

After inhibition of the PI3 kinase/AKT pathway, the binding of mRNA export proteins in nuclear complexes is reduced. The nuclear export of bulk poly(A) RNA and of a subset of specific mRNAs is increased after AKT inhibition. The results show that mRNA export can be regulated by the PI3 kinase/AKT pathway.
Abstract
UAP56, ALY/REF, and NXF1 are mRNA export factors that sequentially bind at the 5′ end of a nuclear mRNA but are also reported to associate with the exon junction complex (EJC). To screen for signal transduction pathways regulating mRNA export complex assembly, we used fluorescence recovery after photobleaching to measure the binding of mRNA export and EJC core proteins in nuclear complexes. The fraction of UAP56, ALY/REF, and NXF1 tightly bound in complexes was reduced by drug inhibition of the phosphatidylinositide 3-kinase (PI3 kinase)/AKT pathway, as was the tightly bound fraction of the core EJC proteins eIF4A3, MAGOH, and Y14. Inhibition of the mTOR mTORC1 pathway decreased the tight binding of MAGOH. Inhibition of the PI3 kinase/AKT pathway increased the export of poly(A) RNA and of a subset of candidate mRNAs. A similar effect of PI3 kinase/AKT inhibition was observed for mRNAs from both intron-containing and intronless histone genes. However, the nuclear export of mRNAs coding for proteins targeted to the endoplasmic reticulum or to mitochondria was not affected by the PI3 kinase/AKT pathway. These results show that the active PI3 kinase/AKT pathway can regulate mRNA export and promote the nuclear retention of some mRNAs.
INTRODUCTION
The export of RNA molecules from the nucleus to the cytoplasm is a critical step in cellular maintenance. The export mechanisms for different classes of RNA share a common approach; RNAs are packaged in messenger ribonucleoprotein (mRNP) complexes that bind export receptors that subsequently dock the complex at nuclear pores for translocation to the cytoplasm (Rodriguez et al., 2004; Stewart, 2007). The export receptor for most mRNAs is NXF1. In this export mechanism, the ATP-dependent RNA helicase UAP56 is deposited on mRNA during splicing. UAP56 recruits the adapter protein ALY/REF (Luo et al., 2001), which in turn binds the NXF1-p15 heterodimer that is required for most mRNA export in species from yeast to humans (Segref et al., 1997; Grüter et al., 1998; Herold et al., 2001).
In one model, this complex of UAP56, ALY/REF, and NXF1-p15 associates with a spliced mRNA at the capped 5′ end of the mRNA (Cheng et al., 2006). UAP56 and ALY/REF are members of the transcription/export (TREX) complex located at the 5′ end (Bensaude, 2011; Nechaev and Adelman, 2011). NXF1 recruited to the complex may subsequently bind at nuclear pores (Forler et al., 2004), and also to transportin-2 (Shamsher et al., 2002), for export of the mRNP complex from the nucleus.
A second complex, the exon junction complex (EJC), forms on the mRNA at sites 24 nucleotides upstream from exon–exon junctions (Singh et al., 2012) and could play a role in export to the cytoplasm. The crystal structure of the core EJC with eIF4A3, MLN51, MAGOH, and Y14 bound to polyU has been solved (Andersen et al., 2006; Bono et al., 2006). UAP56, ALY/REF, and NXF1 are all reported to be members of an extended EJC (Kataoka et al., 2000; Le Hir et al., 2000). In vitro, at least three EJC proteins—UAP56, Y14, and MAGOH—bind to the export receptor NXF1–p15 heterodimer (Kataoka et al., 2001). Proteins interacting with the EJC also mediate the translational repression and nonsense-mediated decay of mRNAs containing premature stop codons (Singh et al., 2007; Isken et al., 2008). RNA export proteins joining the EJC in the nucleus stimulate translation of the mRNA in the cytoplasm (Wiegand et al., 2003; Nott et al., 2004), an effect mediated by the mTOR pathway (Ma et al., 2008).
Some mRNAs leave the nucleus by alternative mechanisms. For example, a family of proliferation-related mRNAs sharing a 50-nucleotide sequence element in their 3′ untranslated region are exported by a mechanism requiring the eukaryotic translation initiation factor eIF4E and the export receptor CRM1 but not NXF1–p15 (Culjkovic et al., 2006). Incorporation of the signal sequence coding region (SSCR) in an mRNA can facilitate efficient nuclear export even in the absence of an intron or 5′ cap structure. SSCR-mediated nuclear export requires NXF1 but not UAP56 or the similar URH49 (Palazzo et al., 2007).
The regulation of mRNA export is inadequately understood. One report identified threonine-219 (T219) on human ALY/REF as an AKT phosphorylation site (Okada et al., 2008). Point mutation of this site has a dominant-negative effect on mRNA export and inhibits cell proliferation. ALY/REF also binds in vitro to phosphatidylinositol-(3,4,5)-tris-phosphate (PI(3,4,5)P3) and phosphatidylinositol-(4,5)-bis-phosphate (PI(4,5)P2). These results linked a TREX complex protein to the phosphatidylinositol (PI) signal transduction pathway.
The PI pathway is a central regulator of cell metabolism, gene expression, and development (Yuan and Cantley, 2008; Chalhoub and Baker, 2009; Skwarek and Boulianne, 2009). PI(4,5)P2 is the key pathway intermediate; it can be further phosphorylated by phosphatidylinositol 3-kinase (PI3 kinase), generating PI(3,4,5)P3, which is a potent activator of several downstream effectors, including AKT (Altomare and Testa, 2005). Surprisingly, PI(4,5)P2 is present in the nucleus at RNA-splicing speckled domains, which are enriched in mRNA-splicing and export factors (Boronenkov et al., 1998; Osborne et al., 2001; Stallings et al., 2005; Bunce et al., 2006). The nucleus of a typical cell has 20–40 RNA-splicing speckled domains around which transcription, RNA splicing, and RNA export from the nucleus are organized. A majority of, but not all, pre-mRNA transcripts are spliced at or near these domains (Smith et al., 1999), and some mRNAs move into the interior of these domains and bind before release to the cytoplasm (Shopland et al., 2002).
In this study we used fluorescence recovery after photobleaching (FRAP) to screen proteins present in mRNA export complexes for changes in binding after inhibition of the PI3K/AKT or mTOR signal transduction pathways. PI3 kinase and AKT did regulate the binding affinity of the essential mRNA export proteins UAP56 and NXF1 in complexes formed at RNA-splicing speckled domains and at sites in the nucleoplasm. The assembly of the core EJC proteins eIF4A3, MAGOH, and Y14 into complexes at speckled domains was also altered by inhibition of PI3 kinase or AKT. MAGOH binding was strongly reduced by the inhibition of mTOR. The activated PI3K/AKT pathway caused increased retention of poly(A) RNA and specific mRNAs in the nucleus, consistent with this signal transduction pathway regulating the export of mRNA.
RESULTS
The binding of mRNA export proteins in live cells is regulated by PI3 kinase and AKT
FRAP measures the relative kinetics and affinities of fluorescent protein binding in complexes that are immobile over the time course of the experiment (Wagner et al., 2004; Lele et al., 2006; Lele and Ingber, 2006; Kota et al., 2008; Nickerson, 2009). To identify mRNA export factors whose assembly into export complexes is regulated by the PI signal transduction pathway, we screened enhanced green fluorescent protein (EGFP)-fusion proteins of known mRNA export factors for altered photobleach recovery kinetics after drug inhibition of the PI pathway in live HeLa cells. EGFP-UAP56 was most concentrated at RNA-splicing speckled domains (Supplemental Figure S1 and Supplemental Movie S1), consistent with previous results (Kota et al., 2008). After photobleaching, EGFP-UAP56 fluorescence recovered at speckled domains with a half-time (t1/2) of 15.6 s (Figure 1 and Table 1). An immobile fraction of 27.7% was measured, which represents the tightly bound fraction that did not exchange during the time course of the experiment. After inhibition of PI3 kinase with LY294002 for 2–3 h, the recovery half-time increased to 21.2 s (Figure 1 and Table 1), and the tightly bound immobile fraction was markedly decreased from 27.7 to 17.1% (Figure 1 and Table 1). This immobile fraction represents the fraction of UAP56 bound in more stable complexes. The immobile fraction of EGFP-UAP56 was similarly reduced from 27.7 to 8.5% by inhibition of AKT with 5 μM Akt inhibitor VIII for 3–4 h (Vlahos et al., 1994; Barnett et al., 2005).
FIGURE 1:

The binding of mRNA export complex proteins, as measured by FRAP, is regulated by PI3 kinase, AKT, and mTOR. HeLa cells were transfected with EGFP-fusion proteins for UAP56, ALY/REF, and NXF1. After 48 h, cells were treated with 20 μM of the PI3 kinase inhibitor LY294002 (green circles) for 2 h, 5 μM of AKT inhibitor VIII (blue triangles) for 3.5 h, and 100 nM of the mTOR inhibitor rapamycin (red squares) for 5 h. Multiple cells were examined by FRAP over a 2-h interval in all treatment groups. The times between drug addition and photobleaching are reported as a mean. Black squares, untreated cells. Normalized fluorescence recovery curves are presented for EGFP-UAP56, EGFP-TAP/NXF1, and EGFP-ALY/REF. Also shown is EGFP-UAP56 K95N, a point mutant that cannot bind ATP. Each colored arrow marks the t1/2 of recovery for the curve of the same color. Statistical analysis of the differences between averaged FRAP curves was by paired one-tailed t test: untreated EGFP-UAP56 vs. LY294002 (p = 0.0083), AKT (p < 0.0001), rapamycin (p = 0.2799); untreated EGFP-ALY/REF vs. LY294002 (p < 0.0001), AKT (p < 0.0001), rapamycin (p < 0.0001); untreated EGFP-NXF1 vs. LY294002 (p = 0.0002), AKT (p = 0.0004), rapamycin (p = 0.4102). Means are plotted with error bars for standard errors. n, number of cells in each treatment group.
TABLE 1:
FRAP measurements for mRNA export proteins.
| EGFP-UAP56 | EGFP-NFX1 | EGFP-ALY/REF | EGFP-UAP56 K95N | ||
|---|---|---|---|---|---|
| Untreated | Half-time (s) | 15.6 | 4.4 | 6.8 | 2.6 |
| Immobile fraction (%) | 27.7 ± 1.2 | 21.4 ± 3.6 | 15.3 ± 2.0 | 5.8 ± 1.1 | |
| LY294002 | Half-time (s) | 21.2 | 7.2 | 5.5 | 2.7 |
| Immobile fraction (%) | 17.1 ± 1.9 | 6.9 ± 1.6 | 6.9 ± 1.4 | 6.0 ± 0.7 | |
| AKT I VIII | Half-time (s) | 15.7 | 8.8 | 7.4 | 2.8 |
| Immobile fraction (%) | 8.5 ± 0.9 | 9.2 ± 1.6 | 4.12 ± 1.8 | 3.5 ± 0.2 | |
| Rapamycin | Half-time (s) | 14.8 | 6.9 | 6.6 | 2 |
| Immobile fraction (%) | 27.8 ± 1.3 | 23.7 ± 3.1 | 10.4 ± 2.2 | 6.6 ± 0.7 |
AKT is activated downstream of PI3 kinase by phosphorylation at threonine 308 (Alessi et al., 1996), but mTOR signaling through mTORC2 can also influence AKT activity (Nave et al., 1999) by phosphorylation of AKT at serine 473 (Sarbassov et al., 2005; Jacinto et al., 2006). Akt inhibitor VIII is an allosteric inhibitor of AKT 1 (IC50 = 58 nM), AKT2 (IC50 = 210 nM), and AKT3 (IC50 = 2119 nM; Lindsley et al., 2005) that inhibits the activating phosphorylations of both T308 and S473 (AKT1 numbering; Calleja et al., 2009). This gives this inhibitor greater specificity for AKT than other inhibitors that target the ATP-binding sites of AKT, sites that have conserved features with other ATP-binding proteins. The crystal structure of Akt inhibitor VIII inhibitor bound to AKT1 was solved (Wu et al., 2010). This compound and its derivatives are candidate agents for cancer therapy (Liu et al., 2009).
Rapamycin, although an mTOR inhibitor, inhibits only TORC1 complex signaling and does not affect the activity of AKT during the time periods of our experiment (Sarbassov et al., 2006). Inhibition of mTOR with 100 nM rapamycin for 4 h did not significantly change the photobleach recovery kinetics or the tightly bound immobile fraction (27.8%) compared with control cells without mTOR inhibition (Figure 1 and Table 1).
UAP56 recruits ALY/REF to mRNA export complexes. EGFP-ALY/REF is also concentrated at the nuclear speckled domains (Supplemental Figure S1 and Supplemental Movie S2). There was little effect of PI3 kinase inhibition on the t1/2 for photobleach recovery of EGFP-ALY/REF (Figure 1 and Table 1). However, inhibition of PI3 kinase, AKT, or mTOR TORC1 decreased the immobile fraction. For example, this tightly bound fraction was reduced from 15.3% in control cells to <5% after AKT inhibition. This in vivo result is consistent with previous evidence of ALY/REF phosphorylation by AKT (Okada et al., 2008). Our data suggest that AKT phosphorylation causes a fraction of ALY/REF to bind more tightly in complexes at speckled domains.
In the export mechanism proposed for mRNA, UAP56 and ALY/REF then recruit the export receptor NXF1 (Grüter et al., 1998; Herold et al., 2001; Luo et al., 2001). EGFP-NXF1 (Supplemental Figure 1 and Supplemental Movie S3) recovered after photobleaching at speckled domains with t1/2 = 4.4 s and an immobile fraction of 21.4%. Inhibition of PI3 kinase with LY294002 or inhibition of AKT with Akt inhibitor VIII increased the t1/2 to 7.2 and 8.8 s, respectively (Table 1). Moreover these two treatments significantly reduced the tightly bound fraction from the 21.4% in control cells to 6.9 and 9.2%, respectively. After inhibition of mTOR with rapamycin the recovery rate was increased to 6.9 s and the immobile fraction increased to 23.7% (Figure 1 and Table 1).
UAP56 is an ATP-dependent RNA helicase. We showed that a point mutant of UAP56 that cannot bind ATP (UAP56 K95N) has a reduced association with complexes at RNA-splicing speckled domains and has a dominant-negative effect on the export of a splicing reporter mRNA (Kota et al., 2008). A similar point mutant in UAP56 of Saccharomyces cerevisiae inhibited its function (Zhang and Green, 2001). In control experiments, EGFP-UAP56K95N was less concentrated at speckled domains and, after a photobleach, recovered with t1/2 = 2.6 s and a very small immobile fraction (5.8%), both consistent with low-affinity binding. For this mutant, there was no significant change in the photobleach recovery kinetics or in the immobile fraction caused by drug treatments (Figure 1 and Table 1).
We verified the effectiveness of the drug treatments used in these FRAP experiments. Cells were treated for 4 h with LY294002 (20 μM), AKT VIII (5 μM), or rapamycin (100 nM). As shown in the Western blot of Supplemental Figure S2, AKT phosphorylation at threonine 308 was inhibited after treatment with LY294002 or AKT VIII, so both drug treatments eliminated AKT activation in the PI pathway. Rapamycin eliminated the activating phosphorylation of mTOR at serine 2448 (Supplemental Figure S2).
The region-of-interest photobleaching results reported in Figure 1 averaged together recovery for the pool of EGFP-UAP56 at speckled domains and at adjacent nucleoplasmic sites. When we compared the fluorescence recovery of EGFP-UAP56 in the nucleoplasm with the recovery at speckled domains, we found that the nucleoplasmic and speckled domain fluorescence had tightly bound immobile fractions of 26.5 and 40.5%, respectively. PI3 kinase inhibition decreased the immobile fraction markedly to 7.6% for nucleoplasmic EGFP-UAP56 and 2.8% for UAP56 in speckled domains (Figure 2). UAP56 binding in both compartments is similarly affected by inhibition of the PI pathway at PI3 kinase or AKT.
FIGURE 2:

EGFP-UAP56 is more tightly bound at nuclear speckled domains than at sites in the nucleoplasm. HeLa cells were transfected with EGFP-UAP56 wild type, and then, after 48 h, cells were treated for 3–5 h with 25 μM LY294002. Normalized fluorescence recovery curves over time were calculated for regions of interest, including either individual speckles or regions of the nucleoplasm devoid of speckles. n, number of cells for each graph. Means were plotted, with error bars showing the standard errors for each collected time point.
The binding of EJC core proteins is also affected by the PI3 kinase/AKT pathway
UAP56, ALY/REF, and NXF1 are recruited to the 5′ end of an mRNA for export (Cheng et al., 2006) but are also reported to associate with the EJC (Le Hir et al., 2000, 2001). Structural studies of the EJC show it to have a stable core consisting of the proteins eIF4A3, MLN51, and the Y14/MAGOH heterodimer (Andersen et al., 2006; Bono et al., 2006). We used FRAP after PI or mTOR pathway inhibition to explore the regulation of EJC core protein binding at RNA-splicing speckled domains.
As shown in Figure 3, fluorescence of EGFP-eIF4A3 (Supplemental Figure S3 and Supplemental Movie S4) recovered at photobleached speckled domains with t1/2 = 6.2 s and a higher immobile fraction than UAP56 (55.4%). EGFP-eIF4A3 showed a significant decrease in the t1/2 of recovery after PI3 kinase, AKT, and mTOR inhibition (Table 2). In addition, the immobile fraction also decreased markedly to 36.4 and 33.7%, respectively, after PI3 kinase or AKT inhibition (Figure 3 and Table 2). eIF4A3 and MAGOH are core proteins of the EJC. EGFP-MAGOH, which also localized to speckled domains (Supplemental Figure S3 and Supplemental Movie 5), recovered with t1/2 = 10.2 s and showed the highest immobile, or tightly bound, fraction (72%) of any protein in this study. Inhibition of PI3 kinase, AKT, or rapamycin did not significantly affect the initial recovery rate (Figure 3 and Table 2). However, the immobile fraction was greatly decreased after all three drug treatments, with inhibition of mTOR having the largest effect and reducing the immobile fraction of MAGOH to 42.2%. Inhibition of PI3 kinase and AKT decreased the tightly bound fraction to 47.7 and 51.2%, respectively (Table 2). These results suggest that the mTOR pathway can regulate the binding of EJC core proteins in complexes at RNA-splicing speckled domains. Further, the effect of mTOR inhibition on the tightly bound, immobile fraction of MAGOH was larger than the effect of AKT or PI3 kinase inhibition, suggesting that mTOR did not act entirely through AKT. We propose that both pathways are regulators of mRNA export protein binding at speckled domains.
FIGURE 3:

The binding of EJC core proteins, as measured by FRAP, is regulated by PI3 kinase, AKT, and mTOR. HeLa cells were transfected with EGFP-fusion proteins. After 48 h, cells were treated with 20 μM PI3 kinase inhibitor LY294002 (green circles) for 2.5 h, 5 μM AKT inhibitor VIII (blue triangles) for 3.5 h, or 100 nM of the mTOR inhibitor rapamycin (red squares) for 4.5 h. Black squares, untreated cells. Normalized fluorescence recovery curves are presented for the EJC fusion proteins EGFP-eIF4A3, EGFP-MAGOH, and EGFP-Y14. Also shown is EGFP-eIF4A3 K88N, a point mutant that cannot bind ATP. Each colored arrow marks the t1/2 of recovery for the curve of the same color. Statistical analysis of the differences between averaged FRAP curves was by paired one-tailed t test: untreated EGFP-eIF4A3 vs. LY294002 (p = 0.0002), AKT (p = 0.0002), rapamycin (p = 0.0002); untreated EGFP-MAGOH vs. LY294002 (p = 0.0001), AKT (p = 0.0001), rapamycin (p = 0.0001); untreated EGFP-Y14 vs. LY294002 (p = 0.0001), AKT (p = 0.0001), rapamycin (p = 0.0527). Means are plotted with standard errors for n cells in each treatment group.
TABLE 2:
FRAP measurements for proteins of the EJC core complex.
| EGFP-eIF4A3 | EGFP-MAGOH | EGFP-Y14 | EGFP-eIF4A3 K88N | ||
|---|---|---|---|---|---|
| Untreated | Recovery half-time (s) | 6.2 | 10.2 | 4.5 | 3.4 |
| Immobile fraction (%) | 55.4 ± 3.0 | 72.0 ± 1.5 | 56.0 ± 2.0 | 16.8 ± 0.8 | |
| LY294002 | Recovery half-time (s) | 3.4 | 12.1 | 5.2 | 2.4 |
| Immobile fraction (%) | 36.4 ± 2.5 | 47.7 ± 4.2 | 43.8 ± 3.6 | 11.0 ± 0.7 | |
| AKT I VIII | Recovery half-time (s) | 3.6 | 12.9 | 4.3 | 1.5 |
| Immobile fraction (%) | 33.7 ± 2.9 | 51.2 ± 3.4 | 37.3 ± 2.2 | 17.0 ± 0.6 | |
| Rapamycin | Recovery half-time (s) | 4.1 | 10.9 | 5.6 | 1.8 |
| Immobile fraction (%) | 51.46 ± 2.5 | 42.2 ± 4.0 | 61.4 ± 2.4 | 16.1 ± 0.6 |
Because MAGOH interacts with Y14, forming a heterodimer, and Y14 is a putative mRNA export factor (Kataoka et al., 2001), we measured the binding of EGFP-Y14 at speckled domains by FRAP (Supplemental Figure S3 and Supplemental Movie S6). EGFP-Y14 recovered very rapidly at speckled domains (t1/2 = 4.5 s) with a high immobile fraction of 56%. Drug treatments did not significantly change the kinetics of recovery for the rapidly exchanging fraction. However, after inhibition of PI3 kinase the immobile fraction of EGFP-Y14 was decreased to 43.8% (Figure 3 and Table 2). Inhibition of AKT had greater effect, reducing the tightly bound fraction to 37.3%. Inhibition of mTOR had no significant effect.
Like UAP56, eIF4A3 is an ATP-dependent RNA helicase of the DExD/H family (Linder, 2008). A point mutant of eIF4A3 that is unable to bind to ATP (Shibuya et al., 2006) was cloned. EGFP-eIF4A3 with this K88N point mutation was less concentrated at speckled domains than was the wild-type eI4A3 but was, instead, more widely distributed in the nucleoplasm, similar to the distribution of the UAP56 K95N point mutant, which cannot bind ATP (Kota et al., 2008). EGFP-eIF4A3K88N was not, however, excluded from speckled domains, where it recovered after a photobleach with t1/2 = 3.4 s and an immobile fraction of 16.8%. This decreased binding compared with the wild-type protein (5.3 s, 27.2%) is consistent with decreased speckled domain localization. The tightly bound immobile fraction of EGFP-eIF4A3 K88N significantly decreased only after PI3 kinase inhibition with LY294002 to 11.4%. The half-time of photobleach recovery was decreased after PI3 kinase, AKT, or mTOR inhibition from 3.4 s to 2.4, 1.4, and 1.8 s, respectively (Figure 3). This mutant eIF4A3, which can still be incorporated in the EJC core complex (Shibuya et al., 2006), retained the ability to be affected by both PI pathway and mTOR pathway signaling.
Inhibition of the PI3 kinase/AKT pathway increases the export of poly(A) RNA
Because the inhibition of PI3K, AKT, or mTOR affected the binding of mRNA export–related proteins in live cells, we reasoned that those changes might have consequences on the export of some mRNAs from the nucleus to cytoplasm. We performed a standard mRNA export assay according to Okada et al. (2008) and Wickramasinghe et al. (2010), quantifying nuclear or cytoplasmic poly(A) RNA with biotinylated oligo(dT) and Alexa 488–streptavidin (Figure 4). Inhibition of PI3 kinase with LY294002 or inhibition of AKT with AKT I VIII caused a significant decrease in nuclear poly(A) RNA and a significant increase in cytoplasmic poly(A) RNA without changing the total signal per cell. Using both inhibitors simultaneously caused a larger effect, increasing our confidence that the PI3 kinase/AKT branch of this complicated pathway is affecting mRNA export (Figure 4).
FIGURE 4:

Inhibition of PI3 kinase or AKT increases cytoplasmic levels and reduces nuclear levels of poly(A) RNA. HeLa cells were treated for 4 h with 20 μM LY294002, 5 μM AKT inhibitor VIII, or 100 nM rapamycin before FISH. Cells were fixed for 10 min on ice and then hybridized with biotinylated 54-mer oligo(dT). The hybridized probe was detected with Alexa 488–conjugated streptavidin. Cells were counterstained with 4′,6-diamidino-2-phenylindole to identify nuclei. (A) Representative field of the poly(A) distribution in untreated HeLa cells. Scale bar, 20 μm. (B) Bar graph of poly(A) in the nucleus or cytoplasm. Values are plotted as the percentage of the total cellular poly(A). Statistical analysis of the significance of differences between untreated and treated samples is by t test. Means are plotted with SDs. ***p ≤ 0.001.
The drug treatments did not change the poly(A) distribution within the nucleus, where it was most concentrated in RNA-splicing speckled domains. In the cytoplasm, however, AKT inhibition caused a decreased perinuclear localization of poly(A). This change was not seen after PI3 kinase or mTOR inhibition.
Inhibition of AKT increases the export of specific endogenous mRNAs
Measurement of nuclear and cytoplasmic poly(A) levels by fluorescence in situ hybridization (FISH) has been a standard assay for assessing nuclear export of mRNA (Strasser et al., 2002; Okada et al., 2008; Wickramasinghe et al., 2010). There are limitations to this approach; the results present an average nuclear-to-cytoplasmic ratio for a very heterogeneous population that includes many nuclear poly(A) RNA species that are noncoding and not exported (Brown et al., 1992; Rinn et al., 2007; Amaral et al., 2011). For mRNAs the export of different species may be regulated differently and, therefore, differentially affected by the experimental manipulation.
For these reasons, we quantified changes in the nuclear-to-cytoplasmic ratio of specific mRNAs by quantitative real-time (qRT)-PCR assay. In initial experiments, drug inhibition of PI3 kinase and AKT treatments changed the nuclear-to-cytoplasmic ratio of several housekeeping gene mRNAs that are commonly used for normalization. Normalization by cell number was an improvement, but there were experiment-to-experiment variations that we attributed to the varying yield in RNA isolation from nuclear and cytoplasmic fractions.
To overcome this problem, we designed an assay with improved quantification for measuring the nuclear and cytoplasmic fractions of individual mRNA species by normalization to a synthetic internal standard RNA added to the nucleus and to the cytoplasm in exactly the same amount before RNA isolation. In vitro–transcribed miniUAA1 mRNA, an RNA not present in human cells, was used as the synthetic internal standard (Wong and Medrano, 2005; Amrani et al., 2008). Normalization to this internal standard corrected for any nuclear-to-cytoplasmic or sample-to-sample variation in protocol steps, including TRIzol mRNA isolation, RNA precipitation, DNase I digestion, and reverse transcription to generate cDNAs. The use of an internal standard also allowed the calculation of real rather than relative nuclear-to-cytoplasmic ratios.
RNA from whole cells, as well as from the separated cytoplasmic and nuclear fractions, each spiked with a constant and known mass of the internal standard, was extracted and isolated before qRT-PCR to quantify individual species. The quality of our separation of nucleus and cytoplasm was measured by RT-PCR. The nuclear noncoding RNA 7SK was used as a nuclear marker, and the mitochondrial-encoded and restricted cytochrome B mRNA was used as a cytoplasmic marker. As seen in Figure 5A, there was little contamination of nuclear RNA by cytochrome B mRNA and little contamination of cytoplasmic RNA by the nuclear-restricted 7SK. Drug treatment of cells to inhibit PI3 kinase, AKT, or mTOR did not affect the separation. The quality of the separation of nuclear and cytoplasmic material was also assessed by Western blotting for the nuclear protein RNA polymerase II and the cytoplasmic protein γ-tubulin (Figure 5B). There was little cross-contamination of the fractions.
FIGURE 5:

The separation of nuclear and cytoplasmic RNA. (A) The fidelity of nuclear and cytoplasmic RNA separation was assessed by qRT-PCR quantification of RNAs known to be restricted to the nucleus or to the cytoplasm. The small nuclear RNA 7SK was the control for the nuclear fraction. Cytochrome B mRNA is a mitochondrially encoded transcript and not present in the nucleus, making it a good cytoplasmic RNA marker. Ct values were normalized using miniUAA1 mRNA from yeast added as an internal standard to each fraction after the separation of nuclei and cytoplasm. There was very little nuclear contamination of cytoplasmic RNA and very little cytoplasmic contamination of nuclear RNA. Treatment of cells with 20 μM PI3 kinase inhibitor LY294002, 5 μM AKT inhibitor VIII, or 100 nM mTOR inhibitor rapamycin did not affect the partitioning of either 7SK RNA or cytochrome B mRNA. Each bar represents the average of two independent experiments in which triplicates were analyzed. Error bars represent the SE of the mean. ns, not significant. (B) The quality of the nuclear–cytoplasmic fractionation was also assessed by Western blotting using the large subunit of RNA polymerase II as a nuclear control and γ-tubulin as a cytoplasmic control. The results confirmed that nuclear and cytoplasmic material was cleanly separated.
Using these validated methods, we quantified the endogenous intron-containing mRNAs for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), Brahma-related gene 1 (BRG1), and heterogeneous RNP (hnRNP) Q in the nucleus and cytoplasm of HeLa cells (Figure 6A). Drug treatment to inhibit PI3 kinase had no significant effect on the nuclear-to-cytoplasmic partitioning of these mRNAs. Inhibition of AKT, however, caused a large decrease in the nuclear-to-cytoplasmic ratios. This result is consistent with an increased rate of nuclear export after AKT inhibition but not PI3 kinase inhibition. Active AKT may be increasing the nuclear retention of these mRNAs. mTOR inhibition did not significantly alter the nuclear-to-cytoplasmic ratios.
FIGURE 6:

Inhibition of AKT increases cytoplasmic levels and reduces nuclear levels of endogenous mRNAs without changing total cellular levels. Cells were treated with LY294002 (20 μM), AKT inhibitor VIII (5 μM), and rapamycin (100 nM), or were untreated, for 4 h at 37°C. Specific mRNAs were quantified in the nuclear and cytoplasmic fractions by qRT-PCR. Ct values were normalized using miniUAA1 mRNA from yeast added as an internal standard to each fraction after the separation of nuclei and cytoplasm. (A) Quantitative analysis of intron-containing mRNAs in the nucleus and cytoplasm: GAPDH, BRG1, and hnRNP Q. (B) Analysis of histone mRNAs whose genes lack introns: histones H2B, H4, and H3A. (C) Analysis of spliced histone mRNAs. The genes for these transcripts contain introns: histones H1F0, H1FX, and H3F3. Each point represents the mean of two independent experiments in which triplicates were analyzed. Error bars, SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ns, not significant.
Intronless mRNAs are exported efficiently by mechanisms using UAP56, ALY/REF, and NXF1 (Huang et al., 1999, 2003; Huang and Steitz, 2001; Herold et al., 2001; Erkmann et al., 2005; Farny et al., 2008; Taniguchi and Ohno, 2008; Read and Digard, 2010). The nuclear export of intron-containing mRNAs and intronless mRNAs might be differently regulated by signal transduction pathways. Histone mRNAs present a matched set of RNAs suitable for examining splicing-dependent differences. Although most histone mRNAs are intronless, a few are spliced. We performed an analysis of nuclear-to-cytoplasmic partitioning for the intronless histone mRNAs—H2B, H4, and H3A (Figure 6B)—and for the spliced histone mRNAs—H1F0, H1FX, and H3F3 (Figure 6C). Once again drug inhibition of AKT decreased the nuclear-to-cytoplasmic ratio of all histone mRNAs, consistent with an increased rate of nuclear export. The presence of an intron did not change the regulation by AKT. Of interest, the inhibition of PI3 kinase increased the nuclear retention of spliced histone H1FX and H3F3 mRNAs but did not significantly affect the nuclear export of spliced histone H1F0. The nuclear export of all histone mRNAs was unaffected by rapamycin treatment to inhibit mTOR.
Although the drug treatments were short, we checked to see whether they were able to affect the transcription of the mRNAs used in Figure 6A. There were some modest drug-specific and transcript-specific changes in total cellular levels of these mRNAs (Supplemental Figure S4), but these changes could not account for the differences in nuclear-to-cytoplasmic ratios.
Overexpression of the mRNA splicing factor SRm160 causes the appearance of intron-containing mRNAs in the cytoplasm (McCracken et al., 2002). The PI3 kinase/AKT signaling pathway can change the alternative splicing of fibronectin mRNA (Blaustein et al., 2005). We wondered, therefore, whether the effects on mRNA partitioning seen after AKT inhibition might involve increased export of unspliced mRNA. Quantification of spliced and unspliced GAPDH (Supplemental Figure S5) showed that drug inhibition of AKT inhibition did significantly increase the amount of unspliced mRNA in the cytoplasm. The effect was small, however, and could not account for the enhanced export of GAPDH mRNA. In addition, based on the ΔΔCt values, the ratio of spliced to intron-containing RNA in the cytoplasm was, on average, 13, a level insignificant relative to the difference in partitioning after AKT inhibition (Figure 5A).
mRNAs encoding either endoplasmic reticulum (ER)–targeted or mitochondrial proteins have, respectively, a SSCR or mitochondrial signal coding region (MSCR) that appropriately targets the protein in cells. These RNA elements can be found near the 5′ end of the open reading frame of most mRNAs that encode secreted, membrane-bound, or mitochondrial-targeted proteins, and they encode peptide domains that target the newly translated protein to the ER or mitochondria (Palazzo et al., 2007). These sequences also promote nuclear export of these mRNAs by a novel mechanism that might be less dependent on the TREX complex for export than most mRNAs or even be TREX independent (Palazzo et al., 2007; Cenik et al., 2011). For both SSCR- and MSCR-containing mRNAs the first 69 nucleotides after the start codon tend to be adenine poor, whereas at the protein level both sequences are leucine rich, as well as rich in hydrophobic amino acids (Palazzo et al., 2007; Cenik et al., 2011). A comprehensive genome analysis showed that genes encoding either ER-targeting or mitochondrial proteins tend to lack introns in their 5′ untranslated regions (5′-UTRs) when compared with other protein-coding genes (Cenik et al., 2011). From this analysis a model to explain the alternative export mechanism has been proposed (Cenik et al., 2011). If the first element emerging from RNA polymerase II is an intron, the mRNA will be exported by a splicing-dependent pathway using TREX complex assembly. If, on the other hand, the first element is an SSCR or MSCR, then this mRNA will be exported by the alternative pathway (Cenik et al., 2011).
Because AKT inhibition decreased the nuclear and increased the cytoplasmic fractions of the selected mRNAs coding for cytoplasmic and nuclear proteins (Figure 6), we decided to determine whether this signaling pathway would also regulate the nuclear export of mRNAs encoding either ER-targeted or mitochondrial proteins. For this experiment we selected mRNAs without an intron at their 5′-UTR and examined the mRNA and protein sequences to determine their fit to consensus SSCR and MSCR motifs (Palazzo et al., 2007; Cenik et al., 2011)
The ER-targeted protein mRNAs were calreticulin (CALR, NM_004343.3), histocompatibility (minor) 13 (HM13, NM_178582), and olfactory receptor, family 10, subfamily A, member 3 (OR10A3, NM_001003745) mRNAs, which have 5, 15, and 19 adenines, respectively, in their first 70 nucleotides (Figure 7A). At the level of amino acid sequence, first 25 residues of CALR and OR10A3 are rich in leucine and hydrophobic amino acids. On the other hand, the first 25 residues of HM13 are poor in leucine and hydrophobic amino acids (Figure 7). The drug inhibition of AKT and mTORC1 had no significant effect on the nuclear export of these mRNAs (Figure 7A), which suggests that AKT specifically regulates the nuclear export of only a subset of mRNAs requiring the TREX complex. Surprisingly, inhibition of PI3 kinase slowed the nuclear export of these mRNAs (Figure 7).
FIGURE 7:

The AKT signaling pathway does not affect the nuclear export of mRNAs encoding either ER-targeted or nuclear-encoded mitochondrial proteins. Cells were treated with LY294002 (20 μM), AKT inhibitor VIII (5 μM), and rapamycin (100 nM), or were untreated, for 4 h at 37°C. Specific mRNAs were quantified in the nuclear and cytoplasmic fractions by qRT-PCR. Ct values were normalized using miniUAA1 mRNA from yeast added as an internal standard to each fraction after the separation of nuclei and cytoplasm. All mRNAs selected for analysis lacked an intron in their 5′ untranslated regions. Each point represents the mean of two independent experiments in which triplicates were analyzed. Sequence features that have been linked to the ALREX pathway are also presented (Palazzo et al., 2007; Cenik et al., 2011). These are the number of adenines in the first 70 nucleotides after the start codon and the number of leucine and hydrophobic amino acids coded for in the same region. (A) mRNAs encoding ER-targeted proteins were CALR (GO:0006984), OR10A3 (GO:0004984), and HM13 (GO:0004190). (B) Nuclear-encoded mRNAs for mitochondrial proteins were F1-ATP synthase (ATP5B; GO:0000275), FDX1 (GO:0005506), IDH3B (GO:0005962), and FH (GO:0004333). Error bars, SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ns, not significant.
The nuclear export of selected nuclear mRNAs that encode mitochondrial proteins was also analyzed. F1 ATP synthase (F1; ATP5B), ferredoxin 1 (FDX1), and fumarate hydratase (FH) were chosen because these mRNAs have few adenines in the first 70 nucleotides (5, 2, and 6, respectively) and because these proteins are rich in hydrophobic amino acids (Figure 7B). A fourth mitochondrial protein mRNA, isocitrate dehydrogenase 3 (NAD+; IDH3B), was chosen because it has 10 adenines in the first 70 nucleotides. The drug inhibition of PI3 kinase, AKT, and mTORC1 had no significant effect on the nuclear export of IDH3B, FDX1, and FH mRNAs (Figure 7B). The inhibition of mTORC1 decreased the nuclear export of ATP5B mRNA (Figure 7B). This profile of signal transduction effects was very different from what was observed for mRNAs lacking both MSCRs and SSCRs (Figure 6).
Nuclear and cytoplasmic mRNA levels are at a steady state determined by the rates of transcription and RNA processing and degradation, as well as of mRNA export to the cytoplasm. Measurements of mRNA export that depend on nuclear-to-cytoplasmic ratios can be confounded by these additional variables. First, we decided to test whether the drug inhibition of PI3 kinase, AKT, or mTOR would affect the degradation rates or the transcription rates of the candidate mRNAs used in the export assays of Figure 6. We used actinomycin D at a concentration that inhibits RNA polymerases I and II (Trask and Muller, 1988). HeLa cells were treated (or not) with actinomycin D (0.5 μg/ml) for 7 h to inhibit all transcription. Over the last 3 h, cells were also treated with the PI3 kinase, AKT, and mTORC1 inhibitors (Supplemental Figure S6). The rate of mRNA degradation was not affected by drug inhibition of PI3 kinase, AKT, and mTORC1, suggesting that the PI3 kinase/AKT and mTORC1 signaling pathways did not affect the degradation of the candidate mRNAs of Figure 6.
Recovery of transcription after actinomycin D inhibition requires hours (Schluederberg et al., 1971), but recovery from 5,6-dichloro-1-β-ribofuranosyl benzimidazole (DRB), an inhibitor of RNA polymerase II elongation (Sehgal et al., 1976; Bensaude, 2011; Nechaev and Adelman, 2011), is rapid. Methods using recovery from DRB have been developed to measure rates of transcription and transcriptional elongation (Singh and Padgett, 2009).
This method (Singh and Padgett, 2009) was used to reversibly block transcription in HeLa cells and measure transcription rates after a chase to remove DRB. After 100 μM DRB treatment of HeLa cells for 7 h, the recovery of pre-mRNA levels was measured by qRT-PCR using intron–exon primer pairs (Supplemental Figure S7). Drug inhibition of PI3 kinase, AKT, and mTORC1 had some transcript-specific effects on transcription rate. These changes could not, however, account for the changes in nuclear-to-cytoplasmic ratios of the mRNAs in our mRNA export assays (Figure 6). Taken together, our data suggest that the changes in the nuclear-to-cytoplasmic ratio of candidate mRNAs after the drug inhibition AKT were not due to decreases in their transcription rates or increases in their degradation rates. The effects of AKT inhibition on nuclear/cytoplasmic partitioning were due to mRNA export. AKT inhibition increases the rate of mRNA export to the cytoplasm for mRNAs that lack an SSCR or MSCR motif.
DISCUSSION
mRNA export has been considered a constitutive process; a transcript that is spliced and processed would automatically be sent to the cytoplasm. On reflection, it would be surprising if export were the only unregulated step in the life cycle of an mRNA. Export to the cytoplasm, and not processing, may be the rate-limiting step in the delivery of some mRNAs to the cytoplasm; nuclear dwell times of 1–20 min (histone H4 and β-actin, respectively) after processing have been calculated for a few selected mRNA species (Gondran et al., 1999). Some mature mRNA molecules do not leave the nucleus at all (Weil et al., 2000).
Regulation of the rate of nuclear export or the extent of retention for mRNAs could be an especially important mechanism for modulating translatable, cytoplasmic levels of messages with short half-lives. RNA half-lives in human cells vary from 10 min for c-myc (Dani et al., 1984) to >50 h for β-globin (Volloch and Housman, 1981). The mean half-life for 5000 profiled mRNAs in mouse fibroblasts is 9 h (Schwanhausser et al., 2011). Global profiling of mRNA turnover rates in human HepG2 cells shows that mRNAs for many of the most important proteins—for example, 15% of transcription factors—have half-lives of <2 h (Yang et al., 2003).
The combined use of FRAP with the experimental manipulation of signal transduction has great potential for identifying the pathways that regulate the assembly of specific macromolecular complexes. Using this screening approach, we showed that the inhibition of either PI3 kinase or AKT in live cells changes the binding affinity of mRNA export–related proteins in macromolecular complexes at mRNA-splicing speckled domains. These domains are large enough to serve as practical FRAP targets and are enriched in mRNA export factors and transcripts.
One branch of the phosphatidylinositol signal transduction pathway, downstream from PI3 kinase, operates through AKT (Barnett et al., 2005; Franke, 2008). AKT has been reported to phosphorylate the mRNA export linker protein ALY/REF in vitro (Okada et al., 2008). Point mutants that cannot be AKT phosphorylated inhibit bulk poly(A) export (Okada et al., 2008). In vitro studies show that UAP56 binds to mRNA before recruiting ALY/REF and then NXF1. In our speckled-domain photobleaching experiments (Figure 1), EGFP-tagged versions of UAP56, ALY/REF, and NXF1 had a significant decrease in the tightly bound “immobile” fraction after either PI3 kinase or AKT inhibition. This suggests that the PI3 kinase/AKT pathway increases the fraction of macromolecular complexes in which these proteins are tightly bound at speckled domains. Rapamycin, which inhibits mTOR but not the PI3 kinase pathway, did not have this effect, establishing specificity to this regulation.
A second complex of proteins assembling on a nuclear mRNA is the EJC (Kataoka et al., 2000; Le Hir et al., 2000). Three proteins of the EJC core complex (Tange et al., 2005)—eIF4A3, Y14, and MAGOH—also had a decrease in tight binding at speckled domains after PI3 kinase and/or AKT inhibition. MAGOH showed a larger decrease in its tightly bound immobile fraction after mTOR inhibition. It has been reported that Y14 and MAGOH form a stable heterodimer (Ballut et al., 2005; Le Hir and Seraphin, 2008). However, because the binding kinetics of the two proteins, as measured by FRAP, are different, we conclude that they either must be exchanging within the heterodimer or are not always dimerized (Figure 3).
We used FRAP as a screening technique to connect mRNA export to specific signal transduction pathways. Mechanistically, fluorescence recovery in our experiments is the result of multiple processes. These include the steps of equilibrium binding and diffusion (Lele et al., 2004; Nickerson, 2009): 1) the unbinding of bleached molecules, 2) the unbinding of still-fluorescent molecules outside of the bleach zone, 3) the exchange of these two pools of molecules by diffusion, and 4) the binding of fluorescent molecules in the bleach zone. FRAP recovery for an mRNA transcript might also depend on the movement of a completed and processed mRNA transcript away from the transcription site, with recovery coming from unbleached molecules binding to a new mRNA being transcribed (Ben-Ari et al., 2010; Brody et al., 2011). For screening purposes we used the complex nuclear region around single RNA-splicing speckled domains as the photobleach target in each cell. Although the transcription and splicing of protein-coding genes are enriched at and around these speckled domains (Smith et al., 1999; Shopland et al., 2002), there may be complexes of mRNA export proteins present that are not yet on mRNA and may have unique binding constants. Reported rates of protein diffusion in the nucleus (Kruhlak et al., 2000; Carrero et al., 2003) are much faster than the recoveries we report here. FRAP was previously used to explore the complex mechanisms of transcription and RNA processing at short reporter genes (Ben-Ari et al., 2010; Yunger et al., 2010; Brody et al., 2011). Based on these studies, on measured rates of transcription at endogenous genes (Singh and Padgett, 2009; Wada et al., 2009), and on other studies of transcriptional dynamics (reviewed in Palangat and Larson, 2012), we expect the rates of mRNA elongation, movement away from the bleach zone, and recovery by additional transcription to be slow relative to the photobleach recovery rates we report here. We conclude that the changes we observed in photobleach recovery rates caused by PI3 kinase, AKT, and mTOR inhibition were effects on the binding of mRNA export complex proteins in complexes at transcripts and were not measuring steady-state transcript exchange.
We proceeded with experiments to directly measure the effects that the same drugs had on the nuclear export of specific transcripts—first, by a standard assay (Okada et al., 2008; Wickramasinghe et al., 2010) that evaluates export by measuring FISH localization of poly(A) mRNA in the nucleus and the cytoplasm. Inhibition of either PI3 kinase or AKT caused a significant decrease in the nuclear-to-cytoplasmic ratio of poly(A) RNA without changing total poly(A) levels (Figure 4). Simultaneous inhibition of PI3 kinase and AKT caused a slightly larger and more significant change. One explanation for these changes is that the PI3 kinase/AKT branch of the PI pathway induces the nuclear retention of some poly(A) molecules, whereas inhibition of the pathway increases the kinetics of their export.
When specific mRNA molecules were quantified in the nucleus and cytoplasm after drug treatment, the nuclear-to-cytoplasmic ratio of all species was significantly decreased by inhibition of AKT but not by inhibition of PI3 kinase or mTOR (Figure 6). This effect was seen for a subset of spliced and polyadenylated mRNAs, as well as for mRNAs from intronless genes and mRNAs lacking a poly(A) tail. These results confirmed the global result obtained by measuring poly(A) after AKT inhibition (Figure 4). Decreased transcription combined with decreased cytoplasmic turnover for a specific mRNA could theoretically contribute to reduced nuclear-to-cytoplasmic ratios without changes in total cellular levels. We ruled out this possibility by measuring transcription rates (Supplemental Figure 7) and turnover rates (Supplemental Figure 6) for each of the mRNAs of Figure 6 after drug treatments. After AKT inhibition, there were no significant changes in mRNA turnover, and the changes in transcription rate were transcript specific, variable in direction, and could not account for the altered nuclear/cytoplasmic partitioning. We conclude that the export of a subset of mRNAs from the nucleus to the cytoplasm is regulated by the PI3 kinase/AKT signal transduction pathway.
Not all mRNAs were subject to AKT regulation of nuclear export. mRNAs targeted to the endoplasmic reticulum by an SSCR motif or to mitochondria by an MSCR did not have altered export after drug inhibition of the AKT branch of the PI3 kinase pathway (Figure 7).
The differential effects of PI3 kinase versus AKT on the mRNA export of specific mRNAs (Figure 6) might result from the branching of the PI pathway downstream of PI3 kinase. Our experiments measure effects of the AKT branch of the downstream pathway. Mutations in budding yeast that reduce inositol hexakis-phosphate (IP6) production in the alternative branch of the PI pathway compromise mRNA export to the cytoplasm (York et al., 1999). Mutants in plc1, ipk2, or ipk1—all required to produce IP6—cause defects in mRNA export. IP6 and the Nup42-binding protein Gle1 cooperate to stimulate the ATPase activity of the DExD/H helicase Dbp5 (Alcazar-Roman et al., 2006; Weirich et al., 2006). Gle1- and IP6-activated Dbp5 remodels the mRNP to promote transit across nuclear pores to the cytoplasm (Tran et al., 2007). These are effects of the PI signaling pathway that are downstream from PI(4,5)P2 but do not involve PI3 kinase or AKT. Both branches of the PI pathway downstream from PI3 kinase affect mRNA export, and these effects may be opposing.
A growing body of work has localized many enzymes and inositol lipid intermediates of the PI pathway to sites in the nuclear interior and most often to speckled domains (reviewed in Neri et al., 2002; Irvine, 2003; Martelli et al., 2006; Okada and Ye, 2009; Barlow et al., 2010). These include PI(4,5)P2, the substrate of PI3 kinase (Boronenkov et al., 1998; Osborne et al., 2001; Stallings et al., 2005; Bunce et al., 2006), and several PI3 kinases (Didichenko and Thelen, 2001; Resnick et al., 2005). All three isoforms of AKT are observed in both the nucleus and the cytoplasm (Brazil et al., 2004; Martelli et al., 2006). All share a conserved CRM1-interacting nuclear export signal, suggesting that they can shuttle between nucleus and cytoplasm (Saji et al., 2005). The results we report here do not distinguish effects of the nuclear versus the cytoplasmic PI pathway on mRNA export factor binding or on bulk nuclear poly(A) export. Still, the spatial juxtaposition of mRNA export factors and of the PI signal transduction pathway that regulates their binding and function is unlikely to be a coincidence.
MATERIALS AND METHODS
Constructs
Standard procedures for cloning fluorescent fusion proteins were as previously described (Herbert et al., 2002; Wagner et al., 2003, 2004; Kota et al., 2008). Point mutations were generated using a Stratagene (La Jolla, CA) QuikChange Site-Directed Mutagenesis Kit (Kota et al., 2008).
Transfections and treatments
HeLa cells were grown on coverslips and then transfected with plasmids using Lipofectamine 2000 (Life Technologies, Paisley, United Kingdom) in serum-free medium for 4 h. After 48 h, transfected cells were treated for 4 h with drugs inhibiting PI3 kinase, AKT, or mTOR. PI3 kinase was inhibited with LY294002 (Cayman Chemical, Ann Arbor, MI) at a final concentration of 20 μM (Vlahos et al., 1994). mTOR was inhibited with rapamycin (Calbiochem, San Diego, CA) at a final concentration of 100 nM (Hosoi et al., 1999). AKT inhibitor VIII (Calbiochem) was used to inhibit AKT1, AKT2, and AKT3 at a final concentration of 5 μM. mRNA degradation rates were measured after transcription was inhibited for 7 h using actinomycin D at a final concentration of 0.5 μg/ml. Transcription rates were measured by qRT-PCR using intron–exon primer pairs in cells pretreated with DRB at final concentration of 100 μM and then at time intervals after removal of DRB.
Western blots
Proteins from HeLa cell lysates were separated by 10% SDS–PAGE and transferred to nitrocellulose membranes. Western blots were probed with primary antibodies, including rabbit antibodies to mTOR and phospho-mTOR serine 2448 (both from Millipore, Billerica, MA); rabbit antibodies to AKT and AKT-P threonine 308 (both from Cell Signaling, Danvers, MA); or mouse monoclonal antibody to actin (clone 4; ICN/MP, Solon, OH). Secondary anti-rabbit or anti-mouse horseradish peroxidase–conjugated secondary antibodies were from GE Healthcare (Little Chalfont, United Kingdom), and the detection system used the SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL).
Fluorescence recovery after photobleaching
FRAP assays were performed at 37°C as previously described (Wagner et al., 2004; Kota et al., 2008). Leica Confocal Software (Leica Microsystems, Exton, PA) was used to measure the intensity of fluorescence in the bleached region of interest and in the whole nuclear profile at each time point. We analyzed those data using Excel spreadsheets (Microsoft, Redmond, WA). Any remaining fluorescence in the bleached area after the bleach was normalized to zero. To calculate the relative fluorescence intensity (Irel) in the bleached area, we used three alternative equations. The first one (Phair and Misteli, 2000) used the equation Irel,t = N0It/NtI0. The second approach used the equation Irel,t = ItN0/Nt − (IpblN0/Npbl)/(I0 − IpblN0/Npbl). The third equation is derived from the second one by subtracting background fluorescence values measured outside of the cell from each fluorescence intensity measurement. For all three equations N0 is the total nuclear fluorescence before bleaching, Npbl is the total nuclear fluorescence in the first image taken after the bleach, Nt is the total nuclear fluorescence at time t, I0 is the fluorescence in the bleach zone before the bleach, It is the fluorescence in the bleach zone at time t, and Ipbl is the fluorescence in the bleach zone in the first image taken after the bleach. Recovery curves were drawn using Prism 5 (GraphPad Software, La Jolla, CA). The percentage of immobile protein (immobile fraction) was determined after normalization of fluorescence in the bleached area to 0 in the first postbleach image and 1 in the prebleached image, and it was calculated as the percentage difference between the relative fluorescence asymptote of the recovery curve and a relative recovery of 1, a value that would reflect complete recovery without an immobile fraction. Individual time points are presented as means, with error bars showing standard errors. The best fit for these photobleach recoveries was obtained using an exponential association curve: F(t) = Fmax(1 – e−kt). All half-times of recovery and immobile fractions were calculated from a best fit to this equation.
Multiple cells were examined by FRAP over a 2-h interval for each drug treatment in each experiment. The times between drug addition and photobleaching are reported as a mean. Three independent experiments were done for each EGFP-fusion protein paired with every drug. The recovery data averaged together in one recovery curve were from the same confocal session. Drug-treated and control cells were always from the same transfection and photobleached in the same confocal session. The first two sets of independent photobleaching experiments were performed on a Leica SP1 laser scanning confocal microscope (Leica, Wetzlar, Germany), and the last set (Figures 1 and 3) was performed on a Leica SP5 AOBS system. Results were independent of the system used. The results presented were from the Leica SP5 AOBS, using a 40×/numerical aperture 1.3 water immersion objective. EGFP fluorescence was photobleached using the 488-nm line of an argon laser for 200 ms at 20 mW. Nuclear speckled domains were spot photobleached. EGFP-NXF1, which was less concentrated in speckled domains, was region-of-interest photobleached.
Fluorescent in situ hybridization
FISH was performed by a modification of Johnson et al. (1991). A 54-mer oligo(dT) (Qiagen, Valencia, CA) was biotinylated with a Roche kit (Roche, Indianapolis, IN). HeLa cells were fixed in 4% paraformaldehyde for 10 min on ice and then permeabilized using 0.5% Triton X-100 in CSK buffer for 5 min at room temperature. After dehydration with ethanol to 70% and then 100%, cells were probed overnight the biotinylated oligo(dT) at 37°C in the presence of 15% formamide. After several washes in SSC buffer (4×, 2×, and 1×) cells were stained for 3 h (at 37°C) with Alexa 488–conjugated streptavidin in a buffer with 4× SSC buffer and 1% of RNase-free bovine serum albumin (Invitrogen/Life Technologies, Carlsbad, CA).
Nuclear/cytoplasmic fractionation
Nuclear and cytoplasmic fractions were separated by the protocol developed by Lamond and colleagues for proteomic studies (Andersen et al., 2002). Briefly, equal number of HeLa cells (5 × 105) were plated in 15-cm dishes and after 24 h were treated using LY294002 (20 μM), AKT inhibitor VIII (5 μM), or rapamycin (100 nM) for 4 h. After treatment and trypsinization, cells were centrifuged at 1000 rpm for 5 min and rinsed with ice-cold phosphate-buffered saline. Cell pellets were gently resuspended in 1 ml of lysis buffer A + VRC (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 2 mM vanadylriboside complex, pH 7.9) and immediately split into two tubes labeled Total and CN, respectively, and containing 500 μl each. Cells from both tubes were kept on ice for 5 min and broken to release their nuclei with a prechilled Dounce homogenizer (20 gentle strokes with a tight pestle). Dounced cells from the CN tubes were centrifuged at 228 × g for 5 min at 4°C. The supernatant was retained as the cytoplasmic fraction and kept on ice. The nuclear pellet was washed once again with buffer A + VRC, centrifuged, resuspended in 500 μl of buffer S1 + VRC (250 mM sucrose, 10 mM MgCl2, 2 mM vanadylriboside complex), and layered over a 500-μl cushion of buffer S3 + VRC (880 mM sucrose, 0.5 mM MgCl2, 2 mM vanadylriboside complex). Nuclei were then centrifuged at 2800 × g for 10 min at 4°C and resuspended in 500 μl of buffer A + VRC. All buffers were made using water pretreated with 0.1% (vol/vol) diethylpyrocarbonate.
Quantitative real-time PCR
A 120-pg amount of a synthetic, capped, and poly(A)-containing mRNA (miniUAA1; Amrani et al., 2008) was in vitro transcribed, purified, and added to each tube before TRIzol (Invitrogen) extraction. This synthetic mRNA worked as an internal standard or calibrator in our qRT-PCR measurement of endogenous mRNAs. Total, cytoplasmic, and nuclear RNA fractions—now in the presence of 120 pg of the synthetic miniUAA1 mRNA—were isolated using TRIzol reagent, digested with DNase I recombinant enzyme (Roche), and purified with DNA-free RNA kit (Zymo Research, Irvine, CA). cDNAs were then synthesized using random primers (Roche) and reverse transcription SuperScript III (Invitrogen). GoTaq mix (Promega, Madison, WI) was used to amplify cDNAs.
All curves were normalized by comparative Ct (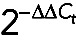 ) method (Wong and Medrano, 2005). Briefly, Ct values were corrected for the mean Ct value of miniUAA1 mRNA as follows: First, raw Ct values of GAPDH mRNA (Ct,GAPDH) were corrected using Mean Ct,miniUAA1, generating a corrected curve for GAPDH mRNA (Ct,GAPDH,C). Then Ct,GAPDH – Mean Ct,miniUAA1 = Ct,GAPDH,C. Because an isolation was performed separating cytoplasmic and nuclear fractions, we created two corrected curves: N-Ct,GAPDH,C for the nuclear fraction of GAPDH mRNA and C-Ct,GAPDH,C for the cytoplasmic fraction of GAPDH mRNA. The two curves were corrected by C-Mean Ct,miniUAA1 and N-Mean Ct,miniUAA1, respectively. Although we added the same amount of the synthetic mRNA miniUAA1 (120 pg) in every fraction, its Ct values were slightly different, so an additional correction was performed using C-Mean Ct,miniUAA1 – N-Mean Ct,miniUAA1.
) method (Wong and Medrano, 2005). Briefly, Ct values were corrected for the mean Ct value of miniUAA1 mRNA as follows: First, raw Ct values of GAPDH mRNA (Ct,GAPDH) were corrected using Mean Ct,miniUAA1, generating a corrected curve for GAPDH mRNA (Ct,GAPDH,C). Then Ct,GAPDH – Mean Ct,miniUAA1 = Ct,GAPDH,C. Because an isolation was performed separating cytoplasmic and nuclear fractions, we created two corrected curves: N-Ct,GAPDH,C for the nuclear fraction of GAPDH mRNA and C-Ct,GAPDH,C for the cytoplasmic fraction of GAPDH mRNA. The two curves were corrected by C-Mean Ct,miniUAA1 and N-Mean Ct,miniUAA1, respectively. Although we added the same amount of the synthetic mRNA miniUAA1 (120 pg) in every fraction, its Ct values were slightly different, so an additional correction was performed using C-Mean Ct,miniUAA1 – N-Mean Ct,miniUAA1.
Eventually two corrected set of data were created: the cytoplasmic versus nuclear fold change (C/N), represented by 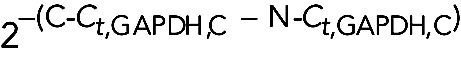 , and the miniUAA1-corrected fold change, represented by
, and the miniUAA1-corrected fold change, represented by 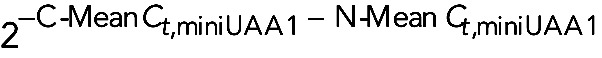 , which must be as close to 1 as possible. Combining both sets of data, we have the cytoplasmic versus nuclear corrected and normalized fold change, q(C/N):
, which must be as close to 1 as possible. Combining both sets of data, we have the cytoplasmic versus nuclear corrected and normalized fold change, q(C/N):
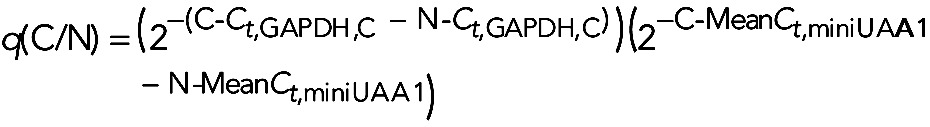 |
The following primers were used for the quantitative PCR: miniUAA1 forward, 5′-GAGAAAAGTTAAGTCGACGCCC-3′; miniUAA1 reverse, 5′-GGATCTATCGATTTCAATTC-3′; GAPDH forward, 5′-GGTGGTCTCCTCTGACTTCAACA-3′; GAPDH reverse, 5′-GTTGCTGTAGCCAAATTCGTTGT-3′; BRG1 forward, 5′-AAGGACGACGAGAGCAAGAA-3′; BRG1 reverse, 5′-GGATGAAGACCTCGCTGA-3′; hnRNP Q forward, 5′-TGCATTGGCAGTTCTTCAAC-3′; hnRNP Q reverse, 5′-TCTGCTACTTTGGTCCCTTG-3′; H2B forward, 5′-CGCTGTTTTTCCTTTTCGTT-3′; H2B reverse, 5′-CTTAGTCACCGCCTTCTTGG-3′; H4 forward, 5′-AGCTGTCTATCGGGCTCCAG-3′; H4 reverse, 5′-CCTTTGCCTAAGCCTTTTCC-3′; H3a forward, 5′-GTTCCGCTGTGCTGTTTTTC-3′; H3a reverse, 5′-GTAGCGGTGGGGCTTTTTC-3′; H1F0 forward, 5′-GTGAAACCCAAAGCAAAGTCC-3′; H1F0 reverse, 5′-AAATAGGAGACAGGAGGGAGTGT-3′; H1FX forward, 5′-CAATCCTCTTGCTACCATGTCC-3′; H1FX reverse, 5′-GCTATTCTTCCTCTTCTTAGATGGG-3′; H3F3 forward, 5′-TGCGGAGACGTAAAGCATT-3′; H3F3 reverse, 5′-CCGCTGAAACTTGTTCACTG-3′; CALR forward, 5′-CTTGGATCCACCCAGAAATTG-3′; CALR reverse, 5′-CCTCAGCGTATGCCTCATCG-3′; HM13 forward, 5′-CATCAACCTCCTGCTGTCCATG-3′; HM13 reverse, 5′-GTACTGGTTCCCGAGAAAGCTG-3′; OR10A3 forward, 5′-GGAGCGATGGCTTATGACCG-3′; OR10A3 reverse, 5′-CAAGTGGTCTGCACAGTAGCC-3′; F1-ATP synthase forward, 5′-TGTCGGCTACGGCCTCGGAT-3′; F1-ATP synthase reverse, 5′-GGGTCGGCGGAGCAACAGAG; FDX1 forward, 5′-CAGACAGATCACGGTTGGGC-3′; FDX1 reverse, 5′-GGAGGTCTTGCCCACATCAATG-3′; IDH3B forward, 5′-GAGCAGGTGCTGAGTTCCATG-3′; IDH3B reverse, 5′-GTCCAACTTACGCCTCAGCC-3′ ; FH forward, 5′-GCTCGAATGGCAAGCCAAAATTC-3′; FH reverse, 5′-CATGCGTTCTGTCACACCTCC-3′; GAPDH (exon 1–intron 2) forward, 5′- GGAAGGTGAAGGTCGGAGTCAAC-3′; GAPDH (exon 1–intron 2) reverse, 5′-GCTCTCCTTGCGGGGAACAGCTAC-3′; BRG1 (exon 1–intron 1) forward, 5′-GCTTCTTTGTTTCGTGAAGAGAAG-3′; BRG1 (exon 1–intron 1) reverse, 5′-CGGCTCCATTCAAACCGCCCTC-3′; hnRNP Q (intron 1–exon 2) forward, 5′-CTTCAGTACCATTTCCATTAAC-3′; hnRNP Q (intron 1–exon 2) reverse, 5′-GGTATAGGCTAGCTGGTGTAG-3′; H3F3A (exon 1–intron 2) forward, 5′-CATAATTTCCAGATTTGGGGAGG-3 ′; H3F3A (exon 1–intron 2) reverse, 5′-GATTTGCGGGCAGTCTGCTTTG-3′.
Supplementary Material
Acknowledgments
We thank Stefan Wagner, Simion Chiosea, Mariya Ivshina, and Krishna Kota for help with cloning and site-directed mutagenesis. Benjamin Blencowe (University of Toronto, Toronto, Canada) kindly provided cDNA clones. Jeanne Lawrence, Anthony Imbalzano, Allan Jacobson, Nadja Amrani, and colleagues (University of Massachusetts Medical School, Worcester, MA) generously provided methods, reagents, and advice for FISH experiments. We thank Maria Carmo-Fonseca (Faculdade de Medicina da Universidade de Lisboa, Lisbon, Portugal) for helpful discussion. Andre vanWijnen and Ricardo Medina (University of Massachusetts Medical School) provided histone reagents and expertise. This work was supported by NASA (CSEMC NNJ06HA28G), the National Cancer Institute (PO1 CA82834), and the National Institute of Biomedical Imaging and Bioengineering (R01 EB014869).
Abbreviations used:
- CSK
cytoskeletal buffer
- EGFP
enhanced green fluorescent protein
- EJC
exon junction complex
- FISH
fluorescence in situ hybridization
- FRAP
fluorescence recovery after photobleaching
- IP6
inositol hexakis-phosphate
- mRNP
messenger ribonucleoprotein
- MSCR
mitochondrial signal coding region
- mTOR
mammalian target of rapamycin
- PI
phosphatidylinositol
- PI3 kinase
phosphatidylinositol 3-kinase
- PI(3,4,5)P3
phosphatidylinositol-(3,4,5)-tris-phosphate
- PI(4,5)P2
phosphatidylinositol-(4,5)-bis-phosphate
- SSC
saline-sodium citrate
- SSCR
signal sequence coding region
- TREX
transcription/export
- VRC
vanadyl ribonucleoside complexes
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0450) on February 20, 2013.
REFERENCES
- Alcazar-Roman AR, Tran EJ, Guo S, Wente SR. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol. 2006;8:711–716. doi: 10.1038/ncb1427. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Ghosh S, Mangus DA, Jacobson A. Translation factors promote the formation of two states of the closed-loop mRNP. Nature. 2008;453:1276–1280. doi: 10.1038/nature06974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CB, et al. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI. Directed proteomic analysis of the human nucleolus. Curr Biol. 2002;12:1–11. doi: 10.1016/s0960-9822(01)00650-9. [DOI] [PubMed] [Google Scholar]
- Ballut L, Marchadier B, Baguet A, Tomasetto C, Seraphin B, Le Hir H. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat Struct Mol Biol. 2005;12:861–869. doi: 10.1038/nsmb990. [DOI] [PubMed] [Google Scholar]
- Barlow CA, Laishram RS, Anderson RA. Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol. 2010;20:25–35. doi: 10.1016/j.tcb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett SF, Bilodeau MT, Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation. Curr Top Med Chem. 2005;5:109–125. doi: 10.2174/1568026053507714. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Brody Y, Kinor N, Mor A, Tsukamoto T, Spector DL, Singer RH, Shav-Tal Y. The life of an mRNA in space and time. J Cell Sci. 2010;123:1761–1774. doi: 10.1242/jcs.062638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O. Inhibiting eukaryotic transcription: which compound to choose? how to evaluate its activity. Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein M, et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol. 2005;12:1037–1044. doi: 10.1038/nsmb1020. [DOI] [PubMed] [Google Scholar]
- Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Boronenkov IV, Loijens JC, Umeda M, Anderson RA. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol Biol Cell. 1998;9:3547–3560. doi: 10.1091/mbc.9.12.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Brody Y, Neufeld N, Bieberstein N, Causse SZ, Bohnlein EM, Neugebauer KM, Darzacq X, Shav-Tal Y. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 2011;9:e1000573. doi: 10.1371/journal.pbio.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- Bunce MW, Bergendahl K, Anderson RA. Nuclear PI(4,5)P(2): a new place for an old signal. Biochim Biophys Acta. 2006;1761:560–569. doi: 10.1016/j.bbalip.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero G, McDonald D, Crawford E, de Vries G, Hendzel MJ. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods. 2003;29:14–28. doi: 10.1016/s1046-2023(02)00288-8. [DOI] [PubMed] [Google Scholar]
- Cenik C, Chua HN, Zhang H, Tarnawsky SP, Akef A, Derti A, Tasan M, Moore MJ, Palazzo AF, Roth FP. Genome analysis reveals interplay between 5’UTR introns and nuclear mRNA export for secretory and mitochondrial genes. PLoS Genet. 2011;7:e1001366. doi: 10.1371/journal.pgen.1001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Dufu K, Lee CS, Hsu JL, Dias A, Reed R. Human mRNA export machinery recruited to the 5’ end of mRNA. Cell. 2006;127:1389–1400. doi: 10.1016/j.cell.2006.10.044. [DOI] [PubMed] [Google Scholar]
- Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol. 2006;175:415–426. doi: 10.1083/jcb.200607020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P. Extreme instability of myc mRNA in normal and transformed human cells. Proc Natl Acad Sci USA. 1984;81:7046–7050. doi: 10.1073/pnas.81.22.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didichenko SA, Thelen M. Phosphatidylinositol 3-kinase c2alpha contains a nuclear localization sequence and associates with nuclear speckles. J Biol Chem. 2001;276:48135–48142. doi: 10.1074/jbc.M104610200. [DOI] [PubMed] [Google Scholar]
- Erkmann JA, Sanchez R, Treichel N, Marzluff WF, Kutay U. Nuclear export of metazoan replication-dependent histone mRNAs is dependent on RNA length and is mediated by TAP. RNA. 2005;11:45–58. doi: 10.1261/rna.7189205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farny NG, Hurt JA, Silver PA. Definition of global and transcript-specific mRNA export pathways in metazoans. Genes Dev. 2008;22:66–78. doi: 10.1101/gad.1616008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forler D, Rabut G, Ciccarelli FD, Herold A, Kocher T, Niggeweg R, Bork P, Ellenberg J, Izaurralde E. RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol Cell Biol. 2004;24:1155–1167. doi: 10.1128/MCB.24.3.1155-1167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- Gondran P, Amiot F, Weil D, Dautry F. Accumulation of mature mRNA in the nuclear fraction of mammalian cells. FEBS Lett. 1999;458:324–328. doi: 10.1016/s0014-5793(99)01175-8. [DOI] [PubMed] [Google Scholar]
- Grüter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol Cell. 1998;1:649–659. doi: 10.1016/s1097-2765(00)80065-9. [DOI] [PubMed] [Google Scholar]
- Herbert A, Wagner S, Nickerson JA. Induction of protein translation by ADAR1 within living cell nuclei is not dependent on RNA editing. Mol Cell. 2002;10:1235–1246. doi: 10.1016/s1097-2765(02)00737-2. [DOI] [PubMed] [Google Scholar]
- Herold A, Klymenko T, Izaurralde E. NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila. RNA. 2001;7:1768–1780. [PMC free article] [PubMed] [Google Scholar]
- Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, Germain GS, Abraham RT, Houghton PJ. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11:837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Huang Y, Steitz JA. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol Cell. 2001;7:899–905. doi: 10.1016/s1097-2765(01)00233-7. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wimler KM, Carmichael GG. Intronless mRNA transport elements may affect multiple steps of pre-mRNA processing. EMBO J. 1999;18:1642–1652. doi: 10.1093/emboj/18.6.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. Nuclear lipid signalling. Nat Rev Mol Cell Biol. 2003;4:349–360. doi: 10.1038/nrm1100. [DOI] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Johnson CV, Singer RH, Lawrence JB. Fluorescent detection of nuclear RNA and DNA: implications for genome organization. Methods Cell Biol. 1991;35:73–99. [PubMed] [Google Scholar]
- Kataoka N, Diem MD, Kim VN, Yong J, Dreyfuss G. MAGOH, a human homolog of Drosophila mago nashi protein, is a component of the splicing-dependent exon-exon junction complex. EMBO J. 2001;20:6424–6433. doi: 10.1093/emboj/20.22.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka N, Yong J, Kim VN, Velazquez F, Perkinson RA, Wang F, Dreyfuss G. Pre-mRNA splicing imprints mRNA in the nucleus with a novel RNA-binding protein that persists in the cytoplasm. Mol Cell. 2000;6:673–682. doi: 10.1016/s1097-2765(00)00065-4. [DOI] [PubMed] [Google Scholar]
- Kota KP, Wagner SR, Huerta E, Underwood JM, Nickerson JA. Binding of ATP to UAP56 is necessary for mRNA export. J Cell Sci. 2008;121:1526–1537. doi: 10.1242/jcs.021055. [DOI] [PubMed] [Google Scholar]
- Kruhlak MJ, Lever MA, Fischle W, Verdin E, Bazett-Jones DP, Hendzel MJ. Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J Cell Biol. 2000;150:41–52. doi: 10.1083/jcb.150.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Seraphin B. EJCs at the heart of translational control. Cell. 2008;133:213–216. doi: 10.1016/j.cell.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Lele T, Oh P, Nickerson JA, Ingber DE. An improved mathematical approach for determination of molecular kinetics in living cells with FRAP. Mech Chem Biosyst. 2004;1:181–190. [PubMed] [Google Scholar]
- Lele T, Wagner SR, Nickerson JA, Ingber DE. Methods for measuring rates of protein binding to insoluble scaffolds in living cells: histone H1-chromatin interactions. J Cell Biochem. 2006;99:1334–1342. doi: 10.1002/jcb.20997. [DOI] [PubMed] [Google Scholar]
- Lele TP, Ingber DE. A mathematical model to determine molecular kinetic rate constants under non-steady state conditions using fluorescence recovery after photobleaching (FRAP) Biophys Chem. 2006;120:32–35. doi: 10.1016/j.bpc.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Linder P. mRNA export: RNP remodeling by DEAD-box proteins. Curr Biol. 2008;18:R297–R299. doi: 10.1016/j.cub.2008.02.027. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Zhou Z, Magni K, Christoforides C, Rappsilber J, Mann M, Reed R. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature. 2001;413:644–647. doi: 10.1038/35098106. [DOI] [PubMed] [Google Scholar]
- Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell. 2008;133:303–313. doi: 10.1016/j.cell.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Martelli AM, Faenza I, Billi AM, Manzoli L, Evangelisti C, Fala F, Cocco L. Intranuclear 3’-phosphoinositide metabolism and Akt signaling: new mechanisms for tumorigenesis and protection against apoptosis. Cell Signal. 2006;18:1101–1107. doi: 10.1016/j.cellsig.2006.01.011. [DOI] [PubMed] [Google Scholar]
- McCracken S, Lambermon M, Blencowe BJ. SRm160 splicing coactivator promotes transcript 3’-end cleavage. Mol Cell Biol. 2002;22:148–160. doi: 10.1128/MCB.22.1.148-160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta. 2011;1809:34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri LM, Borgatti P, Capitani S, Martelli AM. The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta. 2002;1584:73–80. doi: 10.1016/s1388-1981(02)00300-1. [DOI] [PubMed] [Google Scholar]
- Nickerson JA. The biochemistry of RNA metabolism studied in situ. RNA Biol. 2009;6:25–30. doi: 10.4161/rna.6.1.7563. [DOI] [PubMed] [Google Scholar]
- Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 2004;18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Jang SW, Ye K. Akt phosphorylation and nuclear phosphoinositide association mediate mRNA export and cell proliferation activities by ALY. Proc Natl Acad Sci USA. 2008;105:8649–8654. doi: 10.1073/pnas.0802533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Ye K. Nuclear phosphoinositide signaling regulates messenger RNA export. RNA Biol. 2009;6:12–16. doi: 10.4161/rna.6.1.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SL, Thomas CL, Gschmeissner S, Schiavo G. Nuclear PtdIns(4,5)P2 assembles in a mitotically regulated particle involved in pre-mRNA splicing. J Cell Sci. 2001;114:2501–2511. doi: 10.1242/jcs.114.13.2501. [DOI] [PubMed] [Google Scholar]
- Palangat M, Larson DR. Complexity of RNA polymerase II elongation dynamics. Biochim Biophys Acta. 2012;1819:667–672. doi: 10.1016/j.bbagrm.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Springer M, Shibata Y, Lee CS, Dias AP, Rapoport TA. The signal sequence coding region promotes nuclear export of mRNA. PLoS Biol. 2007;5:e322. doi: 10.1371/journal.pbio.0050322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- Read EK, Digard P. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J Gen Virol. 2010;91:1290–1301. doi: 10.1099/vir.0.018564-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH, Saiardi A. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci USA. 2005;102:12783–12788. doi: 10.1073/pnas.0506184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Dargemont C, Stutz F. Nuclear export of RNA. Biol Cell. 2004;96:639–655. doi: 10.1016/j.biolcel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Saji M, Vasko V, Kada F, Allbritton EH, Burman KD, Ringel MD. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem Biophys Res Commun. 2005;332:167–173. doi: 10.1016/j.bbrc.2005.04.109. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schluederberg A, Hendel RC, Chavanich S. Actinomycin D; renewed RNA synthesis after removal from mammalian cells. Science. 1971;172:577–579. doi: 10.1126/science.172.3983.577. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Segref A, Sharma K, Doye V, Hellwig A, Huber J, Luhrmann R, Hurt E. Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 1997;16:3256–3271. doi: 10.1093/emboj/16.11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal PB, Derman E, Molloy GR, Tamm I, Darnell JE. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits initiation of nuclear heterogeneous RNA chains in HeLa cells. Science. 1976;194:431–433. doi: 10.1126/science.982026. [DOI] [PubMed] [Google Scholar]
- Shamsher MK, Ploski J, Radu A. Karyopherin beta 2B participates in mRNA export from the nucleus. Proc Natl Acad Sci USA. 2002;99:14195–14199. doi: 10.1073/pnas.212518199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya T, Tange TO, Stroupe ME, Moore MJ. Mutational analysis of human eIF4AIII identifies regions necessary for exon junction complex formation and nonsense-mediated mRNA decay. RNA. 2006;12:360–374. doi: 10.1261/rna.2190706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland LS, Johnson CV, Lawrence JB. Evidence that all SC-35 domains contain mRNAs and that transcripts can be structurally constrained within these domains. J Struct Biol. 2002;140:131–139. doi: 10.1016/s1047-8477(02)00507-5. [DOI] [PubMed] [Google Scholar]
- Singh G, Jakob S, Kleedehn MG, Lykke-Andersen J. Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol Cell. 2007;27:780–792. doi: 10.1016/j.molcel.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skwarek LC, Boulianne GL. Great expectations for PIP: phosphoinositides as regulators of signaling during development and disease. Dev Cell. 2009;16:12–20. doi: 10.1016/j.devcel.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Smith KP, Moen PT, Wydner KL, Coleman JR, Lawrence JB. Processing of endogenous pre-mRNAs in association with SC-35 domains is gene specific. J Cell Biol. 1999;144:617–629. doi: 10.1083/jcb.144.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings JD, Tall EG, Pentyala S, Rebecchi MJ. Nuclear translocation of phospholipase C-delta1 is linked to the cell cycle and nuclear phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2005;280:22060–22069. doi: 10.1074/jbc.M413813200. [DOI] [PubMed] [Google Scholar]
- Stewart M. Ratcheting mRNA out of the nucleus. Mol Cell. 2007;25:327–330. doi: 10.1016/j.molcel.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Strasser K, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- Tange TO, Shibuya T, Jurica MS, Moore MJ. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA. 2005;11:1869–1883. doi: 10.1261/rna.2155905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi I, Ohno M. ATP-dependent recruitment of export factor Aly/REF onto intronless mRNAs by RNA helicase UAP56. Mol Cell Biol. 2008;28:601–608. doi: 10.1128/MCB.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran EJ, Zhou Y, Corbett AH, Wente SR. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol Cell. 2007;28:850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Trask DK, Muller MT. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc Natl Acad Sci USA. 1988;85:1417–1421. doi: 10.1073/pnas.85.5.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Volloch V, Housman D. Stability of globin mRNA in terminally differentiating murine erythroleukemia cells. Cell. 1981;23:509–514. doi: 10.1016/0092-8674(81)90146-x. [DOI] [PubMed] [Google Scholar]
- Wada Y, et al. A wave of nascent transcription on activated human genes. Proc Natl Acad Sci USA. 2009;106:18357–18361. doi: 10.1073/pnas.0902573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Chiosea S, Nickerson JA. The spatial targeting and nuclear matrix binding domains of SRm160. Proc Natl Acad Sci USA. 2003;100:3269–3274. doi: 10.1073/pnas.0438055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Chiosea S, Ivshina M, Nickerson JA. In vitro FRAP reveals the ATP-dependent nuclear mobilization of the exon junction complex protein SRm160. J Cell Biol. 2004;164:843–850. doi: 10.1083/jcb.200307002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil D, Boutain S, Audibert A, Dautry F. Mature mRNAs accumulated in the nucleus are neither the molecules in transit to the cytoplasm nor constitute a stockpile for gene expression. RNA. 2000;6:962–975. doi: 10.1017/s1355838200000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol. 2006;8:668–676. doi: 10.1038/ncb1424. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe VO, et al. mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol. 2010;20:25–31. doi: 10.1016/j.cub.2009.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Lu S, Cullen BR. Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc Natl Acad Sci USA. 2003;100:11327–11332. doi: 10.1073/pnas.1934877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39:75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
- Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE., Jr Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York JD, Odom AR, Murphy R, Ives EB, Wente SR. A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science. 1999;285:96–100. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunger S, Rosenfeld L, Garini Y, Shav-Tal Y. Single-allele analysis of transcription kinetics in living mammalian cells. Nat Methods. 2010;7:631–633. doi: 10.1038/nmeth.1482. [DOI] [PubMed] [Google Scholar]
- Zhang M, Green MR. Identification and characterization of yUAP/Sub2p, a yeast homolog of the essential human pre-mRNA splicing factor hUAP56. Genes Dev. 2001;15:30–35. doi: 10.1101/gad.851701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.