

Abstract
S-trans, trans-farnesylthiosalicylic acid (FTS) is a synthetic small molecule that acts as a potent and especially nontoxic Ras antagonist. It inhibits both oncogenically activated Ras and growth factor receptor-mediated Ras activation, resulting in the inhibition of Ras-dependent tumor growth. In this work, a FTS conjugate with polyethylene glycol (PEG) through a labile ester linkage, PEG5K-FTS2(L), was developed. PEG5K-FTS2 conjugate readily forms micelles in aqueous solutions with a critical micelle concentration of 0.68 μM and hydrophobic drugs such as paclitaxel (PTX) could be effectively loaded into these particles. Both drug-free and PTX- loaded micelles were spherical in shape with a uniform size of 20 ~ 30 nm. The release of PTX from PTX-loaded PEG5K-FTS2 micelles was significantly slower than that from Taxol formulation. In vitro cytotoxicity studies with several tumor cell lines showed that PEG5K-FTS2(L) was comparable to FTS in antitumor activity. Western immunoblotting showed that total Ras levels were downregulated in several cancer cell lines treated with FTS or PEG5K-FTS2(L). The micellar formulation of PTX exhibited more in vitro cytotoxic activity against several tumor cell lines compared with free PTX, suggesting a possible synergistic effect between the carrier and the codelivered drug. The anti-tumor activity of the PTX loaded PEG5K-FTS2(L) micelles in a syngeneic murine breast cancer model was found to be significantly higher than that of Taxol, which may be attributed to their preferential tumor accumulation and a possible synergistic effect between PEG5K-FTS2 carrier and loaded PTX.
INTRODUCTION
Clinical application of PTX in cancer therapy is limited by problems such as low water solubility, lack of tissue-specificity and high toxicity.1,2 A number of macromolecular delivery systems are under investigation to circumvent these limitations and improve the potential of the anticancer drug.3,4 However, it remains a challenge to design a vehicle that can carry sufficient amounts of drugs and efficiently overcome various physiological barriers to reach the tumor tissues.5 Micelles with a nanoscopic supramolecular core-shell structure, are one such carrier of choice. It has been established that a nanocarrier with the size of 20-100 nm preferentially extravasates into solid tumor tissues owing to the leaky tumor vasculature.6-8 Additionally, a hydrophilic polymer (PEG) grafted onto the surface prolongs blood circulation times of the micelles due to the inhibition of nonspecific uptake by reticuloendothelial system (RES).9,10 However, for most delivery systems, vehicles themselves rarely possess pharmacological activity. The use of “inert” excipients that lack therapeutic activity not only adds to the cost but also potentially imposes safety issue.11
An interesting strategy in the design of multifunctional nanocarriers is the use of highly water-insoluble drug itself as the hydrophobic region of polymeric micelles. One example is pegylated vitamin E, D-α-tocopheryl poly(ethylene glycol) succinate (Vitamin E TPGS or TPGS).12-14 Vitamin E itself shows antitumor effect against different types of cancers and vitamin E TPGS is effective in solubilizing various hydrophobic drugs. Synergistic effects between the TPGS-based carriers and delivered anti-cancer drugs have been demonstrated.15
Recently our group reported another dual-functional carrier that is based on PEG-derivatized embelin.16,17 Embelin shows antitumor activity in various types of cancers through various mechanisms such as the inhibition of the activity of X-linked inhibitor of apoptosis protein (XIAP).18 PEG-modified embelin shows significantly increased solubility (> 200 mg/mL) in aqueous solution. More importantly, PEG-embelin forms micelles that effectively solubilize various types of hydrophobic drugs such as PTX. Significantly improved antitumor was demonstrated for PTX formulated in PEG-embelin micelles both in vitro and in vivo.16,17
In this study, we design a self-assembling nanomicellar system that is based on PEG-derivatized farnesylthiosalicylic acid (FTS) for targeted delivery of PTX. FTS is a first-in-class direct Ras antagonist designed to inhibit overactive cell growth in cancers caused by various Ras proteins.19,20 Permanently active Ras caused by mutation in the Ras family of proto-oncogenes, is present in one-third of human cancers, with the highest incidence of mutational activation of Ras being detected in pancreatic (90%) and colon (50%) cancers.21,22 FTS has demonstrated significant reduction of Ras levels in a wide array of established cancer models without observable side effects.23 One major mechanism involves affecting membrane interaction by inducing dislodgement of ras protein from its anchorage domains, facilitating its degradation, and thus disrupting ras protein to signal in the plasma membrane.24,25 FTS is poorly water-soluble26 and PEGylation is designed to improve its solubility. At the same time, PEG-FTS conjugate forms micelles, which could be applied as an ideal carrier for hydrophobic drugs to improve the efficiency and safety profile. Additionally, the carrier materials can potentially promote synergistic effects with the codelivered anti-cancer drug. The PEG5K-FTS2 conjugate was synthesized and characterized for its structure and molecular weight. We further characterized the biophysical properties of drug-loaded micelles including particle size, loading capacity, and in vitro drug release kinetics. Finally, the in vitro and in vivo antitumor activity of PTX/PEG-FTS was investigated.
EXPERIMENTAL PROCEDURES
Materials
Paclitaxel (98%) was purchased from AK Scientific Inc. (CA, USA). FTS was synthesized and purified following a published literature.20 Pan Ras Ab (Ab-3) was purchased from Calbiochem (La Jolla, CA). HRP-labeled goat anti-mouse IgG and the ECL chemiluminescence kit were purchased from Amersham Biosciences (Piscataway, NJ, USA). Dulbecco’s phosphate buffered saline (DPBS) was purchased from Lonza (MD, USA). Poly(ethylene glycol) methyl ether (MeO-PEG-OH, Mw=5000 kDa), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), trypsin-EDTA solution, Triton X-100, and Dulbecco’s Modified Eagle’s Medium (DMEM) were all purchased from Sigma-Aldrich (MO, USA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were from Invitrogen (NY, USA).
Synthesis of PEG5K-FTS2(L) and PEG5K-FTS2(S) conjugate
PEG5K-FTS2(L) conjugate with a labile linkage was prepared via solution phase condensation reactions from MeO-PEG-OH with a molecular weight of 5000 (Fig. 1). We started with synthesizing carboxyl terminated PEG monomethyl ether (MeO-PEG5k-COOH) from MeO-PEG5k-OH (1 eq.) by a facile chemical reaction with succinic anhydride (5 eq.) and 4-(dimethylamino) pyridine (DMAP 5 eq.) in pyridine according to a reported method.27 To obtain two hydroxyl group terminated PEG monomethyl ether (MeO-PEG5k-(OH)2), diethanolamine (3 eq.) was coupled onto the carboxylic group of MeO-PEG5k-COOH (1 eq.) using N-hydroxysuccinimide (NHS 3.6 eq.)/dicyclohexylcarbodiimide (DCC 3.6 eq.) as coupling agent in chloroform overnight. The polymer was precipitated and washed by ice-cold ether three times, and concentrated under vacuum. MeO-PEG5k-(OH)2, FTS, DCC and DMAP were then dissolved in chloroform with a molar ratio of 1: 6: 3: 0.3 and allowed to react overnight at room temperature. The solution was filtered and precipitated in ice-cold diethyl ether and ethanol twice respectively, and concentrated under vacuum. The powder was then dissolved in water and filtered through a filter with a pore size of 0.2 μm. The final product was obtained by lyophilizing the filtrate. At the same time, we also prepared a PEG5K-FTS2(S) conjugate (Fig. S1) with a stable amide linkage. MeO-PEG5k-OH and (S)-2,6-Bis-tert-butoxycarbonylaminohexanoic acid (Boc-Lys(Boc)-OH) were dissolved in chloroform together with DCC and DMAP with a molar ratio of 1:3:1.5:0.3 and the mixture was left to react overnight at room temperature. PEGylated molecules were recovered from the mixture through three cycles of dissolution/reprecipitation with chloroform and ether, respectively. Boc groups were removed via the treatment with 50% (v/v) trifluoroacetic acid (TFA) in chloroform, and PEGylated molecules were precipitated and washed with ice-cold ether. Finally, FTS was coupled onto the N terminal group of MeO-PEG5k-Lys-(NH2)2 (1 eq.) via NHS (3.6 eq.)/DCC (3.6 eq.) as described above. After the reaction was completed, the solution was precipitated in cold ether. PEG5K-FTS2(S) was similarly purified as that for PEG5K-FTS2(L).
Figure 1.
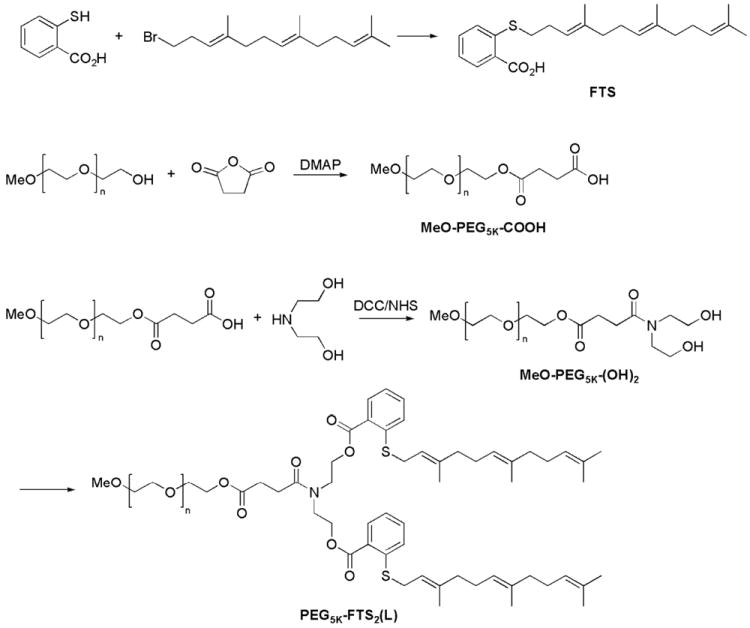
Synthesis scheme of PEG5K-FTS2(L) conjugate.
Preparation of PTX loaded and drug-free micelles
PTX (10 mM in chloroform) and PEG5K-FTS2 (10 mM in chloroform) were mixed with various carrier/drug ratios. The organic solvent was removed by nitrogen flow to form a thin film of drug/carrier mixture. The film was dried under vacuum for 1 h to remove the remaining solvent. DPBS was added to hydrate the thin film and the drug-loaded micelles were formed. Unincorporated PTX (precipitate) was removed by filtering with a syringe filter (pore size: 220 μm). The drug-free micelles were similarly prepared as described above.
Characterizations of PTX-loaded and drug-free micelles
The particle size and zeta potential of micelles were measured by a Zetasizer (DLS) (Zetasizer Nano ZS instrument, Malvern, Worcedtershire, UK). The micelle concentrations were kept at 1 mg/mL. The morphology and size distribution of drug-free or PTX-loaded PEG5K-FTS2 micelles were observed using transmission electron microscopy (TEM). A copper grid with Formvar was used. The copper grid was immersed in a drop of sample solution and stained with 1% uranyl acetate. Imaging was performed at room temperature on JEOL JEM-1011.
The critical micelle concentration (CMC) of PEG5K-FTS2 was determined using pyrene as a fluorescence probe.28 PEG5K-FTS2 was prepared in chloroform at 1.2 mg/mL and various amounts were added to 9 separate vials, respectively. Then 10 μl of 1.8 × 10−4 M of pyrene in chloroform was added to each vial and the solution was mixed well. The organic solvent was removed by nitrogen flow to form a thin film. Finally, 3 ml Milli-Q water was added to each vial and the final pyrene concentration was 6×10-7 M with PEG5K-FTS2 concentrations ranging from 0.0001 to 0.5 mg/mL. The vials were kept on a shaker at 37°C for 24 h to reach equilibrium prior to fluorescence measurement. The excitation spectra were scanned from 310 to 350 nm at a fixed emission wavelength of 390 nm by Synergy H1 Hybrid Multi-Mode Microplate Reader (Winooski, VT). The pyrene fluorescence intensity ratio I338/I334 in the excitation spectra was analyzed for calculation of CMC.
The drug loading efficiency was quantified by high performance liquid chromatography (HPLC) (Alliance 2695-2998 system). The reverse phase Lichrospher® 100 RP-18 (5 μm) column was used and the mobile phase consisted of methanol/water (80: 20 v/v). The flow rate was set at 0.8 mL/min and the column effluent was detected at 227 nm with a UV/Vis detector. Drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated according to the following equation:
In vitro drug release study
Release of PTX from PTX-loaded PEG5K-FTS2 micelles was performed following a method described previously.16 Briefly, 3 mL of PTX-loaded PEG5K-FTS2 micelles or Taxol (1 mg PTX/mL) solution was placed in a dialysis bag (MWCO = 12 KDa, Spectrum Laboratories) that was incubated in 200 mL DPBS (PH = 7.4) containing 0.5% (w/v) Tween 80 at 37 °C with gentle shaking. The concentration of PTX remaining in the dialysis tubes at designated time points was measured by HPLC with the detection wavelength at 227 nm. Values were reported as the means from triplicate samples.
Hemolytic effect of PEG5K-FTS2 micelles
Hemolysis assay was performed using fresh blood collected through cardiac puncture from rats. The red blood cells (RBCs) were collected by centrifugation at 1500 rpm for 10 min at 4 °C. After washing three times with PBS, RBCs were diluted at a final concentration of 2% w/v in PBS. One mL of diluted RBC suspension was mixed with various concentrations (0.2 and 1.0 mg/mL) of PEG5k-FTS2 and PEI, respectively, and then incubated at 37 °C in an incubator shaker for 4 h. The mixtures were centrifuged at 1500 rpm for 10 min at 4 °C, and 100 μL of supernatant from each sample was transferred into a 96-well plate. The release of hemoglobin was determined at 540 nm absorbance using a microplate reader. RBCs incubated with Triton X-100 (2%) and PBS were used as the positive and negative controls, respectively. The percentage of hemolysis of RBCs was calculated as (ODsample-ODnegative control)/ (ODpositive control-ODnegative control) × 100%
Cell culture
DU145 and PC-3 are two androgen-independent human prostate cancer cell lines. 4T1.2 is a mouse metastatic breast cancer cell line. HCT-116 is a human colon carcinoma cell line. All cell lines were cultured in DMEM containing 5% FBS and 1% penicillin-streptomycin at 37 °C in a humidified 5 % CO2 atmosphere.
In vitro cell cytotoxicity
For cytotoxicity measurement, DU145 (2000 cells/well), PC-3 (2000 cells/well), 4T1.2 (1000 cells/ell) or HCT-116 (1000 cells/well) were seeded in 96-well plates. After 24 h of incubation in DMEM with 5% FBS and 1% streptomycin-penicillin, the old medium was removed and the cells were incubated for 72 h in the presence of indicated concentrations of PTX (free or formulated in PEG5K-FTS2 micelles). Twenty μL of 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) in DPBS (5 mg/mL) was added to each well and cells were further incubated for 4 h. MTT formazan was solubilized by DMSO. The absorbance in each well was measured by a microplate reader with wavelength at 550 nm and reference wavelength at 630 nm. Untreated groups were used as controls. Cell viability was calculated as [(ODtreat-ODblank)/ (ODcontrol-ODblank) × 100%]. The cytotoxicity of PEG5K-FTS2(L) or PEG5K-FTS2(S) alone was similarly examined.
Western immunoblotting
Cells were grown in dishes in medium containing 5 % FBS, in the presence of 0.1 % DMSO (control) or indicated concentrations of FTS, PEG5K-FTS2(L), and PEG5K-FTS2(S) respectively. Following treatment for 48 h, cells were washed with ice-cold PBS twice and solubilized in lysis buffer. The lysates were centrifuged at 14000 for 10 min after a 10-min incubation on ice. Samples with equal amounts of total cellular proteins were subjected to 15% sodium dodecylsulfate polyacryl amide gel electrophoresis (SDS-PAGE) before proteins were transferred to nitrocellulose membranes. The membranes were incubated with primary antibody in 5% non fat dry milk dissolved in DPBS with 0.1% Tween 20 (PBST) overnight at 4°C. After washing three times with PBST, the membranes were incubated with secondary antibody at a final dilution of 1:10,000 in PBST for 1 h at room temperature. After washing three times with PBST, bound antibodies were detected by chemiluminescence. β-actin was used as a loading control.
Animals
Female BALB/c mice, 10-12 weeks in age, were purchased from Charles River (Davis, CA). All animals were housed under pathogen-free conditions according to AAALAC guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
In vivo therapeutic study29,30
A syngeneic murine breast cancer model (4T1.2) was used to examine the therapeutic effect of different formulations of PTX. 1 × 105 4T1.2 cells in 200 μL PBS were inoculated s.c. at the right flank of female BALB/c mice. Treatments were started when tumors in the mice reached a tumor volume of ~50 mm3 and this day was designated as day 1. On day 1, these mice were randomly divided into six groups (n=5) and administered i.v. with PBS (control), free PEG5K-FTS2(S) micelles, Taxol (10 mg PTX/kg), PTX-loaded PEG5K-FTS2(L) (10 and 20 mg PTX/kg), and PTX-loaded PEG5K-FTS2(S) (10 mg PTX/kg), respectively on days 1, 4, 6, 8, 11 and 13. Free PEG5K-FTS2(L) micelles were given at the equivalent dosage of the carrier in the group of PTX-loaded PEG5K-FTS2(L) micelles (10 mg PTX/kg). Tumor sizes were measured with digital caliper three times a week and calculated by the formula: (L×W2)/2, where L is the longest, W is the shortest in tumor diameters (mm). To compare between groups, relative tumor volume (RTV) was calculated at each measurement time point (where RTV equals to the tumor volume at a given time point divided by the tumor volume prior to first treatment). Mice were sacrificed when tumor reached 2000 mm3.
Statistical analysis
In all statistical analysis, the significance level was set at a probability of P < 0.05. All results were reported as the mean ± standard error (SEM) unless otherwise indicated. Statistical analysis was performed by Student’s t-test for two groups, and one-way ANOVA for multiple groups, followed by Newman-Keuls test if P < 0.05.
RESULTS
Synthesis of PEG5K-FTS2(L) and PEG5K-FTS2(S) conjugates
PEG5K-FTS2(L) conjugate, containing two molecules of FTS coupled to one molecule of PEG via a labile ester linkage, was developed by solution phase condensation reactions. 1H NMR spectra of PEG5K-FTS2(L) conjugate are shown in Fig. S2A, with signals at 3.63 ppm attributable to the methylene protons of PEG. Carbon chain signals and benzene ring signals of FTS were located at 1.5-2.2 ppm and 7-8 ppm, respectively. A peak appeared at δ 4.5 from −CH2CH2N group confirmed the conjugation of FTS with PEG. The signal ratio of the protons on PEG chain to the protons on -C=C(CH3)2 group of FTS is around 1:8 (Fig. S2A), which suggested that two FTS molecules were attached onto the PEG chain. The molecular weight of the PEG5K-FTS2(L) conjugate measured by MALDI-TOF Mass Spectra is close to the theoretical value (5867) (Fig. S3A), further confirming that two FTS molecules were conjugated to one molecule of PEG. HPLC shows that the purity of the final product [PEG5K-FTS2(L)] is 94.52% (Fig. S4A). We also synthesized PEG5K-FTS2(S) conjugate in which FTS was coupled onto PEG chain via a stable amide linkage. 1H NMR spectra, MALDI-TOF Mass Spectra and HPLC (Figs. S2B, S3B, & S4B) confirmed the successful synthesis of PEG5K-FTS2(S) conjugate.
Characterizations of micelles
The PEG5K-FTS2 conjugate readily formed micelles in aqueous solution. Dynamic light scattering (DLS) measurements showed that these micelles had hydrodynamic sizes around 22 nm at the concentration of 20 mg/mL (Fig. 2A). TEM revealed spherical particles with uniform size distribution (Fig. 2B). The size observed by TEM shows good agreement with that determined by DLS. PTX could be effectively loaded into PEG5K-FTS2 micelles. The size and size distribution were not significantly affected when PTX was loaded into micelles at a drug concentration of 1 mg/mL and a carrier/drug ratio of 5/1 (m/m) (Fig. 2C & Fig. 2D).
Figure 2.
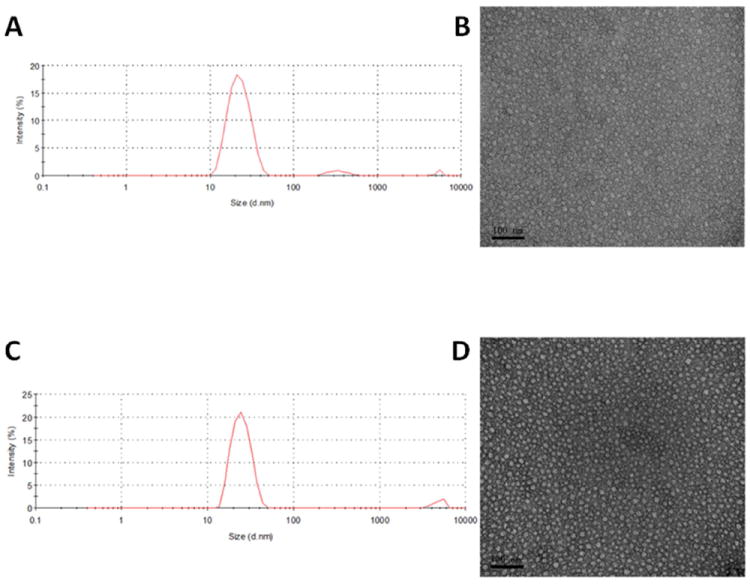
Particle size distribution of drug-free PEG5K-FTS2 (A) and PTX-loaded PEG5K-FTS2 micelles (C). TEM images of drug-free PEG5K-FTS2 (B) and PTX-loaded PEG5K-FTS2 micelles (D).
The CMC of PEG5K-FTS2 micelles was measured using pyrene as a fluorescence probe. When the concentration of the PEG5K-FTS2 reached the CMC, there is a major change for the I338/I334 value due to the transfer of pyrene from polar micro-environment to non-polar micro-environment caused by the formation of micelles. The CMC of the PEG5K-FTS2 micelles was determined to be 0.68 μM (Fig. 3).
Figure 3.
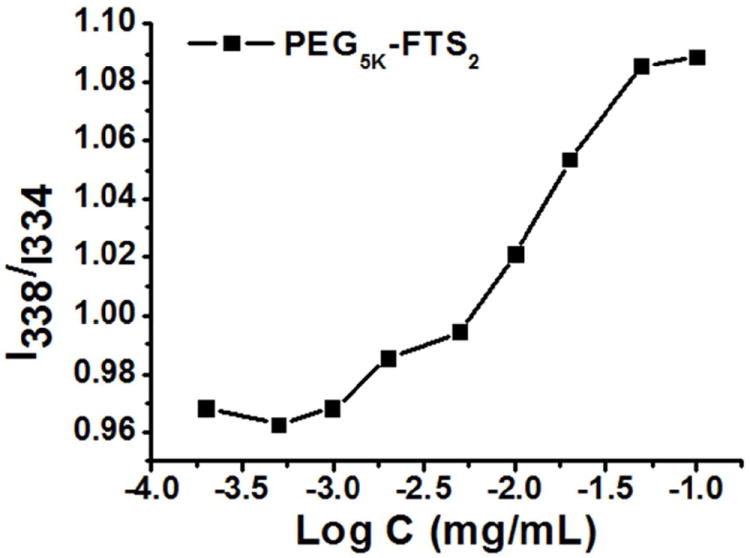
Plot of the ratio of I338/I334 from fluorescence spectra as a function of logarithm concentration of PEG5K-FTS2 micelles. Pyrene was used as molecular probe ([Pyrene]=6.0×10-7 M).
Tab. 1 shows the sizes of PTX-loaded micelles at different carrier/drug molar ratios. All of them had small size of around 20 nm. With increases in the input molar ratio of PEG5K-FTS2/PTX, the sizes of the PTX-loaded micelles were more close to that of drug-free micelles. The drug loading efficiency of PEG5K-FTS2 micelles was over 90% at various carrier/drug ratios examined (Tab. 1).
Table 1.
Physicochemical Characterization of free drug and PTX-loaded PEG5K-FTS2 Micelles
| Micelle | Molar ratio | Conc. of PTXa (mg/mL) | Sizeb (nm) | PDIc | DLEd (%) | DLCe (%) |
|---|---|---|---|---|---|---|
| PEG5K-FTS2 | - | - | 17.61± 0.9 | 0.201 | - | - |
| PEG5K-FTS2 :PTX | 3.75 :1 | 1 | 24.93± 1.2 | 0.354 | 90.6 | 3.73 |
| PEG5K-FTS2 :PTX | 5 :1 | 1 | 25.63± 3.4 | 0.234 | 97.6 | 2.82 |
| PEG5K-FTS2 :PTX | 7.5 :1 | 1 | 22.27± 0.6 | 0.268 | 98.0 | 1.90 |
| PEG5K-FTS2 :PTX | 10 :1 | 1 | 20.17± 0.2 | 0.189 | 96.2 | 1.43 |
PTX concentration in micelle was kept at 1 mg/mL. Blank micelle concentration was 20 mg/mL. Values repotted are the mean ± SD for triplicate samples.
Measured by dynamic light scattering particle sizer.
PDI = polydispersity index.
DLE = drug loading efficiency.
DLC = drug loading capacity.
In vitro drug release study
The profile of PTX release was examined by a dialysis method and compared to that of Taxol. For the initial 8 h, about 41.2 % of PTX was released from the Taxol formulation while only 16.7 % of PTX was released from the PTX-loaded PEG5K-FTS2 micelles (Fig. 4). PTX release from PEG5K-FTS2 micelle formulation was significantly slower compared to Taxol formulation during the entire experimental period. The T1/2 of PTX release is 95.4 h for PEG5K-FTS2 formulation, which is significantly longer than that for Taxol formulation (17.6 h).
Figure 4.
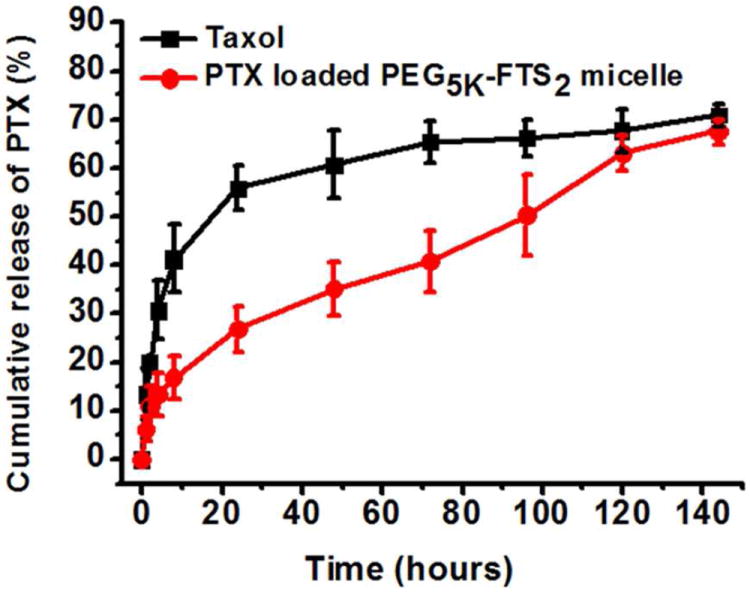
Cumulative PTX release profile from Taxol and PEG5K-FTS2 micelles.
Hemolytic effect of PEG5K-FTS2 micelles
Detrimental interaction of micelles with blood constituents such as RBCs must be avoided when these micelles are injected into the blood circulation as a carrier for drug delivery.31 Fig. 5 shows the hemolytic activities of drug free PEG5K-FTS2 micelles and polyethylenimine (PEI), a cationic polymer known to have significant hemolytic effect.31 PEI induced hemolysis in a dose-dependent manner. In contrast, free PEG5K-FTS2 micelles did not show any detectable hemolytic activities at the same experimental concentrations, suggesting the safe profile of PEG5K-FTS2 micelles.
Figure 5.
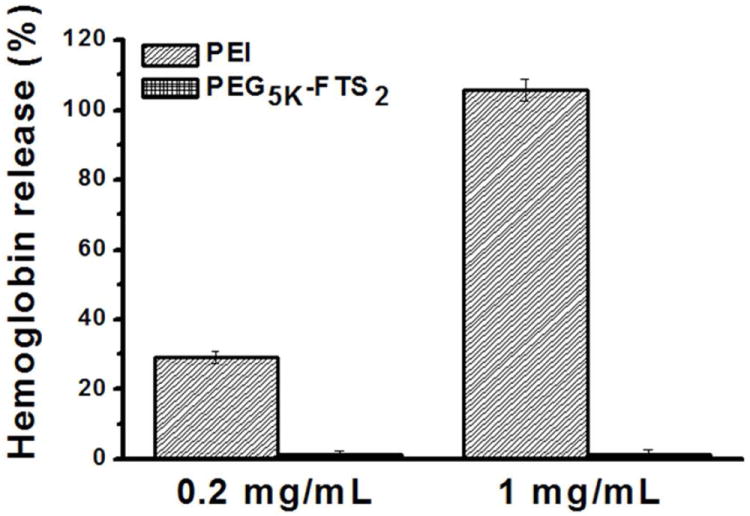
In vitro hemolysis assay of empty PEG5K-FTS2 micelles compared with PEI at two different concentration (0.2 and 1 mg/mL). Values are reported as the mean ± SD for triplicate samples.
Western immunoblotting
We incubated HCT-116 or 4T1.2 cells with fixed concentrations of FTS, PEG5K-FTS2(L) and PEG5K-FTS2(S) for 48 h, respectively, and the protein expression levels of Ras in the cells were examined by Western immunoblotting with pan anti-Ras Ab. As shown in Fig. 6, FTS treatment caused a reduction in the total amount of cellular Ras in both cell lines. These results are consistent with the known action of FTS as a Ras inhibitor. PEG5K-FTS2(L) was comparable to free FTS in reducing the protein levels of Ras in the treated cells. In contrast, PEG5K-FTS2(S) showed minimal effect at the same FTS concentration.
Figure 6.
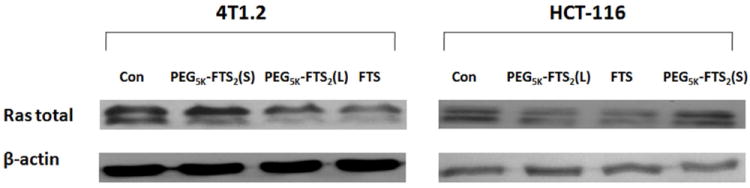
Effect of FTS, PEG5K-FTS2(L) and PEG5K-FTS2(S) on total Ras expression by Western blot analysis. Cells grown in medium containing 5% FBS, were treated with 0.1% DMSO (control), PEG5K-FTS2(L), PEG5K-FTS2(S), and free FTS (at a FTS concentration of 10 μM), respectively, for 48 h. Total cell lysate was subjected to Western blot analysis. Anti-Ras antibody was used to determine the total Ras levels in the cells.
In vitro cytotoxicity
Several cancer cell lines were included in the cytotoxicity studies including murine breast cancer cells 4T1.2, human colon carcinoma cell line HCT-116, and two human prostate cancer cell lines PC-3 and DU145. Fig.7A shows the cytotoxicity of two PTX-free micelles, PEG5K-FTS2(L) and PEG5K-FTS2(S), in comparison with free FTS in 4T1.2 tumor cells. Free FTS inhibited the tumor cell growth in a concentration-dependent manner. Conjugation of FTS to PEG via a labile ester linkage resulted in only a slight decrease in antitumor activity. In contrast, a similar conjugate with a stable amide linkage [PEG5K-FTS2(S)] was significantly less active compared to both free FTS and PEG5K-FTS2 (L). A similar result was obtained in human colon cancer cells, HCT-116 (Fig. 7B).
Figure 7.
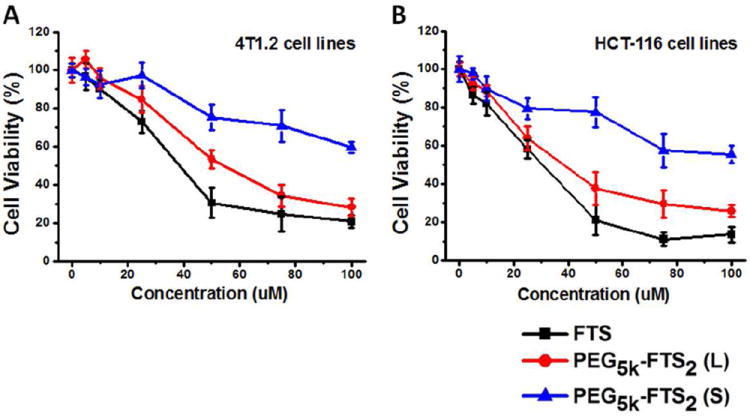
Cytotoxicity of free FTS, PEG5K-FTS2(L) and PEG5K-FTS2(S) in 4T1.2 mouse breast cancer cell line (A) and HCT-116 human colon carcinoma cell line (B).
Fig. 8A shows the cytotoxicity of free PTX (in DMSO) and PTX formulated in PEG5K-FTS2(L) or PEG5K-FTS2(S) micelles in 4T1.2 tumor cells. Free PTX inhibited the tumor cell growth in a concentration dependent manner. Delivery of PTX via PEG5K-FTS2(L) micelles led to a significant increase in the cytotoxicity. Interestingly, PTX formulated in PEG5K-FTS2(S) was less active than free PTX. Similar results were obtained in other tumor cell lines including DU145 (Fig. 8B), PC-3 (Fig. 8C), and HCT-116 (Fig. 8D).
Figure 8.
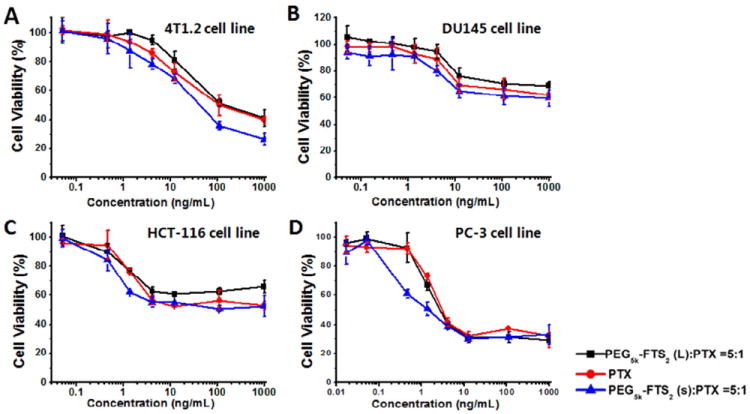
Cytotoxicity of free PTX, PTX/PEG5K-FTS2(L) and PTX/PEG5K-FTS2(S) against a mouse breast cancer cell line 4T1.2 (A), two androgen-independent human prostate cancer cell lines DU145 (B) and PC-3 (D), and a human colon carcinoma cell line HCT-116 (C). Cells were treated for 72 h and cytotoxicity was determined by MTT assay. Values are reported as the means ± SD for triplicate samples.
In vivo therapeutic study
The in vivo therapeutic activity of PTX formulated in PEG5K-FTS2 micelles was investigated in a syngeneic murine breast cancer model (4T1.2). As shown in Fig. 9A, free PEG5K-FTS2 micelles alone showed no effects in inhibiting the tumor growth at the concentration used. The other groups treated with different PTX formulations showed varied levels of antitumor effects and they were ranked as PTX/PEG5K-FTS2(L) > PTX/PEG5K-FTS2(S) > Taxol at the same dose of 10 mg PTX/kg. By day 13, the relative tumor volume (RTV) was 15.1 for Taxol, while the RTVs for PTX/PEG5K-FTS2(S) and PTX/PEG5K-FTS2(L) were 13.3 and 9.6, respectively. Increasing the PTX dosage to 20 mg/kg in PTX-loaded PEG5K-FTS2(L) formulation resulted in a further improvement in antitumor activity with a RTV of 5.2. No significant changes in body weight were noticed in all treatment groups compared to PBS control group (Fig. 9B).
Figure 9.
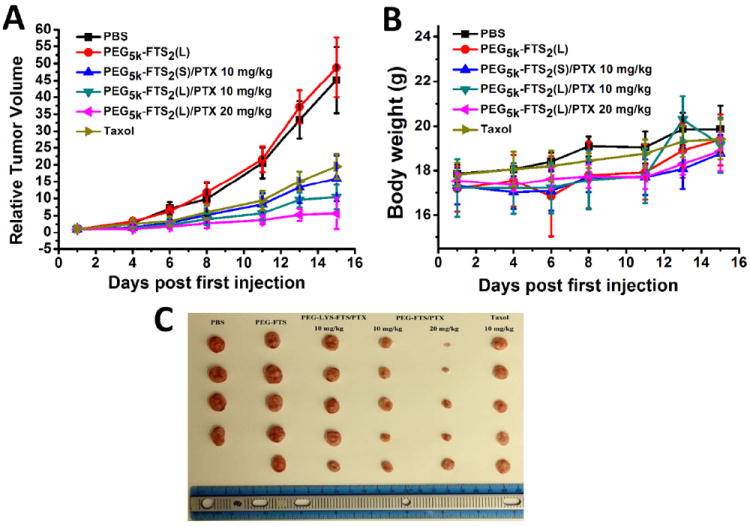
Enhanced antitumor activity of PTX formulated in PEG5K-FTS2(L) and PEG5K-FTS2(S) micelles. BABL/c mice were inoculated s.c. with 4T1.2 cells (1 × 105 cells/mouse). Five days later, mice received various treatments twice a week and tumor growth was monitored and plotted as relative tumor volume (mm3). (A) The changes of relative tumor volume. (B) Body weight after administration. (C) The mice were sacrificed and excised tumor form 4T1.2-bearing mice in each group. P < 0.001 (PTX/PEG5K-FTS2 (L) vs. Taxol). N = 5.
DISCUSSION
In this study, we have developed a micellar system that is based on PEG-derivatized FTS. FTS is an antagonist of Ras and is currently being evaluated as a new therapy for the treatment of various types of cancers that are associated with Ras mutations and elevated levels of Ras activity. A major feature with FTS is its excellent safety profile compared to other chemotherapeutic agents.21,32,33 This will render PEG-FTS micelles a safe delivery system for in vivo delivery of various types of anticancer agents including FTS itself.
One major advantage of PEG-FTS micellar system is its very small size (~ 20 nm). It is generally regarded that particles in the size of 100 ~ 200 nm can be passively targeted to tumors via the leaky tumor vasculature.34-36 However, recent studies have shown that the size of the particles needs to be below 50 ~ 60 nm for them to effectively reach the poorly vascularized tumors.30,37 The small size of PEG-FTS micelles will allow effective passive targeting to various types of cancers including those with poorly developed tumor vasculature.
In vitro release study showed significantly slower release kinetics for PTX formulated in PEG5K-FTS2 micelles compared to Taxol formulation (Fig. 4). This might be due to a more effective drug/carrier interaction for PTX/PEG5K-FTS2 mixed micelles. FTS has a benzene ring and an acyl chain. In addition to hydrophobic interaction with PTX, the π-π stacking and the hydrogen bonding also contribute to the overall carrier/PTX interaction. The close proximity of two FTS molecules in PEG5K-FTS2 conjugate shall enhance the interaction of the carrier with PTX. Indeed, a PEG-FTS conjugate of 1:1 molar ratio was much less active than PEG5K-FTS2 in forming stable mixed micelles with PTX (data not shown). More studies are needed to better define the mechanism of drug/carrier interactions for PTX/PEG5K-FTS2 mixed micelles.
PEG5K-FTS2(L) alone was much more active than PEG5K-FTS2(S) in antitumor activity in both 4T1.2 and HCT-116 cell lines (Fig. 7). It is unlikely that this is attributed to differences in the surface activity of the two conjugates as both showed minimal hemolytic activity at the concentrations that were much higher than those used in the cytotoxicity study. It is likely that FTS is much more readily released from PEG5K-FTS2(L) by esterases following intracellular delivery. The cytotoxicity data agree with Western blotting in which PEG5K-FTS2(L) was more active than PEG5K-FTS2(S) in reducing the protein expression levels of Ras in treated tumor cells (Fig. 6).
In addition to more potent antitumor activity by itself, PEG5K-FTS2(L) was also more active than PEG5K-FTS2(S) in mediating PTX cytotoxicity to tumor cells (Fig. 8). At the concentrations used for PTX delivery, both PEG5K-FTS2(L) and PEG5K-FTS2(S) were not active in killing tumor cells by themselves. This is consistent with published works 19, 38-40 and our own data (Fig. 7) that FTS works at high μM concentrations. It remains to be determined if, at suboptimal concentrations as those in cells treated with PEG5K-FTS2(L), the released FTS could synergize with PTX in the overall antitumor activity. It is also possible that PTX is more readily released from PEG5K-FTS2(L) micelles due to the disintegration of the micellar system following the cleavage and release of FTS from PEG5K-FTS2(L) conjugate. More studies are needed to better understand the mechanism by which PTX/PEG5K-FTS2(L) mediated the cytotoxicity to tumor cells in vitro.
In vivo, PTX formulated in both PEG5K-FTS2(L) and PEG5K-FTS2(S) micelles showed better antitumor activity than Taxol formulation although it is not statistically significant between Taxol and PTX/PEG5K-FTS2(S). This is likely due to a better stability of PEG5K-FTS2 micellar formulation as demonstrated in release study (Fig. 4). Again, PTX/PEG5K-FTS2(L) was more effective than PTX/PEG5K-FTS2(S) in antitumor activity in vivo. It remains to be determined if similar mechanisms are involved in the improved antitumor activity for PTX/PEG5K-FTS2(L) both in vitro and in vivo.
In summary, a conjugate of PEG with two molecules of FTS forms small-sized micelles that effectively solubilize PTX. PTX formulated in this micellar system shows a PTX release kinetics that is significantly slower than that of Taxol. PEG5K-FTS2(L) conjugate well retains the biological activity of FTS and PTX formulated in PEG5K-FTS2(L) micelles is more active in cytotoxicity than free PTX in vitro. In vivo, PTX/PEG5K-FTS2(L) shows an antitumor activity that is more potent than that of Taxol or PTX/PEG5K-FTS2(S). PEG5K-FTS2(L) may represent a promising micellar system that could effectively deliver anticancer agents to tumors. Furthermore it may potentially synergize with codelivered drugs in the overall antitumor activity.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants (R21CA128415 and R21CA155983) and a DOD grant (BC09603).
Footnotes
Supporting Information Available: Synthesis scheme of PEG5K-FTS2(S) conjugate, 1HNMR spectra (400MHz) of PEG5K-FTS2(L) and PEG5K-FTS2(S) conjugates in CDCl3, MADLI-TOF of PEG5K-FTS2(L) and PEG5K-FTS2(S) conjugates, and HPLC trace of PEG5K-FTS2(L) (A) and PEG5K-FTS2(S) (B) conjugates.
References
- 1.Goldspiel BR. Clinical overview of the taxanes. Pharmacotherapy. 1997;17:110–125. [PubMed] [Google Scholar]
- 2.Xie Z, Guan HL, Chen X, Lu C, Chen L, Hu X, Shi Q, Jing X. A novel polymer-paclitaxel conjugate based on amphiphilic triblock copolymer. J Controlled Release. 2007;117:210–216. doi: 10.1016/j.jconrel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 4.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 5.Tong R, Cheng J. Anticancer polymeric nanomedicines. Polymer Reviews. 2007;47:345–381. [Google Scholar]
- 6.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 7.Trubetskoy VS. Polymeric micelles as carriers of diagnostic agents. Adv Drug Deliv Rev. 1999;37:81–88. doi: 10.1016/s0169-409x(98)00100-8. [DOI] [PubMed] [Google Scholar]
- 8.Riley T, Stolnik S, Heald CR, Xiong CD, Garnett MC, Illum L, Davis SS, Purkiss SC, Barlow RJ, Gellert PR. Physicochemical evaluation of nanoparticles assembled from poly(lactic acid)-poly(ethylene glycol) (PLA-PEG) block copolymers as drug delivery vehicles. Langmuir. 2001;17:3168–3174. [Google Scholar]
- 9.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai HJ. PEG branched polymer for functionalization of nanomaterials with ultralong blood circulation. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 11.Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12:4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Feng SS. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27:262–270. doi: 10.1016/j.biomaterials.2005.05.104. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Lee SH, Gan CW, Feng SS. In vitro and in vivo investigation on PLA-TPGS nanoparticles for controlled and sustained small molecule chemotherapy. Pharm Res. 2008;8:1925–1935. doi: 10.1007/s11095-008-9611-6. [DOI] [PubMed] [Google Scholar]
- 14.Prashant C, Dipak M, Yang CT, Chuang KH, Jun D, Feng SS. Superparamagnetic iron oxide--loaded poly(lactic acid)-D-alpha-tocopherol polyethylene glycol 1000 succinate copolymer nanoparticles as MRI contrast agent. Biomaterials. 2010;21:5588–5597. doi: 10.1016/j.biomaterials.2010.03.070. [DOI] [PubMed] [Google Scholar]
- 15.Mi Y, Liu Y, Feng SS. Formulation of docetaxel by folic acid-conjugated D-a-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS2k) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32:4058–4066. doi: 10.1016/j.biomaterials.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 16.Huang YH, Lu JQ, Gao X, Li J, Zhao WC, Sun M, Stolz DB, Venkataramanan R, Rohan LC, Li S. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjugate Chem. 2012;23:1443–1451. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu JQ, Huang YH, Zhao WC, Marquez RT, Meng XJ, Li J, Gao X, Wang Z, Venkataramanan R, Li S. EG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials. 2012 doi: 10.1016/j.biomaterials.2012.10.073. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikolovska-Coleska Z, Xu L, Hu Z, Tomita Y, Li P, Roller PP, Wang R, Fang X, Guo R, Zhang M, Lippman ME, Yang D, Wang S. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three dimensional structure database. J Med Chem. 2004;47:2430–2440. doi: 10.1021/jm030420+. [DOI] [PubMed] [Google Scholar]
- 19.Haklai R, Weisz MG, Elad G, Paz A, Marciano D, Egozi Y, Ben-Baruch G, Kloog Y. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37:1306–1314. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 20.Marciano D, Ben-Baruch G, Marom M, Egozi Y, Haklai R, Kloog Y. Selective inhibition of ras-dependent cell growth by farnesylthiosalicylic acid. J Med Chem. 1995;38:1267–1272. doi: 10.1021/jm00008a004. [DOI] [PubMed] [Google Scholar]
- 21.Blum R, Kloog Y. Tailoring Ras-pathway—Inhibitor combinations for cancer therapy. Drug Resistance Updates. 2005;8:369–380. doi: 10.1016/j.drup.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Kloog Y, Cox AD. RAS inhibitors: potential for cancer therapeutics. Mol Med Today. 2000;6:398–402. doi: 10.1016/s1357-4310(00)01789-5. [DOI] [PubMed] [Google Scholar]
- 23.Gana-Weisz M, Halaschek-Wiener J, Jansen B, Elad G, Haklai R, Kloog Y. The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid chemosensitizes human tumor cells without causing resistance. Clin Cancer Res. 2002;8:555–565. [PubMed] [Google Scholar]
- 24.Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: A potential treatment for cancer. Methods in Enzymology. 2008;439:467–489. doi: 10.1016/S0076-6879(07)00432-6. [DOI] [PubMed] [Google Scholar]
- 25.Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001;51:7486–7493. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 26.Kraitzer A, Kloog Y, Haklai R, Zilberman M. Composite fiber structures with antiproliferative agents exhibit advantageous drug delivery and cell growth inhibition in vitro. J Pharm Sci. 2011;100:133–149. doi: 10.1002/jps.22238. [DOI] [PubMed] [Google Scholar]
- 27.Zhang P, Ye H, Min T, Zhang C. Water soluble polyethylene glycol prodrug of silybin: Design synthesis and characterization. J Appl Polym Sci. 2008;107:3230–3235. [Google Scholar]
- 28.Duan K, Zhang XL, Tang XX, Yu JH, Liu SY, Wang DX, Li YL, Huang J. Fabrication of cationic nanomicelle from chitosan-graft-polycaprolactone as the carrier of 7-ethyl-10-hydroxy-camptothecin. Colloids Surf B: Biointerfaces. 2010;76:475–482. doi: 10.1016/j.colsurfb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, Kato J, Dong TA, Lam KS. Well-defined, reversible disulfide cross-linked micelles for on-demand paclitaxel delivery. Biomaterials. 2011;32:6633–6645. doi: 10.1016/j.biomaterials.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo JT, Xiao K, Li YP, Lee JS, Shi LF, Tan YH, Xing L, Cheng RH, Liu GY, Lam KS. Well-defined, size-tunable, multi-functional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjug Chem. 2010;21:1216–1224. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reul R, Nguyen J, Kissel T. Amine-modified hyperbranched polyesters as nontoxic, biodegradable gene delivery systems. Biomaterials. 2009;30:5815–5824. doi: 10.1016/j.biomaterials.2009.06.057. [DOI] [PubMed] [Google Scholar]
- 32.Kloog Y, Cox AD, Sinensky M. Concepts in Ras-directed therapy. Expert Opin Investig Drugs. 1998;8:2121–2140. doi: 10.1517/13543784.8.12.2121. [DOI] [PubMed] [Google Scholar]
- 33.Haklai R, Elad-Sfadia G, Egozi Y, Kloog Y. Orally administered FTS (salirasib) inhibits human pancreatic tumor growth in nude mice. Cancer Chemotherapy and Pharmacology. 2008;61:89–96. doi: 10.1007/s00280-007-0451-6. [DOI] [PubMed] [Google Scholar]
- 34.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumouritropic accumulation of proteins and the antitumour agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 35.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 36.Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9:253–266. doi: 10.1007/BF00046364. [DOI] [PubMed] [Google Scholar]
- 37.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 38.Clarke HC, Kocher HM, Khwaja A, Kloog Y, Cook HT, Hendry BM. Ras antagonist farnesylthiosalicylic acid (FTS) reduces glomerular cellular proliferation and macrophage number in rat thy-1 nephritis. J Am Soc Nephrol. 2003;14:848–854. doi: 10.1097/01.asn.0000057543.55318.8b. [DOI] [PubMed] [Google Scholar]
- 39.Weisz B, Giehl K, Gana-Weisz M, Egozi Y, Ben-Baruch G, Marciano D, Gierschik P, Kloog Y. A new functional Ras antagonist inhibits human pancreatic tumor growth in nude mice. Oncogene. 1999;18:2579–2588. doi: 10.1038/sj.onc.1202602. [DOI] [PubMed] [Google Scholar]
- 40.Jansen B, Schlagbauer-Wadl H, Kahr H, Heere-Ress E, Mayer BX, Eichler H, Pehamberger H, Gana-Weisz M, Ben-David E, Kloog Y, Wolff K. Novel Ras antagonist blocks human melanoma growth. Proc Natl Acad Sci. 1999;96:14019–14024. doi: 10.1073/pnas.96.24.14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.