

Abstract
Epstein-Barr virus (EBV), a human oncogenic herpesvirus that establishes a lifelong latent infection in the host, occasionally enters lytic infection to produce progeny viruses. The EBV oncogene latent membrane protein 1 (LMP1), which is expressed in both latent and lytic infection, constitutively activates the canonical NF-κB (p65) pathway. Such LMP1-mediated NF-κB activation is necessary for proliferation of latently infected cells and inhibition of viral lytic cycle progression. Actually, canonical NF-κB target gene expression was suppressed upon the onset of lytic infection. TRAF6, which is activated by conjugation of polyubiquitin chains, associates with LMP1 to mediate NF-κB signal transduction. We have found that EBV-encoded BPLF1 interacts with and deubiquitinates TRAF6 to inhibit NF-κB signaling during lytic infection. HEK293 cells with BPLF1-deficient recombinant EBV exhibited poor viral DNA replication compared with the wild type. Furthermore, exogenous expression of BPLF1 or p65 knockdown in cells restored DNA replication of BPLF1-deficient viruses, indicating that EBV BPLF1 deubiquitinates TRAF6 to inhibit NF-κB signal transduction, leading to promotion of viral lytic DNA replication.
INTRODUCTION
Epstein-Barr virus (EBV), a human lymphotropic gammaherpesvirus with a linear double-stranded DNA, 172 kb in length (1), infects resting B lymphocytes, inducing their continuous proliferation without production of virus particles, this being termed latent infection. In the latent phase, a limited number of viral genes are expressed, and the expression pattern of viral latent genes varies depending on the tissue origin and the state of the cells/tumors. Productive (lytic) infection, which occurs spontaneously or can be induced artificially, is triggered by BZLF1 immediate-early protein and characterized by the expression of a number of lytic genes, leading to virus production. The EBV genome is thereby amplified several-hundred-fold by viral replication machinery.
In lymphocytes that are latently infected with EBV, latent membrane protein 1 (LMP1) is expressed to promote survival and proliferation of the cells. LMP1 is uniformly expressed in latency III EBV infection of human B lymphoblastoid cell lines (LCLs), and also in latent II EBV infection in Hodgkin's disease B lymphocytes and in nasopharyngeal carcinoma (NPC) epithelial cells (2). It is a transmembrane protein consisting of a short cytoplasmic N-terminal domain, six transmembrane domains, and a long cytoplasmic C-terminal domain (3, 4). Two subdomains within the C-terminal domain, C-terminal activating region 1 (CTAR1) and CTAR2, associate with tumor necrosis factor receptor-associated factors (TRAFs) which are critical for LMP1 signaling (3, 5, 6). LMP1 is a functional mimic of the tumor necrosis factor receptor superfamily member CD40, an activating receptor constitutively expressed on B cells, macrophages, and dendritic cells (7, 8). As a result, LMP1 causes constitutive activation of cellular signaling, with upregulation of factors such as NF-κB, mitogen-activated protein kinase (MAPK), JAK/STAT, and Akt (9–13). Of several transcriptional activators targeted by LMP1, NF-κB is most important for LMP1-stimulated gene expression (14–18).
The canonical NF-κB, consisting of p65/RelA and p50, plays an important role in regulation of a variety of genes involved in host immune responses and in different features of carcinogenesis, including proliferation, enhanced survival, inflammation, and angiogenesis (19). NF-κB is usually under tight regulation, being kept inactive in the cytoplasm by certain mechanisms, including binding of inhibitors of kappa B (IκBs). A series of NF-κB-activating stimuli converge on the activation of IκB kinase (IKK) complexes composed of a IKKγ regulatory subunit or the NF-κB essential modulator (NEMO), and two kinases, IKKα and IKKβ. The IKK complexes phosphorylate and promote proteasomal degradation of IκB, resulting in release of NF-κB from the inhibitor complex. It was recently demonstrated that TRAF6 associates with the CTAR1 subdomain of LMP1 and is critical for LMP1-mediated activation of NF-κB signaling (5, 20). TRAF6 activates IKK in a K63-ubiquitin (Ub) chain-dependent manner. Ub chains conjugated to signaling molecules during activation of the NF-κB pathway can be inactivated by cellular deubiquitination enzymes (DUBs) such as A20, CYLD, and DUBA (21–23), suggesting that ubiquitin modification enzymes and DUBs play critical roles in the NF-κB response, leading to modulation of immune responses.
High levels of NF-κB protect the cell from the cytopathic effects by viral protein synthesis and promote the establishment of a latent infection. In contrast, EBV lytic reactivation requires downregulation of NF-κB because basal or LMP1-stimulated NF-κB activity suppresses the expression and function of lytic transactivator BZLF1 (also known as ZEBRA and EB1), resulting in inhibition of lytic cycle induction (24, 25). However, LMP1 is paradoxically expressed during the lytic cycle in EBV-positive B cells (26).
EBV-encoded BPLF1 protein is a lytic gene product with DUB activity. Whitehurst et al. (2009) showed that its N-terminal fragment deubiquitinates viral ribonucleotide reductase (RR), resulting in downregulation of viral RR activity (27). Also, Gastaldello et al. (2010) showed that a 325-amino-acid (aa)-length N-terminal fragment of BPLF1 cleaves ubiquitin and NEDD8 conjugates and promotes EBV replication (28). More recently, Whitehurst et al. reported that BPLF1 deubiquitinates the cellular DNA polymerase processivity factor PCNA and attenuates Polη to DNA damage sites (29). In this study, we demonstrated that BPLF1 interacts, directly or indirectly, with and deubiquitinates TRAF6 to block cellular NF-κB signal responses during lytic replication. Cells harboring BPLF1-deficient EBV exhibited poor viral lytic DNA replication, and exogenous expression of BPLF1 restored it. Thus, DUB activity of BPLF1 is required for efficient viral genome replication.
MATERIALS AND METHODS
Cells.
AGS cells transduced with CR2/CD21, the receptor for the EBV expression vector, and infected with enhanced green fluorescent protein (EGFP)-EBV (30) (AGS-EBV cells) were established previously (31) and maintained at 37°C in RPMI 1640 supplemented with 10% fetal calf serum and 150 μg/ml hygromycin B. B95-8 and Namalwa cells were maintained at 37°C in RPMI 1640 supplemented with 10% fetal calf serum. HEK293 cells and derivatives were grown and maintained at 37°C in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal calf serum. HEK293 cells latently infected with recombinant EBV-bacmid (293-EBV) were maintained as previously reported (32).
BAC mutagenesis and transfection.
Wild-type (WT) EBV-bacmid (EBV-WT) has been described previously (33). The region between nucleotides (nt) 1 and 975 of the BPLF1 open reading frame (ORF) was replaced with tandemly arranged neomycin resistance and streptomycin sensitivity (NeoSt+) genes using homologous recombination to construct a BPLF1-deficient EBV-bacmid (EBV-dBPLF1/NeoSt) (34). DNA fragments for recombination were generated by PCR with the following primers: 5′-GCG TAA GAC CCC GGA CCA GAA GGG GGG CGA CAA GGC GTC CTC CCC GCC CCA CCG CCG AAG GGC CTG GTG ATG ATG GCG GGA TC-3′ (forward) and 5′-GGG CCG CAG AGG CCG GGG CCG CAG AGG CCG GAG ACG ACG GCG GGG AGT TGG TCT TTG CAG TCA GAA GAA CTC GTC AAG AAG G-3′ (reverse). Electroporation of Escherichia coli was performed using Gene Pulser III (Bio-Rad). DNAs of EBV-WT and EBV-ΔBPLF1/NeoSt were purified using NucleoBond Bac 100 (Macherey-Nagel, Germany) and transfected into HEK293 cells using Lipofectamine 2000 reagent (Invitrogen) to establish HEK293 cells latently infected with either EBV-WT (293-EBVwt) or EBV-dBPLF1/NeoSt (293-EBVΔ).
Plasmids.
pcDNA-Flag/TRAF6 (pFlag-TRAF6) was a kind gift from E. Harhaj (University of Miami), and pcDNA-BZLF1 (pBZLF1) was generously donated by K. Kuzushima and R. Ohta (Aichi Cancer Center Research Institute). pcDNA-HA-Ub (pHA-Ub) was prepared as described previously (35). To prepare the expression vector for the Flag-tagged N-terminal fragment of BPLF1, pFlag-BPLF1, a portion of the BPLF1 ORF sequence (nt 1 to 975, which is sufficient for deubiquitinase activity), was cloned into EcoRI and XhoI sites of pcDNA3 with a Flag tag (28). Primers used for BPLF1 amplification were as follows: 5′-GAC GAC GAT GAC AAG GAA TTC ATG AGT AAC GGC GAC TGG GGG-3′ (forward) and 5′-AGA TGC ATG CTC GAG TCA AGG ACT ATA CCT GGC GGC AGG GAA TGA GTC-3′ (reverse). A BPLF1 point mutation (C61A, which is a catalytically inactive mutation) was introduced to make pFlag-BPLF1C61A by PCR using the following primers: 5′-ACT GCG TCC TCT ACC TGG TCA AGA G-3′ (forward) and 5′-TGC TGA CTG CCT GGA TGC CG-3′ (reverse) (36).
Antibodies and reagents.
Primary antibodies were purchased from Cell Signaling Technology (IKKβ, phosphorylated-IKKα/β, IκBα, α/β-tubulin, TRAF6), Chemicon (EBV BMRF1-R3, GAPDH [glyceraldehyde 3-phosphate dehydrogenase]), Roche Applied Science (hemagglutinin [HA]-3F10), and Sigma (Flag-M2). The antibodies to BZLF1, BALF2, BALF5, BGLF4, BBLF2 and -3 (BBLF2/3), and LMP1 have been described previously (37–42). Human p65-targeted small interfering (siRNA) was purchased from Santa Cruz. Control siRNA sequence (siRNA-DsRed) was 5′-GCA GAG CUG GUU UAG UGA AdT dT-3′ and 5′-UUC ACU AAA CCA GCU CUG CdT dT-3′, where dT means deoxythymidine.
Transfection and luciferase assays.
Plasmid DNA was transfected into HEK293, 293-EBVwt, or 293-EBVΔ cells using a MP-100 microporator (Digital Bio). The total amounts of plasmid DNA were standardized by addition of an empty vector. Proteins were extracted from cells with the lysis buffer supplied in a dual-luciferase reporter assay system kit (Promega), and luciferase activities were measured using the kit. The counts of firefly luciferase were normalized to those of Renilla luciferase. The protein samples were then subjected to SDS-PAGE followed by immunoblotting.
Immunoprecipitation.
To detect ubiquitinated forms of TRAF6 or physical interaction between BPLF1 and TRAF6, HEK293, 293-EBVwt, or 293-EBVΔ, cells transfected with expression plasmids were lysed 24 h posttransfection (hpt) in 100 μl of TX-100mCSK buffer (10 mM PIPES [pH 6.8], 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 0.1% Triton X-100, and protease inhibitor mixture [Roche]). Cell lysates were then diluted with the same buffer. Immunoprecipitation under stringent conditions was carried out as described previously (43). In brief, denaturing lysis buffer (50 mM Tris-HCl [pH 7.5], 2% SDS) was used in place of TX-100mCSK buffer, and the lysate was subsequently incubated at 95°C for 10 min followed by dilution with the dilution buffer (950 mM Tris-HCl [pH 7.5], 2% bovine serum albumin [BSA]). Diluted cell lysates were precleared with protein G-Sepharose (Amersham Biosciences). Supernatants were then mixed with anti-Flag antibodies and incubated overnight at 4°C. Immunocomplexes were recovered by incubating protein G-Sepharose for 1 h, and the resin was washed five times with the same buffer. The immunoprecipitates were then subjected to SDS-PAGE followed by immunoblotting.
Immunoblotting.
Cells were suspended in 1× sample buffer (65 mM Tris-HCl [pH 6.8], 3% SDS, 10% glycerol, 2% 2-mercaptoethanol) and then sonicated. The debris was removed by centrifugation, and the supernatants were applied for SDS-PAGE and immunoblotting, carried out as described previously (37).
qrt-PCR and PCR analysis.
Lytic replication-induced 293-EBV-WT or 293-EBVΔ cells (1 × 106 cells) were harvested, and total cellular RNA was purified using TriPure isolation reagent (Roche) followed by conversion to cDNA using a SuperScript III first-strand synthesis system (Invitrogen). Quantitative real-time PCR (qrt-PCR) was performed with SYBR Premix Ex Taq II Tli RNaseH Plus (TaKaRa Bio), an ABI Prism 7300 machine (Applied Biosystems), and 3-step cycling conditions (95°C for 30 s, followed by 50 cycles of 95°C for 5 s, 55°C for 30 s, and 72°C for 1 min). Dissociation curves were recorded after each run. Cycle threshold (CT) values were determined by automated threshold analysis with ABI Prism version 1.0 software. qrt-PCR assays were performed in triplicate. The value for an arbitrary RNA in the isolated RNAs was set to 1.0, and a standard curve was constructed using serial dilutions of cDNA from the RNA set to 1.0. A constant amount of RNAs was quantitated based on the standard curve. qrt-PCR with GAPDH primers was also performed to serve as an internal control for input RNA. Primer sequences used were as follows: for interleukin-8 (IL-8), 5′-CAA ACC TTT CCA CCC CAA AT-3′ (forward) and 5′-CTC TGC ACC CAG TTT TCC TT-3′ (reverse); for intercellular adhesion molecule 1 (ICAM-1), 5′-CAA CCG GAA GGT GTA TGA AC-3′ (forward) and 5′-CAG CGT AGG GTA AGG TTC-3′ (reverse); for AGT, 5′-GGA TGA GAG AGA GCC CAC AG-3′ (forward) and 5′-CTC ACT CCA TGC AGC ACA CT-3′ (reverse); for CCL2, 5′-CAT TGT GGC CAA GGA GAT CTG-3′ (forward) and 5′-CTT CGG AGT TTG GGT TTG CTT-3′ (reverse); and for GAPDH, 5′-GGG AAG GTG AAG GTC GGA GT-3′ (forward) and 5′-AAG ACG CCA GTG GAC TCC AC-3′ (reverse). Quantification of viral DNA synthesis during lytic replication was essentially conducted as described previously (44).
PCR analysis was performed with GoTaq Green Master Mix (Promega) and a Veriti thermal cycler (Applied Biosystems), and the PCR conditions used were 94°C for 30 s, 35 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 1 min. Primer sequences used in reverse transcription-PCR (RT-PCR) analysis were as follows: for BPLF1, 5′-GGA CCA TGG ATG TGA ATG C-3′ (forward) and 5′-GAG TCG GAT GTG AAA GAT CG-3′ (reverse); for BZLF1, 5′-AAC AGC CAG AAT CGC TGG AG-3′ (forward) and 5′-GGC ACA TCT GCT TCA ACA GG-3′ (reverse); and for GAPDH, 5′-TGC ACC ACC AAC TGC TAG C-3′ (forward) and 5′-GGC ATG GAC TGT GGT CAT GAG-3′ (reverse) (45).
Statistical analysis.
Results are expressed as means ± standard deviations (SD). Values were compared between groups using analysis of variance (ANOVA) and Fisher's protected-least-significance-difference test. Results were considered statistically significant at a P of <0.05.
RESULTS
Ectopic expression of BPLF1 decreases NF-κB-responsive promoter activity in latently EBV-infected cells.
Ubiquitination is involved in multiple steps of the NF-κB signaling pathway. Therefore, we first tested whether the EBV-encoded deubiquitinating enzyme BPLF1 inhibits NF-κB-dependent promoter activity in latently EBV-infected cells expressing LMP1. B95-8, AGS-EBV, and 293-EBVwt cells were transfected with reporter plasmids (pNF-κB-Fluc and pCMV-Rluc) and pBPLF1wt or pBPLF1C61A expression vectors, and luciferase assays were performed. Ectopic expression of the 325-aa-length N-terminal domain of BPLF1 exhibiting DUB activity decreased NF-κB-dependent promoter activity in these cells (Fig. 1). However, the EBV BPLF1C61A mutant, a mutant that is enzymatically defective due to the mutation of cysteine 61 to alanine (36), showed no significant inhibition (Fig. 1). The result suggests that DUB activity is essential for BPLF1 to suppress LMP1-induced NF-κB-dependent promoter activity.
Fig 1.
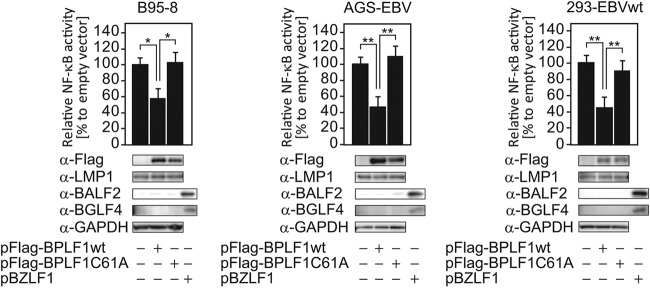
Ectopic expression of BPLF1 decreases NF-κB-dependent promoter activity in cells latently infected with EBV. Latently infected B95-8, AGS-EBV, and 293-EBVwt cells were transfected with the NF-κB-Fluc reporter plasmid (0.2 μg/well), along with the pCMV-Rluc plasmid (0.02 μg/well) and either pBPLF1wt or pBPLF1C61A (0.1 μg/well), in 24-well plates. Luciferase assays were performed at 24 hpt. Firefly luciferase activity was normalized to Renilla reniformis luciferase, and the value obtained by transfecting an empty-vector control was set to 100%. Data are shown as means ± SD of the results of 3 biological replicates. **, P < 0.001; *, P < 0.005. Sample lysates were subsequently subjected to immunoblotting with the specific antibodies indicated, and representative results are presented below the graph. In addition, sample lysates of cells transfected with BZLF1 were also included as controls for lytic replication.
In addition, we tested if overexpression of BPLF1 alone could induce EBV lytic cycle, because BPLF1 reduced NF-κB activity (Fig. 1) and because it was previously reported that inhibition of NF-κB by specific inhibitors causes spontaneous lytic gene expression in EBV-positive cells (46–49). While expression of immediate-early BZLF1 enhanced expression of early genes, including BALF2 and BGLF4, ectopic expression of BPLF1, either wild type or C61A, did not induce expression of the lytic genes (Fig. 1). Therefore, it is likely that, whereas BPLF1 inhibits NF-κB signaling, its expression alone is not sufficient for induction of EBV lytic replication.
Construction of BPLF1-deficient recombinant virus.
We then constructed a BPLF1-deficient recombinant virus to determine the effect of BPLF1 on the NF-κB signaling pathway in EBV lytic replication. A marker cassette was inserted into the BPLF1 gene (nt 1 to 975, encoding its catalytic domain) of EBV-WT to construct dBPLF1/NeoSt, and, as a result, nt 181 to 183 encoding the C61 residue, crucial for deubiquitinase activity, were disrupted (Fig. 2A). The DNA of recombinant EBV bacmid was analyzed by digestion with BamHI or EcoRI (Fig. 2B and C) and PCR (Fig. 2D). Restriction enzyme digestion of wild-type and recombinant bacterial artificial chromosome (BAC) DNAs verified that no large deletions or rearrangements of the EBV genome occurred during recombination and that the BamHI-P and EcoRI-H fragments were of the expected sizes in the wild type (Fig. 2B and C, open arrowheads) and were increased in size by the insertion of a NesSt cassette into the deletion mutant (Fig. 2B and C, closed arrowheads). PCR analysis performed with BPLF1-specific primers amplified an DNA fragment of the expected size in the case of EBV-WT DNA, but not in the case of dBPLF1/NeoSt DNA (Fig. 2D). DNAs of the wild type and dBPLF1/NeoSt were introduced into HEK293 cells, and hygromycin-resistant cell colonies were cloned for further analysis. HEK293 cells containing EBV-WT and dBPLF1/NeoSt DNAs were designated 293-EBVwt and 293-EBVΔ, respectively. For RT-PCR, total RNAs were prepared from the HEK293 cells containing the wild-type or the recombinant EBV genome at 48 hpt with the pBZLF1. While comparable amounts of BZLF1 and GAPDH mRNA were detected in the two cell lines, as expected, BPLF1 mRNA was detected only in 293-EBVwt (Fig. 2E). The 293-EBVwt and 293-EBVΔ cells maintain about 1.9 and 2.1 copies of EBV-BAC DNA, respectively (Fig. 2F). Western blotting with anti-LMP1 antibody verified that 293-EBVwt and 293-EBVΔ cells express similar levels of LMP1 (Fig. 2G). Thus, induction of lytic replication could be started from the similar genome copy and similar latency backgrounds. Expression of BPLF1 mRNA was detected at least by 18 h postinduction (hpi) and continued at least until 48 hpi (Fig. 2H).
Fig 2.

Recombinant EBV-BAC genome structures. (A) Schematic arrangement of recombination of the EBV genome using the neomycin resistance and streptomycin sensitivity genes. The region between nucleotides 1 and 975 of the BPLF1 ORF was replaced with tandemly arranged neomycin resistance and streptomycin sensitivity (NeoSt+) genes to make dBPLF1/NeoSt. (B and C) Electrophoresis of wild-type and recombinant EBV-BAC DNAs. EBV-BAC DNAs were digested with BamHI (B) or EcoRI (C) and separated in a 0.8% agarose gel. The sizes of BamHI-P fragment and a corresponding EcoRI fragment of the EBV-BAC DNAs (open arrows) were shifted by integration of the marker cassettes (closed arrows). Sizes (kbp) for molecular mass markers are indicated at the left side of the panels. (D) PCR analysis of the wild-type and the recombinant BAC DNAs with BPLF1 ORF-specific primers. The PCR product was detected by 1.5% agarose gel electrophoresis. (E) RT-PCR analysis of BPLF1 expressed in pBZLF1-transfected 293-EBVwt and 293-EBVΔ cells. Total RNAs were extracted at 48 hpi, and cDNAs were synthesized as described in Materials and Methods. PCR was performed on cDNA templates with specific primers. BZLF1 was used as an induction marker and GAPDH as an internal control. (F) Total DNAs prepared from 293-EBVwt and 293-EBVΔ cells were applied to qrt-PCR using BALF2-specific primers to quantify intracellular EBV-BAC DNA copies. The values were normalized to that of Namalwa cells, which maintain 2 EBV genomes per cell. (G) Western blotting using anti-LMP1 antibody was performed using whole-cell lysate prepared from 293-EBVwt and 293-EBVΔ cells to confirm that comparable amounts of the latent protein are expressed in both cells. (H) 293-EBVwt cells transfected with 1 μg of pBZLF1 were cultured for indicated periods, and expression levels of BPLF1 mRNA were measured by RT-PCR. Three biological replicates were carried out for the time-course analysis. Data from one representative experiment are shown.
DUB activity is essential for BPLF1 to block activation of the NF-κB pathway in EBV lytic replication.
We then examined whether EBV BPLF1 is involved in regulation of NF-κB signaling in EBV lytic replication. 293-EBVwt or 293-EBVΔ cells were transfected with pBZLF1 and reporter plasmids (pNF-κB-Fluc and pCMV-Rluc). The intrinsic NF-κB reporter activity (normalized with RLuc expression driven by a cytomegalovirus [CMV] promoter) in 293-EBVΔ cells was almost the same as that in 293-EBVwt cells. Transfection of pBZLF1 resulted in downregulation of NF-κB activity in 293-EBVwt compared with cells transfected with a control vector (Fig. 3A, lanes 1 and 2), consistent with a previous report that viral lytic reactivation requires downregulation of NF-κB (50). In contrast, transfection of pBZLF1 into 293-EBVΔ did not decrease NF-κB activity (Fig. 3A, lanes 3 and 4). We then examined the effect of the BPLF1 expression on NF-κB-dependent promoter activity in 293-EBVΔ. Cotransfection of pBPLF1wt together with pBZLF1 into 293-EBVΔ decreased the NF-κB promoter activity to a level comparable to that seen with pBZLF1-transfected 293-EBVwt (Fig. 3A, lane 5), while transfection of enzyme-dead mutant pBPLF1C61A did not (Fig. 3A, lane 6). In the parental EBV-negative HEK293 cells, transfection of BZLF1 did not significantly affect NF-κB activity under our assay conditions (Fig. 3B, lanes 1 and 2). Up- or downregulation of NF-κB activity observed in 293-EBVwt and 293-EBVΔ would be dependent on BZLF1-induced lytic gene expression (Fig. 3A, lanes 2 and 4). These data suggest that EBV BPLF1 is required to downregulate NF-κB-dependent promoter activity during EBV lytic replication, too, and that DUB activity is critical for BPLF1 to antagonize NF-κB functions.
Fig 3.
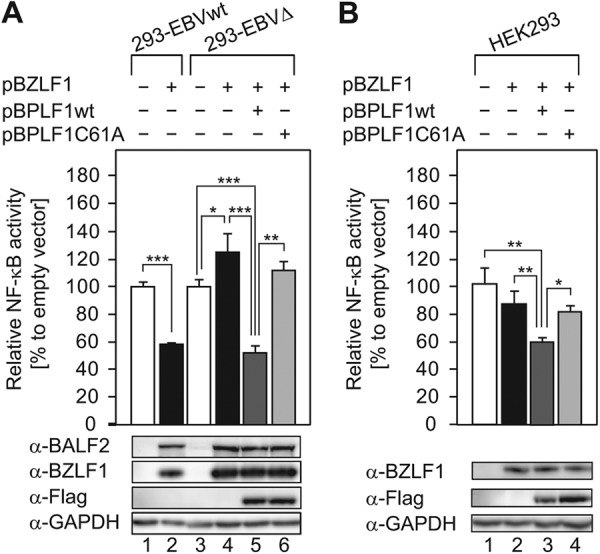
DUB activity is essential for BPLF1 to block activation of the NF-κB pathway during EBV lytic replication. Wild-type and recombinant BPLF1 expression vectors (0.1 μg each) were cotransfected into 293-EBVwt and 293-EBVΔ cells along with the pBZLF1 plasmid (1 μg), the pNF-κB-Fluc reporter plasmid (0.2 μg), and pCMV-Rluc (0.02 μg) using an MP-100 electroporator. An empty vector (pcDNA3) was used as a control. Cell extracts were collected at 24 hpt and analyzed for firefly and Renilla luciferase expression. Firefly luciferase activity was normalized to the Renilla reniformis luciferase, and the values obtained by transfecting the empty-plasmid control into 293-EBVwt or 293-EBVΔ were set to 100%. Sample lysates were subsequently subjected to immunoblotting with specific antibodies, and a representative result is presented below the graph. BZLF1 (immediate-early) and BALF2 (early) were used as induction markers. Data are shown as means ± SD of the results of 5 biological replicates. ***, P < 0.001; **, P < 0.005; *, P < 0.01.
Moreover, since exogenous expression of BPLF1 could attenuate the NF-κB activity even in parental HEK293, which is devoid of EBV (Fig. 3B, lane 3), we speculate that suppression of NF-κB activity by BPLF1 is not specific to LMP1 or to EBV.
BPLF1 suppresses canonical NF-κB-regulated genes during the EBV lytic life cycle.
Expression of canonical NF-κB-regulated genes, including AGT, CCL2 (monocyte chemoattractant protein-1 [MCP-1]), ICAM-1, and IL-8 (51–54), was conducted to confirm that BPLF1 actually inhibits NF-κB target gene expression during EBV lytic replication (Fig. 4). Total RNA was extracted, reverse transcribed into cDNA, and analyzed by qrt-PCR. Induction of EBV lytic replication in 293-EBVwt cells resulted in downregulation of a series of NF-κB-regulated genes, such as AGT (0.034-fold), CCL2 (0.104-fold), and ICAM-1 (0.528-fold) (Fig. 4A, top panel). Despite IL-8 expression being upregulated by the canonical NF-κB (55), IL-8 expression remained unchanged. It was earlier reported that BZLF1 induces IL-8 expression at both the protein and mRNA levels by directly binding to BZLF1-responsive elements in the IL-8 promoter (56), suggesting that the level of IL-8 expression in 293-EBVwt was compensated by BZLF1 during lytic replication. In contrast, the expression of AGT, CCL2, ICAM-1, and IL-8 was markedly elevated (4.7-fold, 4.1-fold, 2.7-fold, and 3.4-fold, respectively) in 293-EBVΔ (Fig. 4A, bottom panel). In addition, we confirmed, by RT-PCR, that BPLF1 was induced by BZLF1 in the wild type and that no BPLF1 signal was obtained in the knockout virus (Fig. 4B). Thus, BPLF1 appears to prevent canonical NF-κB-regulated gene expression in EBV lytic replication.
Fig 4.
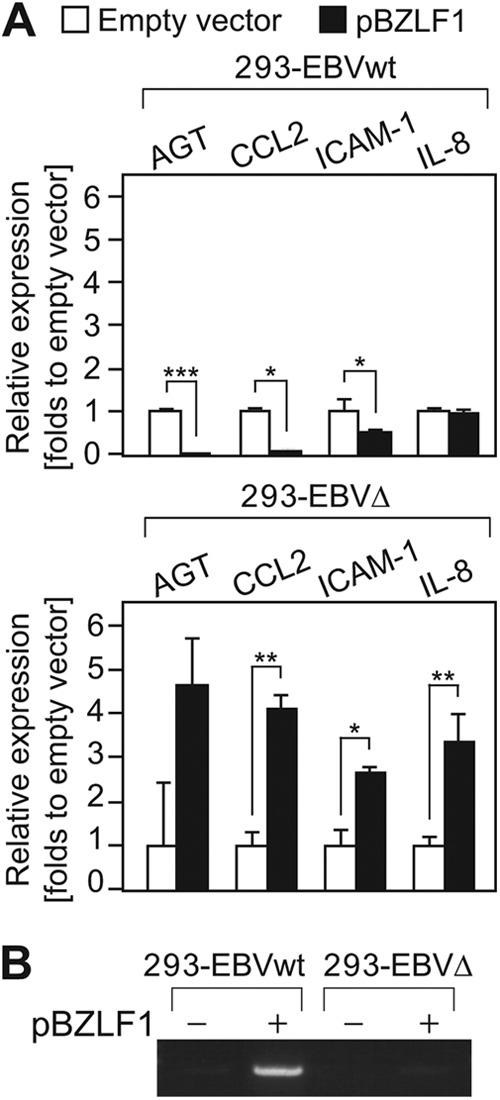
BPLF1 suppresses expression of NF-κB-regulated genes during the EBV lytic life cycle. (A) 293-EBVwt and 293-EBVΔ cells were transfected with control or BZLF1 expression plasmids. At 24 hpi, cells were subjected to qrt-PCR to measure the mRNA levels of NF-κB-dependent genes. Values were normalized to GAPDH mRNA, and the value obtained by transfecting an empty-plasmid control into 293-EBVwt or 293-EBVΔ was set to 1. Data are shown as means ± SD of the results of 3 biological replicates. ***, P < 0.001; **, P < 0.005; *, P < 0.05. (B) RT-PCR was carried out in order to detect BPLF1 mRNA in the same samples described for panel A, followed by an agarose electrophoresis.
Inhibition of NF-κB signaling by BPLF1 correlates with TRAF6 deubiquitination and increased IκBα.
Ubiquitination or deubiquitination of key signaling molecules is an important regulatory mechanism in NF-κB signaling. It is known that TRAF6 is an especially critical host factor for LMP1-mediated B cell activation, and its ubiquitination activates NF-κB signaling in latently infected cells (20). Therefore, we set out to examine whether BPLF1 could target TRAF6 to suppress NF-κB signaling. Ubiquitination assays performed by means of a heterologous expression system with 293 cells demonstrated that overexpressed TRAF6 became polyubiquitinated (Fig. 5A, lane 3). The assays also revealed that ubiquitinated TRAF6 was deubiquitinated by a coexpressed wild-type BPLF1 in a dose-dependent manner (Fig. 5A, lanes 4 to 6), but not by the enzymatically defective BPLF1C61A mutant (Fig. 5A, lanes 7 to 9). Since there is a possibility that TRAF6 may interact with other protein(s) that may also be polyubiquitinated, we then performed immunoprecipitation under stringent conditions to avoid that possibility (Fig. 5B). Similar results were obtained under the stringent conditions, supporting the result shown in Fig. 5A.
Fig 5.

BPLF1 interacts with and inhibits ubiquitination of TRAF6. (A) HEK293 cells cultured in 6-well plates were cotransfected with hemagglutinin (HA)-tagged Ub (2 μg/well) and TRAF6 (3 μg/well) expression plasmids and increasing quantities (0.1, 0.2, or 0.5 μg/well) of the designated BPLF1 expression plasmid. Cell lysates were prepared at 24 hpi and immunoprecipitated (IP) with anti-Flag antibodies, and ubiquitin conjugation of the TRAF6 protein was verified by immunoblotting with anti-HA antibodies. Production of exogenously expressed tagged proteins was verified with the indicated antibodies. The experiment shown is a representative of three independent experiments. (B) The conditions were basically the same as described for panel A except that cells were lysed with the denaturing lysis buffer containing 2% SDS followed by a 10-min incubation at 95°C. The amount of transfected BPLF1 expression plasmid was 0.1 or 0.5 μg/well. The experiment shown is a representative of three independent experiments. (C) HEK293 cells cultured in 6-well plates were transfected with an empty plasmid or designated BPLF1 (0.5 μg/well) expression plasmids. Cell lysates were prepared at 24 hpi and immunoprecipitated with anti-Flag antibodies, followed by immunoblot analysis with anti-TRAF6 antibodies. Production of exogenously expressed BPLF1 proteins was verified with anti-Flag antibody. The experiment shown is a representative of two independent experiments.
A coimmunoprecipitation assay revealed that endogenously expressed TRAF6 protein was coprecipitated with Flag-tagged BPLF1 protein (Fig. 5C). Similar amounts of TRAF6 were also coprecipitated with enzymatically defective BPLF1. The result indicates that BPLF1 interacts with TRAF6, directly or indirectly, independently of its catalytic activity.
We further investigated whether the ability of BPLF1 to antagonize NF-κB is associated with deubiquitination of TRAF6 in EBV lytic replication. Ubiquitination states of TRAF6 in 293-EBVwt and 293-EBVΔ cells were compared when they were induced to perform lytic replication. When 293-EBVwt cells were transfected with pBZLF1, the TRAF6 polyubiquitination was markedly inhibited (Fig. 6A, lanes 1 and 2). In contrast, when 293-EBVΔ cells were transfected with pBZLF1, reduction of the TRAF6 ubiquitination state was much less significant compared to the case of 293 WT cells (Fig. 6A, compare lane 2 with lane 4). Furthermore, coexpression of wild-type BPLF1 in 293-EBVΔ diminished ubiquitination of TRAF6 (Fig. 6A, lane 5), while coexpression of the BPLF1C61A mutant did not show such an effect (Fig. 6A, lane 6). Partial reduction in TRAF6 ubiquitination in lytic replication-induced 293-EBVΔ cells might be due to other EBV-encoded deubiquitinating proteins such as BSLF1 and BXLF1 (36). We suggest that BPLF1 mainly deubiquitinates TRAF6 in the lytic phase of EBV replication, although BSLF1 and/or BXLF1 might also be involved in the deubiquitination of TRAF6.
Fig 6.
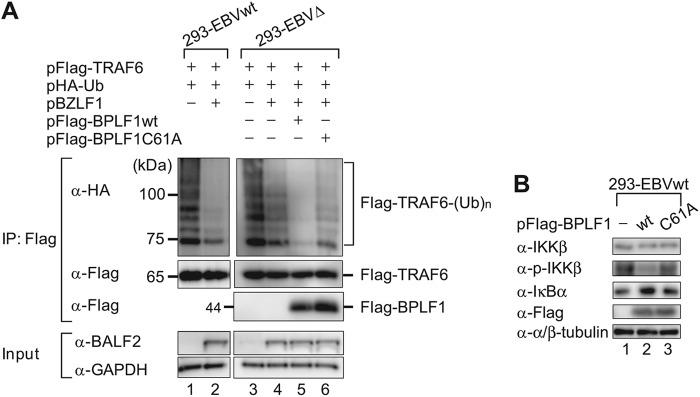
Endogenous BPLF1 deubiquitinates TRAF6. (A) 293-EBVwt and 293-EBVΔ cells cultured in 6-well plates were cotransfected with HA-tagged Ub (2 μg/well), TRAF6 (3 μg/well), and BZLF1 (1 μg/well) expression plasmids. Four hours after the initial transfection, the cells were further transfected with either wild-type or enzyme-dead BPLF1 expression plasmids (0.5 μg/well). Cell lysates were prepared at 24 h after initial transfection, and immunoprecipitation experiments were performed in the same fashion as described for Fig. 5A. The experiment shown is a representative of three independent experiments. (B) 293-EBVwt cells were transfected with empty plasmid or designated BPLF1 (0.5 μg/well) expression plasmid. Cell lysates were prepared at 24 hpi, and immunoblot analysis was performed using indicated antibodies. The experiment shown is a representative of two independent experiments.
Upon activation of the canonical NF-κB signaling pathway, IKK (IκB kinase) is activated by phosphorylation of β subunit (IKKβ), and then active IKK phosphorylates IκB, resulting in its proteasomal degradation (57). This liberates NF-κB, which translocates to the nucleus and binds to promoters of NF-κB-regulated genes. Overexpression of BPLF1 resulted in repression of IKKβ phosphorylation and accumulation of IκBα protein in 293-EBVwt cells in which the canonical NF-κB signaling is constitutively activated (Fig. 6B, lane 2). Collectively, the findings indicate that BPLF1 blocks ubiquitination of TRAF6, leading to inhibition of IκBα degradation to prevent NF-κB target gene expression.
BPLF1 promotes EBV genome replication.
Regarding the effects of BPLF1 DUB activity on EBV lytic DNA replication, immunoblotting revealed that the protein levels of viral early genes (BALF2, BBLF2/3, BGLF4, and BMRF1) were not affected by disrupting BPLF1 expression at 48 hpi, although the level of BALF5 DNA polymerase was to some extent lower (Fig. 7A). However, qrt-PCR using EBV genome DNA-specific primers revealed that viral DNA synthesis in 293-EBVΔ cells at 48 hpi was significantly impaired, comparing with the 293-EBVwt case (Fig. 7B), suggesting that BPLF1 promotes viral genome replication. In addition, when lytic replication was induced with the smaller amount of pBZLF1 (0.1 μg), lytic gene expression in 293-EBVΔ appeared to be lower than in 293-EBVwt at 21, 24, and 27 hpi, although after 30 hpi, comparable levels of expression were observed, suggesting that BPLF1 affects early gene expression under conditions of lower levels of BZLF1 expression (Fig. 7C).
Fig 7.
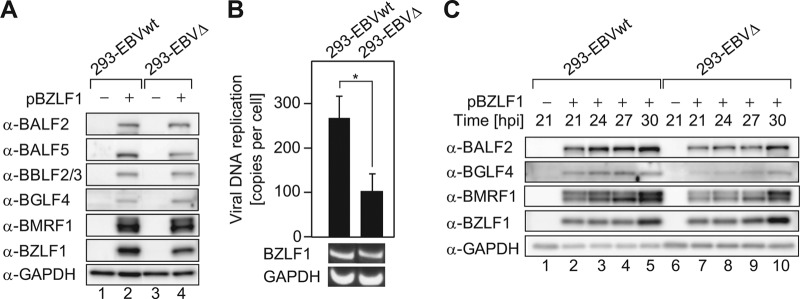
BPLF1 promotes EBV genome replication. (A) 293-EBVwt and 293-EBVΔ cells were transfected with pBZLF1 (1 μg) to induce lytic replication, harvested at 48 hpt, and washed with PBS (−), and then whole-cell lysates were extracted. Protein levels of viral early genes (BALF2, BALF5, BBLF2/3, BGLF4, and BMRF1) and the BZLF1 immediate-early gene were analyzed in 293-EBVwt and 293-EBVΔ cells by immunoblotting. GAPDH was used as an internal control. (B) At 48 h after pBZLF1 (1 μg) transfection, cells were washed with PBS (−), and total DNAs were extracted. qrt-PCR analysis was performed with BALF2- and GAPDH-specific primers. Intracellular viral DNA copy numbers were calculated as follows: BALF2 values were normalized to each GAPDH value, and the BALF2/GAPDH values were further compared to those for Namalwa cells, which maintain 2 EBV genomes per cell. RT-PCR data from one representative experiment are shown. Data are expressed as fold increase in comparison to untransfected cells and means ± SD of the results of 5 biological replicates. *, P < 0.005. (C) The threshold necessary amount (0.1 μg) of pBZLF1-transfected 293-EBVwt and 293-EBVΔ cells was cultured for the indicated periods. Protein levels of viral early genes (BALF2, BGLF4, BMRF1) were analyzed by immunoblotting. The experiment shown is representative of two independent experiments.
Since ectopically expressed BPLF1 is sufficient for at least inactivation of cellular NF-κB activity (Fig. 1 and 3) and deubiquitination of TRAF6 (Fig. 5 and 6), we tested whether ectopic expression of the BPLF1 DUB domain in 293-EBVΔ cells might promote viral genome replication. The expression of pBPLF1wt in 293-EBVΔ distinctly restored viral DNA synthesis, while expression of the BPLF1 mutant did not (Fig. 8A).
Fig 8.
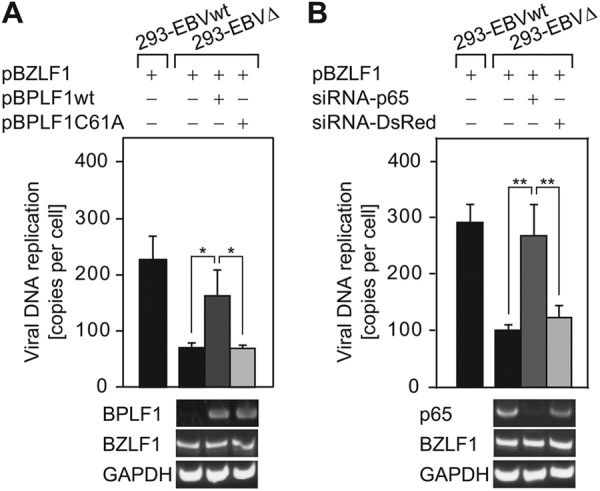
Exogenous expression of BPLF1 deubiquitinase or p65 knockdown restores viral DNA replication of the BPLF1-deficient virus. (A) The BZLF1 expression plasmid (0.5 μg/well) was transfected into 293EBVΔ cells using an electroporator, and 4 h after the initial transfection, the cells were further transfected with wild-type or enzyme-dead BPLF1 expression plasmids (0.5 μg/well) using Lipofectamine 2000. At 48 h after the initial transfection, cells were washed with PBS (−), and total DNA was extracted. qrt-PCR analysis was performed with the same method as described for Fig. 7B. cDNAs were prepared from the mRNAs extracted in parallel with the total DNAs. RT-PCR data from one representative experiment are shown below the graph. (B) p65-targeted or control siRNA (0.2 μg/well) was cotransfected with the BZLF1 expression plasmid (0.2 μg/well) into 293-EBVΔ cells using an electroporator and cultured for 48 h and processed similarly to the method described for panel A. Data are expressed as fold increase in comparison to untransfected cells and means ± SD of the results of 3 biological replicates. **, P < 0.001; *, P < 0.01.
Knockdown of p65 promotes viral DNA replication.
We further examined whether inhibition of NF-κB signaling actually promotes viral DNA replication in 293-EBVΔ cells. Cotransfection of p65-targeting siRNA together with pBZLF1 resulted in increased numbers of copies of the EBV genome that are comparable to the level seen with the EBV genome in lytic replication-induced 293 WT cells, while cotransfection of control siRNA (DsRed) had no significant effect (Fig. 8B). Taking our results together, BPLF1 appears to promote lytic viral genome replication, at least partly by blocking the canonical NF-κB signaling via DUB activity.
DISCUSSION
Our present study clearly demonstrated that the DUB activity of BPLF1 is involved in the downregulation of NF-κB signaling in the context of viral lytic replication. The immediate-early BZLF1 protein, a key initiator of EBV lytic replication, is known to interact with the NF-κB family member p65/RelA to inhibit its transcriptional activity, and p65/RelA in turn inhibits the transcriptional activity of BZLF1 (58). Also, Brown et al. reported that overexpression of p65 inhibits EBV lytic replication, and they predicted that cells expressing a high level of active NF-κB would hardly enter the lytic life cycle (24). It was also reported that NF-κB activation inhibits lytic cycle induction (25). Furthermore, in some cell lines such as B95-8 and HH514 (59), lyLMP1, an amino-terminally truncated and late-lytic-cycle-associated form of LMP1, is expressed to function as a dominant-negative regulator of NF-κB signaling by LMP1 (59, 60). Thus, EBV appears to utilize various strategies to downregulate NF-κB activity during lytic replication, highlighting the biological relevance of NF-κB inhibition by BPLF1. Our results, together with those of previous reports (24, 25), strongly support the idea that BPLF1 is necessary to establish cellular circumstances with decreased NF-κB activity for the lytic life cycle to proceed.
It has been reported that upregulation of NF-κB promotes host cell survival but inhibits the initiation of lytic replication in EBV-infected cells (61). There are several reports demonstrating that NF-κB inhibitors cause spontaneous apoptosis and lytic gene expression in EBV-positive B-lymphocytes and in nasopharyngeal and Burkitt's lymphoma cells (46–49). We showed here that expression of the N-terminal 325-aa region of BPLF1 carrying DUB activity was sufficient to suppress NF-κB activity in latently EBV-infected cells. However, expression of BPLF1 itself did not induce early lytic gene expression in B95-8, AGS-EBV, and 293-EBVwt cells (Fig. 1). Also, no death was observed in cells transfected with pFlag-BPLF1. Our results indicate that BPLF1 downregulates NF-κB signaling in a DUB activity-dependent manner, but it likely causes neither spontaneous apoptosis nor lytic gene expression in latently infected cells, at least under our conditions.
The known deubiquitinases of other viruses, including PLP2 of murine hepatitis virus A59 (62), ORF64 of Kaposi's sarcoma-associated herpesvirus (63), and Lpro of foot-and-mouth disease virus (64), were reported to provide an opportunity for effective virus invasion into a new host by downregulating beta interferon (IFN-β) activity. Downregulation of IFN-β activity may result in suppression of numerous IFN-stimulated genes, including important antiviral molecules such as PKR, MX1, OAS1, ISG15, and TRIM5 (65). Meanwhile, whether downregulation of canonical NF-κB target genes such as AGT, CCL-2, and ICAM-1 is physiologically advantageous to EBV lytic replication has yet to be elucidated. The AGT gene encodes angiotensinogen, a precursor of angiotensin II, which conversely activates such transcription factors, including the canonical NF-κB (66). Monocyte chemoattractant protein-1 (MCP-1; also known as CCL2) has been shown to mediate recruitment of monocytes to inflamed sites (67–69). Intercellular adhesion molecule 1 (ICAM-1) is a glycosylated, integral membrane protein that plays an important role in inflammatory responses by promoting cell-cell interactions (70). It also serves as a counterreceptor for lymphocyte function-associated antigen 1, which is found on all types of leukocytes and has been implicated in migration of leukocytes to sites of inflammation (71–73). A variety of viral proteins are expressed in lytic replication-induced cells, and they should be targeted by the host immune system. EBV may downregulate the expression of molecules that otherwise recruit monocytes and leukocytes to the infected cells, which would be disadvantageous to EBV productive replication.
The BPLF1 protein is conserved among members of the herpesvirus family and has been classified as a potential tegument protein by theoretical computer analysis using the Swiss-Prot database (74). Many details concerning functional domains have been elucidated, primarily through studies on BPLF1 homologs such as herpes simplex virus 1 (HSV-1) pUL36 and pseudorabies virus (PrV) pUL36. The results of these studies collectively suggest that the C-terminal part of the protein containing multiple binding sites for the capsid protein is critical for the virus production. In cells infected with HSV-1 lacking UL36 or a mutant encoding only the first 361 aa of pUL36, the capsids reach the cytosol, but there is no secondary envelopment, no cell egress, and no plaque formation (75, 76). Also, the PrV pUL36 is essential for production of virus particles (77, 78). Whitehurst et al., however, have previously reported that knockdown of BPLF1 expression with short hairpin RNA resulted in decreased viral particle production but that it did not completely inhibit the production (27). We also confirmed that induction of lytic replication in cells harboring the BPLF1-deleted EBV genome produced infectious viruses, although the yield of BPLF1-deleted EBV was 0.65-fold lower than that of wild-type virus.
Since Brown et al. reported that the canonical NF-κB inhibits activation of the early lytic BHLF1 promoter harboring BZLF1- and BRLF1-responsive elements (24), we speculate that NF-κB-mediated inhibition of early lytic genes is released by BPLF1 at the beginning of lytic replication. They also indicated that inhibition of lytic promoters by NF-κB is reversible: overexpression of BZLF1 and BRLF1 restored lytic promoter activation. In accordance with the literature cited above, early lytic protein expression in 293-EBVΔ appeared to be lower than that in 293-EBVwt when the lytic replication was induced with a smaller amount of pBZLF1 (Fig. 7C). The decreased levels of lytic proteins were observed until 27 hpi, but comparable levels were observed at 30 hpi. Our results suggest that the impairment of viral DNA synthesis in 293-EBVΔ cells could be partly due to the decreased expression of early genes at around 27 hpi, although the impairment was observed even with the higher BZLF1 expression. A study on human CMV (HCMV) UL48, the counterpart of BPLF1, demonstrated that the mutant virus, which has full-length but catalytically inactive UL48(C24S), replicated more slowly than the wild type and with lower yields of extracellular virus (79). Our results, together with those of the study on UL48, suggest that the loss of viral DUB activity was partly attributable to the decreased and delayed expression of early genes. Interestingly, the growth kinetics of the UL48(C24S) mutant virus were similar to those of the wild type at a multiplicity of infection (MOI) of 3, whereas the mutant virus infection produced about 10-fold-fewer progeny virions than did wild-type virus at an MOI of 0.1. In addition, slightly reduced levels of viral immediate-early, early, and late proteins were observed in Western blot analysis in the mutant virus compared to the wild type at a low MOI.
It is reported that EBV BILF1 and BLLF3 proteins expressed in later stages of lytic replication again upregulate NF-κB signaling (80, 81). EBV G-protein-coupled receptor (EBV BILF1) also appeared to activate the NF-κB pathway in COS-7 and Burkitt's lymphoma cells (82). Furthermore, activation of NF-κB by the EBV dUTPase (EBV BLLF3) through TLR-2 has been previously described (80). These proteins might protect the host cell from death caused by cytopathic effects of viral infection in later phases of lytic replication. We propose that BPLF1 is an accelerating agent of the latent-to-lytic switch that antagonizes NF-κB function at the earlier phase of lytic replication (Fig. 9). BPLF1 may reduce the biological threshold of NF-κB activity required for switching from the latent to the lytic life cycle of EBV.
Fig 9.
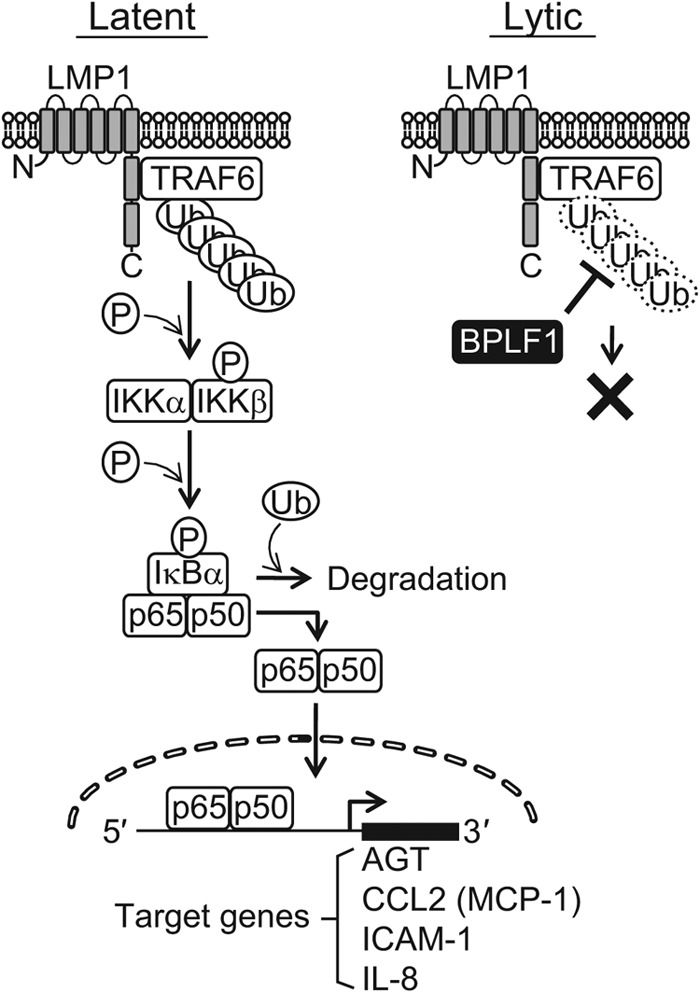
A schematic model demonstrating the inhibition of NF-κB signaling by BPLF1 in the EBV life cycle. In EBV latent infection, NF-κB is activated by viral LMP1 protein; TRAF6 associates with LMP1 and is constitutively polyubiquitinated. Activation of NF-κB confers cell survival (83) and inhibition of spontaneous lytic replication as well (24). Changes in the host cell microenvironment or other unknown triggers can downregulate the NF-κB activity and disrupt the balance between the latent cycle and the lytic cycle of EBV (61). Once lytic replication is induced, BPLF1 then deubiquitinates and inactivates TRAF6 to further block NF-κB signaling, promoting efficient viral genome replication.
ACKNOWLEDGMENTS
This work was supported by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, Culture and Technology of Japan (no. 23114512, 3390118, and 24659213 to T.T.), by the Ministry of Health, Labor and Welfare (T.T.), and partly by the Takeda Foundation (T.T.). A.S. is a Research Fellow of the Japanese Society for the Promotion of Science for Young Scientists.
We are grateful to W. Hammerschmidt (German Research Center for Environment and Health) and H. J. Delecluse (German Cancer Research Center) for providing the EBV Bac system. We also express our appreciation to E. Harhaj (University of Miami) for providing the Flag-TRAF6 expression plasmid. In addition, we thank K. Kuzushima and R. Ohta (Aichi Cancer Center Research Institute) for pcDNA-BZLF1 and Y. Kawaguchi (University of Tokyo) for the anti-BGLF4 antibody.
Footnotes
Published ahead of print 30 January 2013
REFERENCES
- 1. Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Séguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211 [DOI] [PubMed] [Google Scholar]
- 2. Morrison JA, Gulley ML, Pathmanathan R, Raab-Traub N. 2004. Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res. 64:5251–5260 [DOI] [PubMed] [Google Scholar]
- 3. Wu S, Xie P, Welsh K, Li C, Ni CZ, Zhu X, Reed JC, Satterthwait AC, Bishop GA, Ely KR. 2005. LMP1 protein from the Epstein-Barr virus is a structural CD40 decoy in B lymphocytes for binding to TRAF3. J. Biol. Chem. 280:33620–33626 [DOI] [PubMed] [Google Scholar]
- 4. Xie P, Bishop GA. 2004. Roles of TNF receptor-associated factor 3 in signaling to B lymphocytes by carboxyl-terminal activating regions 1 and 2 of the EBV-encoded oncoprotein latent membrane protein 1. J. Immunol. 173:5546–5555 [DOI] [PubMed] [Google Scholar]
- 5. Schultheiss U, Puschner S, Kremmer E, Mak TW, Engelmann H, Hammerschmidt W, Kieser A. 2001. TRAF6 is a critical mediator of signal transduction by the viral oncogene latent membrane protein 1. EMBO J. 20:5678–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu L, Nakano H, Wu Z. 2006. The C-terminal activating region 2 of the Epstein-Barr virus-encoded latent membrane protein 1 activates NF-kappaB through TRAF6 and TAK1. J. Biol. Chem. 281:2162–2169 [DOI] [PubMed] [Google Scholar]
- 7. Bishop GA, Hostager BS. 2001. Signaling by CD40 and its mimics in B cell activation. Immunol. Res. 24:97–109 [DOI] [PubMed] [Google Scholar]
- 8. Xie P, Hostager BS, Bishop GA. 2004. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J. Exp. Med. 199:661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. 1999. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol. 73:1023–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eliopoulos AG, Gallagher NJ, Blake SM, Dawson CW, Young LS. 1999. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 274:16085–16096 [DOI] [PubMed] [Google Scholar]
- 11. Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, Zeidler R, Scheffer B, Ueffing M, Hammerschmidt W. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 18:3064–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Higuchi M, Kieff E, Izumi KM. 2002. The Epstein-Barr virus latent membrane protein 1 putative Janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J. Virol. 76:455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sylla BS, Hung SC, Davidson DM, Hatzivassiliou E, Malinin NL, Wallach D, Gilmore TD, Kieff E, Mosialos G. 1998. Epstein-Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-kappaB through a pathway that includes the NF-kappaB-inducing kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc. Natl. Acad. Sci. U. S. A. 95:10106–10111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devergne O, Cahir McFarland ED, Mosialos G, Izumi KM, Ware CF, Kieff E. 1998. Role of the TRAF binding site and NF-kappaB activation in Epstein-Barr virus latent membrane protein 1-induced cell gene expression. J. Virol. 72:7900–7908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Z, Xin B, Yang X, Chan C, Cao L. 2000. Nuclear factor-kappaB activation is involved in LMP1-mediated transformation and tumorigenesis of rat-1 fibroblasts. Cancer Res. 60:1845–1848 [PubMed] [Google Scholar]
- 16. Mehl AM, Floettmann JE, Jones M, Brennan P, Rowe M. 2001. Characterization of intercellular adhesion molecule-1 regulation by Epstein-Barr virus-encoded latent membrane protein-1 identifies pathways that cooperate with nuclear factor kappa B to activate transcription. J. Biol. Chem. 276:984–992 [DOI] [PubMed] [Google Scholar]
- 17. Pai S, O'Sullivan BJ, Cooper L, Thomas R, Khanna R. 2002. RelB nuclear translocation mediated by C-terminal activator regions of Epstein-Barr virus-encoded latent membrane protein 1 and its effect on antigen-presenting function in B cells. J. Virol. 76:1914–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Wu L, Hong K, Pagano JS. 2001. Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J. Virol. 75:12393–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bassères DS, Baldwin AS. 2006. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 25:6817–6830 [DOI] [PubMed] [Google Scholar]
- 20. Arcipowski KM, Stunz LL, Graham JP, Kraus ZJ, Vanden Bush TJ, Bishop GA. 2011. Molecular mechanisms of TNFR-associated factor 6 (TRAF6) utilization by the oncogenic viral mimic of CD40, latent membrane protein 1 (LMP1). J. Biol. Chem. 286:9948–9955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. 2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O'Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM. 2007. DUBA: a deubiquitinase that regulates type I interferon production. Science 318:1628–1632 [DOI] [PubMed] [Google Scholar]
- 23. Wertz IE, Dixit VM. 2010. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2:a003350 doi:10.1101/cshperspect.a003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prince S, Keating S, Fielding C, Brennan P, Floettmann E, Rowe M. 2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J. Virol. 77:5000–5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rowe M, Lear AL, Croom-Carter D, Davies AH, Rickinson AB. 1992. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 66:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitehurst CB, Ning S, Bentz GL, Dufour F, Gershburg E, Shackelford J, Langelier Y, Pagano JS. 2009. The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J. Virol. 83:4345–4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gastaldello S, Hildebrand S, Faridani O, Callegari S, Palmkvist M, Di Guglielmo C, Masucci MG. 2010. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat. Cell Biol. 12:351–361 [DOI] [PubMed] [Google Scholar]
- 29. Whitehurst CB, Vaziri C, Shackelford J, Pagano JS. 2012. Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates Pol{square} recruitment to DNA damage sites. J. Virol. 86:8097–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maruo S, Yang L, Takada K. 2001. Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J. Gen. Virol. 82:2373–2383 [DOI] [PubMed] [Google Scholar]
- 31. Noda C, Murata T, Kanda T, Yoshiyama H, Sugimoto A, Kawashima D, Saito S, Isomura H, Tsurumi T. 2011. Identification and characterization of CCAAT enhancer-binding protein (C/EBP) as a transcriptional activator for Epstein-Barr virus oncogene latent membrane protein 1. J. Biol. Chem. 286:42524–42533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81 [DOI] [PubMed] [Google Scholar]
- 33. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Isomura H, Tsurumi T, Stinski MF. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 78:12788–12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murata T, Shimotohno K. 2006. Ubiquitination and proteasome-dependent degradation of human eukaryotic translation initiation factor 4E. J. Biol. Chem. 281:20788–20800 [DOI] [PubMed] [Google Scholar]
- 36. Sompallae R, Gastaldello S, Hildebrand S, Zinin N, Hassink G, Lindsten K, Haas J, Persson B, Masucci MG. 2008. Epstein-Barr virus encodes three bona fide ubiquitin-specific proteases. J. Virol. 82:10477–10486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwahori S, Murata T, Kudoh A, Sato Y, Nakayama S, Isomura H, Kanda T, Tsurumi T. 2009. Phosphorylation of p27Kip1 by Epstein-Barr virus protein kinase induces its degradation through SCFSkp2 ubiquitin ligase actions during viral lytic replication. J. Biol. Chem. 284:18923–18931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J. Virol. 78:7004–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kudoh A, Fujita M, Kiyono T, Kuzushima K, Sugaya Y, Izuta S, Nishiyama Y, Tsurumi T. 2003. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J. Virol. 77:851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsurumi T, Kobayashi A, Tamai K, Daikoku T, Kurachi R, Nishiyama Y. 1993. Functional expression and characterization of the Epstein-Barr virus DNA polymerase catalytic subunit. J. Virol. 67:4651–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsurumi T, Kobayashi A, Tamai K, Yamada H, Daikoku T, Yamashita Y, Nishiyama Y. 1996. Epstein-Barr virus single-stranded DNA-binding protein: purification, characterization, and action on DNA synthesis by the viral DNA polymerase. Virology 222:352–364 [DOI] [PubMed] [Google Scholar]
- 42. Yokoyama N, Fujii K, Hirata M, Tamai K, Kiyono T, Kuzushima K, Nishiyama Y, Fujita M, Tsurumi T. 1999. Assembly of the Epstein-Barr virus BBLF4, BSLF1 and BBLF2/3 proteins and their interactive properties. J. Gen. Virol. 80(Pt 11):2879–2887 [DOI] [PubMed] [Google Scholar]
- 43. Harlow E, Lane D. 2006. Immunoprecipitation: denaturing lysis. CSH Protoc. 2006:pdb.prot4534. doi:10.1101/pdb.prot4534 [DOI] [PubMed] [Google Scholar]
- 44. Nakayama S, Murata T, Yasui Y, Murayama K, Isomura H, Kanda T, Tsurumi T. 2010. Tetrameric ring formation of Epstein-Barr virus polymerase processivity factor is crucial for viral replication. J. Virol. 84:12589–12598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. 2012. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 86:4752–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. 2000. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. U. S. A. 97:6055–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kurokawa M, Ghosh SK, Ramos JC, Mian AM, Toomey NL, Cabral L, Whitby D, Barber GN, Dittmer DP, Harrington WJ., Jr 2005. Azidothymidine inhibits NF-kappaB and induces Epstein-Barr virus gene expression in Burkitt lymphoma. Blood 106:235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu SF, Wang H, Lin XC, Xiang H, Deng XY, Li W, Tang M, Cao Y. 2008. NF-kappaB inhibitors induce lytic cytotoxicity in Epstein-Barr virus-positive nasopharyngeal carcinoma cells. Cell Biol. Int. 32:1006–1013 [DOI] [PubMed] [Google Scholar]
- 49. Miyake A, Dewan MZ, Ishida T, Watanabe M, Honda M, Sata T, Yamamoto N, Umezawa K, Watanabe T, Horie R. 2008. Induction of apoptosis in Epstein-Barr virus-infected B-lymphocytes by the NF-kappaB inhibitor DHMEQ. Microbes Infect. 10:748–756 [DOI] [PubMed] [Google Scholar]
- 50. Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. 2001. DNA binding by Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ledebur HC, Parks TP. 1995. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J. Biol. Chem. 270:933–943 [DOI] [PubMed] [Google Scholar]
- 52. Li J, Brasier AR. 1996. Angiotensinogen gene activation by angiotensin II is mediated by the rel A (nuclear factor-kappaB p65) transcription factor: one mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol. Endocrinol. 10:252–264 [DOI] [PubMed] [Google Scholar]
- 53. Rahman A, Anwar KN, True AL, Malik AB. 1999. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J. Immunol. 162:5466–5476 [PubMed] [Google Scholar]
- 54. Stylianou E, Nie M, Ueda A, Zhao L. 1999. c-Rel and p65 trans-activate the monocyte chemoattractant protein-1 gene in interleukin-1 stimulated mesangial cells. Kidney Int. 56:873–882 [DOI] [PubMed] [Google Scholar]
- 55. Hoberg JE, Yeung F, Mayo MW. 2004. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol. Cell 16:245–255 [DOI] [PubMed] [Google Scholar]
- 56. Hsu M, Wu SY, Chang SS, Su IJ, Tsai CH, Lai SJ, Shiau AL, Takada K, Chang Y. 2008. Epstein-Barr virus lytic transactivator Zta enhances chemotactic activity through induction of interleukin-8 in nasopharyngeal carcinoma cells. J. Virol. 82:3679–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. 1994. Tumor necrosis factor alpha-induced phosphorylation of I kappa B alpha is a signal for its degradation but not dissociation from NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 91:12740–12744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morrison TE, Kenney SC. 2004. BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 328:219–232 [DOI] [PubMed] [Google Scholar]
- 59. Erickson KD, Berger C, Coffin WF, 3rd, Schiff E, Walling DM, Martin JM. 2003. Unexpected absence of the Epstein-Barr virus (EBV) lyLMP-1 open reading frame in tumor virus isolates: lack of correlation between Met129 status and EBV strain identity. J. Virol. 77:4415–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pandya J, Walling DM. 2006. Oncogenic activity of Epstein-Barr virus latent membrane protein 1 (LMP-1) is down-regulated by lytic LMP-1. J. Virol. 80:8038–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Oliveira DE, Ballon G, Cesarman E. 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 18:248–257 [DOI] [PubMed] [Google Scholar]
- 62. Wang G, Chen G, Zheng D, Cheng G, Tang H. 2011. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS One 6:e17192 doi:10.1371/journal.pone.0017192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. 2011. Inhibition of RIG-I-mediated signaling by Kaposi's sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85:10899–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85:3758–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1:519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brasier AR, Li J. 1996. Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension 27:465–475 [DOI] [PubMed] [Google Scholar]
- 67. Burns MJ, Sellati TJ, Teng EI, Furie MB. 1997. Production of interleukin-8 (IL-8) by cultured endothelial cells in response to Borrelia burgdorferi occurs independently of secreted [corrected] IL-1 and tumor necrosis factor alpha and is required for subsequent transendothelial migration of neutrophils. Infect. Immun. 65:1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tam FW, Karkar AM, Smith J, Yoshimura T, Steinkasserer A, Kurrle R, Langner K, Rees AJ. 1996. Differential expression of macrophage inflammatory protein-2 and monocyte chemoattractant protein-1 in experimental glomerulonephritis. Kidney Int. 49:715–721 [DOI] [PubMed] [Google Scholar]
- 69. Wenzel U, Schneider A, Valente AJ, Abboud HE, Thaiss F, Helmchen UM, Stahl RA. 1997. Monocyte chemoattractant protein-1 mediates monocyte/macrophage influx in anti-thymocyte antibody-induced glomerulonephritis. Kidney Int. 51:770–776 [DOI] [PubMed] [Google Scholar]
- 70. Boyd AW, Wawryk SO, Burns GF, Fecondo JV. 1988. Intercellular adhesion molecule 1 (ICAM-1) has a central role in cell-cell contact-mediated immune mechanisms. Proc. Natl. Acad. Sci. U. S. A. 85:3095–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dustin ML, Springer TA. 1991. Role of lymphocyte adhesion receptors in transient interactions and cell locomotion. Annu. Rev. Immunol. 9:27–66 [DOI] [PubMed] [Google Scholar]
- 72. Rothlein R, Dustin ML, Marlin SD, Springer TA. 1986. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J. Immunol. 137:1270–1274 [PubMed] [Google Scholar]
- 73. van de Stolpe A, van der Saag PT. 1996. Intercellular adhesion molecule-1. J. Mol. Med. (Berl) 74:13–33 [DOI] [PubMed] [Google Scholar]
- 74. Schmaus S, Wolf H, Schwarzmann F. 2004. The reading frame BPLF1 of Epstein-Barr virus: a homologue of herpes simplex virus protein VP16. Virus Genes 29:267–277 [DOI] [PubMed] [Google Scholar]
- 75. Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Böttcher S, Granzow H, Maresch C, Mohl B, Klupp BG, Mettenleiter TC. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 81:13403–13411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim ET, Oh SE, Lee YO, Gibson W, Ahn JH. 2009. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 83:12046–12056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. 2009. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182:851–859 [DOI] [PubMed] [Google Scholar]
- 81. Nijmeijer S, Leurs R, Smit MJ, Vischer HF. 2010. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J. Biol. Chem. 285:29632–29641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Beisser PS, Verzijl D, Gruijthuijsen YK, Beuken E, Smit MJ, Leurs R, Bruggeman CA, Vink C. 2005. The Epstein-Barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 79:441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Soni V, Cahir-McFarland E, Kieff E. 2007. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597:173–187 [DOI] [PubMed] [Google Scholar]