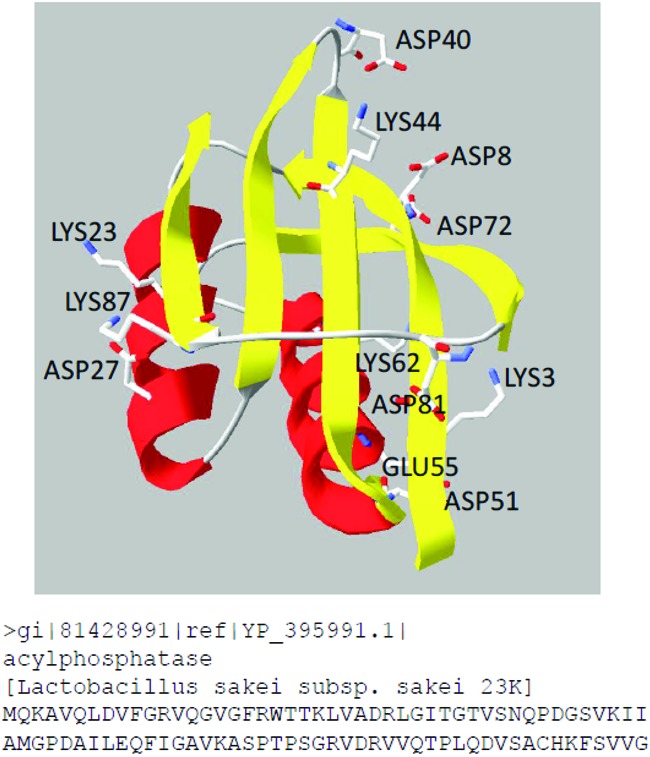
Structure of AcP homology model and amino acid sequence of AcP. The SWISS-MODEL workspace identified Protein Data Bank entry 1w2iB (40% sequence identity) as the most suitable template (52). The protein under entry 1w2iB is annotated as a monomer. The five lysines, six aspartates, and one glutamate are highlighted.
The awareness that proteins have multiple functions is an issue that is only beginning to be appreciated in genomic analysis. When genomes lack typical genes for classical pathways, this does not necessarily mean that the organism is unable to produce the product of the pathway. Although catalytic promiscuity is particularly frequent among PLP-dependent enzymes, due to their common mechanistic feature, we have shown that hydrolases can also recruit PLP to perform aminotransferase reactions, which might be of biological relevance. Thus, our results have significant implications for gene annotation projects and system biology studies.
In conclusion, without complete knowledge of the moonlighting activities of proteins, the metabolic capacity of microbial strains used for food production can only hardly be deduced from their genomic sequences. Additional biochemical efforts are needed to predict the potential of novel strains for the food industry.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Hoffmann for his support with LC-MS analysis.
This research project was supported by the German Ministry of Economics and Technology and the FEI (Forschungskreis der Ernährungsindustrie E.V., Bonn, Germany) (project AiF 15458 N).
Footnotes
Published ahead of print 25 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03762-12.
REFERENCES
- 1. Dainty R, Blom H. 1995. Flavour chemistry of fermented sausages, p 176–193 In Fermented meats. Blackie Academic and Professional, London, United Kingdom. [Google Scholar]
- 2. Ordóñez JA, Hierro EM, Bruna JM, de la Hoz L. 1999. Changes in the components of dry-fermented sausages during ripening. CRC Crit. Rev. Food Sci. 39: 329– 367 [DOI] [PubMed] [Google Scholar]
- 3. Hugas M, Garriga M, Aymerich T, Monfort JM. 1993. Biochemical characterization of lactobacilli from dry fermented sausages. Int. J. Food Microbiol. 18: 107– 113 [DOI] [PubMed] [Google Scholar]
- 4. Montel M-C, Masson F, Talon R. 1998. Bacterial role in flavour development. Meat Sci. 49: 111– 123 [PubMed] [Google Scholar]
- 5. Trüpler HG, De'Clari L. 1997. Taxonomic note: necessary correction of specific epithets formed as substantive nouns “in apposition.” Int. J. Syst. Bacteriol. 47: 908– 909 [Google Scholar]
- 6. Hammes WP, Bantleon A, Min S. 1990. Lactic acid bacteria in meat fermentation. FEMS Microbiol. Rev. 87: 165– 174 [Google Scholar]
- 7. Smit BA, Engels WJM, Smit G. 2009. Branched chain aldehydes: production and breakdown pathways and relevance for flavour in foods. Appl. Microbiol. Biotechnol. 81: 987– 999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ardö Y. 2006. Flavour formation by amino acid catabolism. Biotechnol. Adv. 24: 238– 242 [DOI] [PubMed] [Google Scholar]
- 9. Smit G, Smit BA, Engels WJM. 2005. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol. Rev. 29: 591– 610 [DOI] [PubMed] [Google Scholar]
- 10. Meynier A, Novelli E, Chizzolini R, Zanardi E, Gandemer G. 1999. Volatile compounds of commercial Milano salami. Meat Sci. 51: 175– 183 [DOI] [PubMed] [Google Scholar]
- 11. Chaillou S, Champomier-Vergès Cornet M-CM, Crutz-Le Cop Dudez A-M, Martin A-MV, Beaufils S, Darbon-Rongère E, Bossy R, Loux V, Zagorec M. 2005. The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 23K. Nat. Biotechnol. 23: 1527– 1533 [DOI] [PubMed] [Google Scholar]
- 12. Freiding S, Gutsche KA, Ehrmann MA, Vogel RF. 2011. Genetic screening of Lactobacillus sakei and Lactobacillus curvatus strains for their peptidolytic system and amino acid metabolism, and comparison of their volatilomes in a model system. Syst. Appl. Microbiol. 34: 311– 320 [DOI] [PubMed] [Google Scholar]
- 13. Liu M, Nauta A, Francke C, Siezen RJ. 2008. Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl. Environ. Microbiol. 74: 4590– 4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larrouture C, Ardaillon V, Pépin M, Montel MC. 2000. Ability of meat starter cultures to catabolize leucine and evaluation of the degradation products by using an HPLC method. Food Microbiol. 17: 563– 570 [Google Scholar]
- 15. Sinz Q, Schwab W. 2012. Metabolism of amino acids, dipeptides and tetrapeptides by Lactobacillus sakei. Food Microbiol. 29: 215– 223 [DOI] [PubMed] [Google Scholar]
- 16. Freiding F, Ehrmann MA, Vogel RF. 2012. Comparison of different IlvE aminotransferases in Lactobacillus sakei and investigation of their contribution to aroma formation from branched chain amino acids. Food Microbiol. 29: 205– 214 [DOI] [PubMed] [Google Scholar]
- 17. De Man JC, Rogosa M, Sharpe ME. 1960. A medium for the cultivation of lactobacilli. J. Appl. Bacteriol. 23: 130– 135 [Google Scholar]
- 18. Stolz P, Böcker G, Hammes WP, Vogel RF. 1995. Utilization of electron acceptors by lactobacilli isolated from sourdough. I. Lactobacillus sanfrancisco. Z. Lebensm. Unters. Forsch. 201: 91– 96 [Google Scholar]
- 19. Luria SE, Adams JN, Ting RC. 1960. Transduction of lactose-utilizing ability among strains of E. coli and S. dysenteriae and the properties of the transducing phage particles. Virology 12: 348– 390 [DOI] [PubMed] [Google Scholar]
- 20. Bradford MM. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248– 254 [DOI] [PubMed] [Google Scholar]
- 21. Blum H, Beier H, Gross HJ. 1987. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8: 93– 99 [Google Scholar]
- 22. Thage BV, Rattray FP, Laustsen MW, Ardö Y, Barkholt V, Houlberg U. 2004. Purification and characterization of a branched-chain amino acid aminotransferase from Lactobacillus paracasei subsp. paracasei CHCC 2115. J. Appl. Microbiol. 96: 593– 602 [DOI] [PubMed] [Google Scholar]
- 23. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 24. Albina JE, Mastrofrancesco B, Reichner JS. 1999. Acyl phosphatase activity of NO-inhibited glyceraldehyde-3-phosphate dehydrogenase GAPDH: a potential mechanism for uncoupling glycolysis from ATP generation in NO-producing cells. Biochem. J. 341: 5– 9 [PMC free article] [PubMed] [Google Scholar]
- 25. Lipmann F, Tuttle LC. 1945. A specific micromethod for the determination of acyl phosphates. J. Biol. Chem. 159: 21– 28 [Google Scholar]
- 26. Pappin DJ, Hojrup P, Bleasby AJ. 1993. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3: 327– 332 [DOI] [PubMed] [Google Scholar]
- 27. de Carvalho KG, Bambirra FH, Kruger MF, Barbosa MS, Oliveira JS, Santos AM, Nicoli JR, Bemquerer MP, de Miranda A, Salvucci EJ, Sesma FJ, Franco BD. 2010. Antimicrobial compounds produced by Lactobacillus sakei subsp. sakei 2a, a bacteriocinogenic strain isolated from a Brazilian meat product. J. Ind. Microbiol. Biotechnol. 37: 381– 390 [DOI] [PubMed] [Google Scholar]
- 28. Holmgren A. 1985. Thioredoxin. Annu. Rev. Biochem. 54: 237– 271 [DOI] [PubMed] [Google Scholar]
- 29. Viana R, Monedero V, Dossonnet V, Vadeboncoeur C, Pérez-Martínez G, Deutscher J. 2000. Enzyme I and HPr from Lactobacillus casei: their role in sugar transport, carbon catabolite repression and inducer exclusion. Mol. Microbiol. 36: 570– 584 [DOI] [PubMed] [Google Scholar]
- 30. van de Guchte M, Serror P, Chervaux C, Smokvina T, Ehrlich SD, Maguin E. 2002. Stress response in lactic acid bacteria. Antonie Van Leeuwenhoek 82: 187– 216 [PubMed] [Google Scholar]
- 31. Champomier-Vergès M-C, Maguin E, Mistou M-Y, Anglade P, Chich JF. 2002. Lactic acid bacteria and proteomics: current knowledge and perspectives. J. Chromatogr. B 771: 329– 342 [DOI] [PubMed] [Google Scholar]
- 32. Rosano C, Zuccotti S, Bucciantini M, Stefani M, Ramponi G, Bolognesi M. 2002. Crystal structure and anion binding in the prokaryotic hydrogenase maturation factor HypF acylphosphatase-like domain. J. Mol. Biol. 321: 785– 796 [DOI] [PubMed] [Google Scholar]
- 33. Corazza A, Rosano C, Pagano K, Alverdi V, Esposito G, Capanni C, Bemporad F, Plakoutsi G, Stefani M, Chiti F, Zuccotti S, Bolognesi M, Viglino P. 2006. Structure, conformational stability, and enzymatic properties of acylphosphatase from the hyperthermophile Sulfolobus solfataricus. Proteins 62: 64– 79 [DOI] [PubMed] [Google Scholar]
- 34. Burns P, Sánchez B, Vinderola G, Ruas-Madiedo P, Ruiz L, Margolles A, Reinheimer J, de los Reyes-Gavilán CG. 2010. Inside the adaptation process of Lactobacillus delbrueckii subsp. lactis to bile. Int. J. Food Microbiol. 142: 132– 141 [DOI] [PubMed] [Google Scholar]
- 35. Söllner K, Schieberle P. 2009. Decoding the key aroma compounds of a Hungarian-type salami by molecular sensory science approaches. J. Agric. Food Chem. 57: 4319– 4327 [DOI] [PubMed] [Google Scholar]
- 36. Imoto T, Yamada H, Okazaki K, Ueda T, Kuroki R, Yasukochi T. 1987. Modifications of stability and function of lysozyme. J. Protein Chem. 6: 95– 107 [Google Scholar]
- 37. Wang NC, Lee CY. 2006. Molecular cloning of the aspartate 4-decarboxylase gene from Pseudomonas sp. ATCC19121 and characterization of the bifunctional recombinant enzyme. Appl. Microbiol. Biotechnol. 73: 339– 348 [DOI] [PubMed] [Google Scholar]
- 38. Stevenson DE, Akhtar M, Gani D. 1990. l-Methionine decarboxylase from Dryopteris filix-mas: purification, characterization, substrate specificity, abortive transamination of the coenzyme, and stereochemical courses of substrate decarboxylation and coenzyme transamination. Biochemistry 29: 7631– 7647 [DOI] [PubMed] [Google Scholar]
- 39. Yorifuji T, Soda K. 1971. Arginine racemase of Pseudomonas graveolens. II. Racemization and transamination of ornithine catalyzed by arginine racemase. J. Biol. Chem. 246: 5093– 5101 [PubMed] [Google Scholar]
- 40. Ahmed SA, Esaki N, Tanaka H, Soda K. 1985. Mechanism of inactivation of α-amino-ε-caprolactam racemase by α-amino-δ-valerolactam. Agric. Biol. Chem. 49: 2991– 2997 [Google Scholar]
- 41. Kurokawa Y, Watanabe A, Yoshimura T, Esaki N, Soda K. 1998. Transamination as a side-reaction catalyzed by alanine racemase of Bacillus stearothermophilus. J. Biochem. 124: 1163– 1169 [DOI] [PubMed] [Google Scholar]
- 42. Häring D, Lees MR, Banaszak LJ, Distefano MD. 2002. Exploring routes to stabilize a cationic pyridoxamine in an artificial transaminase: site-directed mutagenesis versus synthetic cofactors. Protein Eng. 15: 603– 610 [DOI] [PubMed] [Google Scholar]
- 43. Tanizawa K, Soda K. 1979. Inducible and constitutive kynureninases. Control of the inducible enzyme activity by transamination and inhibition of the constitutive enzyme by 3-hydroxyanthranilate. J. Biochem. 86: 499– 508 [DOI] [PubMed] [Google Scholar]
- 44. Baker AS, Ciocci MJ, Metcalf WW, Kim J, Babbitt C, Wanner BL, Martin BM, Dunaway-Mariano D. 1998. Insights into the mechanism of catalysis by the P-C bond-cleaving enzyme phosphonoacetaldehyde hydrolase derived from gene sequence analysis and mutagenesis. Biochemistry 37: 9305– 9315 [DOI] [PubMed] [Google Scholar]
- 45. Engels WJM, Alting AC, Arntz MMTG, Gruppen H, Voragen AGJ, Smit G, Visser S. 2000. Partial purification and characterization of two aminotransferases from Lactococcus lactis subsp. cremoris B78 involved in the catabolism of methionine and branched-chain amino acids. Int. Dairy J. 10: 443– 452 [Google Scholar]
- 46. Yvon M, Thirouin S, Rijnen L, Fromentier D, Gripon JC. 1997. An aminotransferase from Lactococcus lactis initiates conversion of amino acids to cheese flavor compounds. Appl. Environ. Microbiol. 63: 414– 419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Powell JT, Morrison JF. 1978. The purification and properties of the aspartate aminotransferase and aromatic amino acid aminotransferase from Escherichia coli. Eur. J. Biochem. 87: 391– 400 [DOI] [PubMed] [Google Scholar]
- 48. Liu SQ, Holland R, Crow VL. 2003. The potential of dairy lactic acid bacteria to metabolise amino acids via non-transaminating reactions and endogenous transamination. Int. J. Food Microbiol. 86: 257– 269 [DOI] [PubMed] [Google Scholar]
- 49. Häring D, Distefano MD. 2001. Enzymes by design: chemogenetic assembly of transamination active sites containing lysine residues for covalent catalysis. Bioconjug. Chem. 12: 385– 390 [DOI] [PubMed] [Google Scholar]
- 50. Kirsch JF, Eichele G, Ford G, Vincent MG, Jansonius JN, Gehring H, Christen P. 1984. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J. Mol. Biol. 174: 497– 525 [DOI] [PubMed] [Google Scholar]
- 51. Yano T, Kuramitsu S, Tanase S, Morino Y, Kagamiyama H. 1992. Role of Asp222 in the catalytic mechanism of Escherichia coli aspartate aminotransferase: the amino acid residue which enhances the function of the enzyme-bound coenzyme pyridoxal 5′-phosphate. Biochemistry 31: 5878– 5887 [DOI] [PubMed] [Google Scholar]
- 52. Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a Web-based environment for protein structure homology modeling. Bioinformatics 22: 195– 201 [DOI] [PubMed] [Google Scholar]
- 53. Pagano K, Ramazzotti M, Viglino P, Esposito G, Degl'Innocenti D, Taddei N, Corazza A. 2006. NMR solution structure of the acylphosphatase from Escherichia coli. J. Biomol. NMR 36: 199– 204 [DOI] [PubMed] [Google Scholar]
- 54. Percudani R, Peracchi A. 2003. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 4: 850– 854 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.