

Abstract
Phase II metabolic enzymes are a battery of critical proteins that detoxify xenobiotics by increasing their hydrophilicity and enhancing their disposal. These enzymes have long been studied for their preventative and protective effects against mutagens and carcinogens and for their regulation via the Keap1 (Kelch-like ECH associated protein 1) / Nrf2 (Nuclear factor erythroid 2 related factor 2) / ARE (antioxidant response elements) pathway. Recently, a series of studies have reported the altered expression of phase II genes in postmortem tissue of patients with various neurological diseases. These observations hint at a role for phase II enzymes in the evolution of such conditions. Furthermore, promising findings reveal that overexpression of phase II genes, either by genetic or chemical approaches, confers neuroprotection in vitro and in vivo. Therefore, there is a need to summarize the current literature on phase II genes in the central nervous system (CNS). This should help guide future studies on phase II genes as therapeutic targets in neurological diseases. In this review, we first briefly introduce the concept of phase I, II and III enzymes, with a special focus on phase II enzymes. We then discuss their expression regulation, their inducers and executors. Following this background, we expand our discussion to the neuroprotective effects of phase II enzymes and the potential application of Nrf2 inducers to the treatment of neurological diseases.
Keywords: phase II genes, Keap1/Nrf2/ARE, inducers, effectors, acute neurological diseases, neurodegenerative diseases
1 Introduction
Once they enter cells or tissues, drugs or xenobiotics induce a series of compensatory cellular reactions. The purpose of these endogenous reactions is to reduce the potential injury caused by these compounds via their metabolism and excretion. These procedures occur in a stepwise fashion and are mediated by a group of enzymes known as drug metabolizing enzymes (DMEs). Based on the sequential nature of catalysis, DEMs are categorized into three groups, the phase I, II, and III enzymes, respectively. Each group of the DMEs serves distinct roles. Briefly, phase I enzymes oxidize drugs or xenobiotics, while phase II enzymes conjugate products of phase I reactions. In contrast, phase III enzymes transport or extrude the final metabolites out of cells. The distribution and functions of phase I, II, and III enzymes are summarized in Table 1 (Nakata et al., 2006; Xu et al., 2005).
Table 1.
Summary of drug metabolizing enzymes
| Categories | Enzymes | Locations | Functions |
|---|---|---|---|
| Phase I | Aldo-keto reductase (AKR), Carboxylesterases (CES), Cytochrome P450 monooxygenase (CYP), Epoxide hydrolase. | Liver, lung, GI tract, and kidney | Oxidize, reduce or hydrolyze xenobiotics and drugs. |
| Phase II | γ-glutamylcysteine synthetase (GCL) Glutathione peroxidase (GPX) Glutathione S-transferase (GST), Heme oxygenase 1 (HO-1), Menadione reductase (NMO), N-acetyltransferase (NAT), NADPH quinine oxidoreductase 1 (NQO-1), Peroxiredoxin (PRX), Sulfiredoxin (SRXN), Sulfotransferase (SULT), Thioredoxin (Trx), Thioredoxin reductase (TrxR), UDP-glucuronosyltransferase (UGT). | Varies dependingon the specific gene and its subfamily. | Conjugate drug metabolites or endobiotics via acetylation, glucuronidation, glutathionylation, methylation, and sulphation, making them more hydrophilic; or degrade heme or quinone. |
| Phase III | Multidrug resistance associated protein (MRP), Organic anion transporting polypeptide 2 (OATP2), P-glycoprotein (P-gp), Transporters. | Brain, liver, intestine, and kidney. | Transport or excrete drug metabolites out of cells. |
The expression of phase I genes is governed by a number of nuclear receptors, including aryl hydrocarbon receptors (AhR) and orphan nuclear receptors such as constitutive androstane receptors and pregnane X receptors. Each of these receptors binds a particular consensus sequence on its target genes (Xu et al., 2005). For instance, AhR regulates its target genes mainly by interacting with the xenobiotic response element (XRE). The major regulator of phase II genes is nuclear factor erythroid 2 related factor 2 (Nrf2), which binds the antioxidant response elements (ARE) consensus (Itoh et al., 1997). Transcriptional regulation of phase III genes is not fully understood. On the other hand, there is some overlap between the expression regulation of phase I, II, and III enzymes. For example, 3-methylcholanthrene, a carcinogen, induces not only phase I genes, but also phase II and phase III genes (Rushmore and Kong, 2002). One explanation is that the intermediate products of phase I enzymes serve as potential inducers of Nrf2 pathway/phase II enzymes. Another study suggests that DME genes share some common cis-acting elements or trans-acting factors. For instance, AhR can bind both XRE and ARE consensus sequences (Kohle and Bock, 2009). The regulation of phase I, II, and III genes is generally complicated and beyond the scope of this review. Herein, we will mainly focus on phase II enzymes and the Nrf2 pathway, as it is the main regulator of phase II genes (Jancova et al., 2010).
2 Regulation of Nrf2/ARE pathway
The Nrf2/ARE pathway is a major determinant of phase II gene induction. The importance of Nrf2 is evident from reports showing that the levels of phase II genes, such as glutathione s-transferase (GST) and NAD(P)H: quinone oxidoreductase 1 (NQO1), are significantly reduced in Nrf2-deficient mice and that the induction of phase II genes is abolished by Nrf2 disruption (Ramos-Gomez et al., 2001). It has long been known that Keap1 is a negative regulator of the Nrf2/ARE pathway by the formation of a Keap1/Nrf2 complex in the cytoplasm. This complex sequesters Nrf2 away from the nucleus and prevents its downstream effects on phase II genes. However, recent studies also implicate Keap1-independent means of regulating Nrf2/ARE. Therefore, we will discuss both Keap1-dependent and -independent Nrf2/ARE pathways.
2.1 Keap1 dependent Nrf2 pathway
2.1.1 Structural features of Keap1/ Nrf2 in the cytoplasm
Nrf2 belongs to the basic leucine zipper transcription factor family, which also contains NF-E2, Nrf1, Nrf3, Bach1, and Bach2 (Motohashi et al., 2002). Nrf2 is composed of six functional domains known as Nrf2-ECH homologies (Neh) and designated as Neh1-6, respectively. Each Neh domain serves its own function and the details have been well summarized by Baird and colleagues (Baird and Dinkova-Kostova, 2011).
Keap1 (Kelch-like ECH associated protein 1) is composed of three functional domains: a bric-a-brac (BTB) domain, an intervening region (IVR), and a Kelch domain (also named DGR domain). Keap1 forms a homodimer and each dimer binds one molecule of Nrf2 via its two Kelch domains, with one weak affinity binding site (DLG motif, residues 24-31, latch) and one high affinity binding site (ETGF motif, residues 78-82, hinge). Both motifs are located in the Neh2 domain of Nrf2 (Tong et al., 2007) (Figure 1). The ETGF motif (KD ≈ 1000 nmol/L) has a higher affinity for Keap1 than the DLG motif (KD ≈ 5.3 nmol/L) (Tong et al., 2006). This is the so-called “hinge-and-latch” model. The Keap1-Nrf2 complex is linked to a functional E3 ubiquitin ligase complex (Rbx1) via an adaptor protein, Cullin3. Conjugating Nrf2 with Keap1 by the two DLG and ETGF motifs aligns the seven lysine residues of Nrf2 between the two motifs and facilitates Rbx1 mediated ubiquitination of Nrf2 (McMahon et al., 2006). Ubiquitinated Nrf2 is subsequently degraded by the 26S proteasome. As a consequence, the binding of Keap1 homodimers to Nrf2 can be considered inhibitory to Nrf2 function because it effectively facilitates Nrf2 degradation.
Figure 1. Classic model of Keap1/Nrf2/ARE signaling.
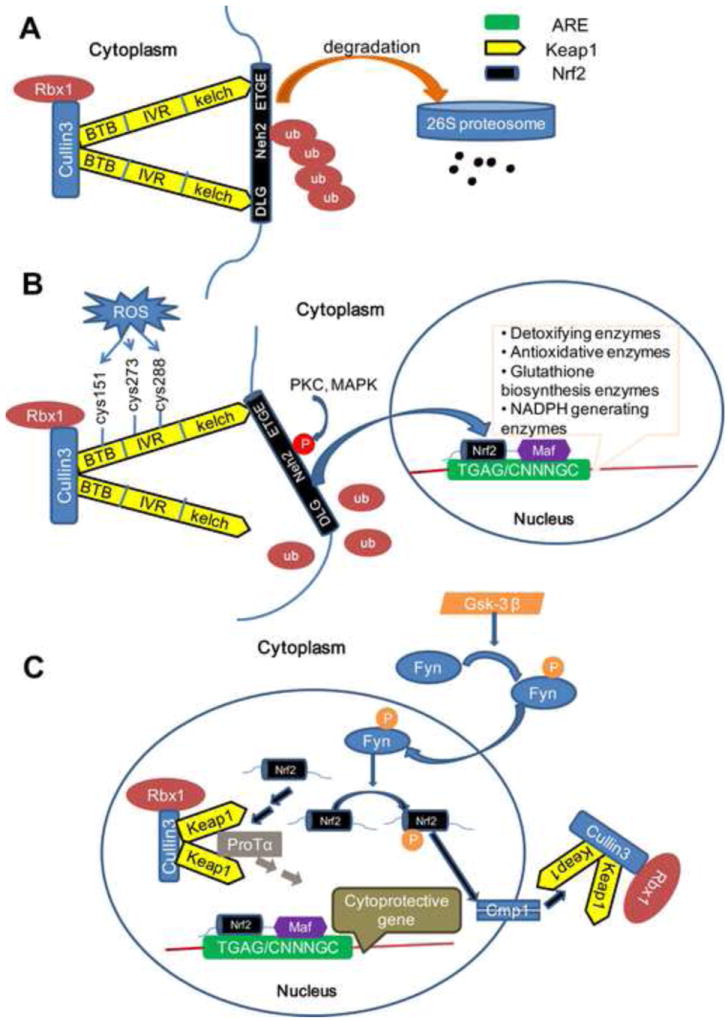
(A) Under basal conditions, the Cul3-Keap1 complex sequesters Nrf2 in the cytosol by binding its ETGF and DLG motifs. This facilitates the ubiquitination and proteasomal degradation of Nrf2. (B) The DLG motif of Nrf2 is loosened from the Cul3-Keap1 complex when cells are exposed to ROS which blocks the ubiquitination and degradation of Nrf2. Following an intricate series of phosphorylations by several kinases, Nrf2 translocates into the nucleus and subsequently binds to the ARE elements by forming a heterodimer with Maf protein and initiating the transcription of phase II genes. (C) Nuclear Nrf2 can be phosphorylated by Fyn and be extruded back to the cytoplasm through the Cmp1 system. On the other hand, nuclear Nrf2 may also be sequestered by several Cul3-Keap1 complexes in the nucleus that are imported by ProTα. Both of these mechanisms help cells return back to basal conditions.
2.1.2 Dissociation of Nrf2-Keap1 complex
One widely accepted mechanism of Nrf2/ARE activation involves the dissociation of the Nrf2-Keap1 complex in the cytoplasm. Endowed with high reactive cysteine residues, Keap1 can modulate Nrf2 dependent gene expression by serving as a sensor of various chemical signals. Specifically, two cysteine residues (Cys273, Cys288) in the IVR domain are essential for Keap1 to bind and suppress the activity of Nrf2, whereas C151 in the BTB domain exerts the opposite effect (Yamamoto et al., 2008). The oxidation of these cysteine residues affects the conformation of Keap1 and thereby initiates the dissociation of Keap1 from the DLG motif of Nrf2. In this way, oxidative products enhance the stability of Nrf2 and increase the expression of phase II genes (McMahon et al., 2006).
2.1.3 Nuclear translocation of Nrf2 and activation of Nrf2 pathway
As a transcription factor, it is essential that Nrf2 translocates to the nucleus in order to transactivate. Given its critical role in sensing stress, it is not surprising that this translocation is quite rapid. Indeed, Nrf2 accumulates in the nucleus within 15 min after tert-Butylhydroquinone (t-BHQ) treatment (Jain et al., 2005). However, it is still not fully clear how Nrf2 translocates to nucleus. The key mediators that regulate nuclear import and export of transcription factors are the nuclear localization signals (NLS) and nuclear export sequences (NES). These sequences interact with the nuclear pore complex. A number of such nuclear shuttling signals have been identified on Nrf2, including three NLS motifs, named NLS1, NLS2 and NLS3, and two NES motifs, named NES1, NES2 and NES3 based on their location (Figure 2). In murine Nrf2, NLS1 contain residues 42–53 (RQKDYELEKQKK) in the Neh2 domain (Theodore et al., 2008), NLS2 contains residues 494-511 (RRRGKQKVAANQCRKRK) in the Neh6 domain (Jain et al., 2005) and NLS3 contains residues 587–593 (PKSKKPD) in the Neh3 domain (Theodore et al., 2008). On the nuclear export side, NES1 contains residues 175-186 (LLSIPELQCLNI) in the Neh5 domain, which is redox sensitive as it has a cysteine residue (Cys183) (Li et al., 2006), and NES2 contains residues 545-554 (LKRRLSTLYL) in Neh1 (Jain et al., 2005). This may correspond to residues 537-546 (LKKQLSTLYL) in human Nrf2 (Li et al., 2005).
Figure 2. Structure of Nrf2: domain, phosphorylation sites and nuclear shuttling signals.
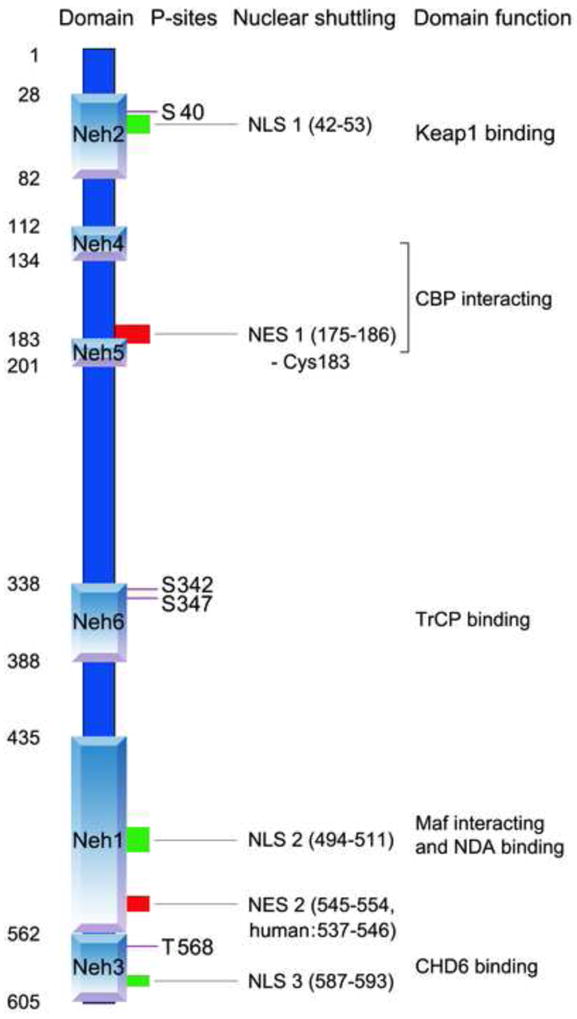
Nrf2 has six domains. From N-terminus to C-terminus, these include Neh2, Neh4, Neh5, Neh6, Neh1 to Neh3. Four phosphorylation sites have been pinpointed - Ser40 in Heh2 domain by PKC, Ser342/Ser347 in Neh6 by GSK3! and Tyr 568 in Neh3 by Fyn. Three nuclear localization signals (NLS) have been identified - NLS1 located in Neh2, NLS2 located in Neh1, and NLS3 located in Neh 3. Two nuclear export sequences (NES) have also been identified - NES1 partially overlapped with Heh5, which contains a cysteine (Cys183) and NES2 located in Neh1. Purple line: phosphorylation sites; green box: NLS; red box: NES. CBP: CREB-binding protein; CHD6: chromo-ATPase/helicase DNA binding protein family member 6 (both CREB? and CHD6 are transcriptional co-activators); Maf: musculo-aponeurotic fibrosarcoma; TrCP: beta-transducin repeats-containing proteins, an E3 ligase.
The direction of Nrf2 movement seems to be determined by a homeostatic balance between import and export driving forces. It has been proposed that redox sensitive NES1 may play an important role in this equilibrium (Li and Kong, 2009; Li et al., 2006). Under normal conditions, the import force is less than the export force while NES1 is functional, and Nrf2 stays in the cytosol; under oxidative or electrophilic stressors, Cys183 of NES1 is adducted and NES1 becomes inoperative. This import force then overwhelms the export force, leading to Nef2 nuclear translocation (Li and Kong, 2009). Additionally, Nrf2 phosphorylation may also contribute to Nrf2 nuclear transportation. Nrf2 is the substrate of several protein kinases, including protein kinase C (PKC) (Huang et al., 2002; Numazawa et al., 2003), phosphatidylinositol 3-kinase (PI3K) (Lee et al., 2001), glycogen synthase kinase-3 (GSK3! ! (Rada et al., 2012), casein kinase 2 (CK2) (Apopa et al., 2008), extracellular signal-regulated kinase (ERK) (Yu et al., 1999), p38 (Yu et al., 2000), PKR-like endoplasmic reticulum kinase (PERK) (Cullinan et al., 2003) and Fyn (Jain and Jaiswal, 2006). Among them, the phosphorylation sites of PKC, GSK3! and Fyn have been identified. PKC phosphorylates Ser40 in Neh2 domain (Huang et al., 2002) but its role in Nrf2 nuclear shuttling is controversial. It has been reported that the nuclear translocation of a Nrf2 S40A mutant protein was decreased compared to wild-type protein after electrophilic stress, indicating a role for Ser40 phosphorylation in nuclear import (Numazawa et al., 2003). Another report showed that Ser40 was necessary for the release of Nrf2 from Keap1, although it was not required for Nrf2 accumulation in the nucleus (Bloom and Jaiswal, 2003). GSK3β phosphorylates Nrf2 at Ser342 and Ser347, mediating Nrf2 degradation via another E3 ligase, beta-transducin repeats-containing protein (TrCP) (Rada et al., 2012). Fyn phosphorylates Tyr568 in the Neh3 domain, faciliating nuclear export of Nrf2 (Jain and Jaiswal, 2006; Rada et al., 2011). The activity of Fyn itself is controlled by GSK3β (Jain and Jaiswal, 2007; Rojo et al., 2008), suggesting an important role of GSK3β in the regulation of Nrf2 nuclear shuttling.
In summary, the Cul3-Keap1 complex sequesters Nrf2 in the cytoplasm and facilitates the ubiquitination and proteasomal degradation of Nrf2 under quiescent conditions (Kobayashi et al., 2004). Indeed, the half-life of Nrf2 in non-stressed, physiological conditions is only about 20 min (Itoh et al., 2003). When exposed to oxidants or electrophiles, Nrf2 is rapidly liberated from the Cul3-Keap1 complex and translocated into the nucleus, where it forms a heterodimer with a small musculo-aponeurotic fibrosarcoma (Maf) protein through its Neh1domain and subsequently binds ARE (Itoh et al., 1997) (Figure 1). ARE is a cis-acting DNA response sequence located in the regulatory regions of phase II genes with the consensus of TGAG/CNNNGC (N represents any base) (Nguyen et al., 2003). In this way, oxidants or electrophiles activate Nrf2 and up-regulate phase II genes in order to compensate against their damaging effects (Baird and Dinkova-Kostova, 2011).
2.2 Keap1 independent Nrf2 pathway
Recent studies have also proposed a Keap1 independent ubiquitination model of Nrf2 degradation (Rada et al., 2011; Rojo et al., 2012). In this model, GSK- 3β phosphorylates the Neh6 domain of Nrf2 at Ser342 and 347 (Rada et al., 2012); phosphorylated Neh6 on Nrf2 can be recognized by and act as a bait for β-TrCP, an E3 ubiquitin ligase. β-TrCP is a scaffolding protein that directly links Nrf2 to the Cullin1/Rbx1 ubiquitination complex. Therefore, GSK-3β mediated phosphorylation of Neh6 causes the ubiquitination and degradation of Nrf2 via β-TrCP in place of Keap1. This model is supported by the stabilization of Nrf2 by GSK-3β inhibitors in Keap1 -/- mouse embryo fibroblasts (MEFs) (Rada et al., 2011). Additionally, cancer-chemopreventive agent nordihydroguaiaretic acid can activate Nrf2 and increase HO-1 protein levels through inhibiting GSK-3β phosphorylation in Keap1-/- MEFs (Rojo et al., 2012). However, this pathway needs to be better examined, as current studies are only limited to cultured cell lines. Efforts should be extended to physiological and pathological animal models to further evaluate this regulatory mechanism.
2.3 Inactivation of Nrf2 pathway
When challenged with oxidative products, the activated Nrf2/ARE pathway boosts the expression of phase II genes either by Keap1 dependent or independent means. Under these circumstances, cells also initiate endogenous regulatory mechanisms to quench this pathway to prevent excessive activation. Several contributors participate in this negative feedback loop. First, AREs are located in the promoter region of Cul3, Rbx1 and Keap1 genes. Studies show that these inhibitory proteins are subject to transcriptional regulation by Nrf2. In other words, activation of Nrf2 enhances the expression of Rbx1-Cul3-Keap1 complex, which acts to sequester Nrf2 and mediate its rapid degradation (Kaspar and Jaiswal, 2011; Lee et al., 2007). This negative feedback loop is known as an autoregulatory arm of the Nrf2/ARE pathway. Second, prothymosinα (ProTα), a Keap1 binding protein with a nuclear localization signal, can mediate the nuclear import of the Keap1-Cul3-Rbx1 complex. As a result, 10-15% of Keap1-Cul3-Rbx1 complex is localized in the nucleus. Once it enters the nucleus, the Keap1-Cul3-Rbx1 complex releases ProTα and binds Nrf2, leading to the ubiquitination and degradation of nuclear Nrf2 (Niture and Jaiswal, 2009). Third, oxidative stressors such as hydrogen peroxide promote the activation of GSK-3β by phosphorylating its tyrosine 216 residue. Activated GSK-3β subsequently phosphorylates Fyn (p-Fyn, a member of Src family) at a threonine residue(s), leading to the accumulation of p-Fyn in the nucleus and subsequent phosphorylation of Nrf2 at tyrosine 568. Ultimately, phosphorylated Nrf2 interacts with Crm1 or exportin 1 and is exported out of nucleus (Jain and Jaiswal, 2006, 2007) (Figure 1C). These compensatory factors all complement each other and guide the cell back towards homeostasis.
2.4 Proteins that directly regulate Nrf2/ARE signaling
Multiple proteins are involved in the regulation of the Nrf2/ARE signaling pathway. Some proteins exert their functions by directly modifying the Keap1-Nrf2 complex in the cytoplasm, others function in the nucleus. One example of cytosolic regulation involves p21Cip1/WAF1, an essential protein that protects cells from oxidative stress. Recent findings suggest that p21Cip1/WAF1 directly binds the DLG motif of Nrf2 through its C-terminal KRR motif. The DLG motif is also the binding site of Keap1. The competition between p21Cip1/WAF1 and Keap1 for Nrf2 binding compromises Keap1 mediated ubiquitination of Nrf2 (Chen et al., 2009b). This has been confirmed in p21 deficient mice, which demonstrate reduced expression of Nrf2 and Nrf2 target genes (Chen et al., 2009b; Toledano, 2009). Separately from p21Cip1/WAF1, another protein sequestosome-1(also known as p62) binds and inactivates Keap1 and thus also augments the expression of genes regulated by Nrf2 (Komatsu et al., 2010). Specifically, the Ser349 of sequestosome-1 is the key residue in regulating binding activity of sequestosome-1 to Keap1 (Hancock et al., 2011).
Other proteins affect Nrf2 activity in the nucleus. For instance, BACH1 has been identified as a repressor of Nrf2. Under basal conditions, BACH1 forms a heterodimer with the Maf protein and this complex subsequently occupies the ARE sequences and negatively regulates several phase II genes such as heme oxygenase 1 (HO-1). When cells are challenged by oxidative insults, BACH1 is phosphorylated and exported to the cytoplasm. Free Maf protein then forms a heterodimer with Nrf2 and triggers the expression of downstream genes (Dhakshinamoorthy et al., 2005; Sun et al., 2004).
2.5 Kinases involved in modulating Nrf2 transcriptional activity
Phosphorylation is another key mechanism that regulates Nrf2 dependent gene expression, and as discussed above, several protein kinases can phosphorylate Nrf2 (Figure 2). As will be discussed below, some kinases such as PKC and PI3K increase Nrf2/ARE transcription; other kinases such as the mitogen-activated protein kinase (MAPK) family play different roles in Nrf2/ARE activation depending on cell type.
It has been shown that the PKC activator phorbol 12-myristate 13-acetate (PMA) increases Nrf2 phosphorylation as well as ARE transcriptional activity, whereas the PKC inhibitor staurosporine down-regulates these effects in HepG2 cells (Huang et al., 2000). PKC phosphorylates Nrf2 at Ser40. Although this does not affect Nrf2 binding with ARE, it promotes its dissociation from Keap1 (Huang et al., 2002). This phosphorylated residue is located in the Neh2 domain of Nrf2, which is critical for the interaction with Keap1. As a result, it has been suggested that a conformational change of Nrf2 secondary to its phosphorylation may lead to its dissociation from Keap1 (Bloom and Jaiswal, 2003). In addition to PKC, studies attempting to address the relationship between PI3K signaling and the Nrf2 pathway showed that PI3K lies upstream of Nrf2 and positively regulates its transcriptional activity in IMR-32 human neuroblastoma cells (Lee et al., 2001) and primary cortical cultures (Kraft et al., 2004), although the detailed mechanisms are not yet fully understood.
Because the MEK inhibitor PD98059 impairs ARE-dependent phase II gene expression in HepG2 cells, ERKs are also thought to upregulate phase II genes (Yu et al., 1999). On the other hand, modulation of ERK1/2 activity does not affect Nrf2-dependent ARE activation in IMR-32 human neuroblastoma cells (Lee et al., 2001). These discrepancies may be explained by the different cell types used in those studies. Another MAPK, p38, demonstrates opposite effects on Nrf2 activation depending on the model (Correa et al., 2011). Although it functions as a negative regulator in human hepatoma HepG2 and murine hepatoma Hepa1c1c7 cells (Yu et al., 2000), it acts as a positive regulator in MCF-7 mammary epithelial cells (Alam et al., 2000). MAPK seems to regulate Nrf2-dependent gene transcription by post-translational modification of CBP (CREB-binding protein) and/or other unidentified transacting factors or co-activators that bind to the Nrf2 transcription machinery, in addition to directly phosphorylating Nrf2 (Shen et al., 2004).
In aggregate, ubiquitination of Nrf2 either by Keap1 dependent or independent means is a fundamental mechanism to inhibit Nrf2. Activation of Nrf2 is initiated by the dissociation of Nrf2 from Keap1, preventing its ubiquitination, and causing its translocation into the nucleus. Through its exquisitely coordinated control by multiple kinases and proteins, Nrf2 ultimately binds with ARE and triggers phase II gene expression.
3 Inducers
As will be discussed below, the upregulation of phase II enzymes is protective against several models of neurological diseases. Therefore, it is important to identify and classify inducers of the Nrf2 pathway for subsequent translation of these compounds to the clinic. As described above, Keap1 is the major inhibitor of Nrf2 activation; variation in the structure of Keap1 or/and dissociation of Keap1 from Nrf2 results in the release and activation of Nrf2 (Figure 1 and 3A). This is the target mechanism of nearly all known Nrf2 inducers and might involve modulating the cysteine residues of Keap1 or blocking their binding sites.
Figure 3.
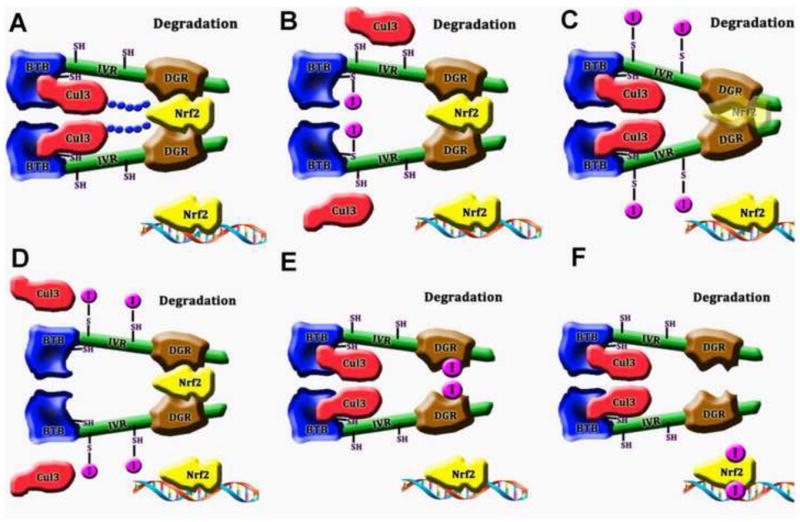
Mechanisms of Nrf2 activation by different classes of inducers (
 : Inducers
: Inducers
 : Ubiquitin)
: Ubiquitin)
(A) Without inducers, Keap1 binds Nrf2 and facilitates the degradation of the Nrf2-Keap1 complex via Cul3. Under these circumstances, Nrf2 remains bound to Keap1 and is not free to translocate to the nucleus. (B) Inducers that act on the BTB domain. These interactions lead to structural changes of Keap1, blocking the binding of Cul3 to Keap1. Nrf2 is thereby protected from ubiquitinization. Consequently, the available pool of Nrf2 increases in size. (C) Inducers that act on IVR and interrupt the association of Keap1 with Nrf2. Translucent Nrf2 indicates that it cannot bind to Keap1. (D) Inducers that also act on IVR but interrupt the association of Keap1 with Cul3. (E) Inducers that act on the DGR domain and block the binding site of Nrf2. (F) Inducers that phosphorylate Nrf2.
The inducers of phase II enzymes can be classified into three ways based on their origin, chemical structure, and the reaction cascades that they ignite. Based on their origin, inducers can be divided into two classes, exogenous and endogenous. Exogenous inducers include xenobiotics, drugs, and heavy metals; endogenous inducers include lipid peroxidation products, nitric oxide and derivatives as well as prostaglandins and derivatives (Kobayashi et al., 2009; McMahon et al., 2010; Yu et al., 2006). The second method to classify the inducers is based on chemical structure. Holtzclaw and co-workers divide them into 10 groups, which include oxidizable phenols, Michael reaction acceptors, isothiocyanates, thiocarbamates, trivalent arsenicals, 1,2-dithiole-3-thiones, hydroperoxides, vicinal dimercaptans, heavy metals, and polyenes (Holtzclaw et al., 2004).
Based on their effects on the Keap1-Nrf2 complex, Nrf2 inducers have also been divided into 6 classes as suggested by Kobayashi and colleagues (Kobayashi et al., 2009). This is a useful way to group Nrf2 inducers as it combines both structural and mechanistic aspects. However, with advances in this field, this taxonomy can be further improved. We propose a four-class categorization, based on the Keap1 domains that the inducers react with (Table 2).
Table 2.
Classification of Nrf2 inducers
| Class | I | II | III | IV |
|---|---|---|---|---|
| Target domain | BTB | IVR | DGR | IVR &DGR |
| Target amino acids | C151 | C273, C288 | Y334, N382, S363, S602 | H225, C226, C613 |
| Chemical characteristics | Multiple: NO and small molecules with a sulfydryl | Alkenals, prostaglandin and derivatives | Proteins | Heavy metal |
| Sources | Mostly exogenesis | Endogenesis | Endogenesis | Exogenesis |
| Mechanism | Free from Cul3 | Free from Cul3 or Keap1 | Free from Keap1 | Unknown |
3.1 Inducers that act on the BTB domain
The BTB domain of Keap1 plays two important roles. It is thought to serve as a dimerization domain, maintaining the dimer structure of Keap1 because mutation of Ser140 in BTB leads to the dedimerization of Keap1 and subsequent release of Nrf2 (Zipper and Mulcahy, 2002). Furthermore, the BTB domain is also the binding site for Cul3 (Eggler et al., 2009).
A critical amino acid residue of Keap1 is Cys151. As a nucleophile, Cys151 is sensitive to many inducers and is therefore considered to be a stress sensor (McMahon et al., 2010; Zhang and Hannink, 2003). Modulation of Cys151 leads to structural changes in Keap1, separating it from Cul3 (Eggler et al., 2009). As a result, Nrf2 ubiquitination is prevented, and isolated Nrf2 is then free to translocate to the nucleus and induce phase II genes (Figure 3B).
Inducers that act on the BTB domain include tBHQ, 1,2-dithiole-3-thiones (D3T), Michael reaction acceptors such as 4-HNE and ebselen, oxidizable phenols such diethylmaleate (DEM) and naphthoquinone (1,2-NQ), and isothiocyanates such as sulforaphane (SFN) (Kobayashi et al., 2009; McMahon et al., 2010). Additionally, some small molecular inducers, such as H2O2, NO, and HOCl, all demonstrate C151-dependent inhibition of Keap1 (Fourquet et al., 2010). Co2+ can replace Zn2+ in the BTB domain and also reconfigure Keap1 (Dinkova-Kostova et al., 2005). Dihydro-CDDO-trifluoroethyl amide (CDDO-TFEA), a newly identified potent inducer, also belongs to this group (Ichikawa et al., 2009).
3.2 Inducers that act on the IVR domain
The IVR domain is enriched with cysteines. It is therefore a potential site for modulation by inducers. Two cysteines, Cys273 and Cys288 are indispensable for Keap1 activity (McMahon et al., 2010; Yamamoto et al., 2008; Zhang and Hannink, 2003). Oxidation of these cysteines changes the structure of Keap1 and reduces its affinity for Nrf2 (Wakabayashi et al., 2004) (Figure 3C). Additionally, Keap1 structural changes caused by oxidization of these two cysteines may also dissociate Cul3 from Keap1, as the N-terminal of the IVR domain is also a Cul3 binding site (Kobayashi et al., 2006) (Figure 3D).
Most inducers in this group are Michael reaction acceptors, including 5d-PGJ2, PGA2 (Kobayashi et al., 2004), and alkenals such as 4-HNE and acrolein (McMahon et al., 2010). They represent two kinds of endogenous inducers and all of them are derived from polyunsaturated fatty acids.
3.3 Inducers that act on the DGR domain
The DGR domain is composed of 6 Kelch repeats. It binds the Neh2 domain of Nrf2 and also associates with cytoskeletal actin. Keap1 with a mutated DGR domain fails to bind either Nrf2 or actin (Kang et al., 2004).
Inducers in this group interrupt the binding of Nrf2 to Keap1. Two members, both of which are proteins, have been identified. One is p62 (Komatsu et al., 2010) and another is p21 (Chen et al., 2009b). P62 binds the DGR motif of Keap1, leading to the separation of Nrf2 and Keap1 (Komatsu et al., 2010). Eight amino acids of the DGR motif, especially Y334, S363, N382, and S602, are important for the interaction of Keap1 with p62 (Komatsu et al., 2010). Mechanistically, p21 competes with Keap1 to directly bind the DLG and ETGE motifs of Nrf2 (Chen et al., 2009b). Additionally, some kinases can also be considered inducers in this group. For example, MAPK, PI3K, PKC can phosphorylate Nrf2 amino acids and change its conformation to prevent the association with Keap1 (Taguchi et al., 2011) (Figure 3). It is not surprising that the PI3K and MAP kinase families of proteins lie upstream of Nrf2 activation in some models, because these proteins are highly sensitive to cellular stress and often determine the balance of pro-survival or apoptotic signaling cascades. By collaring Nrf2 and phase II enzymes into the cellular response to stress, these proteins can help determine cellular fate under conditions of injury.
3.4 Inducers acting on multiple domains
Heavy metal such as Hg2+, Cd2+, Zn2+, As3+, and Se4+ can also activate Nrf2 (McMahon et al., 2010; Prestera et al., 1993). A recent study showed that at least three amino acids from two domains are simultaneously necessary to sense the presence of heavy metals. These include the His225/Cys226 dyad from the IVR domain and Cys613 from the DGR domain (McMahon et al., 2010).
To summarize, Nrf2 inducers mainly target Keap1 or target its binding with Nrf2. They can be divided into 4 groups based on the Keap1 domains that they react with. However, this taxonomy is not perfect because some inducers can be classified into more than one group, such as 4-HNE and 15d-PGJ2 (Eggler et al., 2009; McMahon et al., 2010). Thus, the classification of inducers needs to be further explored.
4 Effectors
Currently, over two hundred Nrf2/ARE driven genes are exploited for detoxification and antioxidant defense. Obviously, a description of the roles of each of these effectors is beyond the scope of this review. Phase II genes initially caught the interest of scientists for their preventative action against carcinogens (Zhang et al., 1992). Furthermore, their function has recently been expanded to neuroprotection (Kraft et al., 2006; Shih et al., 2005). Below, we will focus our discussion on the phase II enzymes that participate in neuroprotection against models of common neurological diseases.
4.1 Thioredoxin enzyme system
Thioredoxin (Trx) enzyme systems are fundamental guards in defense against oxidative stressors. They reduce disulfide bridges of various proteins and remove H2O2 or peroxides by utilizing NADPH as the electron donor (Patenaude et al., 2005). This system is mainly comprised of Trx, Trx reductase (TrxR), peroxiredoxin (Prx), and sulfiredoxin (Srxn). The central core of this system is Trx, while other members act to keep Trx functional. The graphic depiction of this cascade is illustrated in Figure 4. Molecules subjected to regulation by Nrf2 are highlighted in red.
Figure 4. The impact of Nrf2 on the antioxidant activities of Trx system.
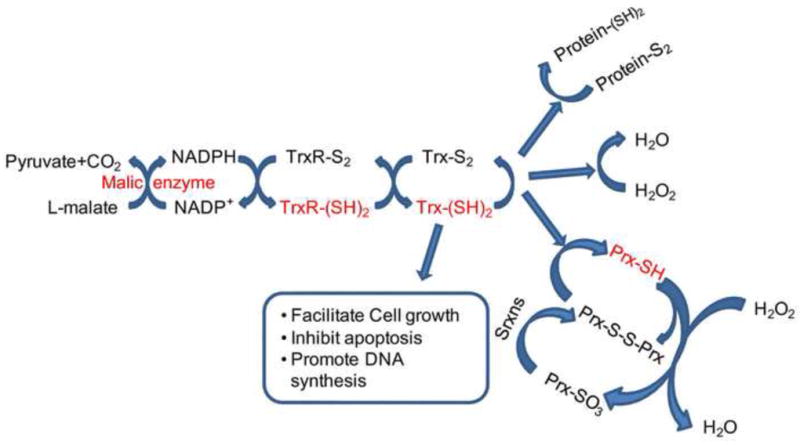
Trx can be oxidized to Trx-S2. This helps to reduce Protein-S2, H2O2 and Prx-S-S-Prx. Because Prx-SO3 cannot be reduced by Trx, Sfx restores Prx-SO3 back into the Trx cycle. Trx-S2 is reduced by TrxRs, with NADPH as the electron donor. The hydrogen and electron necessary for NADPH restoration come from pyruvate and the L-malate reaction cycle, which is catalyzed by the malic enzyme. The molecules that are subject to the control of Nrf2 are highlighted in red and include malic enzyme, TrxR-(SH)2,Trx-(SH)2, Prx-SH, Srxns. Abbreviations: GSH: glutathione; Prx: peroxiredoxin; Srxn: sulfiredoxin; Trx: thioredoxins; TrxR: thioredoxin reductase.
4.1.1 Thioredoxins(Trxs)
Trxs are 12-kDa enzymes initially reported in 1964 as hydrogen donors for ribonucleotide reductase in Escherichia coli (Laurent et al., 1964). Subsequent research demonstrated that mammalian Trxs contain a conserved Cys-X-X-Cys motif in their active center, and established their role in reducing oxidized proteins via the exchange between cysteine thiol and protein disulfides (Powis and Montfort, 2001). Currently, two isoforms of Trxs have been identified – Trx1 and Trx2. Trx1 localizes to the cytosol and Trx2 localizes to the inner membrane of mitochondria (Rybnikova et al., 2000). Both of them are widely distributed in the central nervous system, including piriform cortex, the dentate gyrus, the CA3/CA4 region of the hippocampal formation, the locus coeruleus, as well as the paraventricular hypothalamic nucleus and the nucleus of the solitary tract in rat brain (Lippoldt et al., 1995). This distribution pattern is probably related to the high metabolic activity of these brain regions and consequent oxidative stress.
Evidence shows that the induction of Trxs by heme or tBHQ is dependent on the activation of Nrf2/ARE in cultured cells (Kim et al., 2003; Nakaso et al., 2003). Trx1 is subject to the regulation of other transcriptional elements such as SP1, GCF, AP-1 (Masutani et al., 1996). In contrast, the transcriptional regulation of Trx2 is remains to be elucidated.
Trxs have been shown to confer neuroprotection in vitro and in vivo. Trxs in the submicromolar range prevent the apoptosis of neuronal SH-SY5Y cells invoked by serum deprivation or by MPP+(Andoh et al., 2002; Chen et al., 2006). Overexpression of either Trx1 or Trx2 significantly reduces retinal ganglion cell death in rat models of glaucoma (Caprioli et al., 2009; Munemasa et al., 2009). Transgenic mice over-expressing Trxs have attenuated focal ischemic brain damage (Takagi et al., 1999). Promising findings of translational relevance reveal that intravenously infused human Trx (rhTrx) permeates the blood-brain barrier in the ischemic hemisphere and exerts neuroprotective effects in the mouse middle cerebral artery occlusion (MCAO) model (Hattori et al., 2004).
In addition to their direct roles in attenuating oxidative damage, Trxs also perform several other biological functions which may contribute to their neuroprotective effects. For example, reduced Trxs form a complex with apoptosis signal-regulating kinase 1 (ASK1) and negatively regulate the ASK1/JNK/p38 apoptotic pathway (Hu et al., 2011). Trx1 can be translocated into the nucleus and enhance the biosynthesis of Mn-superoxide dismutase (Mn-SOD) and mitochondrial anti-apoptotic Bcl-2 (Andoh et al., 2002). Moreover, recent data demonstrated that Trx1 and Trx2 are able to facilitate ischemia-induced angiogenesis, which may promote recovery from ischemic brain injury by increasing reperfusion (Dunn et al., 2010; Takagi et al., 2011).
4.1.2 Thioredoxin reductases (TrxRs)
TrxRs are a group of selenocysteine-containing enzymes that reduce oxidized Trxs by consuming NADPH (Mustacich and Powis, 2000; Patenaude et al., 2005) (Figure 4). A remarkable upregulation of Trxs and TrxRs is detected in the rat retina following exposure to bright-cyclic-light-reared stimulation. This is coupled to increased nuclear translocation of Nrf2 and binding with the ARE sequence. Conversely, TrxR-deficient animals showed increased accumulation of nuclear Nrf2 protein, which may be due to a compensatory antioxidative effect (Suvorova et al., 2009). Furthermore, Nrf2 dependent expression of Trxs and TrxRs is enhanced by sublethal doses of HNE (5uM), which protect 661W cells (Tanito et al., 2007). Collectively, these studies support the concept that TrxRs are subject to regulation by Nrf2/ARE and are indispensable in neuroprotection.
4.1.3 Peroxiredoxins (Prxs)
Prxs, also known as thioredoxin peroxidases, catalyze the reduction of peroxides by utilizing Trxs (Figure 4). Six isoforms of Prxs (Prx 1-6) exist in the CNS and can be classified into two groups: 2-Cys Prxs (Prx1-Prx5) and 1-Cys Prx (Prx1) (Rhee and Woo, 2011). Nrf2 was identified as a critical transcription factor for induction of Prx 6 in primary murine bone marrow-derived macrophages (Erttmann et al., 2011) as well as in a human lung-derived cell line (Chowdhury et al., 2009). Similarly, induction of Prx1 is also subject to Nrf2 regulation in other models (Ishii et al., 2000). However, Nrf2 has not been implicated in the induction of the other isoforms. Several reports suggest that Prxs are highly neuroprotective. For example, recent studies showed that enhancement of Prx1 expression by icariin confers neuroprotection against H2O2 in primary cortical neurons (Zhang et al., 2010b) and that transgenic overexpression of Prx2 protects brain against ischemic injury (Gan et al.) and models of Parkinson’s disease (Hu et al., 2011). Prx2 also contributes to the protective effects of probucol and atorvastatin against stroke (Du et al., 2012).
4.1.4 Sulfiredoxin (Srxn)
Srxn is an enzyme that works upstream of 2-Cys Prxs (Woo et al., 2005). Srxn reduces the sulfinic acid phosphoric ester on oxidized Prxs in an ATP-dependent manner, thereby reactivating Prxs (Jonsson et al., 2008; Rhee and Woo, 2011). In other words, Srxn is a partner of Prxs. Since Trxs cannot reduce Prx-SO3, Srxns are very important in restoring Prx-SO3 back to the thioredoxin cycle and preventing permanent oxidative inactivation of Prxs after exposure to strong oxidation (Figure 4) (Jonsson et al., 2008). Both D3T and SFN up-regulate Srxn in primary cortical neurons and glia, and Nrf2 directly regulates Srxn1 expression via a cis-acting ARE, as reported in a Srxn-Luc reporter gene study (Bae et al., 2009; Soriano et al., 2008). Furthermore, a recent study show that ischemic preconditioning increases the transcription of Srxn, suggesting a neuroprotective role for Srxn (Bell et al., 2011)
4.2 Glutathione system (GSH)
GSH is composed of three animo acids- glutamic acid, cysteine and glycine, and is well known for its anti-oxidant role in the CNS. In fact, GSH is so important to redox homeostasis that it is present in the millimolar range in many cell types. GSH scavenges multiple oxidative species such as superoxide, NO, hydroxyl radical, and ONOO (Aoyama et al., 2008); it also serves as a reservoir for cysteine to protect against toxicity secondary to high cysteine concentrations (Janaky et al., 2000). In addition to GSH itself, the GSH redox system contains three groups of enzymes that catalyze its biosynthesis, transfer GSH to its substrates, and catalyze the reduction of oxidized GSH. Nrf2 governs the expression of these GSH-related enzymes. Because GSH is so fundamental for cellular self-defense, Nrf2 plays an important protective role in the maintenance of cellular redox state.
The synthesis of GSH requires the sequential action of two enzymes. The first enzyme, γ-glutamylcysteine synthetase (GCL) ligases glutamic acid to cysteine, the rate-limiting step in GSH biosynthesis. The second enzyme, glutathione synthetase, adds glycine to form the final GSH product. In primary glial and neuronal cultures, overexpressing Nrf2 via adenovirus increases GSH synthesis as well as the expression of GSH synthetase (Shih et al., 2003). Nrf2 also controls the expression of some membrane transporters that transport the raw materials required for GSH synthesis. One example is the excitatory amino acid carrier 1 (EAAC1), which transfers cysteine, the rate limiting substrate for GSH biosynthesis, into neurons. The transcription of EAAC1 is subject to regulation by Nrf2 both in vitro and in vivo (Escartin et al., 2011). Furthermore, disruption of either EAAC1 or Nrf2 perturbs the synthesis of neuronal GSH (Escartin et al., 2011).
The transfer of GSH to its substrates is mediated by several enzymes. Two specific enzymes that catalyze this process are glutathione peroxidase (Gpx) (Cho et al., 2005; Singh et al., 2006) and glutathione S-transferases (GST) (Shih et al., 2003). There are eight isoenzymes of Gpx, known as Gpx1 to Gpx 8; they play a role in reducing levels of hydrogen peroxide or oxidized lipids. GST is an abundant protein that has a number of isoenzymes; their role is to conjugate GSH to electrophiles and xenobiotics (Raza, 2011). Some of these GST isoenzymes participate in neuroprotection. For example, glutathione-S-transferase pi 1 (GSTP1), the most abundant member of the GST family, has been identified as a negative regulator of cyclin dependent kinase-5, which is implicated in many neurological disorders (Sun et al., 2011). Other studies have proposed that GSTP1 suppresses pro-apoptotic c-Jun N-terminal kinases (JNK) activation by stress (Elsby et al., 2003). Glutathione reductase (GR) plays an important role in recycling GSH by converting oxidized GSH back to reduced GSH. This recycling process consumes NADPH. The protective role and expression regulation of GR is not fully understood. In short, many enzymes that utilize GSH to scavenge ROS are subjected to regulation by Nrf2 and are depicted in red in Figure 5, which illustrates GSH synthesis and utilization.
Figure 5. The impact of Nrf2 on the collaboration between neurons and astrocytes in GSH synthesis and function.
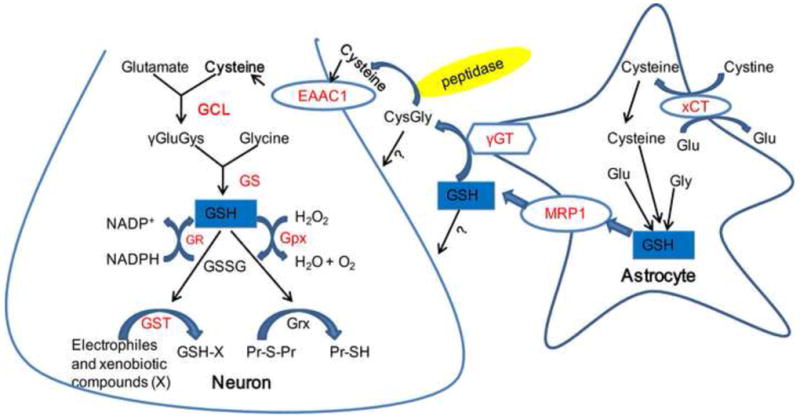
GSH is synthesized from glutamate, cysteine and glycine, a reaction which is catalyzed by GCL and GS. Cysteine is the rate limiting substrate and GCL is the rate limiting biosynthetic enzyme in neurons. Cystine imported from xCT participates in astrocytic GSH synthesis. Synthesized GSH is then extruded by MRP1 and further cleaved by γGT and peptidases to generate free cysteine in the extracellular space. Cysteine then enters neurons through EAAC1 and facilitates neuronal GSH production and function. GSH exerts antioxidant effects by detoxifying H2O2, endogenous toxic and xenobiotic compounds and Pr-S-Pr. The molecules controlled by Nrf2 are highlighted in red. Abbreviations: Glu: glutamate; Cys: cysteine; Gly: glycine; γGluCys: γ-glutamylcysteine; CysGly: cysteinylglycine; GSSG: glutathione disulfide; GCL: γ-glutamylcysteine ligase; GS: glutathione synthase; Gpx: glutathione peroxidase; GR: glutathione reductase; GST: glutathione-S-transferase; γGT: γ-glutamyltransferase; Grx: glutaredoxin; Pr: protein; MRP1: multidrug resistance protein 1.
Glia are well known for their ability to support redox homeostasis in neurons. For example, neurons are somewhat dependent on glia for GSH synthesis. Specifically, astrocytes provide neurons directly with glutathione (Anderson et al., 2003; Dringen et al., 2000), or provide the precursors for GSH synthesis, such as Cys-Gly and γGlu-Cys from the hydrolysis of GSH (Dringen et al. , 1999; Qin et al., 2006). Nrf2-overexpressing glia release GSH and can protect neurons from glutamate toxicity in neuron-glia co-cultures (Shih et al., 2003). The collaboration between neurons and neighboring astrocytes in GSH homeostasis is described in Figure 5.
4.3 Transferases
Transferases are a group of enzymes that transfer various functional groups to the polar groups of their acceptors. Based on the functional groups they transfer, the transferases can be divided into several subtypes, including 1) glutathione S-transferase, which transfers a GSH, methyltransferase that, in turn, transfers a methyl group, 2) N-acetyltransferase, which transfers an acetyl group, 3) sulfotransferase, which transfers a sulfate group, and 4) UDP-glucuronosyltransferase (UGT), which transfers a glycosyl group (Nakata et al., 2006; Xu et al., 2005). Except for GST, the neuroprotective roles of other transferases are relatively unexplored, either because they are not protective or because their function has not yet been investigated. The major function of the transferases is to conjugate drug metabolites, making them more hydrophilic and excretable. As a result, they need specific substrates, which may or may not be present in cells. In addition, transferases possess little, if any, of the anti-oxidative function that characterizes other phase 2 enzymes and that is critical for neuroprotection. Despite these observations, their roles still need to be further investigated for a better understanding of their evolutionary roles.
4.3 Detoxifying enzymes
Heme and quinone both transfer electrons and are therefore direct sources of free radicals and ROS. As a result, overall oxidative stress is effectively reduced by the degradation of heme and quinone.
4.4.1 Heme oxygenase 1 (HO-1)
Heme oxygenase catalyzes the first and rate-limiting step of heme catabolism, the breakdown of heme to carbon monoxide, biliverdin and iron (Ferrandiz and Devesa, 2008; Yoshida and Kikuchi, 1974). There are two isoforms of active HO, HO-1 and HO-2. Whereas HO-2 is expressed constitutively, HO-1 is only expressed in an inducible manner and belongs to the phase II enzymes. The human HO-1 gene (HMOX1) is mapped at 22q12, spanning five exons and four introns (Kuwano et al., 1994). The molecular weight of HO-1 is around 32 kDa. Because of its characteristic induction by stress, it is also called heat shock protein 32 (HSP32). HO-1 has a short half-life, about 3 hr for messenger RNA and 15-21 hr for protein (Dwyer et al., 1992; Leautaud and Demple, 2007; Schipper et al., 2009). The importance of HO-1 in neuroprotection is two-fold: the breakdown of heme and the generation of antioxidants.
Three major classes of proteins contain heme: hemoglobin, oxidase, and peroxidase. The heme in hemoglobin is essential for oxygen transport, whereas the hemes in oxidases and peroxidases play key roles in superoxide generation and electron transfer (Chrissobolis and Faraci, 2008; Everse and Coates, 2009). Heme-containing oxidases include non-mitochondrial NADPH oxidase and cyclooxygenases, as well as mitochondrial succinate dehydrogenase (Complex II) and cytochrome c oxidase (Complex IV). All of these molecules are major sources of superoxide and ROS (Chrissobolis and Faraci, 2008). On the other hand, heme-containing peroxidases catalyze the reactions between hydrogen peroxide and large biomolecules, leading to their damage (Everse and Coates, 2009). By breaking down heme, HO-1 can thus protect cells through a net reduction in superoxide and other ROS.
In addition, the breakdown products of heme possess protective properties. For example, biliverdin and bilirubin are both strong antioxidants (Deguchi et al., 2008; Stocker et al., 1987) that can protect the brain from ischemic injury (Deguchi et al., 2008). CO is similar to NO but much more stable and can activate guanylate cyclase, generating the secondary messenger cyclic 3’,5’-monophosphate (cGMP) (Verma et al., 1993). In turn, cGMP activates protein kinase G, which then decreases intracellular calcium levels, leading to cytoprotection and vasodilation (D’Ascenzo et al., 2002; Lincoln et al., 2006; Takuma et al., 2001). In a positive feedback loop, CO plays an anti-apoptotic role by inducing the expression of HO-1 under conditions of endoplasmic reticulum stress (Kim et al., 2007; Wang et al., 2007b).
4.4.2 NAD(P)H: quinone oxidoreductase 1 (NQO1)
NQO1, also named DT-diaphorase, was first identified by Ernster and his colleagues in 1958 (Smith, 1999). Using either NADPH or NADH as the hydride donor, NQO1 catalyzes the two-electron reduction of quinone to the redox-stable hydroquinone, preventing free radical formation from quinone derivatives (Talalay et al. 1995). The hydroquinone generated from NQO1 reduction can be subsequently converted into glucuronide and sulfate conjugates and ultimately expelled.
NQO1 plays an important role in neuroprotection through its anti-oxidative properties (Lim et al., 2008). For example, in cultured dopaminergic CATH.a cells, the induction of NQO1 by BHA dramatically and dose-dependently blocked METH-induced cytotoxicity by scavenging quino proteins (Miyazaki et al., 2006). Furthermore, deprenyl can protect PC12 cells against MPP+ induced oxidative stress via the upregulation of NQO1 (Xiao et al., 2011). Upregulation of NQO1 by 4-hydroxybenzyl alcohol also reduces cerebral infarct size and improves neurological functions in rats (Yu et al., 2011).
5 Protection against neurological diseases
As will be discussed below, the Nrf2/ARE pathway confers neuroprotection in various models of neurological diseases by the regulation of multiple downstream genes
5.1 Acute Neurological Diseases
5.1.1 Traumatic brain injury (TBI)
TBI is a serious public health problem affecting millions of people annually and remains a leading cause of death and disability (Feeser and Loria, 2011). Oxidative stress plays an integral role in neuronal injury after TBI. Therefore, activating the Nrf2 pathway to battle TBI has generated recent interest.
TBI significantly increases the level of Nrf2 as well as phase II enzymes such as NQO1 and HO-1 (Yan et al., 2009; Yan et al., 2008), suggesting that the Nrf2/ARE pathway is an endogenous compensatory adaptation against TBI. It has also been reported that intraperitoneal administration of SFN is capable of reducing neuronal death, contusion volume, and neurological dysfunction 7d after TBI in rats (Hong et al., 2010). In line with this result, Nrf2-/-mice exhibit exacerbated deficits in neurologic function and oxidative damage. Furthermore, the neuroprotective capacity of SFN is blunted in Nrf2-/- mice. Histone deacetylase inhibitors (Wang et al., 2012a) and tBHQ (Hatic et al., 2011) can also protect against traumatic neuronal injury by activating Nrf2. These results all demonstrate that activation of Nrf2 enhances recovery from TBI.
Nrf2 activation also protects the blood brain barrier (BBB) during TBI. TBI causes a biphasic opening of the BBB. The first opening happens within hours (acute phase) after TBI and the other peaks 1-3 days (secondary phase) after injury. The latter opening is associated with a loss of endothelial cells and tight junction proteins (Zhao et al., 2007a). Enhanced Nrf2 staining can be detected in the blood brain barrier following TBI (Yan et al., 2009). SFN reduces Evans Blue extravasation in the acute phase when applied before injury and also reduces the secondary phase of BBB permeability when administrated 6 h after TBI (Zhao et al., 2007a). This protective effect was abolished in Nrf2-/- mice or in rats pretreated with decoy ARE oligonucleotides containing the binding site of Nrf2 (Zhao et al., 2007a). Independent studies from Jin and his coworkers also reported that mice with Nrf2 disruption exhibit increased severity of brain edema at 24h after TBI (Jin et al., 2009). Further investigations are needed to address which cell type or which tight junction proteins contribute to these conspicuous changes.
Jin’s group has also shown that the neuroprotective mechanism of Nrf2 in TBI may involve anti-inflammatory effects. Increased mRNA and protein expression of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), intercellular adhesion molecule-1 (ICAM-1) and interleukin-6 (IL-6) have all been detected in Nrf2-/- mice after TBI. The inhibitory role of Nrf2 on cerebral NF-κB activity likely contributes to the pro-inflammatory cytokines changes in Nrf2-/- mice (Jin et al., 2008b). Jin’s lab also exploited the systemic effect of TBI on inflammation and detected exacerbated inflammatory responses in the lung (Jin et al., 2008d) as well as the intestines (Jin et al., 2008a; Jin et al., 2008c) in Nrf2 deficient mice. These studies underscore the systemic effects of TBI and support both central and peripheral effects of Nrf2 on inflammatory changes after TBI
5.1.2 Ischemic Stroke
Strokes are the leading cause of disability and the third leading cause of mortality in the world. Ischemic stroke is the most common type of stroke. Multiple pathological processes are involved in the progression of stroke, including excitotoxicity, oxidative stress, inflammation, mitochondrial dysfunction, etc. Oxidative stress represents a potential target for treatment because it is one of the most critical insults in stroke. Similar to the situation in TBI, Nrf2 may play a role as an endogenous compensatory adaptation against stroke. For example, Keap1 is decreased 2 hours after reperfusion following MCAO. The fall in Keap1, not surprisingly, is paralleled by a rise in Nrf2 that starts at 2 hours and peaks at 8 hours after reperfusion. Furthermore, antioxidative proteins that are downstream of Nrf2, including Trxs, GSH, and HO-1 all showed significant increases 24–72 hours after MCAO in the peri-infarct region (Tanaka et al.).
Administration of tBHQ or CDDO through either intracerebroventricular or intraperitoneal routes reduces sensorimotor deficits and infarct size after ischemia in the rodent MCAO model (Shih et al., 2005; Zhang et al., 2012). Consistent with this study, Nrf2-/-mice subjected to 90-min MCAO followed by 24 h reperfusion exhibit worse neurological deficits and larger infarct sizes (Shah et al., 2007). Furthermore, Nrf2-/- mice display an exacerbated outcome 7 days after injury in a focal ischemia model combined with permanent distal middle cerebral artery occlusion (Shih et al., 2005).
Activation of the Nrf2 pathway is critical for scavenging ROS, which contributes to neuroprotection against ischemic brain injury. For example, Nrf2-/- mice produce more ROS species in brain injury (Zhao et al., 2007b). In order to identify the cell type which elicits Nrf2-dependent anti-oxidative effects, Shih and colleagues transfected neuron-glia co-cultures with an adenovirus-based Nrf2 over-expression vector and found that Nrf2-overexpressing astrocytes exhibited more efficient antioxidant properties than neurons. The underlying mechanism may involve GSH homeostasis because Nrf2 overexpressing astrocytes release more GSH to protect neurons against H2O2 (Shih et al., 2003). However, an independent study from Wang and colleagues proposed that the severe outcome in Nrf2-/- mice is caused by ROS released from neutrophils (Wang et al., 2007a). It is quite likely that upregulation of GSH redox systems and suppression of inflammatory mediators both underlie the protection elicited by Nrf2.
It is of interest that tBHQ can not only protect primary cultured neurons from free radicals but also from excitotoxic insults such as NMDA or glutamate (Shah et al., 2007). One might therefore speculate that Nrf2 may also regulate the transcription of glutamate transporters or receptors. Future studies to examine this possibility are warranted.
5.1.3 Hemorrhagic strokes
Depending on where the bleeding occurs and blood accumulates, hemorrhagic strokes can be divided into intracerebral hemorrhage (ICH) when there is bleeding into brain parenchyma and subarachnoid hemorrhage (SAH) when there is bleeding into the subarachnoid space. With hypertension as the major cause, ICH is associated with high mortality due to its acute onset and mass effects of hematoma and edema, which lead to intracranial hypertension and brain herniation (Keep et al., 2012). Subsequent ischemic and oxidative stress also contribute to the brain injury after ICH. The Nrf2 pathway is activated in the brain following ICH, as indicated by increased HO-1 expression. In mice, HO-1 upregulation begins at 24 hr after ICH, peaks at day 5 and subsides on day 8 (Chen and Regan, 2007). HO-1 is predominantly expressed in microglia/macrophage and endothelial cells, modestly in astrocytes and rarely in neurons (Wang and Doré, 2007).
It has been reported that Nrf2-deficient mice demonstrate more severe neurologic deficits and ICH-mediated damage after ICH (Wang et al., 2007a ; Zhao et al., 2007b), suggesting a neuroprotective role of Nrf2 against ICH. In support of this notion, administration of SFN (Zhao et al., 2007b) or curcumin (Sun et al., 2011) protected against ICH by activating Nrf2 and reducing oxidative stress, brain edema and neuroinflammation. Though Nrf2 activation is protective, the responsible enzymes in this model have not been clarified. HO-1 is the only characterized enzyme in the setting of ICH. However, HO-1 is detrimental to ICH, because HO-1 knockout mice exhibit a decreased injury volume after ICH (Wang and Doré, 2007). An increase in free iron may lie behind this observation, as HO-1 degrades heme from the hematoma and generates large amount of free iron, leading to oxidative stress and neuroinflammation (Wang and Doré, 2007). It will be interesting to investigate whether concomitant administration of deferoxamine would reduce free iron overload and thereby protect the brain (Okauchi et al., 2009).
SAH is caused by ruptured arterial aneurysms or arteriovenous malformation (AVM). The major pathophysiological processes of SAH are vasospasm, secondary ischemia and subsequent early brain injury (Zhou et al., 2011). Following experimental SAH in rats, Nrf2 was activated in both endothelial and smooth muscle cells of the basilar artery, as indicated by increased nuclear Nrf2 levels and DNA binding (Wang et al., 2010). HO-1 was also upregulated in cerebral arteries after SAH (Ono et al., 2000), as well as in microglia and astrocytes (Matz et al., 1996).
It has been reported that Nrf2 activation plays a protective role against SAH. For example, the administration of curcumin reduced vascular inflammation and cerebral vasospasm in mice after ICH (Wakade et al., 2009), and decreased both oxidative stress and mortality in rats (Kuo et al., 2011). SFN also activated Nrf2, upregulated downstream enzymes such as HO-1 and NQO-1, and reduced cortical apoptosis, brain edema and BBB impairment (Chen et al., 2011). In addition to these classic Nrf2 inducers, two hormones have also been reported to protect the brain from SAH by activating the Nrf2 pathway - erythropoietin (Zhang et al., 2010a) and melatonin (Wang et al., 2012b). These hormones upregulated phase 2 enzymes such as HO-1 and NQO-1, reduced early brain injury such as cortical apoptosis and protected the BBB (Wang et al., 2012b ; Zhang et al., 2010a). Unlike the detrimental role of HO-1 in ICH, the upregulation of HO-1 is thought to be beneficial in SAH. It has been reported that fusion of HO-1 with an eleven-arginine transduction domain can facilitate HO-1 crossing of the cell membrane (Ogawa et al., 2011). When fused HO-1 was injected into the cisternal space, an increase of HO-1 level and activity was detected in the basilar artery, which attenuated cerebral vasospasm following SAH in rats (Ogawa et al., 2011). Similarly, adenovirus-mediated HO-1 expression also reduced cerebral vasospasm after experimental SAH, indicating a protective role of HO-1 against SAH (Ono et al., 2002).
5.2 Neurodegenerative Diseases
5.2.1 Parkinson’s Disease (PD)
PD is an incurable movement disorder characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and deposition of Lewy bodies across many regions of the brain (Tufekci et al., 2011). Postmortem data from five PD patients reveal that Nrf2 is expressed at higher levels in the nucleus of substantia nigra neurons (Ramsey et al., 2007). This observation is reminiscent of TBI and stroke (see above) and the upregulation of Nrf2 in PD may also be a compensatory attempt to enhance antioxidant defenses in response to oxidative toxicity.
In an acute Parkinson’s model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a greater loss of dopamine transporters was observed in the striatum of Nrf2-/- mice at all MPTP doses used, ranging from 20 to 60 mg/kg. In addition, oral administration of the Nrf2 inducer D3T to wild-type mice is protective against MPTP (Burton et al., 2006). In a subacute model, Nrf2-/- mice display fewer TH positive cells than WT mice (Chen et al., 2009a). In addition to the MPTP studies, cortical neurons are more vulnerable to another neurotoxin, 6-hydroxydopamine (6-OHDA) in Nrf2-/- mice (Jakel et al., 2007). Furthermore, overexpressing Nrf2 or its DNA-binding dimerization partner-Maf, or down-regulating Keap1 can all restore locomotor activity in genetic models of familial PD (Barone et al., 2011).
A protective role of Nrf2 in PD is further supported by the clinical use of Deprenyl (selegiline), a B-type monoamine oxidase inhibitor. Recent studies show that the effect of Deprenyl is dependent on its activation of Nrf2 (Xiao et al., 2011). Nrf2 may also be involved in the neuroprotective effects of DJ-1/PARK7, a gene implicated in 1-2% of early onset familial PD. DJ-1 has 189 amino acids and belongs to the Thi/PfpI protein superfamily. It decreases the ubiquitination of Nrf2 by preventing its association with Keap1. Furthermore, tBHQ can no longer induce the nuclear translocation of Nrf2 in DJ-1 knockout mice (Clements et al., 2006; Gan et al., 2010). DJ-1-deficient patients exhibit reduced expression of Nrf2-dependent genes coupled with increased oxidative stress (Mosser and Edwards, 2008; Zhou and Freed, 2005). Thus, Nrf2 dysfunction may be involved in the pathogenesis of early onset familial PD linked to DJ-1 mutations.
Similar to ischemic stroke, astrocytic Nrf2, rather than neuronal Nrf2, is thought to play a dominant role in neuroprotection against PD models. Using transgenic mice with Nrf2 under control of an astrocyte-specific promoter on both Nrf2+/+ and Nrf2-/- backgrounds, Peichun and colleagues showed that only astrocytes with Nrf2 expression can abolish the neurotoxicity of MPTP (Chen et al., 2009a). Indeed, early studies in rodent primary neuronal cultures already showed that basal expression and activation of the ARE occurred predominantly in astrocytes (Ahlgren-Beckendorf et al., 1999; Eftekharpour et al., 2000). Furthermore, the majority of the protective genes induced by tBHQ are expressed heavily in astrocytes (Kraft et al., 2004). Astrocytes indirectly support neuronal activity through GSH, as described in Section 4. As in the animal models of PD, decreased levels of GSH in astrocytes are considered a hallmark of PD, potentially leading to neuron death (Dawson and Dawson, 2003). In addition to astrocytic expression, Nrf2 expression in meningeal cells can also protect against neurotoxicity from excessive dopamine (Shih et al., 2007).
The mechanism underlying the sensitization of Nrf2 deficient mice to MPTP or 6-OHDA may be two-fold. The first mechanism may involve toxin detoxification. It has been reported that Nrf2 deficient mice demonstrated more severe neuronal loss than HO-1 deficient mice in response to MPTP (Innamorato et al., 2010). This may not be surprising because Nrf2 also upregulates other phase II enzymes in addition to HO-1 that might detoxify MPTP or MPP+. The second mechanism involves the anti-inflammatory effects of Nrf2. Microglia have at least two different phenotypes: the classical activation phenotype (CA-MU), which participates in inflammatory stress and neuronal death under pathological conditions and the alternative activation phenotype (AA-MU), which contributes to the resolution of inflammation and wound healing. Following exposure to MPTP, both wild type and Nrf2 deficient mice show increased levels of COX-2 and iNOS, two markers of classical microglial activation. However, only Nrf2 deficient mice also demonstrate decreased levels of FIZZ-1, Arginase-1 and IL-4, all of which are markers of alternative microglial activation (Rojo et al., 2010). These studies indicate that Nrf2 may contribute to the resolution of inflammation and wound healing via AA-MU and suggest an important role for Nrf2 in the modulation of microglial dynamics.
5.2.2 Alzheimer’s Disease (AD)
AD is a neurodegenerative disorder manifested by a pathological loss of synapses and neurons and the formation of intracellular neurofibrillary tangles and extracellular deposits of amyloid-beta (Aβ). AD patients exhibit a dramatic reduction in nuclear Nrf2 within hippocampal neurons (Ramsey et al., 2007). In addition, the widely used APP/PS1 transgenic mice with significant amounts of Aβ deposits demonstrate a decline in Nrf2/ARE targeted proteins (Kanninen et al., 2008). Because oxidative stress is integral to the pathogenesis of AD and can activate the Nrf2 pathway, it is not clear if the decreases in Nrf2 nuclear translocation and phase II enzymes cause pathology in AD or are the result of Aβ-induced neuron death. To attempt to address this, Nrf2 expression and activation in early stages of AD must be examined. Nevertheless, these findings suggest that the Nrf2/ARE pathway is impaired in AD and may form at least part of the pathology. This appears fundamentally different from the situation in TBI, stroke, and PD, where there are compensatory rises in Nrf2, not a fall.
Recent studies have investigated the role of the Nrf2/ARE pathway and its potential therapeutic value in AD models. Boosting Nrf2 activity by tBHQ or over-expressing Nrf2 through adenovirus-mediated gene delivery confers protection against Aβ1-42 induced neuronal death of cultured hippocampus (Kanninen et al., 2008). In vivo, delivering lentiviral vectors encoding human Nrf2 bilaterally into the hippocampus of APP/PS1 mice alleviates the spatial learning deficits and robustly reduces the infiltration of astrocytes but not microglia. A promising study reported that feeding Aβ-injected rats with tBHQ reduced Aβ accumulation and Aβ induced cell apoptosis (Nouhi et al., 2011). These data confirm a protective role of Nrf2 activation in AD models. Thus, loss of Nrf2 in the human disease may exacerbate amyloid-related pathology.
The Nrf2/ARE pathway may also protect against vascular dementia. In hypobaric hypoxia - induced dementia, ALCAR (acetyl-L-carnitine) was documented to increase TrkA expression and ERK phosphorylation. Phosphorylated ERK then increased translocation of Nrf2 into the nucleus, which in turn ameliorated memory impairment induced by hypobaric hypoxia by combating oxidative stress (Barhwal et al., 2009).
5.2.3 Multiple sclerosis (MS)
Multiple sclerosis is an autoimmune and inflammatory disease with lesions typically located in the white matters of the brain and spinal cord. The precise etiology of MS has not been identified. However, it is generally accepted that the proliferation of CNS-infiltrated immune cells, such as T cells, damage oligodendrocytes and axons via neuroinflammation and oxidative stress (Frohman et al., 2006; Linker et al., 2011). MS may be initiated by the abnormal activation of CD4+ T cells exposed to myelin-like antigenic peptides in the periphery. Subsequently, these sensitized CD4+ T cells cross the blood brain barrier and result in a series of toxic effects (Benedict and Zivadinov, 2011).
CD4+ T cell infiltration leads to excessive activation of macrophages, microglia and astrocytes, generating ROS and directly damaging normal tissues. Interestingly, Nrf2 can modulate autoimmune neuroinflammatory responses in MS models. Experimental autoimmune encephalomyelitis (EAE) is a widely accepted MS animal model. Evidence suggests an enhanced immune cell infiltration (CD4+ T cells, CD19+ B cells) and glial cell activation (astrocytes, microglia) in Nrf2 knockout mice suffering from EAE (Johnson et al., 2010). Furthermore, Nrf2 deficient mice with EAE exhibit increased expression of inflammatory enzymes (iNOS, phox-47, gp91-phox, and phox-67), cytokines (IFN-γ, IL1-β, TNF-α, and IL-12), and chemokines (BLC and MIG) (Johnson et al., 2010). Furthermore, Nrf2 knockout mice are highly sensitive to the neuroinflammation induced by LPS and exhibit increased microglia infiltration and inflammatory mediator expression. These features can be reversed by SFN (Innamorato et al., 2008).
Elegant pathological studies of MS patients’ postmortem tissue demonstrate that Nrf2-mediated transcription occurs mostly in MHC class II-positive infiltrating macrophages and to a lesser extent in reactive astrocytes. In patients with chronic-progressive MS, alpha-motor neurons express higher Nrf2 compared to controls (Linker et al., 2011). Surprisingly, Nrf2 is undetectable in oligodendrocytes in either control white matter or MS brain tissue (van Horssen et al., 2010).
Nrf2 knockout mice suffering from EAE exhibit more severe behavioral dysfunctions and enhanced leukocyte infiltration as well as glial activation in the spinal cord (Hubbs et al., 2007; Johnson et al., 2010). Consistent with these findings, Johnson and co-workers reported that Nrf2 knockout mice displayed pronounced demyelination and axonal loss in the brain (Johnson et al., 2010).
Efforts have been made to treat MS by activating the Nrf2 pathway. This is warranted based on findings that dimethyl fumarate (DMF), a promising drug in clinical trials for MS, can promote Nrf2 activation through direct modification of Keap1 at cysteine residue 151 (Kappos et al., 2008; Linker et al., 2011). Fumarate compounds further upregulate GSH and HO-1 and protect against MS models? or is it really human MS? (Lin et al., 2011; Scannevin et al., 2012). Additionally, CDDO-TFEA, a strong inducer of Nrf2, suppresses neuroinflammation in EAE (Pareek et al., 2011), suggesting anti-oxidative and anti-inflammatory roles of Nrf2 against MS.
5.2.4 Huntington’s Disease (HD)
HD is an autosomal dominantly inherited neurodegenerative disease. HD is caused by excessive trinucleotide CAG repeat expansion in the HD gene coding huntingtin (HTT), resulting in an expanded N-terminal polyglutamine tract. It is characterized by abnormal body movements called chorea, cognitive impairments, and personality changes (Kumar et al., 2010).
Several lines of transgenic HD mice have been generated based on the length of HTT N-terminal fragments. Some knock in mice only over-express part of N-terminal fragment. For instance, mice expressing a 90 amino acid N-terminal fragment are designated as R6/2 mice and mice expressing a 171 amino acid N-terminal fragment are named N171-82Q mice. The mice that over-express full-length HTT are named BAC or YAC, which are more valuable for studies (Ross and Tabrizi). HD transgenic R6/1 mice show increased activity of Cu/Zn SOD, an Nrf2 dependent enzyme, at the age of 19 weeks. However, when these mice reach 35 weeks, the activity of Cu/Zn SOD diminished (Santamaria et al., 2001). In the more severe N171-82Q transgenic HD model, basal levels of striatal Nrf2 were significantly reduced (Chaturvedi et al., 2010). These findings were confirmed in a PC12 HD model (van Roon-Mom et al., 2008).
It has been reported that DMF increases neuronal Nrf2 and promotes recovery in R6/2/YAC128 mice, a well known Huntington’s model in which the HD gene is expressed with 141–157 CAG repeats (Ellrichmann et al., 2011). One notable feature of this study is that Nrf2 is expressed within neurons but not glia (Ellrichmann et al., 2011). In addition, oral administration of triterpenoids upregulates Nrf2/ARE induced genes and reduces striatal atrophy in N171-82Q mice (Stack et al., 2010). Mitochondrial complex II inhibition with 3-nitropropionic acid (3-NP) or malonate leads to a type of striatal degeneration that resembles Huntington’s disease. As expected, Nrf2 -/- mice are more sensitive to the mitochondrial complex II inhibitors 3-NP and malonate (Calkins et al., 2005). Strikingly, transplantation of primary astrocytes infected with Ad-Nrf2-GFP into the striatum protects neurons from the malonate-induced lesions (Calkins et al., 2005). Collectively, these findings show that targeting the Nrf2/ARE pathway shows promise for the treatment of HD.
5.2.5 Amyotrophic lateral sclerosis (ALS)
ALS is an adult-onset motor neuron disease caused by progressive degeneration of upper and lower motor neurons in the spinal cord, brain stem, and motor cortex. Epidemiological studies show that ALS is sporadic in 90–95% of cases and familial in 5–10% of cases, and that approximately 10%–20% of familial ALS cases are caused by the mutations in the ubiquitously expressed Cu/Zn SOD antioxidant protein (Rosen, 1993). The most widely used animal model of ALS exploits the mutation of a glycine to an alanine at position 93 of human SOD1 (SODG93A mice) (Gurney et al., 1994).
Motor neurons over-expressing the SOD1 (G93A) mutation display decreased levels of Nrf2 and the enzymes involved in GSH biosynthesis (Pehar et al., 2007). Similarly, Nrf2 transcriptional genes are also repressed in NSC34 cells exposed to mutant SOD1, as revealed by microarray analysis (Kirby et al., 2005). Studies on human primary motor cortex and the spinal cord of postmortem tissue samples from five ALS patients confirm that mRNA and protein levels of Nrf2 were reduced in ALS motor cortices as well as spinal cord. In addition, there was a trend toward higher Keap1 mRNA signal intensity in the motor cortex of ALS (Sarlette et al., 2008). This rise in mRNA did not, however, translate into higher protein levels. Nonetheless, the situation in ALS thus appears more similar to AD than PD, stroke, and TBI, because in the latter three conditions, Nrf2 levels were raised by the disease process instead of lowered.
Two potent Nrf2 activators - 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) ethylamide and CDDO-TFEA can up-regulate Nrf2 and its downstream enzymes, not only in NSC-34 cells transfected with mutant G93A SOD1 but also in the spinal cord of G93A SOD1 mutant mice (Neymotin et al., 2011). Both compounds significantly attenuate the progression of ALS and extend the survival of G93A SOD1 mice when administrated either at a presymptomatic age or a symptomatic age (Neymotin et al., 2011). Consistent with other neurodegenerative studies, ALS researchers also propose that Nrf2 activation within astrocytes protects the motor neurons against mutant SOD1 toxicity. It is possible that the GSH secreted by astrocytes helps neuronal defense against oxidative insults, because Mrp1-siRNA or MK-571, inhibitors of GSH secretion from astrocytes, abolish the neuroprotection of Nrf2 (Vargas et al., 2008).
6 Conclusions and perspectives
It is evident that significant progress has been made in our understanding of pathways converging on and diverging away from Nrf2. Nrf2 levels can rapidly fluctuate in response to dynamic changes in the environmental milieu, revealing an elaborate and sophisticated machinery designed to preserve homeostasis in mammalian cells. The exquisite sensitivity of Nrf2 to stressors and its upregulation in various neurodegenerative conditions speaks to the fundamental importance of this molecule in intrinsic or endogenous protective responses to injury. However, in some conditions, such as AD and ALS, a drop in Nrf2 may actually contribute to the pathology. The up- or down-regulation of Nrf2 thus appears to closely depend on the nature of the stress or type of disease and is likely to involve its elegant positive and negative feedback loops.
Collectively, Nrf2 and its downstream phase II genes are promising targets for the treatment of neurological diseases (Table 3) owing to their potent ability to detoxify harmful compounds, combat ROS and directly or indirectly modulate the inflammatory response, immunological system, and BBB permeability. Many Nrf2 inducers have been identified and proven effective in animal models of common neurological diseases. However, there is still a dearth of clinical trials of these inducers. This may be attributed to several concerns about Nrf2 modulators that preclude their rapid translation to the clinic. First, some reports have proposed that Nrf2 activation arms the cell with extensive protection against inherently fluctuating microenvironments. This imbalance may affect normal cell growth and apoptosis (Hayes and McMahon, 2009; Shibata et al., 2008). Additional studies to interpret this effect are warranted. Second, current pharmacological inducers of the Nrf2 pathway do not cross the blood brain barrier to a significant degree with the exception of luteolin (Wruck et al., 2007). Further studies directed towards these concerns will help to develop Nrf-2 based strategies for neurological diseases.
Table 3.
Nrf2 and neurological diseases in vivo
| Diseases | Ways to regulate Nrf2 | Animal models | Results | References |
|---|---|---|---|---|
| TBI | Administration of SFN | TBI in rats | Protective | Hong, 2011; Zhao, 2007 |
| Nrf2 KO | Nrf2-/- mice | Exacerbated neurologic deficit | Zhao, 2007 | |
| Spinal cord injury | Nrf2 KO | Nrf2-/- mice | Exacerbated motor dysfunction and neuronal death | Mao, 2011 |
| Ischemia | tBHQ injection (ICV or IP) | MCAO in rats | Protective | Shi, 2003; |
| Nrf2KO | MCAO in Nrf2-/- mice | Increased infarct size and neurologic deficits | Shi, 2005; Shi, 2007 | |
| IP injection of Curcumin | MCAO in rats | Protective | Yang, 2009 | |
| Systemic administration of SFN | MCAO in rats | Protective | Zhao, 2006 | |
| Oral administration of D3T | MPTP in mice | Protective | Burton, 2006 | |
| PD | Nrf2KO | 6-OHDA in Nrf- /- mice | Exacerbated neurologic function | Jakel, 2007 |
| Oral selegiline | PD patients | Protective | Xiao, 2011 | |
| AD | Intrahippocampal injection of a lenti-Nrf2 | APP/PS1 mice | Protective | Kanninen, 2009 |
| Oral tBHQ | Amyloid beta in rats | Protective | Nouhi, 2011 | |
| Oral ALCAR | Hypobaric hypoxia - induced dementia in rats | Protective | Barhwal, 2009 | |
| MS | Nrf2 KO | EAE in Nrf2-/- mice | Impairedbehavior and enhanced pathology | Johnson, 2010 Hubbs, 2007 |
| HD | Oral DMF | R6/2 and YAC128 HD transgenic mice | Protective | Ellrichmann, 2011 |
| Nrf2 -/- mice | 3-NP or malonate in Nrf2-/- mice | Exacerbated neurologic function | Calkins, 2005 | |
| Intrastriatal transplantation of Nrf2- overexpressing astrocytes | Malonate induced HD in mice | Protective | Calkins, 2005 | |
| ALS | Oral CDDO-EA CDDO-TFEA | G93A SOD1 mutated mice | Protective | Vargas, 2008 |
Highlights.
Phase II metabolic enzymes detoxify xenobiotics by increasing their hydrophilicity and enhancing their disposal.
The Nrf2/ARE pathway is the main regulator of phase II genes
Several phase II genes demonstrate neuroprotective properties in vivo and in vitro
Upregulation of Nrf2/ARE, either by genetic or chemical approaches, confers neuroprotection in neurological disease models
Acknowledgments
This work was supported by funds from the National Institutes of Health (NS36736, NS43802 and NS45048 to J.C.), the VA Merit Review Grant (to J.C.) and the American Heart Association (10SDG2560122 to F.Z.). We thank Pat Strickler for secretarial support.
Abbreviations
- Aβ
amyloid-beta
- AA-MU
alternative activation phenotype
- AD
Alzheimer’s disease
- AhR
aryl hydrocarbon receptor
- ALCAR
acetyl-L-carnitine
- ALS
amyotrophic lateral sclerosis
- ARE
antioxidant response elements
- AP-1
activator protein-1
- ASK1
apoptosis signal-regulating kinase
- BBB
blood brain barrier
- BTB
bric-a-brac
- β-TrCP
beta-transducin repeats-containing proteins
- CA-MU
classical activation phenotype
- CBP
CREB-binding protein
- CDDO
2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid
- cGMP
cyclic 3’,5’-monophosphate
- CHD6
chromo-ATPase/helicase DNA binding protein family member 6
- CK2
casein kinase 2
- CNS
central nervous system
- Cul3
Cullin3
- D3T
3H-1,2-dithiole-3-thione
- DMEs
drug metabolizing enzymes
- DMF
dimethyl fumarate
- EAE
experimental autoimmune encephalomyelitis
- ERKs
extracellular signal-regulated kinases
- EAAC1
excitatory amino acid carrier 1
- GCL
γ-glutamylcysteine synthetase
- Gpx
glutathione peroxidase
- GR
glutathione reductase
- GST
glutathione s-transferases
- GSTP1
glutathione-S-transferase pi 1
- GSK-3β
glycogen synthase kinase-3
- 6-OHDA
6-hydroxydopamine
- HD
Huntington’s Disease
- HTT
Huntingtin
- HSP32
heat shock protein 32
- ICH
intracerebral hemorrhage
- MAPK
mitogen-activated protein kinase
- MCAO
middle cerebral artery occlusion
- MEFs
mouse embryo fibroblasts
- MS
multiple sclerosis
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 3-NP
3-nitropropionic acid
- NES
nuclear export sequence
- NLS
nuclear localization signal
- Nrf2
nuclear factor erythroid 2 related factor 2
- Keap1
Kelch-like ECH associated protein 1
- PD
Parkinson’s disease
- PERK
PKR-like endoplasmic reticulum kinase
- ProTα
prothymosinα
- Prx
peroxiredoxin
- PKC
protein kinase C
- PI3K
phosphatidylinositol 3-kinase
- Rbx1
E3 ubiquitin ligase complex
- SAH
subarachnoid hemorrhage
- SNpc
substantia nigra pars compacta
- Srxn
sulfiredoxin
- SFN
sulforaphane
- tBHQ
tert-butylhydroquinone
- TBI
traumatic brain injury
- Trx
thioredoxin
- TrxR
Trx reductase
- XRE
xenobiotic response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlgren-Beckendorf JA, Reising AM, Schander MA, Herdler JW, Johnson JA. Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: role of the antioxidant/electrophile responsive element. Glia. 1999;25:131–142. [PubMed] [Google Scholar]
- Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–27702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- Anderson MF, Blomstrand F, Blomstrand C, Eriksson PS, Nilsson M. Astrocytes and stroke: networking for survival? Neurochem Res. 2003;28:293–305. doi: 10.1023/a:1022385402197. [DOI] [PubMed] [Google Scholar]
- Andoh T, Chock PB, Chiueh CC. The roles of thioredoxin in protection against oxidative stress-induced apoptosis in SH-SY5Y cells. J Biol Chem. 2002;277:9655–9660. doi: 10.1074/jbc.M110701200. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Watabe M, Nakaki T. Regulation of neuronal glutathione synthesis. J Pharmacol Sci. 2008;108:227–238. doi: 10.1254/jphs.08r01cr. [DOI] [PubMed] [Google Scholar]
- Apopa PL, He X, Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol. 2008;22:63–76. doi: 10.1002/jbt.20212. [DOI] [PubMed] [Google Scholar]
- Bae SH, Woo HA, Sung SH, Lee HE, Lee SK, Kil IS, Rhee SG. Induction of sulfiredoxin via an Nrf2-dependent pathway and hyperoxidation of peroxiredoxin III in the lungs of mice exposed to hyperoxia. Antioxid Redox Signal. 2009;11:937–948. doi: 10.1089/ars.2008.2325. [DOI] [PubMed] [Google Scholar]
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- Barhwal K, Hota SK, Jain V, Prasad D, Singh SB, Ilavazhagan G. Acetyl-l-carnitine (ALCAR) prevents hypobaric hypoxia-induced spatial memory impairment through extracellular related kinase-mediated nuclear factor erythroid 2-related factor 2 phosphorylation. Neuroscience. 2009;161:501–514. doi: 10.1016/j.neuroscience.2009.02.086. [DOI] [PubMed] [Google Scholar]
- Barone MC, Sykiotis GP, Bohmann D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis Model Mech. 2011;4:701–707. doi: 10.1242/dmm.007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, Chowdhry S, Patani R, Chandran S, Horsburgh K, Hayes JD, Hardingham GE. Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proceedings of the National Academy of Sciences. 2011;108:E1–E2. doi: 10.1073/pnas.1015229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict RH, Zivadinov R. Risk factors for and management of cognitive dysfunction in multiple sclerosis. Nat Rev Neurol. 2011;7:332–342. doi: 10.1038/nrneurol.2011.61. [DOI] [PubMed] [Google Scholar]
- Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by Protein Kinase C in Response to Antioxidants Leads to the Release of Nrf2 from INrf2, but Is Not Required for Nrf2 Stabilization/Accumulation in the Nucleus and Transcriptional Activation of Antioxidant Response Element-mediated NAD(P)H:Quinone Oxidoreductase-1 Gene Expression. Journal of Biological Chemistry. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102:244–249. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli J, Munemasa Y, Kwong JM, Piri N. Overexpression of thioredoxins 1 and 2 increases retinal ganglion cell survival after pharmacologically induced oxidative stress, optic nerve transection, and in experimental glaucoma. Trans Am Ophthalmol Soc. 2009;107:161–165. [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF. Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington’s disease following chronic energy deprivation. Hum Mol Genet. 2010;19:3190–3205. doi: 10.1093/hmg/ddq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Fang Q, Zhang J, Zhou D, Wang Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res. 2011;89:515–523. doi: 10.1002/jnr.22577. [DOI] [PubMed] [Google Scholar]
- Chen M, Regan RF. Time course of increased heme oxygenase activity and expression after experimental intracerebral hemorrhage: correlation with oxidative injury. Journal of Neurochemistry. 2007;103:2015–2021. doi: 10.1111/j.1471-4159.2007.04885.x. [DOI] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc Natl Acad Sci U S A. 2009a;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009b;34:663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu M, Jones DP, Greenamyre JT, Cai J. Protection against oxidant-induced apoptosis by mitochondrial thioredoxin in SH-SY5Y neuroblastoma cells. Toxicol Appl Pharmacol. 2006;216:256–262. doi: 10.1016/j.taap.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, Feinstein SI. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med. 2009;46:146–153. doi: 10.1016/reeradbiomed.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol Med. 2008;14:495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer-and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M. The Nrf2-inducible antioxidant defense in astrocytes can be both up- and down-regulated by activated microglia:Involvement of p38 MAPK. Glia. 2011;59:785–799. doi: 10.1002/glia.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Molecular and Cellular Biology. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascenzo M, Martinotti G, Azzena GB, Grassi C. cGMP/protein kinase G-dependent inhibition of N-type Ca2+ channels induced by nitric oxide in human neuroblastoma IMR32 cells. J Neurosci. 2002;22:7485–7492. doi: 10.1523/JNEUROSCI.22-17-07485.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Hayashi T, Nagotani S, Sehara Y, Zhang H, Tsuchiya A, Ohta Y, Tomiyama K, Morimoto N, Miyazaki M, Huh NH, Nakao A, Kamiya T, Abe K. Reduction of cerebral infarction in rats by biliverdin associated with amelioration of oxidative stress. Brain Res. 2008;1188:1–8. doi: 10.1016/j.brainres.2007.07.104. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–6899. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Zhang X, Ji H, Liu H, Li S, Li L. Probucol and atorvastatin in combination protect rat brains in MCAO model: Upregulating Peroxiredoxin2, Foxo3a and Nrf2 expression. Neuroscience Letters. 2012;509:110–115. doi: 10.1016/j.neulet.2011.12.054. [DOI] [PubMed] [Google Scholar]
- Dunn LL, Buckle AM, Cooke JP, Ng MK. The emerging role of the thioredoxin system in angiogenesis. Arterioscler Thromb Vasc Biol. 2010;30:2089–2098. doi: 10.1161/ATVBAHA.110.209643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer BE, Nishimura RN, De Vellis J, Yoshida T. Heme oxygenase is a heat shock protein and PEST protein in rat astroglial cells. Glia. 1992;5:300–305. doi: 10.1002/glia.440050407. [DOI] [PubMed] [Google Scholar]
- Eftekharpour E, Holmgren A, Juurlink BH. Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia. 2000;31:241–248. doi: 10.1002/1098-1136(200009)31:3<241::aid-glia50>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Eggler AL, Small E, Hannink M, Mesecar AD. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J. 2009;422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrichmann G, Petrasch-Parwez E, Lee DH, Reick C, Arning L, Saft C, Gold R, Linker RA. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS One. 2011;6:e16172. doi: 10.1371/journal.pone.0016172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J Biol Chem. 2003;278:22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- Erttmann SF, Bast A, Seidel J, Breitbach K, Walther R, Steinmetz I. PGD2 and PGE2 regulate gene expression of Prx 6 in primary macrophages via Nrf2. Free Radic Biol Med. 2011;51:626–640. doi: 10.1016/j.freeradbiomed.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Escartin C, Won SJ, Malgorn C, Auregan G, Berman AE, Chen PC, Deglon N, Johnson JA, Suh SW, Swanson RA. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J Neurosci. 2011;31:7392–7401. doi: 10.1523/JNEUROSCI.6577-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everse J, Coates PW. Neurodegeneration and peroxidases. Neurobiol Aging. 2009;30:1011–1025. doi: 10.1016/j.neurobiolaging.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Feeser VR, Loria RM. Modulation of traumatic brain injury using progesterone and the role of glial cells on its neuroprotective actions. J Neuroimmunol. 2011;237:4–12. doi: 10.1016/j.jneuroim.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Ferrandiz ML, Devesa I. Inducers of heme oxygenase-1. Curr Pharm Des. 2008;14:473–486. doi: 10.2174/138161208783597399. [DOI] [PubMed] [Google Scholar]
- Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Gan L, Johnson DA, Johnson JA. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur J Neurosci. 2010;31:967–977. doi: 10.1111/j.1460-9568.2010.07138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Ji X, Hu X, Luo Y, Zhang L, Li P, Liu X, Yan F, Vosler P, Gao Y, Stetler RA, Chen J. Transgenic Overexpression of Peroxiredoxin-2 Attenuates Ischemic Neuronal Injury Via Suppression of a Redox-Sensitive Pro-Death Signaling Pathway. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hancock R, Bertrand HC, Tsujita T, Naz S, El-Bakry A, Laoruchupong J, Hayes JD, Wells G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic Biol Med. 2011 doi: 10.1016/j.freeradbiomed.2011.10.486. [DOI] [PubMed] [Google Scholar]
- Hatic H, Kane MJ, Saykally JN, Citron BA. Modulation of Transcription Factor Nrf2 in an In Vitro Model of Traumatic Brain Injury. J Neurotrauma. 2011 doi: 10.1089/neu.2011.1806. [DOI] [PubMed] [Google Scholar]
- Hattori I, Takagi Y, Nakamura H, Nozaki K, Bai J, Kondo N, Sugino T, Nishimura M, Hashimoto N, Yodoi J. Intravenous administration of thioredoxin decreases brain damage following transient focal cerebral ischemia in mice. Antioxid Redox Signal. 2004;6:81–87. doi: 10.1089/152308604771978372. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Holtzclaw WD, Dinkova-Kostova AT, Talalay P. Protection against electrophile and oxidative stress by induction of phase 2 genes: the quest for the elusive sensor that responds to inducers. Adv Enzyme Regul. 2004;44:335–367. doi: 10.1016/j.advenzreg.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Hong Y, Yan W, Chen S, Sun CR, Zhang JM. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin. 2010;31:1421–1430. doi: 10.1038/aps.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J, Hastings T, Greenamyre JT, Chen J. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci. 2011;31:247–261. doi: 10.1523/JNEUROSCI.4589-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- Hubbs AF, Benkovic SA, Miller DB, O’Callaghan JP, Battelli L, Schwegler-Berry D, Ma Q. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T, Li J, Meyer CJ, Janicki JS, Hannink M, Cui T. Dihydro-CDDO-trifluoroethyl amide (dh404), a novel Nrf2 activator, suppresses oxidative stress in cardiomyocytes. PLoS One. 2009;4:e8391. doi: 10.1371/journal.pone.0008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorato NG, Jazwa A, Rojo AI, Garcia C, Fernandez-Ruiz J, Grochot-Przeczek A, Stachurska A, Jozkowicz A, Dulak J, Cuadrado A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS One. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Jain AK, Bloom DA, Jaiswal AK. Nuclear Import and Export Signals in Control of Nrf2. Journal of Biological Chemistry. 2005;280:29158–29168. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janaky R, Varga V, Hermann A, Saransaari P, Oja SS. Mechanisms of L-cysteine neurotoxicity. Neurochem Res. 2000;25:1397–1405. doi: 10.1023/a:1007616817499. [DOI] [PubMed] [Google Scholar]
- Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:103–116. doi: 10.5507/bp.2010.017. [DOI] [PubMed] [Google Scholar]
- Jin W, Wang H, Ji Y, Hu Q, Yan W, Chen G, Yin H. Increased intestinal inflammatory response and gut barrier dysfunction in Nrf2-deficient mice after traumatic brain injury. Cytokine. 2008a;44:135–140. doi: 10.1016/j.cyto.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Jin W, Wang H, Yan W, Xu L, Wang X, Zhao X, Yang X, Chen G, Ji Y. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediators Inflamm. 2008b;2008:725174. doi: 10.1155/2008/725174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Wang H, Yan W, Zhu L, Hu Z, Ding Y, Tang K. Role of Nrf2 in protection against traumatic brain injury in mice. J Neurotrauma. 2009;26:131–139. doi: 10.1089/neu.2008.0655. [DOI] [PubMed] [Google Scholar]
- Jin W, Wang HD, Hu ZG, Yan W, Chen G, Yin HX. Transcription Factor Nrf2 Plays a Pivotal Role in Protection Against Traumatic Brain Injury-Induced Acute Intestinal Mucosal Injury in Mice. J Surg Res. 2008c doi: 10.1016/j.jss.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Jin W, Zhu L, Guan Q, Chen G, Wang QF, Yin HX, Hang CH, Shi JX, Wang HD. Influence of Nrf2 genotype on pulmonary NF-kappaB activity and inflammatory response after traumatic brain injury. Ann Clin Lab Sci. 2008d;38:221–227. [PubMed] [Google Scholar]
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson TJ, Johnson LC, Lowther WT. Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature. 2008;451:98–101. doi: 10.1038/nature06415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S, Levonen AL, Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, Polman CH, Schmierer K, Yousry TA, Yang M, Eraksoy M, Meluzinova E, Rektor I, Dawson KT, Sandrock AW, O’Neill GN. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Jaiswal AK. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. J Biol Chem. 2011;285:21349–21358. doi: 10.1074/jbc.M110.121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. The Lancet Neurology. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Pae HO, Zheng M, Park R, Kim YM, Chung HT. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res. 2007;101:919–927. doi: 10.1161/CIRCRESAHA.107.154781. [DOI] [PubMed] [Google Scholar]
- Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–1865. doi: 10.1038/sj.onc.1206369. [DOI] [PubMed] [Google Scholar]
- Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J, Shaw PJ. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–1706. doi: 10.1093/brain/awh503. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohle C, Bock KW. Coordinate regulation of human drug-metabolizing enzymes, and conjugate transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem Pharmacol. 2009;77:689–699. doi: 10.1016/j.bcp.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem. 2006;98:1852–1865. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kalonia H, Kumar A. Huntington’s disease: pathogenesis to animal models. Pharmacol Rep. 2010;62:1–14. doi: 10.1016/s1734-1140(10)70238-3. [DOI] [PubMed] [Google Scholar]
- Kuo CP, Lu CH, Wen LL, Cherng CH, Wong CS, Borel CO, Ju DT, Chen CM, Wu CT. Neuroprotective effect of curcumin in an experimental rat model of subarachnoid hemorrhage. Anesthesiology. 2011;115:1229–1238. doi: 10.1097/ALN.0b013e31823306f0. [DOI] [PubMed] [Google Scholar]
- Kuwano A, Ikeda H, Takeda K, Nakai H, Kondo I, Shibahara S. Mapping of the human gene for inducible heme oxygenase to chromosome 22q12. Tohoku J Exp Med. 1994;172:389–392. doi: 10.1620/tjem.172.389. [DOI] [PubMed] [Google Scholar]
- Laurent TC, Moore EC, Reichard P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B. J Biol Chem. 1964;239:3436–3444. [PubMed] [Google Scholar]
- Leautaud V, Demple B. Regulation of heme oxygenase-1 mRNA deadenylation and turnover in NIH3T3 cells by nitrosative or alkylation stress. BMC Molecular Biology. 2007;8:116. doi: 10.1186/1471-2199-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem. 2001;276:20011–20016. doi: 10.1074/jbc.M100734200. [DOI] [PubMed] [Google Scholar]
- Lee OH, Jain AK, Papusha V, Jaiswal AK. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J Biol Chem. 2007;282:36412–36420. doi: 10.1074/jbc.M706517200. [DOI] [PubMed] [Google Scholar]
- Li W, Jain MR, Chen C, Yue X, Hebbar V, Zhou R, Kong A-NT. Nrf2 Possesses a Redox-insensitive Nuclear Export Signal Overlapping with the Leucine Zipper Motif. Journal of Biological Chemistry. 2005;280:28430–28438. doi: 10.1074/jbc.M410601200. [DOI] [PubMed] [Google Scholar]
- Li W, Kong A-N. Molecular mechanisms of Nrf2-mediated antioxidant response. Molecular Carcinogenesis. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu SW, Kong A-NT. Nrf2 Possesses a Redox-sensitive Nuclear Exporting Signal in the Neh5 Transactivation Domain. Journal of Biological Chemistry. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- Lim JH, Kim KM, Kim SW, Hwang O, Choi HJ. Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacol Res. 2008;57:325–331. doi: 10.1016/j.phrs.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Lin SX, Lisi L, Dello Russo C, Polak PE, Sharp A, Weinberg G, Kalinin S, Feinstein DL. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro. 2011;3 doi: 10.1042/AN20100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci. 2006;11:356–367. doi: 10.2741/1803. [DOI] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Bruck W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- Lippoldt A, Padilla CA, Gerst H, Andbjer B, Richter E, Holmgren A, Fuxe K. Localization of thioredoxin in the rat brain and functional implications. J Neurosci. 1995;15:6747–6756. doi: 10.1523/JNEUROSCI.15-10-06747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani H, Hirota K, Sasada T, Ueda-Taniguchi Y, Taniguchi Y, Sono H, Yodoi J. Transactivation of an inducible anti-oxidative stress protein, human thioredoxin by HTLV-I Tax. Immunol Lett. 1996;54:67–71. doi: 10.1016/s0165-2478(96)02651-x. [DOI] [PubMed] [Google Scholar]
- Matz P, Turner C, Weinstein PR, Massa SM, Panter SS, Sharp FR. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Research. 1996;713:211–222. doi: 10.1016/0006-8993(95)01511-6. [DOI] [PubMed] [Google Scholar]
- McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Diaz-Corrales FJ, Fukuda M, Kitaichi K, Miyoshi K, Ogawa N. Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone-formation-related molecules. FASEB J. 2006;20:571–573. doi: 10.1096/fj.05-4996fje. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009;16:17–25. doi: 10.1038/gt.2008.126. [DOI] [PubMed] [Google Scholar]
- Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003;546:181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- Nakata K, Tanaka Y, Nakano T, Adachi T, Tanaka H, Kaminuma T, Ishikawa T. Nuclear receptor-mediated transcriptional regulation in Phase I, II, and III xenobiotic metabolizing systems. Drug Metab Pharmacokinet. 2006;21:437–457. doi: 10.2133/dmpk.21.437. [DOI] [PubMed] [Google Scholar]
- Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF, Kiaei M. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Niture SK, Jaiswal AK. Prothymosin-alpha mediates nuclear import of the INrf2/Cul3 Rbx1 complex to degrade nuclear Nrf2. J Biol Chem. 2009;284:13856–13868. doi: 10.1074/jbc.M808084200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nouhi F, Tusi SK, Abdi A, Khodagholi F. Dietary supplementation with tBHQ, an Nrf2 stabilizer molecule, confers neuroprotection against apoptosis in amyloid beta-injected rat. Neurochem Res. 2011;36:870–878. doi: 10.1007/s11064-011-0417-2. [DOI] [PubMed] [Google Scholar]
- Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. American Journal of Physiology - Cell Physiology. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Hanggi D, Wu Y, Michiue H, Tomizawa K, Ono S, Matsui H, Date I, Steiger H-J. Protein therapy using heme-oxygenase-1 fused to a polyarginine transduction domain attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:2231–2242. doi: 10.1038/jcbfm.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okauchi M, Hua Y, Keep RF, Morgenstern LB, Xi G. Effects of Deferoxamine on Intracerebral Hemorrhage-Induced Brain Injury in Aged Rats. Stroke. 2009;40:1858–1863. doi: 10.1161/STROKEAHA.108.535765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Komuro T, Macdonald RL. Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. Journal of Neurosurgery. 2002;96:1094–1102. doi: 10.3171/jns.2002.96.6.1094. [DOI] [PubMed] [Google Scholar]
- Ono S, Zhang ZD, Marton LS, Yamini B, Windmeyer E, Johns L, Kowalczuk A, Lin G, Macdonald RL. Heme oxygenase-1 and ferritin are increased in cerebral arteries after subarachnoid hemorrhage in monkeys. J Cereb Blood Flow Metab. 2000;20:1066–1076. doi: 10.1097/00004647-200007000-00006. [DOI] [PubMed] [Google Scholar]
- Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, Meyer C, Liby KT, Miller RH, Sporn MB, Letterio JJ. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci Rep. 2011;1:201. doi: 10.1038/srep00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude A, Murthy MR, Mirault ME. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell Mol Life Sci. 2005;62:1063–1080. doi: 10.1007/s00018-005-4541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Robinson KM, Cassina P, Diaz-Amarilla PJ, Hagen TM, Radi R, Barbeito L, Beckman JS. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–7785. doi: 10.1523/JNEUROSCI.0823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- Prestera T, Zhang Y, Spencer SR, Wilczak CA, Talalay P. The electrophile counterattack response: protection against neoplasia and toxicity. Adv Enzyme Regul. 1993;33:281–296. doi: 10.1016/0065-2571(93)90024-8. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobón-Velasco JC, Devijver H, García-Mayoral MF, Van Leuven F, Hayes JD, Bertho G, Cuadrado A. Structural and functional characterization of Nrf2 degradation by the GSK-3/β-TrCP axis. Molecular and Cellular Biology. 2012 doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS Journal. 2011;278:4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger HO, and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Medina-Campos ON, Rada P, Zuniga-Toala A, Lopez-Gazcon A, Espada S, Pedraza-Chaverri J, Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52:473–487. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Rada P, Egea J, Rosa AO, Lopez MG, Cuadrado A. Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol Cell Neurosci. 2008;39:125–132. doi: 10.1016/j.mcn.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- Rybnikova E, Damdimopoulos AE, Gustafsson JA, Spyrou G, Pelto-Huikko M. Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur J Neurosci. 2000;12:1669–1678. doi: 10.1046/j.1460-9568.2000.00059.x. [DOI] [PubMed] [Google Scholar]
- Santamaria A, Perez-Severiano F, Rodriguez-Martinez E, Maldonado PD, Pedraza-Chaverri J, Rios C, Segovia J. Comparative analysis of superoxide dismutase activity between acute pharmacological models and a transgenic mouse model of Huntington’s disease. Neurochem Res. 2001;26:419–424. doi: 10.1023/a:1010911417383. [DOI] [PubMed] [Google Scholar]
- Sarlette A, Krampfl K, Grothe C, Neuhoff N, Dengler R, Petri S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2008;67:1055–1062. doi: 10.1097/NEN.0b013e31818b4906. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Chollate S, Jung M-y, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, Rhodes KJ. Fumarates Promote Cytoprotection of Central Nervous System Cells against Oxidative Stress via the Nuclear Factor (Erythroid-Derived 2)-Like 2 Pathway. Journal of Pharmacology and Experimental Therapeutics. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem. 2009;110:469–485. doi: 10.1111/j.1471-4159.2009.06160.x. [DOI] [PubMed] [Google Scholar]
- Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, Keum YS, Han J, Gallo MA, Kong AN. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem. 2004;279:23052–23060. doi: 10.1074/jbc.M401368200. [DOI] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Erb H, Murphy TH. Dopamine activates Nrf2-regulated neuroprotective pathways in astrocytes and meningeal cells. J Neurochem. 2007;101:109–119. doi: 10.1111/j.1471-4159.2006.04345.x. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohe R, Kensler TW, Yamamoto M, Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35:639–650. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT. Benzene, NQO1, and genetic susceptibility to cancer. Proc Natl Acad Sci U S A. 1999;96:7624–7626. doi: 10.1073/pnas.96.14.7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Leveille F, Papadia S, Higgins LG, Varley J, Baxter P, Hayes JD, Hardingham GE. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J Neurochem. 2008;107:533–543. doi: 10.1111/j.1471-4159.2008.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic Biol Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A. 2004;101:1461–1466. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun KH, Chang KH, Clawson S, Ghosh S, Mirzaei H, Regnier F, Shah K. Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase activity. J Neurochem. 2011;118:902–914. doi: 10.1111/j.1471-4159.2011.07343.x. [DOI] [PubMed] [Google Scholar]
- Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, Schmidt EE. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4:e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Kikuta K, Moriwaki T, Aoki T, Nozaki K, Hashimoto N, Miyamoto S. Expression of thioredoxin-1 and hypoxia inducible factor-1alpha in cerebral arteriovenous malformations: Possible role of redox regulatory factor in neoangiogenic property. Surg Neurol Int. 2011;2:61. doi: 10.4103/2152-7806.80356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Mitsui A, Nishiyama A, Nozaki K, Sono H, Gon Y, Hashimoto N, Yodoi J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc Natl Acad Sci U S A. 1999;96:4131–4136. doi: 10.1073/pnas.96.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J Biol Chem. 2001;276:48093–48099. doi: 10.1074/jbc.M108622200. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, Matsuura T, Abe K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Tanito M, Agbaga MP, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med. 2007;42:1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–8994. doi: 10.1074/jbc.M709040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledano MB. The guardian recruits cops: the p53-p21 axis delegates prosurvival duties to the Keap1-Nrf2 stress pathway. Mol Cell. 2009;34:637–639. doi: 10.1016/j.molcel.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekci KU, Civi Bayin E, Genc S, Genc K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Parkinsons Dis. 2011:314082. doi: 10.4061/2011/314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, Drexhage JA, Flor T, Gerritsen W, van der Valk P, de Vries HE. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic Biol Med. 2010;49:1283–1289. doi: 10.1016/j.freeradbiomed.2010.07.013. [DOI] [PubMed] [Google Scholar]
- van Roon-Mom WM, Pepers BA, t Hoen PA, Verwijmeren CA, den Dunnen JT, Dorsman JC, van Ommen GB. Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington’s disease. BMC Mol Biol. 2008;9:84. doi: 10.1186/1471-2199-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakade C, King MD, Laird MD, Alleyne CH, Jr, Dhandapani KM. Curcumin attenuates vascular inflammation and cerebral vasospasm after subarachnoid hemorrhage in mice. Antioxid Redox Signal. 2009;11:35–45. doi: 10.1089/ars.2008.2056. [DOI] [PubMed] [Google Scholar]
- Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li R-c, Xu Y, Dore S, Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radical Biology and Medicine. 2012a;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007a;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2007b;282:1718–1726. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen G, Zhu WW, Zhou D. Activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) in the basilar artery after subarachnoid hemorrhage in rats. Ann Clin Lab Sci. 2010;40:233–239. [PubMed] [Google Scholar]
- Wang Z, Ma C, Meng CJ, Zhu GQ, Sun XB, Huo L, Zhang J, Liu HX, He WC, Shen XM, Shu Z, Chen G. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J Pineal Res. 2012b doi: 10.1111/j.1600-079X.2012.00978.x. [DOI] [PubMed] [Google Scholar]
- Woo HA, Jeong W, Chang T-S, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of Cysteine Sulfinic Acid by Sulfiredoxin Is Specific to 2-Cys Peroxiredoxins. Journal of Biological Chemistry. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- Wruck CJ, Claussen M, Fuhrmann G, Romer L, Schulz A, Pufe T, Waetzig V, Peipp M, Herdegen T, Gotz ME. Luteolin protects rat PC12 and C6 cells against MPP+ induced toxicity via an ERK dependent Keap1-Nrf2-ARE pathway. J Neural Transm Suppl. 2007:57–67. doi: 10.1007/978-3-211-73574-9_9. [DOI] [PubMed] [Google Scholar]
- Xiao H, Lv F, Xu W, Zhang L, Jing P, Cao X. Deprenyl prevents MPP(+)- induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology. 2011;290:287–295. doi: 10.1016/j.tox.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Wang HD, Feng XM, Ding YS, Jin W, Tang K. The expression of NF-E2-related factor 2 in the rat brain after traumatic brain injury. J Trauma. 2009;66:1431–1435. doi: 10.1097/TA.0b013e318180f5c7. [DOI] [PubMed] [Google Scholar]
- Yan W, Wang HD, Hu ZG, Wang QF, Yin HX. Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci Lett. 2008;431:150–154. doi: 10.1016/j.neulet.2007.11.060. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kikuchi G. Sequence of the reaction of heme catabolism catalyzed by the microsomal heme oxygenase system. FEBS Lett. 1974;48:256–261. doi: 10.1016/0014-5793(74)80481-3. [DOI] [PubMed] [Google Scholar]
- Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AN. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274:27545–27552. doi: 10.1074/jbc.274.39.27545. [DOI] [PubMed] [Google Scholar]
- Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN. p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J Biol Chem. 2000;275:2322–2327. doi: 10.1074/jbc.275.4.2322. [DOI] [PubMed] [Google Scholar]
- Yu SS, Zhao J, Lei SP, Lin XM, Wang LL, Zhao Y. 4-hydroxybenzyl alcohol ameliorates cerebral injury in rats by antioxidant action. Neurochem Res. 2011;36:339–346. doi: 10.1007/s11064-010-0335-8. [DOI] [PubMed] [Google Scholar]
- Yu X, Egner PA, Wakabayashi J, Wakabayashi N, Yamamoto M, Kensler TW. Nrf2-mediated induction of cytoprotective enzymes by 15-deoxy-Delta12,14-prostaglandin J2 is attenuated by alkenal/one oxidoreductase. J Biol Chem. 2006;281:26245–26252. doi: 10.1074/jbc.M604620200. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang S, Zhang M, Weng Z, Li P, Gan Y, Zhang L, Cao G, Gao Y, Leak RK, Sporn MB, Chen J. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke. 2012;43:1390–1397. doi: 10.1161/STROKEAHA.111.647420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu Y, Zhou D, Wang Z, Chen G. Recombinant human erythropoietin (rhEPO) alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Nrf2-ARE pathway. Cytokine. 2010a;52:252–257. doi: 10.1016/j.cyto.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang S, Chen Y, Wang Z, Li E, Xu Y. Icariin Inhibits Hydrogen Peroxide-Mediated Cytotoxicity by Up-regulating Sirtuin Type 1-Dependent Catalase and Peroxiredoxin. Basic Clin Pharmacol Toxicol. 2010b doi: 10.1111/j.1742-7843.2010.00595.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007a;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007b;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–43158. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Martin RD, Zhang JH. Advances in experimental subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;110:15–21. doi: 10.1007/978-3-7091-0353-1_3. [DOI] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]