

Abstract
Mononuclear cytotrophoblasts of the human placenta proliferate rapidly, subsequently fuse, and differentiate to form multinucleated syncytiotrophoblast with induction of aromatase (hCYP19A1) and chorionic gonadotropin (hCGβ) expression. Using microarray analysis, we identified members of the miR-17∼92 cluster and its paralogs, miR-106a∼363 and miR-106b∼25, that are significantly downregulated upon syncytiotrophoblast differentiation. Interestingly, miR-19b and miR-106a directly targeted hCYP19A1 expression, while miR-19b also targeted human GCM1 (hGCM1), a transcription factor critical for mouse labyrinthine trophoblast development. Overexpression of these microRNAs (miRNAs) impaired syncytiotrophoblast differentiation. hGCM1 knockdown decreased hCYP19A1 and hCGβ expression, substantiating its important role in human trophoblast differentiation. Expression of the c-Myc proto-oncogene was increased in proliferating cytotrophoblasts compared to that in differentiated syncytiotrophoblast. Moreover, c-Myc overexpression upregulated miR-17∼92 and inhibited hCYP19A1 and hCGβ expression. Binding of endogenous c-Myc to genomic regions upstream of the miR-17∼92 and miR-106a∼363 clusters in cytotrophoblasts dramatically decreased upon syncytiotrophoblast differentiation. Intriguingly, we observed higher levels of miR-106a and -19b and lower aromatase and hGCM1 expression in placentas from preeclamptic women than in placentas from gestation-matched normotensive women. Our findings reveal that c-Myc-regulated members of the miR-17∼92 and miR-106a∼363 clusters inhibit trophoblast differentiation by repressing hGCM1 and hCYP19A1 and suggest that aberrant regulation of these miRNAs may contribute to the pathogenesis of preeclampsia.
INTRODUCTION
The multinucleated syncytiotrophoblast of the human placenta is formed by fusion of underlying proliferating cytotrophoblasts. This multinucleated cell layer, which covers the chorionic villi, is bathed in maternal blood and performs several essential functions to ensure growth and survival of the developing embryo. These include transport of O2 and nutrients and synthesis and secretion of syncytiotrophoblast-specific protein and steroid hormones, including estrogen and progesterone. Synthesis of estrogens from C19 steroids is catalyzed by aromatase P450 (P450arom, product of the hCYP19A1 gene). The ability of the human placenta to synthesize estrogens is vastly increased after the ninth week of gestation (1), in association with cytotrophoblast invasion and enlargement of the uterine arterioles, increased blood flow, and O2 availability to the floating chorionic villi (2, 3). Trophoblast stem cells and cytotrophoblasts do not express hCYP19A1/aromatase; however, when cytotrophoblasts fuse to form multinucleated syncytiotrophoblast, aromatase is markedly induced (4, 5). The exceptionally high levels of placental aromatase likely function to metabolize large amounts of C19 steroids produced by the human fetal adrenals (e.g., dehydroepiandrosterone), thus preventing conversion of these steroids to active androgens, which can masculinize the fetus. Biologically active estrogens and their metabolites formed by placental aromatase may also enhance angiogenesis and uteroplacental blood flow and reduce systemic vascular resistance (6–10).
While different genetic programs have been found to be switched on and off during syncytiotrophoblast differentiation, the regulatory mechanisms that affect these events remain incompletely defined. We previously observed that when midgestation human trophoblasts were cultured in a hypoxic (2% O2) environment, syncytiotrophoblast differentiation and induction of hCYP19 gene expression were prevented (3). These inhibitory effects of hypoxia were associated with increased expression and binding of the basic helix-loop-helix (bHLH)-zipper transcription factors USF1 and -2 to two E boxes within placenta-specific hCYP19 exon I.1 and its 5′-flanking region (11). Conversely, increased O2 tension promoted enhanced USF1/2 polyubiquitination and degradation via the proteasome pathway (12). We therefore suggest that with the gestational increase in placental vascularization and increased O2 availability to trophoblast cells, the inhibitory USFs are degraded, which may allow stimulatory transcription factors to bind to these E boxes and/or to other response elements and promote placenta-specific expression. Placental estrogens are believed to play an autocrine role in trophoblast differentiation (13, 14). In recent studies, we have established crucial roles for estrogen receptor α (ERα) (15) and estrogen-related receptor γ (ERRγ) (16) in the O2-mediated induction of CYP19 expression during human trophoblast differentiation.
The transcription factor glial cells missing 1 (GCM1) has been shown to play a fundamental role in trophoblast differentiation and syncytiotrophoblast formation. GCM1 knockout mice die at midgestation due to the failure of a functional placental labyrinth, analogous to the human syncytiotrophoblast layer (17, 18). Human GCM1 (hGCM1) induces the expression of syncytin A, a gene involved in trophoblast fusion (19), and regulates expression of placental growth factor (20). While the hCYP19A1 promoter has been shown to have a GCM1 binding element that is required for reporter activity (21), a critical role for endogenous hGCM1 in the regulation of CYP19A1 expression in human syncytiotrophoblast has yet to be established.
MicroRNAs (miRNAs) are regulatory, small noncoding RNAs of 20 to 25 nucleotides (nt) in length, with a fundamental role in different aspects of cell biology, including cell cycle regulation, differentiation, apoptosis, and maintaining stemness (22). MicroRNAs function to repress gene expression by binding via their seed sequences to complementary sites in the 3′ untranslated regions (3′ UTR) of target mRNAs. This results in degradation of the mRNA target and/or inhibition of translation (23). Individual miRNAs can bind to and regulate networks of mRNAs with related biological functions and can act as rheostats or as on-off switches of gene expression, while multiple miRNAs can target a single mRNA. MicroRNAs have been found to play important roles in cell differentiation (24–26) and cancer (27, 28) and have been found to have particularly important roles in female reproduction (29–31).
Thus, to further define the cellular mechanisms that underlie human trophoblast differentiation and induction of CYP19A1 gene expression, in the present study, we have investigated the potential role of miRNAs. Interestingly, we have uncovered a novel role for the c-Myc-regulated microRNA-17∼92 (miR-17∼92) cluster and its paralog miR-106a∼363 in directly targeting and inhibiting hCYP19A1 and hGCM1 gene expression in cytotrophoblasts and thereby repressing human trophoblast differentiation and aromatase expression. We suggest that dysregulation of this pathway may have an important consequence in the pathogenesis of preeclampsia, a pregnancy-induced hypertensive disorder in which the placenta plays a central role.
MATERIALS AND METHODS
Term human placental samples and primary culture of human trophoblast cells.
Term placental tissues from control normotensive women and preeclamptic women were obtained at the time of cesarean section. The placental tissues were immediately placed in RNAlater (Qiagen, Foster City, CA) solution and snap-frozen at −80°C.
Midtrimester human placental tissues were obtained from Advanced Bioscience Resources (Alameda, CA) in accordance with the Donors Anatomical Gift Act of the State of Texas. Protocols were approved by the Institutional Review Board of the University of Texas Southwestern Medical Center at Dallas. Cytotrophoblasts were isolated from midgestation human placenta and placed in primary culture, as described in detail previously (5, 32). Briefly, the placental tissues were washed with Hanks' balanced salt solution (pH 7.4) (Life Technologies, Grand Island, NY) and then finely minced and digested with 0.125% trypsin in Hanks' balanced salt solution at 37°C for 20 min. At the end of the digestion, the supernatant was collected, layered over 10 ml of serum, and then briefly centrifuged at 1,000 × g. The resulting pellet was suspended in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies), filtered, and layered over a Percoll gradient (70% to 5%). This procedure was repeated three times with the pellet from the original trypsin digestion. The gradients were centrifuged at 1,200 × g for 20 min at room temperature, and cells in the middle layer (density, 1.045 to 1.062 g/ml) were collected, washed, and counted. The cells were then resuspended in DMEM supplemented with 10% fetal bovine serum (FBS) and 1.2% antibiotic/antimycotic solution (Life Technologies) and plated at a density of 2 × 106 cells per 35-mm culture dish or 15 × 106 cells per 100-mm dish. The cells were cultured overnight; the medium was then changed to DMEM containing 2% FBS. For expression of miRNA mimics or miR-19b inhibitor, freshly isolated cytotrophoblasts were transfected with either 10 nM miRNA mimic (Qiagen), LNA hsa-miR-19 family inhibitor (Exiqon, Woburn, MA), LNA scrambled control (Exiqon), or AllStars Negative control (Qiagen) using the HiPerfect transfection reagent (Qiagen). Cells were harvested for RNA and protein analysis 24, 48, and 72 h posttransfection.
Quantitative reverse transcriptase PCR (qRT-PCR).
Total RNA, including small RNAs, from trophoblast cells cultured for 24, 48, or 72 h was extracted using the miRNeasy minikit (Qiagen) according to the manufacturer's instructions. Quantification of miRNAs was carried out by TaqMan real-time PCR (Applied Biosystems). Primer sets specific for the placenta-specific hCYP19A1 transcript, hCYP19I.1 (15), along with constitutively expressed RPLP0, were designed utilizing the Primer Express software program (PE Applied Biosystems, Boston, MA): hCYP19I.1 forward primer, 5′-ACG GAA GGT CCT GTG CTC G-3′; reverse primer, 5′-GTA TCG GGT TCA GCA TTT CCA-3′; RPLP0 forward primer, 5′-TGC ATC AGT ACC CCA TTC TAT CA-3′; reverse primer, 5′-AAG GTG TAA TCC GTC TCC ACA GA-3′.
The relative abundance of each transcript was determined by qRT-PCR using previously published methods (15). All primer sets produced amplicons of the expected size and sequence. The relative fold changes were calculated using the comparative cycle times (CT) method with RPLP0 as the internal reference.
Immunoblot analysis.
Nuclear and cytoplasmic extracts were prepared from human trophoblast cells according to a protocol described previously (15). Protein concentrations were determined by using the Bradford assay (33) (Bio-Rad) and resolved by electrophoresis on 4 to 12% Bis-Tris gels (Invitrogen). Rabbit polyclonal c-Myc antibody (9402) was obtained from Cell Signaling (Beverly, MA). Antiaromatase (A7981) was obtained from Sigma (St. Louis, MO), anti-GCM1 from Aviva Systems Biology (San Diego, CA), anti-lamin A/C (catalog no. 05-714) from Upstate (Lake Placid, NY), and anti-β-actin (ab8227) from Abcam (Cambridge, MA). Horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgGs from GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom) were used as secondary antibodies. The membranes were incubated with enhanced Supersignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL) and exposed to X-ray film.
Cell culture and luciferase assay.
Twelve hundred base pairs of the 3′ UTR of human CYP19A1 and 500 bp of the 3′ UTR of the human GCM1 were amplified from placental genomic DNA and cloned downstream of luciferase in the p-MIR-REPORT miRNA reporter vector (Applied Biosystems). JEG3 cells maintained in RPMI medium supplemented with 10% fetal bovine serum were transfected with either the empty pMIR-REPORT luciferase plasmid (LUC) or with LUC containing the 3′ UTR of the CYP19A1 or hGCM1 gene (LUC-CYP19A1 or LUC-GCM1) with or without the mutated miRNA response element (MRE) (LUC-CYPmut or LUC-GCMmut). Nontargeting control (NTC) or miRNA mimics of miR-19b or miR-106a and a plasmid expressing beta-galactosidase (β-Gal) to correct for transfection efficiency were cotransfected. Forty-eight hours after transfection, cells were harvested and luciferase and β-Gal activities were measured. The mutation of the MRE site in placenta-specific hCYP19 3′ UTR was generated using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). All constructs were confirmed by DNA sequencing.
Adenoviral overexpression and RNA interference.
Recombinant adenoviruses expressing human c-Myc were obtained from Vector Biolabs (Philadelphia, PA). A cytomegalovirus β-gal-containing (CMV-β-gal) adenovirus was kindly provided by Joseph Alcorn (University of Texas Medical School, Houston, TX). Recombinant adenoviruses were used to infect freshly isolated cytotrophoblasts at a multiplicity of infection (MOI) of 2.0. Lentiviral vectors containing short hairpin RNAs (shRNAs) targeting GCM-1 in pGIPZ vectors were obtained from a recombinant lentivirus shRNAmir library (Open Biosystems) (34). Lentiviruses expressing shRNAs for GCM1 were produced in human embryonic kidney (HEK)-293T cells by calcium phosphate transfection of the plasmids pMD2.G and psPAX2 and specific human GIPZ lentiviral shRNA for GCM1, according to a protocol described earlier (16). Lentiviral supernatants were collected after 24, 48, and 72 h of culture. The viral particles were concentrated by ultracentrifugation, and the titer of the viral stock (MOI or number of transducing units per cell) was determined according to the protocol recommended by Open Biosystems (Thermo Scientific). As a control, cells were infected with nonsilencing GIPZ lentivirus shRNA. Two different shRNA vectors were used for silencing of GCM-1. The following GCM1 shRNA clones were used: Open Biosystems catalog no. RHS4430-99166548 and RHS4430-98703843.
Chromatin immunoprecipitation assay.
Human trophoblasts were cultured for 5 h or 72 h in DMEM containing 2% FBS. Utilizing the ChIP kit (Millipore, Temecula, CA), chromatin immunoprecipitation (ChIP) assays were performed as described previously (15). Precleared cross-linked chromatin was immunoprecipitated using either c-Myc antibody or nonimmune rabbit IgG control (sc-2027; Santa Cruz Biotechnology, Santa Cruz, CA). Quantitative RT-PCR using primers that flanked E boxes corresponding to putative Myc-responsive elements (identified by using the Genomatrix MatInspector program; Genomatrix Software GmbH, Munich, Germany) in the genomic regions of the miR-17∼92 and the miR-106a∼363 clusters was used to assess the fold enrichment of the immunoprecipitated protein-DNA complex for endogenous c-Myc. Primers used in this assay were the following: miR-17∼92 E box 1f, AAAGGCAGGCTCGTCGTTG; miR-17∼92 E box 1r, CGGGATAAAGAGTTGTTTCTCCAA; miR-17∼92 E box 2f, CTCGACTCTTACTCTCACAAATGG; miR-17∼92 E box 2r, GCTACTGGTGCAGTTAGGTCC; miR-106A∼3631f, GAGAGGGGGAGTCCAAAATC; miR-106A∼3631r, TGGTTTCAACCAAATCCTGA; miR-106A∼3632f, GACTGGAGCGCGTGGTGTGA; miR-106A∼3632r, CTCCCGTTGAAGTCCGCGAGC.
Quantitative PCR was performed using SYBR green PCR master mix (Applied Biosystems) on a Bio-Rad CFX384 real-time PCR detection system. Signals were normalized to input samples and expressed relative to that for control IgG. Samples from three independent ChIP experiments were analyzed.
Data analysis.
Data are expressed as means ± standard deviations (SD). Differences between groups were analyzed by Student's t test. Statistical significance was set as P values of <0.05, and each experiment was performed at least three times (from three independent cell preparations) in triplicate.
RESULTS
Microarray analysis of miRNAs in trophoblast cells reveals decreased expression of members of the miR-17∼92 cluster and its paralog miR-106a∼363 during human trophoblast differentiation.
To identify miRNAs that are differentially regulated during human trophoblast differentiation, RNA isolated from human cytotrophoblasts before (0 h) and after 24 and 48 h of culture was sent to LC Sciences (Houston, TX), which carried out miRNA microarray using quadruple replicates of the 0-h-, 24-h-, and 48-h-of-incubation time points. We detected 16 miRs that were significantly downregulated and 9 miRs that were significantly upregulated >2-fold at both 24 and 48 h, compared to results at 0 h (a heat map of all significantly regulated miRNAs is shown in Fig. S1 in the supplemental material). Shown in Fig. 1A is a heat map of several members of the miR-17∼92 cluster and its paralogs miR-106a∼363 and miR-106b∼25 that were found to be significantly downregulated during human trophoblast differentiation. In the human genome, the miR-17∼92 cluster encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1), while its paralog, the miR-106a∼363 cluster, also encodes six miRNAs (miR-106a, miR-18b, miR-20b, miR-19b-2, miR-92a-2, and miR-363). Using TargetScan v5.1 analysis, we identified several predicted and confirmed mRNA targets of these miRs (Fig. 1B). ERα and SRC-3 are known targets of the miR-17∼92 and miR-106a∼363 clusters (35, 36). MicroRNAs in these clusters that share similar seed sequences are grouped in Fig. 1C. miR-106a and miR-19b are predicted to target hCYP19A1 mRNA. In previous studies, we observed that hCYP19A1 expression is highly upregulated during syncytiotrophoblast differentiation (5) and the estrogens formed via the action of hCYP19A1/aromatase act in a positive feed-forward manner through ERα to further promote syncytiotrophoblast differentiation and the induction of hCYP19 expression (15). Furthermore, miR-19b, a member of the miR-17∼92 cluster, is also predicted to target hGCM1, a transcription factor critical for trophoblast differentiation (18) and upregulation of hCYP19I.1 promoter activity (21). As hCYP19I.1 expression increased during syncytiotrophoblast differentiation (Fig. 1D), downregulation of several members of the miR-17∼92 cluster and its paralog miR-106a∼363 was confirmed in cultured trophoblasts using TaqMan-based RT-qPCR (Fig. 1E). Interestingly, JEG-3 cells, which express low levels of aromatase and are cytotrophoblast-like, manifested significantly higher expression of these miRNAs than syncytiotrophoblast (Fig. 1D and E). Thus, we have clearly demonstrated that members of the miR-17∼92 cluster and its paralog miR-106a∼363 are highly expressed in cytotrophoblasts and their expression decreases as the cells differentiate to form syncytiotrophoblast.
Fig 1.

Members of the miR-17∼92 cluster and its evolutionary paralogs, miR-106a∼363 and miR-106b∼93, are downregulated during human trophoblast differentiation. (A) Heat map of differentially expressed miRNAs. The heat map shows in green the miRNAs that were significantly downregulated during human trophoblast differentiation. Statistical analysis of the microarray data revealed 6 miRs of the miR-17 and miR-19 families that were downregulated >2-fold at both 24 and 48 h compared to results at 0 h. (B) Table of the conserved putative and known mRNA targets of these miRNAs, identified using the Target Scan v5.1 software tool, that may be of importance in trophoblast differentiation. (C) miR-17, -106a, -20a, -20b, and -93, encoded in clusters on three different chromosomes in humans (in the upper panel, gray areas represent premiRNAs and black areas represent mature miRNAs), are members of the miR-17/20/106 family and thus share the same seed sequence and bind the same mRNA targets. Shown as groups are different miRNA families encoded within these clusters that share common seed sequences and thus common targets. (D) RNA from freshly isolated cytotrophoblasts before (Cyto) and after 24, 48, and 72 h of culture (syncytiotrophoblast) and from JEG-3 cells was analyzed for hCYP19I.1 mRNA expression by SYBR green qRT-PCR. (E) TaqMan qRT-PCR analysis of miRNA expression using RNA from freshly isolated cytotrophoblasts before (Cyto) and after 24, 48, and 72 h of culture (syncytiotrophoblast) and from JEG-3 cells.
miR-106a and miR-19b negatively regulate hCYP19A1 expression via elements in its 3′ UTR.
Since miR-17, -20a, and -106b are predicted to target hCYP19A1 mRNA, contain identical seed sequences, and were significantly downregulated along with miR-19b during syncytiotrophoblast differentiation, we initially analyzed the effects of overexpression of miR-19b and -106a on their predicted target, hCYP19A1. Nontargeting control (NTC) or miRNA mimics of miR-19b or -106a were transfected into freshly isolated placental trophoblasts. After 72 h of culture, the cells were harvested for RNA; although TaqMan qRT-PCR analysis revealed a 100-fold increase in either miR-19b or miR-106a expression (data not shown), a significant decrease in hCYP19I.1 mRNA expression was observed in cells transfected with miR-19b but not in cells transfected with nontargeting control or miR-106a (Fig. 2A). Since miRNAs are also known to inhibit mRNA translation, we performed immunoblot analysis of aromatase and observed that overexpression of either miR-19b and -106a greatly decreased endogenous aromatase protein expression in syncytiotrophoblast (Fig. 2B, top panel). The immunoblot results of three independent experiments quantitated using the software program Image J demonstrated that these miRNAs significantly decreased aromatase protein expression (Fig. 2B, bottom panel). To inhibit endogenous miR-19b expression, LNA scrambled control (Con) or LNA miR-19 inhibitors were transfected into freshly isolated trophoblasts. After 72 h, the cells were analyzed for expression of miRNA by TaqMan qRT-PCR analysis and aromatase protein by immunoblot analysis. Inhibition of miR-19b specifically decreased miR-19b (and had no effect on miR-106a expression) and increased aromatase protein expression compared to results for cells transfected with a LNA scrambled control (Fig. 2C). Using TargetScan prediction software, we identified 2 binding sites for miR-17, -20a, and -106a and one for miR-19b in the 3′ UTR of hCYP19A1 mRNA, as shown in Fig. 2D. To demonstrate that miR-106a and miR-19b directly regulate hCYP19A1 expression, we transfected a reporter construct containing the hCYP19A1 3′ UTR subcloned downstream of the luciferase gene (LUC/CYP19) together with miRNA mimics into JEG-3 cells and observed a significant reduction in luciferase activity compared to results for cells transfected with nontargeting control miRNA (Fig. 2D). Notably, no reduction in luciferase activity was detected when the putative miRNA binding sequence in the hCYP19A1 3′ UTR luciferase reporter construct was mutated (LUC/CYP19mut) (Fig. 2D). These data suggest that hCYP19A1 expression is negatively regulated in human trophoblasts via direct interaction of both miR-106a and miR-19b miRNA response elements in the hCYP19A1 3′ UTR.
Fig 2.

miR-106a and miR-19b negatively regulate hCYP19 expression via elements in its 3′ UTR during trophoblast differentiation. (A and B) Nontargeting control (NTC) or miRNA mimics of miR-19b or -106a were transfected into freshly isolated placental trophoblasts. After culture for 72 h, the cells were analyzed for the expression of hCYP19I.1 mRNA (A) or for the aromatase protein by immunoblot analysis (B, top panel). The immunoblot results of three independent experiments were quantitated using Image J and corrected for loading and transfer using β-actin (B, bottom panel). (C) LNA scrambled control (Con) or LNA miR-19 inhibitors were transfected into freshly isolated trophoblasts. After 48 h, the cells were analyzed for expression of miRNA by TaqMan qRT-PCR analysis and for the aromatase protein by immunoblot analysis. (D) The 3′ UTR of hCYP19A1 mRNA was cloned downstream of the luciferase gene in the pMIR-REPORT luciferase plasmid. Reporters in which the putative miR-19b or miR-106a binding sites, shown within the 3′ UTR, were mutated also were constructed. JEG3 cells were transfected either with an “empty” pMIR-REPORT luciferase plasmid (LUC) or with pMIR-REPORT-LUC containing the 3′ UTR of the hCYP19A1 gene with wild-type (LUC-CYP19) or mutated (LUC-CYP19mut) sequences corresponding to the miR-19b (left panel) or 106a (right panel) binding sites. Nontargeting control (NTC) or miRNA mimics of miR-19b or miR-106a and a plasmid expressing β-Gal to correct for transfection efficiency were cotransfected. Forty-eight hours after transfection, cells were harvested and luciferase and β-Gal activities were measured.
miR-19b negatively regulates hGCM1 expression via elements in its 3′ UTR.
As mentioned, GCM1 is a transcription factor critical for trophoblast differentiation and fusion (18), as well as hCYP19I.1 expression (21). Accordingly, we observed that hGCM1 protein expression increased rapidly in cultured human trophoblasts, reaching peak levels at 24 h. Although GCM1 expression declined at later time points, it remained at markedly higher levels than in the cytotrophoblasts prior to culture (Fig. 3A). Since miR-19b is predicted to target hGCM1, mimics of miR-19b, -106a, or NTC were transfected into freshly isolated human trophoblasts. After 72 h of culture, the cells were harvested and analyzed for hGCM1 mRNA and protein. While qRT-PCR analysis revealed no change in hGCM1 mRNA expression, overexpression of miR-19b and -106a mimics caused a significant decrease in human chorionic gonadotropin β (hCGβ) mRNA, a marker of syncytiotrophoblast differentiation (Fig. 3B). However, immunoblot analysis revealed that overexpression of miR-19b but not miR-106a markedly decreased endogenous hGCM1 protein expression in syncytiotrophoblast (Fig. 3C, top panel). The inhibitory effect of miR-19b on hGCM1 protein expression was confirmed by Image J analysis of immunoblots from three independent experiments (Fig. 3C, bottom panel). In contrast, transfection of cultured human trophoblasts with the LNA miR-19b inhibitor increased GCM1 protein expression in cells compared to that in cells transfected with LNA scrambled control (Con) (Fig. 3D). Thus, miR-19b targets hGCM1 in human trophoblasts. While hCGβ is not a predicted target of these miRNAs, its expression was probably decreased by miR-19b and miR-106a transfection as a consequence of reduced expression of either hGCM1 or hCYP19.
Fig 3.

hGCM1 is rapidly induced during syncytiotrophoblast differentiation; miR-19b negatively regulates hGCM1 expression in cytotrophoblasts via elements in its 3′ UTR. (A) Nuclear proteins extracted from cytotrophoblasts (Cyto) or syncytiotrophoblast after 24, 48, and 72 h of culture were analyzed by immunoblotting using antiserum to hGCM1 or lamin A/C (catalog no 05-714; Upstate, Lake Placid, NY) as a loading control. (B and C) Nontargeting control (NTC) or miRNA mimics of miR-19b or -106a were transfected into freshly isolated placental trophoblasts. After culture for 72 h, the cells were analyzed for the expression of hGCM1 and hCGβ mRNA (B) or for hGCM1 protein by immunoblot analysis (C, upper panel). The immunoblot results of three independent experiments were quantitated using the Image J program (C, lower panel). (D) LNA scrambled control (Con) or LNA miR-19 inhibitors (19b inh) were transfected into freshly isolated trophoblasts. After 48 h, the cells were analyzed for expression of the GCM1 protein by immunoblot analysis. (E) The 3′ UTR of hGCM1 mRNA was cloned downstream of the luciferase gene in the pMIR-REPORT luciferase plasmid (upper panel). A reporter in which the miR-19b binding site within the 3′ UTR was mutated was also constructed. JEG3 cells were transfected with either an “empty” pMIR-REPORT luciferase plasmid (LUC) or with pMIR-REPORT-LUC containing the 3′ UTR of hGCM1 with a wild-type (LUC-GCM1) or mutated (LUC-GCMmut) miR-19b binding site. Nontargeting control (NTC) or miRNA mimics of miR-19b and a plasmid expressing β-Gal to correct for transfection efficiency were cotransfected. Forty-eight hours after transfection, cells were harvested and luciferase and β-Gal activities were analyzed (lower panel).
To demonstrate that miR-19b directly targets hGCM1 mRNA, we transfected a hGCM1 3′ UTR luciferase reporter construct (LUC/GCM) together with miRNA mimics into JEG-3 cells and observed a significant reduction in luciferase activity in cells cotransfected with miR-19b, compared to that in cells cotransfected with nontargeting control (Fig. 3E). On the other hand, no reduction in luciferase activity was detected upon cotransfection of miR-19b when the putative miR binding sequence in the hGCM1 3′ UTR luciferase reporter construct was mutated (LUC/GCMmut) (Fig. 3E). These findings suggest that hGCM1 expression is translationally suppressed in human cytotrophoblasts by increased levels of miR-19b binding to a response element in its 3′ UTR.
hGCM1 is required for hCYP19 and hCGβ expression in trophoblasts.
Having established that miR-19b directly targets hGCM1 expression in human trophoblasts, we further investigated the functional role of endogenous hGCM1. While cotransfection studies in a choriocarcinoma cells line indicated that hCYP19I.1 promoter activity was upregulated by hGCM1 (21), a role for endogenous hGCM1 in the regulation of hCYP19A1 in primary human syncytiotrophoblast has yet to be established. Therefore, we utilized an RNA interference approach to knock down hGCM1. Two different lentivirus constructs carrying hGCM1-targeting shRNA (GCM shRNA) were used to infect freshly isolated human cytotrophoblasts at a multiplicity of infection (MOI) of 0.5 and 1.0. Parallel dishes of cells were infected with lentiviruses expressing a nontargeting shRNA, as a control. After 72 h of culture, a significant knockdown in hGCM1 mRNA and protein expression was observed. This was associated with a corresponding decrease in hCYP19I.1 and hCGβ mRNA expression, compared to results for cells infected with control lentivirus (Fig. 4A). A decrease in endogenous hGCM1 protein in cells infected with lentiviruses expressing hGCM1 shRNA was confirmed by immunoblot analysis (Fig. 4B). These findings clearly identify an essential role for hGCM1 in regulating hCYP19I.1 and hCGβ expression during human trophoblast differentiation and suggest that miR-19b-mediated repression of hGCM1 in cytotrophoblasts contributes to suppression of hCYP19I.1 and hCGβ expression.
Fig 4.
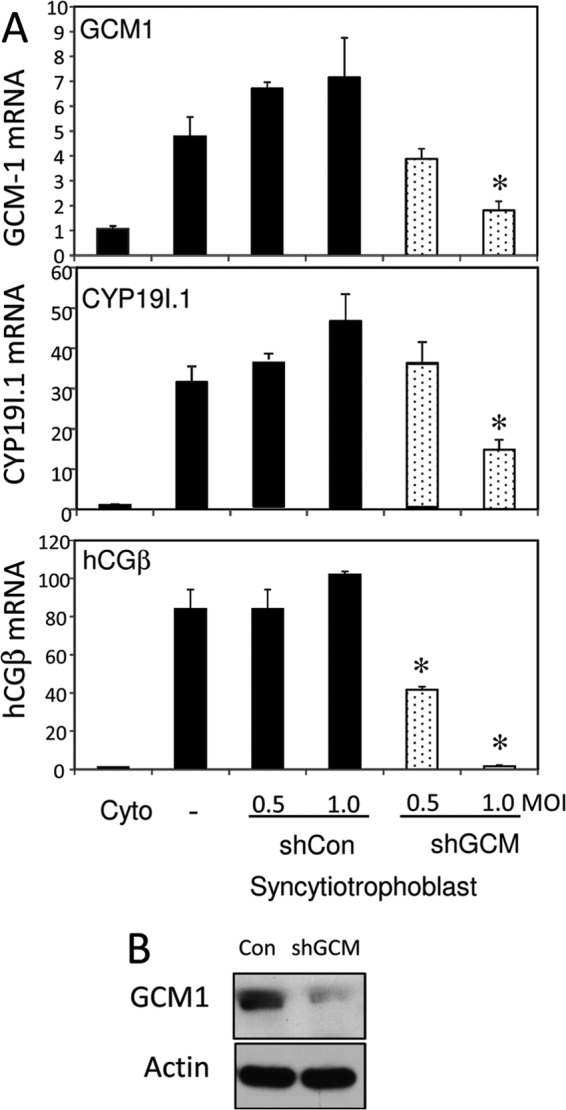
hGCM1 serves an important role in hCYP19 and hCGβ expression during differentiation of human trophoblasts in culture. (A) Freshly isolated human cytotrophoblasts were infected with lentiviral vectors carrying GCM1-targeting shRNA (shGCM) or control shRNA (shCon). Other dishes of cells were untreated (−). RNA was isolated 72 h later, and hCYP19I.1, GCM1, and hCGβ mRNA levels were analyzed by qRT-PCR; RPLP0 was used as the reference. (B) Nuclear proteins (20 μg) extracted from syncytiotrophoblast after 72 h of culture were analyzed for hGCM1 or β-actin by immunoblotting using specific antisera.
c-Myc upregulates miR-17∼92 and miR-106a∼363 expression in cultured human trophoblasts.
c-Myc belongs to a family of helix-loop-helix/leucine zipper transcription factors and together with its obligatory binding partner, Max, regulates cell proliferation, transformation, growth, differentiation and apoptosis (37). Increased c-Myc expression is evident in the proliferative cytotrophoblasts of human placenta (38). Interestingly, studies have shown that c-Myc directly binds and activates expression of the miR-17∼92 cluster in immortalized B lymphocytes (P493-6 cells) stably expressing a tetracycline-regulated c-Myc transgene (39). Thus, we explored a role for c-Myc in differentiation of human primary trophoblast cells in culture. During trophoblast differentiation, c-Myc mRNA (Fig. 5A) and protein expression (Fig. 5B) were markedly decreased. To determine whether c-Myc overexpression in cultured trophoblast cells altered expression of miR-17∼92 and miR-106a∼363 family members and whether this occurred at the transcriptional level, freshly isolated cytotrophoblasts were infected with recombinant adenoviruses expressing c-Myc or β-Gal, as control, and cultured for 24, 48, or 72 h. Effects of c-Myc overexpression on levels of mature miRNAs in the miR-17∼92 and miR-106a∼363 clusters was analyzed, as was expression of pri-miR-17∼92. Expression of hCYP19I.1, hCGβ, and c-Myc mRNA levels was also analyzed by qRT-PCR. As can be seen in Fig. 5C, c-Myc overexpression significantly increased mature miR-17, -19, -20a, and -106a expression after 48 h and 72 h of culture. Notably, increased expression of pri-miR-17∼92 was evident within 24 h of culture in cells overexpressing c-Myc (Fig. 5D), indicating that c-Myc transcriptionally activates expression of the miR-17∼92 cluster. Importantly, this was associated with a marked inhibition of hCYP19I.1 and hCGβ expression (Fig. 5E), suggesting that increased c-Myc expression in cytotrophoblasts upregulates miR-17∼92 and miR-106a∼363 cluster expression, which in turn blocks the induction of hCYP19I.1 and hCGβ. Since hCYP19I.1 mRNA levels also were inhibited within 24 h of culture in cells overexpressing c-Myc, it remains possible that c-Myc also directly represses hCYP19A1 gene expression. However, when the cMyc-transduced cells were cotransfected with the miR-19b inhibitor, aromatase and GCM1 protein expression was greater than that observed with c-Myc alone (Fig. 5F). This suggests that c-Myc repression of GCM1 and aromatase expression in the cultured trophoblasts is mediated in part via induction of the miR-17-92 cluster expression. When human trophoblasts were transfected with c-Myc small interfering RNA (siRNA) to knock down endogenous c-Myc expression, significant decreases in expression of mature miR-17, -19b, -20a, and -106a, as well as pri-miR-17-92, were observed after 72 h of culture (see Fig. S3 in the supplemental material).
Fig 5.

Overexpression of c-Myc in human trophoblast cells increases expression of the miR-17∼92 cluster and decreases expression of hCYP19I.1/aromatase and hCGβ. (A and B) RNA and protein isolated from freshly isolated cytotrophoblasts (Cyto) and after 24, 48, and 72 h of culture (syncytiotrophoblast) were analyzed for c-Myc mRNA expression by SYBR green qRT-PCR (A) or protein by immunoblot analysis (B). (C to E) Freshly isolated human cytotrophoblasts were infected with adenoviruses expressing c-Myc or β-Gal, as a control. Other dishes of cells were untreated (−). RNA was isolated 24, 48, and 72 h later; expression of mature miRNAs in the miR-17∼92 and miR-106a-363 clusters (C) or pri-miR-17∼92 transcripts (D) was analyzed by TaqMan-based qPCR; U6 RNA was used as a reference. hCYP19I.1, hCGβ, and c-Myc mRNA levels were analyzed by SYBR green qRT-PCR; RPLP0 was used as the reference (E). Aromatase and GCM1 protein expression was analyzed by immunoblot analysis in cells infected with c-Myc adenovirus and transfected with either LNA scrambled control or LNA miR19b inhibitor (19b Inh) (F).
To determine whether c-Myc increases miR-17∼92 and miR-106a∼363 expression by directly binding to the genomic regulatory regions, we performed ChIP assays with human trophoblasts before and after differentiation in culture. Chromatin was cross-linked in cells before culture (0 h), after 5 h (a time point at which we previously observed early chromatin modifications associated with differentiation) (15), or after 72 h of culture, when the cells have differentiated to form syncytiotrophoblast. After immunoprecipitation with anti-c-Myc antibody or control IgG, PCR was carried out using primers that amplify two genomic regions containing E boxes within intron I of MIR17HG, the gene coding for the miR-17∼92 cluster (Fig. 6A), that are conserved between human and mouse and that have been previously demonstrated to recruit c-Myc (39). We also identified putative c-Myc binding sites upstream of the miR-106a∼363 genomic cluster (Fig. 6C) and designed primers to amplify two of these regions. Using ChIP, we observed increased binding of endogenous c-Myc (relative to the IgG control) to E box 1 within the miR-17∼92 cluster genomic region (Fig. 6B) in freshly isolated cytotrophoblasts; relative binding activity markedly declined after 5 h of culture and was undetectable by 72 h. No specific c-Myc binding to E-box 2 was observed. Increased binding of endogenous c-Myc (relative to the IgG control) to the MIR106A1 region upstream of the miR-106a∼363 cluster in freshly isolated cytotrophoblasts also was observed (Fig. 6D); again, c-Myc binding declined rapidly and was undetectable after 5 h of culture. Binding of endogenous c-Myc to the MIR106A2 genomic region was undetectable at any time point. These findings provide strong evidence that these miRNAs are directly upregulated by c-Myc in cytotrophoblasts; as c-Myc expression decreases upon trophoblast differentiation, expression of the miR-17∼92 and miR-106a∼363 clusters declines, releasing repression of their target genes.
Fig 6.

Endogenous c-Myc binds to genomic regions of the miR-17∼92 and miR-106a∼363 clusters; binding declines with syncytiotrophoblast differentiation. (A and C) Schematic representations of genomic regulatory regions of the miR-17∼92 (A) or miR-106a∼363 (C) clusters containing putative c-Myc binding sites. (B and D) Human cytotrophoblasts before (0 h) and after culture for 5 h or 72 h were treated with 1% formaldehyde and subjected to ChIP analysis using c-Myc IgG or nonimmune IgG, as a control. The immunoprecipitated complexes were quantified by PCR using specific primers designed to amplify putative c-Myc response elements, as indicated by the arrows in the diagrams in panels A and C. The ChIP data were normalized to input data and expressed relative to nonspecific IgG. Error bars represent standard errors from three independent experiments, each conducted in triplicate. ∗, significantly different (P < 0.05) from results for IgG.
Overexpression of miR-106a and miR-19b in cytotrophoblasts prevents trophoblast differentiation.
Since overexpression of miR-19b and miR-106a in trophoblasts inhibited expression of their direct targets, hGCM1 and hCYP19, as well as hCGβ, a critical marker of trophoblast differentiation, we next examined the morphology of the human trophoblasts in which these miRNAs were overexpressed. Freshly isolated human cytotrophoblasts were transfected with nontargeting control (NTC) or with mimics of miR-19b or -106a. After 72 h of culture, the cells were stained with hematoxylin and eosin and viewed by light microscopy. When the cells were transfected with NTC miRNA, there was clear syncytium formation; however, when cells were transfected with either miR-106a or miR-19b, syncytium formation was markedly reduced (Fig. 7A). The Image J program was used to quantify the percentage of syncytiotrophoblasts compared to the total number of cells; statistical analysis indicated that miR-106a and miR-19b transfection significantly decreased syncytium formation (Fig. 7B). c-Myc overexpression decreased syncytium formation, while in the presence of miR19b inhibitors, this inhibition was prevented (see Fig. S4 in the supplemental material). This effect of miR-19b inhibitor may be due to increased expression of GCM1 in the cultured cells (Fig. 3D).
Fig 7.

Overexpression of miR-106a and miR-19b in cytotrophoblasts prevents trophoblast differentiation. Nontargeting control (NTC) or miRNA mimics of miR-19b or -106a were transfected into freshly isolated placental trophoblasts. (A) After 72 h of culture, the cells were stained with hematoxylin and eosin and viewed by light microscopy to assess syncytiotrophoblast formation. (B) Image J was used to quantify the percentage of syncytiotrophoblasts relative to total cell number in 10 different fields from two independent experiments performed in duplicate. ∗, significantly (P < 0.05) decreased compared to results for cells transfected with NTC.
Expression of miR-106a and miR-19b and of the hCYP19/aromatase protein in placentas from preeclamptic and gestation-matched normotensive women.
In consideration of the potential roles of the miR-17∼92 and miR-106∼363 clusters in human trophoblast differentiation and the impairment of trophoblast differentiation in preeclampsia, we next investigated expression of members of these miRNA clusters and their targets, hCYP19A1 and hGCM1, in placentas of preeclamptic and normotensive women near term. RNA and protein from term placentas of 8 preeclamptic and 8 gestation-matched normotensive women were analyzed for miR-106a and miR-19b expression using TaqMan-based qPCR; aromatase/CYP19A1 and hGCM1 protein expression were analyzed by immunoblotting. We observed significantly higher levels of miR-106a and -19b expression in placentas from preeclamptic women than in placentas from gestation-matched normotensive women (Fig. 8A). Correspondingly, aromatase and hGCM1 protein expression in protein extracts of preeclamptic placentas compared to that in normotensive placentas was significantly decreased (Fig. 8B and C). However, expression of CYP19A1 and hGCM1 mRNA was not significantly altered (see Fig. S5 in the supplemental material). These findings suggest that aberrant expression of members of the miR-17∼92 and miR-106a∼363 clusters may contribute to the pathogenesis of preeclampsia.
Fig 8.

Differential expression of miRNAs, hCYP19, and hGCM1 in placentas from preeclamptic and gestation-matched normotensive women. RNA and protein were isolated from placentas of 8 preeclamptic and 8 term gestation-matched normotensive women and measured for miRNA expression by TaqMan-based PCR (A) or for hCYP19A1/aromatase and hGCM1 protein expression by immunoblotting (B and C, left panels). These immunoblot results were quantitated using Image J and corrected for loading and transfer using GAPDH (B, right panel) or actin (C, right panel). ∗, significantly (P < 0.05) different from results for the normotensive group.
DISCUSSION
In this study, we have analyzed the miRNA profile of cytotrophoblasts versus that of syncytiotrophoblast and demonstrated that members of the miR-17∼92 cluster and its paralogs, the miR-106a∼363 and miR-106b∼25 clusters, are expressed at relatively high levels in rapidly proliferating cytotrophoblasts and that their expression declines in association with syncytiotrophoblast differentiation (Fig. 1). The miR-17∼92 cluster, also known as oncomir 1, is among the most potent of oncogenic miRNAs (28, 40, 41). It is overexpressed in a variety of human cancers, including lung carcinomas, B-cell lymphomas, and retinoblastomas (42–44). Overexpression is frequently due to amplification of its locus (44). These miRNAs are known to contribute to tumorigenesis by targeting the cyclin-dependent kinase inhibitor CDKN1A (p21), a potent negative regulator of the G1-S checkpoint; thus, increased expression of these miRNAs enhances cell cycle progression (45). miR-19 also targets PTEN, a critical negative regulator of the highly oncogenic prosurvival P13K/Akt signaling pathway (46). Thus, it is likely that the relatively high levels of expression of all three miRNA clusters observed in human cytotrophoblasts promotes their proliferation and prevents syncytiotrophoblast differentiation. Notably, a subset of cytotrophoblasts in placental villi have been equated to tumor cells because they rapidly proliferate, invade maternal blood vessels, and form aggregates that attach the fetus to the maternal uterine wall (47). Thus, our findings further support the concept that cytotrophoblasts share properties with tumor cells in their similarly high expression levels of these miRNAs.
Despite these well-studied actions of the miR-17∼92 cluster in tumorigenesis, a role for miRNAs in this cluster during normal development also has been established. Loss of function of the miR-17∼92 cluster in mice resulted in smaller embryos and postnatal death associated with lung hypoplasia, cardiac defects, a block in B-cell development, and abnormal skeletal mineralization and patterning (48). While mice lacking the miR-106a∼363 cluster do not manifest any phenotypic abnormalities, embryos that are doubly deficient in both the miR-17∼92 and miR-106a∼363 clusters die prior to embryonic day 15.5 (E15.5), suggesting that these miRNAs functionally cooperate (48). The miR-17∼92 cluster is highly expressed in embryonic lung, and expression decreases toward term. Moreover, overexpression of these miRNAs in lung epithelium of transgenic mice resulted in severe developmental defects with enhanced cell proliferation and inhibition of epithelial cell differentiation (49). The miR-17∼92 cluster also was found to be downregulated in myeloid progenitors undergoing macrophage differentiation (50), and studies of developing mouse embryos suggest that the miR-17∼92 cluster inhibits stem cell differentiation (51).
In the present study, we have clearly established that members of the miR-17∼92 and miR-106a∼363 clusters are highly expressed in cytotrophoblasts and that expression declines upon terminal differentiation to syncytiotrophoblast. Members of these miR clusters were found to target hCYP19A1/aromatase (Fig. 2) and hGCM1 (Fig. 3) directly, and their overexpression in cytotrophoblasts prevented syncytiotrophoblast differentiation (Fig. 7). Our novel findings indicate that miR-106a and miR-19b can both directly target hCYP19A1, while miR-19b directly targets hGCM1. Since miR-17, -20a, -20b, -106a, -106b, and -93 all share the same seed sequence, whereas miR-19a and miR-19b share a different seed sequence, they all can be expected to target hCYP19A1 and may cooperatively promote hCYP19A1 gene repression. Moreover, we confirmed that miR-17 also exerts an effect similar to that of miR-106a to target aromatase protein expression and inhibit trophoblast differentiation (see Fig. S2 in the supplemental material).
GCM1 functions as an important transcription factor during formation of the mouse labyrinth; fusion of chorion trophoblast cells to form syncytiotrophoblast was blocked when GCM1 was ablated (17). Our results indicate that knockdown of hGCM1 in cultured human trophoblasts inhibits the induction of hCYP19I.1 and hCGβ (Fig. 4), known markers of trophoblast differentiation. Moreover, miRNA overexpression mimicked the effects of hGCM1 knockdown by inhibiting trophoblast differentiation (Fig. 3). Notably, miR-18a and miR-19a also target and repress expression of estrogen receptor α (ERα) and were found to inhibit neuroblast differentiation (52). In previous studies, we observed that ERα is expressed in human trophoblasts and was upregulated during differentiation in culture (15). Furthermore, estrogens acting via ERα upregulated hCYP19A1 expression and contributed to trophoblast differentiation in a positive feed-forward manner (15). Thus, the miR-17∼92 and miR-106a∼363 clusters may also inhibit trophoblast differentiation by inhibiting ERα expression in cytotrophoblasts. Another studied target of the miR-17/20/106 family is steroid receptor coactivator 3 (SRC-3), which serves as a coactivator for several nuclear receptors and other transcription factors, including ERα (35). Although we have yet to establish a functional role for SRC-3 in human trophoblasts, we observed that SRC-3 mRNA levels were upregulated in association with syncytiotrophoblast differentiation and the decline in miR-17∼92 expression (see Fig. S6 in the supplemental material). Moreover, SRC-2 and SRC-3 have been reported to act cooperatively to promote mouse labyrinthine trophoblast development (53). Consequently, multiple miRNAs of the miR-17∼92 and miR-106a∼363 clusters may converge to inhibit trophoblast differentiation by targeting several key mRNA targets.
The c-myc proto-oncogene encodes a basic helix-loop-helix leucine zipper transcription factor that modulates diverse cellular processes, including proliferation, growth, apoptosis, and differentiation (37). In human placenta, c-Myc was observed to peak at 4 to 5 weeks of gestation and to be highly expressed in the proliferating cytotrophoblasts (38). In the present study, we detected relatively high levels of c-Myc mRNA and protein expression in freshly isolated cytotrophoblasts, which declined upon syncytiotrophoblast differentiation in culture (Fig. 5A and B). Elevated expression of the miR-17∼92 and miR-106a∼363 clusters in cytotrophoblasts was associated with increased binding of endogenous c-Myc to a previously identified conserved region in the first intron of the miR-17∼92 cluster (39) (Fig. 6B) and to a genomic region upstream of the miR-106a∼363 cluster identified in the present study (Fig. 6C). Endogenous c-Myc binding to these genomic regions declined rapidly within 5 h of culture. While elevated c-myc mRNA expression was still evident at 24 h (Fig. 5A), a pronounced reduction in c-Myc protein levels was evident at this time point (Fig. 5B). Several posttranslational modifications, including phosphorylation, acetylation, and ubiquitylation of c-Myc, are known to control its transcriptional activity (54); one or more of these modifications may be responsible for the rapid decline in DNA binding activity to the miR-17∼92 and miR-106a∼363 clusters. Notably, targeted deletion of the c-myc gene in mice resulted in apparent placental defects by E10.5, with markedly decreased cell proliferation and inhibition of branching morphogenesis (55).
c-Myc has been shown to directly activate transcription of the miR-17∼92 cluster (39) and thereby promote cell proliferation, survival, angiogenesis, and metabolic reprogramming in a number of tumor cell lines (56). In cultured human trophoblasts, we found that c-Myc overexpression upregulated miR-17∼92 and miR-106a∼363 transcription (Fig. 5C). Thus, our findings support the important role of c-Myc in the increased transcription of the miR-17∼92 and miR-106a∼363 clusters in proliferating cytotrophoblasts, thereby suppressing translation of CYP19A1 and hGCM1 and inhibiting trophoblast differentiation (Fig. 9).
Fig 9.

Model of miRNA regulation in human trophoblasts. Early in human gestation, c-Myc, whose expression is elevated in proliferative mononuclear cytotrophoblasts (Cyto), binds to the genomic regulatory regions of the miR-17∼92 and miR-106a∼363 clusters to increase their expression. These miRs inhibit expression of hCYP19 and hGCM1 and prevent trophoblast differentiation. With increased placental vascularization and O2 availability, differentiation of cytotrophoblasts to multinucleated syncytiotrophoblast (Syn) is enhanced. During this process, a decline in c-Myc expression and consequent decrease in levels of these miRNAs allow upregulation of hCYP19 and hGCM1 expression and further advance human trophoblast differentiation.
Preeclampsia, characterized by the onset of hypertension and proteinuria after 20 weeks of gestation, affects 6 to 8% of all pregnancies in the United States and is a leading cause of maternal and neonatal morbidity and mortality (57–59). In preeclampsia, the placenta is relatively hypoxic and cytotrophoblast differentiation into invasive cells is inhibited. Thus, fewer arterioles are invaded, and O2 availability to the hormone-producing syncytiotrophoblast is reduced (60). Recently it was observed that miR-17, -20a, and -20b, encoded within the miR-17∼92 cluster, were significantly increased in placentas of preeclamptic women and that these miRNAs directly target Eph receptor B4 (EPHB4) and ephrin B2 (61), which are critical for vascular patterning and trophoblast invasion during placentation (62). In another study, miR-18a and -19a were among 22 miRNAs significantly upregulated in preeclamptic placentas (63). Importantly, in the present study, we observed that miR-106a and miR-19b were expressed at higher levels in placentas of preeclamptic women than in placentas of gestation-matched normotensive women (Fig. 8A). Elevated expression of these miRNAs may reflect upregulated expression of the miR-17∼92 and miR-106a∼363 clusters and/or a relative increase in the proportion of undifferentiated cytotrophoblasts in preeclamptic placentas. Since our primary cell culture studies suggest that increased miR-17∼92/106a∼363 expression prevents syncytiotrophoblast differentiation, it may be difficult to clearly establish a cause-effect relationship.
While the molecular basis for placental dysregulation in preeclampsia remains unclear, our findings indicate that increased miR-106a and -19b expression in preeclamptic placentas was associated with decreased expression of aromatase (Fig. 8B). Importantly, placental aromatase deficiency has been found in women with preeclampsia (64), and a hCYP19A1 gene polymorphism associated with decreased aromatase levels has been reported to occur more commonly in preeclamptic patients (65). Notably, hGCM1, which we also found to be a target of miR-19b, was found to be markedly decreased in placentas of women with preeclampsia (66). We also observed decreased GCM1 protein levels in placentas from preeclamptic women (Fig. 8C). Moreover, reduced GCM1 expression in mouse placenta was associated with defective syncytiotrophoblast differentiation and pregnancy outcomes resembling preeclampsia in women (67). In light of our important findings and to further understand the significance of increased miRNA expression in preeclampsia, our future work will include analysis of clinical endpoints of preeclampsia in a transgenic mouse model of miR-17∼92 overexpression.
In summary, the present findings provide evidence for a critical inhibitory role of a c-Myc/miR-17∼92/-106a∼363 signaling pathway in the regulation of syncytiotrophoblast differentiation and the expression of hGCM1 and hCYP19A1 in human placenta (Fig. 9). Early in the first trimester, when the placenta is poorly vascularized and relatively hypoxic, c-Myc expression is elevated, resulting in increased transcription of the miR-17∼92 and miR-106a∼363 clusters. We suggest that elevated expression of c-Myc and these miRNA clusters in cytotrophoblasts promotes “stemness,” exemplified by increased expression levels of the stem cell factor, achaete-scute homologous protein 2 (ASCL2/Mash-2) (3). Increased expression of these miRNA clusters may promote increased cytotrophoblast proliferation via targeting of p21 (45) and suppression of hGCM1 and hCYP19A1 to block syncytiotrophoblast differentiation. After 9 to 10 weeks of gestation, increased cytotrophoblast invasion of the spiral arteries results in increased placental perfusion and enhanced oxygen tension. This is associated with suppression of ASCL2 and c-Myc expression and decreased expression of the miR-17∼92/106a∼363 clusters, which permits upregulation of hGCM1 to promote syncytiotrophoblast differentiation and increased hCYP19A1 expression. We suggest that aberrant regulation of this novel signaling pathway interferes with normal induction of trophoblast differentiation that may contribute to the pathogenesis of preeclampsia.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the expert assistance of Jo Francis Smith in isolation and culture of human placental cells.
This work was supported by NIH grant R01 DK031206.
Footnotes
Published ahead of print 25 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01228-12.
REFERENCES
- 1. Everett RB, MacDonald PC. 1979. Endocrinology of the placenta. Annu. Rev. Med. 30: 473–488 [DOI] [PubMed] [Google Scholar]
- 2. Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. 1996. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J. Clin. Invest. 97: 540–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang B, Kamat A, Mendelson CR. 2000. Hypoxia prevents induction of aromatase expression in human trophoblast cells in culture: potential inhibitory role of the hypoxia-inducible transcription factor Mash-2 (mammalian achaete-scute homologous protein-2). Mol. Endocrinol. 14: 1661–1673 [DOI] [PubMed] [Google Scholar]
- 4. Fournet-Dulguerov N, MacLusky NJ, Leranth CZ, Todd R, Mendelson CR, Simpson ER, Naftolin F. 1987. Immunohistochemical localization of aromatase cytochrome P-450 and estradiol dehydrogenase in the syncytiotrophoblast of the human placenta. J. Clin. Endocrinol. Metab. 65: 757–764 [DOI] [PubMed] [Google Scholar]
- 5. Kamat A, Alcorn JL, Kunczt C, Mendelson CR. 1998. Characterization of the regulatory regions of the human aromatase (P450arom) gene involved in placenta-specific expression. Mol. Endocrinol. 12: 1764–1777 [DOI] [PubMed] [Google Scholar]
- 6. Rosenfeld CR, Morriss FH, Jr, Battaglia FC, Makowski EL, Meschia G. 1976. Effect of estradiol-17β on blood flow to reproductive and nonreproductive tissues in pregnant ewes. Am. J. Obstet. Gynecol. 124: 618–629 [DOI] [PubMed] [Google Scholar]
- 7. Jobe SO, Ramadoss J, Koch JM, Jiang Y, Zheng J, Magness RR. 2010. Estradiol-17β and its cytochrome P450- and catechol-O-methyltransferase-derived metabolites stimulate proliferation in uterine artery endothelial cells: role of estrogen receptor-α versus estrogen receptor-β. Hypertension 55: 1005–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, Strauss JF, Kalluri R. 2008. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 453: 1117–1121 [DOI] [PubMed] [Google Scholar]
- 9. Lee SB, Wong AP, Kanasaki K, Xu Y, Shenoy VK, McElrath TF, Whitesides GM, Kalluri R. 2010. Preeclampsia: 2-methoxyestradiol induces cytotrophoblast invasion and vascular development specifically under hypoxic conditions. Am. J. Pathol. 176: 710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Albrecht ED, Pepe GJ. 2010. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int. J. Dev. Biol. 54: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang B, Mendelson CR. 2003. USF1 and USF2 mediate inhibition of human trophoblast differentiation and CYP19 gene expression by Mash-2 and hypoxia. Mol. Cell. Biol. 23: 6117–6128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang B, Mendelson CR. 2005. O2 enhancement of human trophoblast differentiation and hCYP19 (aromatase) gene expression are mediated by proteasomal degradation of USF1 and USF2. Mol. Cell. Biol. 25: 8824–8833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petraglia F, Florio P, Nappi C, Genazzani AR. 1996. Peptide signaling in human placenta and membranes: autocrine, paracrine, and endocrine mechanisms. Endocr. Rev. 17: 156–186 [DOI] [PubMed] [Google Scholar]
- 14. Cronier L, Guibourdenche J, Niger C, Malassine A. 1999. Oestradiol stimulates morphological and functional differentiation of human villous cytotrophoblast. Placenta 20: 669–676 [DOI] [PubMed] [Google Scholar]
- 15. Kumar P, Kamat A, Mendelson CR. 2009. Estrogen receptor α (ERα) mediates stimulatory effects of estrogen on aromatase (CYP19) gene expression in human placenta. Mol. Endocrinol. 23: 784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar P, Mendelson CR. 2011. Estrogen-related receptor γ (ERRγ) mediates oxygen-dependent induction of aromatase (CYP19) gene expression during human trophoblast differentiation. Mol. Endocrinol. 25: 1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anson-Cartwright L, Dawson K, Holmyard D, Fisher SJ, Lazzarini RA, Cross JC. 2000. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 25: 311–314 [DOI] [PubMed] [Google Scholar]
- 18. Schreiber J, Riethmacher-Sonnenberg E, Riethmacher D, Tuerk EE, Enderich J, Bosl MR, Wegner M. 2000. Placental failure in mice lacking the mammalian homolog of glial cells missing, GCMa. Mol. Cell. Biol. 20: 2466–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu C, Shen K, Lin M, Chen P, Lin C, Chang GD, Chen H. 2002. GCMa regulates the syncytin-mediated trophoblastic fusion. J. Biol. Chem. 277: 50062–50068 [DOI] [PubMed] [Google Scholar]
- 20. Chang M, Mukherjea D, Gobble RM, Groesch KA, Torry RJ, Torry DS. 2008. Glial cell missing 1 regulates placental growth factor (PGF) gene transcription in human trophoblast. Biol. Reprod. 78: 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamada K, Ogawa H, Honda S, Harada N, Okazaki T. 1999. A GCM motif protein is involved in placenta-specific expression of human aromatase gene. J. Biol. Chem. 274: 32279–32286 [DOI] [PubMed] [Google Scholar]
- 22. Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- 23. Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Rooij E, Liu N, Olson EN. 2008. MicroRNAs flex their muscles. Trends Genet. 24: 159–166 [DOI] [PubMed] [Google Scholar]
- 25. Turner ML, Schnorfeil FM, Brocker T. 2011. MicroRNAs regulate dendritic cell differentiation and function. J. Immunol. 187: 3911–3917 [DOI] [PubMed] [Google Scholar]
- 26. Braun T, Gautel M. 2011. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 12: 349–361 [DOI] [PubMed] [Google Scholar]
- 27. Garzon R, Calin GA, Croce CM. 2009. MicroRNAs in cancer. Annu. Rev. Med. 60: 167–179 [DOI] [PubMed] [Google Scholar]
- 28. Mendell JT. 2008. miRiad roles for the miR-17-92 cluster in development and disease. Cell 133: 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hawkins SM, Buchold GM, Matzuk MM. 2011. Minireview: the roles of small RNA pathways in reproductive medicine. Mol. Endocrinol. 25: 1257–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Renthal NE, Chen CC, Williams KC, Gerard RD, Prange-Kiel J, Mendelson CR. 2010. miR-200 family and targets, ZEB1 and ZEB2, modulate uterine quiescence and contractility during pregnancy and labor. Proc. Natl. Acad. Sci. U. S. A. 107: 20828–20833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams KC, Renthal NE, Condon JC, Gerard RD, Mendelson CR. 2012. MicroRNA-200a serves a key role in the decline of progesterone receptor function leading to term and preterm labor. Proc. Natl. Acad. Sci. U. S. A. 109: 7529–7534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang B, Mendelson CR. 2006. Adenoviral-mediated gene delivery to trophoblast cells. Methods Mol. Med. 121: 451–461 [DOI] [PubMed] [Google Scholar]
- 33. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254 [DOI] [PubMed] [Google Scholar]
- 34. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- 35. Hossain A, Kuo MT, Saunders GF. 2006. MiR-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol. Cell. Biol. 26: 8191–8201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castellano L, Giamas G, Jacob J, Coombes RC, Lucchesi W, Thiruchelvam P, Barton G, Jiao LR, Wait R, Waxman J, Hannon GJ, Stebbing J. 2009. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. U. S. A. 106: 15732–15737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eilers M, Eisenman RN. 2008. Myc's broad reach. Genes Dev. 22: 2755–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rydnert J, Pfeifer-Ohlsson S, Goustin AS, Ohlsson R. 1987. Temporal and spatial pattern of cellular myc oncogene expression during human placental development. Placenta 8: 339–345 [DOI] [PubMed] [Google Scholar]
- 39. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843 [DOI] [PubMed] [Google Scholar]
- 40. Olive V, Jiang I, He L. 2010. miR-17-92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 42: 1348–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Concepcion CP, Bonetti C, Ventura A. 2012. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. 18: 262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, MacPherson D. 2011. miR-17∼92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 25: 1734–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. 2005. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65: 9628–9632 [DOI] [PubMed] [Google Scholar]
- 44. Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. 2004. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 64: 3087–3095 [DOI] [PubMed] [Google Scholar]
- 45. Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA. 2008. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol. 28: 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D'Andrea A, Sander C, Ventura A. 2009. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 23: 2806–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou Y, Genbacev O, Fisher SJ. 2003. The human placenta remodels the uterus by using a combination of molecules that govern vasculogenesis or leukocyte extravasation. Ann. N. Y. Acad. Sci. 995: 73–83 [DOI] [PubMed] [Google Scholar]
- 48. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. 2008. Targeted deletion reveals essential and overlapping functions of the miR-17∼92 family of miRNA clusters. Cell 132: 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. 2007. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev. Biol. 310: 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A, Necas E, Zavadil J, Laslo P, Stopka T. 2011. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J. 30: 4450–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Foshay KM, Gallicano GI. 2009. miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 326: 431–443 [DOI] [PubMed] [Google Scholar]
- 52. Loven J, Zinin N, Wahlstrom T, Muller I, Brodin P, Fredlund E, Ribacke U, Pivarcsi A, Pahlman S, Henriksson M. 2010. MYCN-regulated microRNAs repress estrogen receptor-α (ESR1) expression and neuronal differentiation in human neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 107: 1553–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen X, Liu Z, Xu J. 2010. The cooperative function of nuclear receptor coactivator 1 (NCOA1) and NCOA3 in placental development and embryo survival. Mol. Endocrinol. 24: 1917–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vervoorts J, Luscher-Firzlaff J, Luscher B. 2006. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 281: 34725–34729 [DOI] [PubMed] [Google Scholar]
- 55. Dubois NC, Adolphe C, Ehninger A, Wang RA, Robertson EJ, Trumpp A. 2008. Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 135: 2455–2465 [DOI] [PubMed] [Google Scholar]
- 56. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. 2006. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 38: 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Redman CW, Sargent IL. 2005. Latest advances in understanding preeclampsia. Science 308: 1592–1594 [DOI] [PubMed] [Google Scholar]
- 58. Ness RB, Sibai BM. 2006. Shared and disparate components of the pathophysiologies of fetal growth restriction and preeclampsia. Am. J. Obstet. Gynecol. 195: 40–49 [DOI] [PubMed] [Google Scholar]
- 59. Pineles BL, Romero R, Montenegro D, Tarca AL, Han YM, Kim YM, Draghici S, Espinoza J, Kusanovic JP, Mittal P, Hassan SS, Kim CJ. 2007. Distinct subsets of microRNAs are expressed differentially in the human placentas of patients with preeclampsia. Am. J. Obstet. Gynecol. 196: 261–266 [DOI] [PubMed] [Google Scholar]
- 60. Lunell NO, Nylund LE, Lewander R, Sarby B. 1982. Uteroplacental blood flow in pre-eclampsia measurements with indium-113m and a computer-linked gamma camera. Clin. Exp. Hypertens. B 1: 105–117 [DOI] [PubMed] [Google Scholar]
- 61. Wang W, Feng L, Zhang H, Hachy S, Satohisa S, Laurent LC, Parast M, Zheng J, Chen DB. 2012. Preeclampsia up-regulates angiogenesis-associated microRNA (i.e., miR-17, -20a, and -20b) that target ephrin-B2 and EPHB4 in human placenta. J. Clin. Endocrinol. Metab. 97: E1051–E1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Red-Horse K, Kapidzic M, Zhou Y, Feng KT, Singh H, Fisher SJ. 2005. EPHB4 regulates chemokine-evoked trophoblast responses: a mechanism for incorporating the human placenta into the maternal circulation. Development 132: 4097–4106 [DOI] [PubMed] [Google Scholar]
- 63. Ishibashi O, Ohkuchi A, Ali MM, Kurashina R, Luo SS, Ishikawa T, Takizawa T, Hirashima C, Takahashi K, Migita M, Ishikawa G, Yoneyama K, Asakura H, Izumi A, Matsubara S, Takeshita T, Takizawa T. 2012. Hydroxysteroid (17-β) dehydrogenase 1 is dysregulated by miR-210 and miR-518c that are aberrantly expressed in preeclamptic placentas: a novel marker for predicting preeclampsia. Hypertension 59: 265–273 [DOI] [PubMed] [Google Scholar]
- 64. Hertig A, Liere P, Chabbert-Buffet N, Fort J, Pianos A, Eychenne B, Cambourg A, Schumacher M, Berkane N, Lefevre G, Uzan S, Rondeau E, Rozenberg P, Rafestin-Oblin ME. 2010. Steroid profiling in preeclamptic women: evidence for aromatase deficiency. Am. J. Obstet. Gynecol. 203: 477–479 [DOI] [PubMed] [Google Scholar]
- 65. Shimodaira M, Nakayama T, Sato I, Sato N, Izawa N, Mizutani Y, Furuya K, Yamamoto T. 2012. Estrogen synthesis genes CYP19A1, HSD3B1, and HSD3B2 in hypertensive disorders of pregnancy. Endocrine 42: 700–707 [DOI] [PubMed] [Google Scholar]
- 66. Chen CP, Chen CY, Yang YC, Su TH, Chen H. 2004. Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta 25: 413–421 [DOI] [PubMed] [Google Scholar]
- 67. Bainbridge SA, Minhas A, Whiteley KJ, Qu D, Sled JG, Kingdom JC, Adamson SL. 2012. Effects of reduced Gcm1 expression on trophoblast morphology, fetoplacental vascularity, and pregnancy outcomes in mice. Hypertension 59: 732–739 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.