

Abstract
The low-density lipoprotein receptor (LDLR) is a critical determinant of plasma cholesterol levels that internalizes lipoprotein cargo via clathrin-mediated endocytosis. Here, we show that the E3 ubiquitin ligase IDOL stimulates a previously unrecognized, clathrin-independent pathway for LDLR internalization. Real-time single-particle tracking and electron microscopy reveal that IDOL is recruited to the plasma membrane by LDLR, promotes LDLR internalization in the absence of clathrin or caveolae, and facilitates LDLR degradation by shuttling it into the multivesicular body (MVB) protein-sorting pathway. The IDOL-dependent degradation pathway is distinct from that mediated by PCSK9 as only IDOL employs ESCRT (endosomal-sorting complex required for transport) complexes to recognize and traffic LDLR to lysosomes. Small interfering RNA (siRNA)-mediated knockdown of ESCRT-0 (HGS) or ESCRT-I (TSG101) components prevents IDOL-mediated LDLR degradation. We further show that USP8 acts downstream of IDOL to deubiquitinate LDLR and that USP8 is required for LDLR entry into the MVB pathway. These results provide key mechanistic insights into an evolutionarily conserved pathway for the control of lipoprotein receptor expression and cellular lipid uptake.
INTRODUCTION
The low-density lipoprotein receptor (LDLR), a plasma membrane protein, is essential for regulation of plasma lipoprotein levels. Mutations in this receptor are the main cause for familial hypercholesterolemia, a disease characterized by elevated plasma cholesterol levels and accelerated atherosclerosis (1–3). LDLR levels on the cell surface are modulated by transcriptional and posttranscriptional pathways. The primary transcriptional regulator of LDLR is sterol regulatory element-binding protein 2 (SREBP-2) (4). Two proteins regulate LDLR levels at the posttranscriptional level: IDOL (inducible degrader of the LDLR) and PCSK9 (proprotein convertase subtilisin/kexin type 9).
IDOL is an E3-ubiquitin ligase and promotes ubiquitination of the LDLR, thereby marking it for degradation (5). Expression of the IDOL gene is induced by the sterol-activated transcription factors liver X receptor α (LXRα) and LXRβ. IDOL-deficient cells exhibit markedly elevated levels of the LDLR protein under basal and sterol-depleted growth conditions and also manifest increased rates of LDL uptake. In addition, IDOL-null cells are unable to downregulate LDLR levels in response to synthetic LXR ligands (6). PCSK9 is a secreted factor that binds to the extracellular domain of LDLR and triggers its intracellular degradation (7–12). Although IDOL and PCSK9 share the same protein substrates (5, 13–15), PCSK9 retains its ability to induce LDLR degradation in IDOL-null cells, suggesting that IDOL and PCSK9 act in complementary but independent pathways (6).
The molecular mechanism by which IDOL accomplishes LDLR degradation is incompletely understood. IDOL interacts directly with the cytoplasmic tails of its target proteins in a sequence-specific manner and promotes their ubiquitination in cooperation with the UBE2D family of E2-ubiquitin-conjugating enzymes (16–18). However, the mechanism whereby ubiquitinated LDLR is recognized, the endocytic route that it follows to the lysosome, and whether IDOL and PCSK9 utilize common or distinct downstream degradation pathways are unknown. In this study, we define the cellular pathway for IDOL-mediated internalization and intracellular sorting of the LDLR.
MATERIALS AND METHODS
Reagents.
GW3965 was provided by T. Wilson (GlaxoSmithKline). Lipoprotein-deficient fetal bovine serum (LPDS) was from Intracell. MG132, bafilomycin A1, dynasore, filipin, and 5-(N,N-dimethyl)amiloride hydrochloride (DMA) were from Sigma. Reactive fluorescent dyes and antibiotics were from Invitrogen. Purified PCSK9 was provided by Jay Horton (University of Texas Southwestern Medical Center [UTSW], Dallas, TX).
Cell culture.
HeLa, HepG2, HEK293, mouse embryonic fibroblasts (MEFs), and peritoneal macrophages were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Omega Scientific) unless otherwise specified.
Primary peritoneal macrophages were obtained from thioglycolate-treated wild-type and IDOL knockout (KO) mice 4 days after injection (6). T-Rex CHO A7 cells were cultured as previously described (19). The embryonic stem (ES) cells were cultured in Glasgow minimum essential medium (GMEM) as previously described (6). GW3965, simvastatin, mevalonic acid, PCSK9 protein, and doxycycline were added to the medium for the times indicated in the figure legends.
Plasmids and constructs.
Human IDOL, human IDOL with a mutation in the RING domain (IDOL C387A), human FERM (residues 1 to 344), and ΔFERM (residues 345 to 445) were cloned into tagged vectors using Gateway technology (Invitrogen). All other constructs were previously described (5, 16, 19). Mutations were introduced using a QuikChange site-directed mutagenesis kit (Stratagene) and verified by DNA sequencing. Transfections were performed using Fugene (Roche Diagnostics) according to the manufacturer's instructions. Cells were harvested 24 to 48 h after transfection.
Antibodies.
Hybridomas expressing a rat monoclonal antibody against mouse IDOL (mIDOL), clone 10E7F10 (1:1,000 for Western blotting [WB]), were isolated and cultured in serum/protein-free medium, and immunoglobulins in the medium were purified on a GammaBind Plus Sepharose column (GE Healthcare). The purity of immunoglobulins was documented with Coomassie blue-stained SDS-polyacrylamide gels. Commercial IDOL antibodies were from Abcam (ab74562; 1:1,000 for WB) and Novus Biologicals (NBP1-56017; 1:500 for WB). Monoclonal rabbit anti-LDLR, clone EP1553Y (ab52818; 1:5,000 for WB) was from Abcam. Mouse anti-V5 antibody (R960-25; 1:5,000 for WB), horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (65-6120; 1:4,000 for WB), and mouse anti-TFRC (13-6800; 1:3,000 for WB) were from Invitrogen. HRP-conjugated goat anti-mouse IgG (115-035-146; 1:5,000 for WB) was obtained from Jackson ImmunoResearch. Mouse antihemagglutinin (anti-HA) antibody (MMS-101R-500; 1:5,000 for WB) was from Covance. Mouse anti-CHC (610499; 1:5,000 for WB) was from BD Transduction Laboratories. Rabbit anti-TSG101 (GTX118736), AMSH (GTX62646), and HGS (GTX101738) were purchased from Genetex (dilution of 1:1,000 for WB). Rabbit anti-USP8 (U2385; 1:1,000 for WB), HRP-conjugated rabbit anti-rat IgG (A5795; 1:2,000 for WB), and rabbit antiactin (A2066; 1:10,000 for WB) were from Sigma. Rabbit anti-ABCA1 (NB 400-105; 1:1,000 for WB) was from Novus Biologicals. Commercial antibodies were used according to the manufacturers' instructions.
Immunoblot analysis and immunoprecipitation.
Total cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 100 mM Tris-HCl, pH 7.4) supplemented with protease inhibitors (Roche Molecular Biochemicals). Lysates were cleared by centrifugation at 4°C for 10 min at 10,000 × g. Protein concentration was determined with a bicinchoninic acid (BCA) protein assay (Pierce) with bovine serum albumin (BSA) as a reference. Samples (10 to 60 μg) were separated on NuPAGE Bis-Tris gels (Invitrogen) and transferred to nitrocellulose. The membranes were probed sequentially with primary and secondary antibodies diluted in milk solution, and the bands were visualized with an ECL kit (GE Healthcare) using an ImageQuant LAS4000 (GE Healthcare). To immunoprecipitate LDLR-V5, lysates were prepared as described above, and equal amounts of protein of cleared lysate were incubated with 1 μg of anti-V5 rabbit monoclonal and protein G-Sepharose overnight. Subsequently, beads were washed four times with RIPA buffer supplemented with protease inhibitors. Proteins were eluted from the beads by boiling in protein sample buffer for 5 min. The biotinylation of membrane proteins was performed as described previously (6).
siRNA interference.
For HGS, USP8, and CHC silencing, HeLa cells were treated twice over 96 h with either control small interfering RNA (siRNA) duplex or specific siRNA duplex pool (Dharmacon) at a concentration of 45 nM or 80 nM, respectively, using Dharmafect-1 (Dharmacon). For TSG101 and AMSH and IDOL siRNA experiments, HeLa cells were transfected over 48 h with either 45 nM or 80 nM control siRNA duplex or specific siRNA duplex pools using Dharmafect-1 (Dharmacon).
In the last 12 to 24 h, the medium was replaced with lipoprotein-deficient medium containing simvastatin (5 μM) and mevalonic acid (100 μM) in presence or absence of GW3965 (1 μM).
Immunofluorescence microscopy.
T-Rex CHO A7 cells, HeLa cells, and HepG2 cells were plated in chamber slides (LabTekII; Thermo Scientific) and transfected as previously described (19) or treated with 10% lipoprotein-deficient serum (LPDS) and GW3965. For LDLR imaging, the cells were processed as previously described (19) using streptavidin-Alexa Fluor 568 (Invitrogen). The cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) and washed with PBS. Cells were permeabilized with 0.1% Triton and blocked with 5% normal goat serum and 1% BSA. Cells were incubated with antibodies in 5% normal goat serum and 1% BSA for 1 h, washed, and incubated with antibodies conjugated to Alexa Fluor 488 (A-11008 at 1:1,000; Invitrogen) or Alexa Fluor 555 (A-21434 at 1:1,000; Invitrogen) for 1 h. After samples were washed, cells were mounted in the presence of ProLong Gold Antifade Reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen). Images were collected with an LSM 510 confocal laser scanning microscope (Carl Zeiss) with a 63× oil objective with a 1.4 numerical aperture (63×/1.4). The frame size was 1,024 by 1,024 pixels. The manufacturer's software was used for data acquisition and for fluorescence profiles. The weighted colocalization coefficients were calculated using AIM (Carl Zeiss). Transferrin-Alexa Fluor 488 (Invitrogen) internalization was performed as previously described (20). The cells were visualized with an Axiovert 25 instrument (Carl Zeiss) with a 10×/0.25 or 32×/0.40 objective.
Time-lapse video microscopy.
HepG2 cells were plated in 35-mm glass-bottom dishes (P35G-1.0-14C; MatTek). Full-serum DMEM was changed to DMEM with 0.5% FBS, 5 μM simvastatin, and 100 μM mevalonic acid for 5 h. Cells were then treated overnight with dimethyl sulfoxide (DMSO) or 1 μM GW3965. Each sample was then treated with fluorescently labeled LDL (DiI-LDL) (L3482; Molecular Probes) at a final concentration of 5 μg/ml at the time of imaging. Images of live cells were recorded at room temperature (25°C) on a Leica DMI6000 inverted microscope with a 60× Leica objective with a Yokogawa CSU-X1 spinning-disk confocal scanner unit. The detector used for experiments was an electron-multiplying charge-coupled-device (EMCCD) camera (Andor), and fluorescently stained cells were excited with a 640-nm laser and a 405-nm laser. Image acquisition and analysis were carried out with custom routines written using ImageJ and MatLab. Confocal spinning-disk microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at the University of California, Los Angeles (UCLA).
Single-particle imaging and tracking.
The procedures for quantum dot (QD) labeling and live imaging have been described previously (21). Briefly, living cells previously exposed to tetracycline and biotin were incubated in phenol red-free Leibovitz's L-15 medium containing 10% FBS at 37°C with streptavidin QDs (Invitrogen) in QD binding buffer for 1 min. QDs emitting at 655 nm were used at a 1:10,000 dilution. Cells were monitored with a custom-made wide-field epifluorescence microscope equipped with an oil immersion objective (Nikon Plan Apo TIRF 60×/1.45), a Reliant 150 Select argon ion laser (excitation line, 488 nm), and a custom-made heating chamber. FF499-Di01-25 dichroic, FF01-655/15-25 (for QDs), and FF01-530/43-25 (for green fluorescent protein [GFP]) emission filters (Semrock) were used. A total of 250 consecutive frames were acquired with an integration time of 100 ms with an iXon X3 (Andor) electron-multiplying charge-coupled-device camera with the Andor Solis software (Andor). Tracking of single QDs was performed with MatLab (MathWorks, Natick, MA) using a custom-made macro that accounts for blinking in the fluorescence signal (21–23). First, fluorescent spots were detected by cross-correlating the image with a Gaussian model of the point spread function. A least-squares Gaussian fit was applied (around the local maximum above a threshold) to determine the center of each spot with a spatial accuracy of 10 to 20 nm (depending on the signal-to-noise ratio). Second, QD trajectories were assembled automatically by linking, from frame to frame, the centers of the fluorescent spots likely coming from the same QD. The association criterion was based on the assumption of free Brownian diffusion and took into account short blinking events. After completion of the process, a manual association step was performed in which QD trajectories of maximal length were assembled from smaller fragments separated by longer blinking events that were not taken into account by the automatic linking procedure. The trajectories showed in the movies were obtained with ImageJ (24). The concentration of streptavidin (strep)-QDs is largely in excess with respect of biotinylated LDLR (biotin-LDLR). A large number of accessible biotin-LDLR molecules on the plasma membrane are therefore expected to be labeled. In our analysis we monitored mainly the dynamics of blinking QDs, indicative of single LDLR molecules, and included only few permanently “on” QDs, possibly deriving from dimerization of two labeled LDLR molecules.
Quantitative analysis of diffusion coefficient.
The mean square displacement (MSD) analysis allows the calculation of the initial diffusion coefficient (D) of each particle (21, 23). Briefly, physical parameters can be extracted from each trajectory (x(t), y(t)) at time t by computing the MSD (25), determined from the following formula:
where xi and yi are the coordinates of a particle on frame i, dt is the time between two successive frames, N is the total number of frames of the trajectory, and n is the number of frames used to define the time interval ndt over which the displacement is averaged. This function enables the analysis of the lateral dynamics on short (initial diffusion coefficient) and long (types of motion) time scales. Different types of motion can be distinguished from the time dependence of the MSD. The initial diffusion coefficient (D) is determined by fitting the initial two to five points of the MSD against time plot with the equations MSD(t) = 4D2-5 t + b and b = 4σx2, where σx is the spot localization accuracy in one direction. The cumulative probability, C(d), of the diffusion coefficient D is defined as the probability that a random D is less a specific value d and can be expressed as CD(d) = P(D < d). We compared cumulative probability distributions and median instead of mean values because D values were spread over several orders of magnitude. The position vector (r) for frame i is defined as follows: ri = (xi2 + yi2)1/2.
Velocity (v) values for actively transported molecules were obtained by fitting the MSD against time plot with the equation for directed motion (25): MSD(t) = 4Dt + (vt)2. Comparisons between the different cumulative distributions were performed by the nonparametric Kolmogorov-Smirnov test. A P value of <0.05 was considered statistically significant.
Electron microscopy.
HepG2 cells were transfected with plasmids expressing a biotin acceptor peptide fused to LDLR with the mutation Y807C (AP-Y807C LDLR), endoplasmic reticulum-localized biotin ligase (BirA-ER) (19), and pSLIK-hygromycin expressing mIdol (ratio of 1:1:1). After 24 h cells were treated with 10 μM biotin in lipoprotein-deficient medium in the presence of simvastatin and mevalonic acid. After 12 h the cells were labeled at 4°C with streptavidin 10-nm colloidal gold conjugate (5 μg/ml; Molecular Probes) in Dulbecco's PBS (DPBS) containing 1% (wt/vol) BSA for 10 min. At the end of incubation, excess labeling reagent was removed by gently washing cells three times with warm DPBS. Thereafter, cells were treated with 2 μg/ml doxycycline for the times indicated in the figure legends and fixed. After fixation in 2.5% glutaraldehyde and 2% formaldehyde in 100 mM sodium cacodylate at 4C overnight, 1% osmium tetroxide was added for 1 h at room temperature, followed by three washes with distilled water and then 2% uranyl acetate at 4°C in the dark overnight. The next day uranyl acetate was removed, and the cells were rinsed three times with distilled water before undergoing sequential dehydration with acetone (20%, 30%, 50%, 70%, and 100% for 30 min each time). Cells were gradually infiltrated with Spurr's resindiluted in acetone (25%, 50%, and 75% for 1 h each) and finally in 100% Spurr's resin for 1 h. Next day, the plastic coverslips were transferred to fresh 100% Spurr's resin and incubated at 70°C overnight to polymerize the resin. Sections were cut using a diatome diamond knife with a Leica UCT Ultramicrotome in the California NanoSystems Institute (UCLA). Sections (50 nm thick) were placed on 200-mesh copper grids. Sections were then stained with Reynolds lead citrate for 5 min. Sections were imaged using a 100CX JEOL electron microscope at 80 kV in the Brain Research Institute (UCLA).
Statistical analysis.
Colocalization analysis results are expressed as means ± standard deviations. Statistical analyses were performed with one-way analysis of variance using GraphPad Prism, version 4.0 (GraphPad Software). Comparisons between the different cumulative distributions were performed by a Kolmogorov-Smirnov test. A P value of <0.05 was considered statistically significant.
RESULTS
IDOL blocks LDL association with the plasma membrane.
Previous work has shown that activation of LXR inhibits LDL uptake (6). To visualize this process in cells, we treated HepG2 cells that stably express an LDLR-GFP fusion protein with vehicle or LXR agonist overnight and then incubated them with DiI-labeled LDL. The association of LDL with the cells was followed for 2.5 h with live-cell imaging (Fig. 1). Much of the LDLR-GFP in the stably transformed HepG2 cells is intracellular, due to supraphysiological levels of expression. However, after 2.5 h of incubation, cells treated with LXR agonist had no LDLR-GFP at the cell surface and virtually no LDL association. These data indicate that the LXR-IDOL pathway has a major effect on plasma membrane LDLR levels.
Fig 1.

The LXR-IDOL pathway blocks LDL association with LDLR at the plasma membrane in living cells. (A) HepG2 cells stably overexpressing GFP-LDLR were treated with GW3965 (1 μM) overnight and then incubated with DiI-LDL. The association of DiI-LDL was determined in living cells by spinning-disc confocal microscopy. Still images at time zero and 2.5 h are shown. The entire movie shown in Video S1 in the supplemental material covers 3 h. (B) Immunoblot analysis of IDOL protein expression in transiently transfected 293T cells with monoclonal antibody (10E7F10). Cells were treated with or without MG132 for 4 h prior to harvest. (C) Immunoblot analysis of endogenous IDOL protein expression with antibody 10E7F10 in wild-type (WT) and IDOL-deficient cells. Primary mouse peritoneal macrophages were starved in lipoprotein-deficient medium and then treated with GW3965 overnight and MG132 for 5 h. T, time.
Detection of endogenous IDOL with a monoclonal antibody.
IDOL is a rare and unstable protein due to autoubiquitination and proteasomal degradation (16), and the lack of an effective IDOL antibody has been a major limitation. Although we previously described polyclonal antibodies capable of detecting adenovirally expressed IDOL (5), we have been unable to consistently visualize endogenous or transfected protein with these reagents. Other IDOL antibodies have been described that recognize bands of ∼51 kDa (26, 27); however, the major proteins detected by these reagents are not responsive to LXR agonist or MG132 and were present in IDOL-null cells (see Fig. S1 in the supplemental material). We developed a monoclonal antibody that detected transfected IDOL in 293T cells and T-Rex CHO A7 cells (Fig. 1B; see also Fig. S2 in the supplemental material), as well as endogenous IDOL in murine macrophages and human HepG2 cells (Fig. 1C; see also Fig. S2). Endogenous IDOL has an apparent molecular mass of 47 kDa, is responsive to LXR agonist (GW3965), and is stabilized by MG132. IDOL has been reported to respond to fibroblast growth factor 21 (FGF-21) (27); however, we did not observe an effect of FGF-21 on endogenous IDOL protein in our system. Most importantly, the 47-kDa protein is absent in IDOL-null cells (Fig. 1C).
LDLR-IDOL membrane interaction.
The localization of native IDOL in intact cells has not been established. To address this issue, we used a biotin ligase (BirA)/streptavidin-labeling technique to visualize the cell-surface pool of LDLR tagged with biotin acceptor peptide (AP-LDLR) (19). We verified construct expression by immunoblotting and confirmed that both wild-type IDOL and a carboxyl-terminal IDOL-GFP fusion protein were able to degrade AP-LDLR (see Fig. S2B and C in the supplemental material). Amino-terminal GFP-IDOL fusion proteins were inactive. As expected, the IDOL C387A RING mutant (5) was unable to degrade the LDLR (see Fig. S2B). To determine localization, we transfected T-Rex CHO A7 cells (which lack functional LDLRs) with AP-LDLR and IDOL C387A-GFP. We predicted that IDOL C387A would retain the ability to interact with LDLR through its FERM domain but not degrade it, thereby allowing us to detect colocalization. In the absence of LDLR, IDOL C387A was uniformly distributed in the cytoplasm (Fig. 2A). However, when LDLR was coexpressed, IDOL was almost exclusively localized to the plasma membrane. This result was confirmed with V5-IDOL C387A (Fig. 2B). The prominent intracellular structure observed using IDOL C387A-GFP was likely an artifact of GFP overexpression since it was not observed with IDOL containing a V5 tag.
Fig 2.

LDLR recruits IDOL to the plasma membrane in a FERM-dependent manner. T-Rex cells were transfected with BirA-ER and the indicated constructs and then labeled with streptavidin-Alexa Fluor 568 (10 μg/ml), fixed, and stained with V5 antibody. Nuclei were counterstained with DAPI (blue). Representative confocal images are shown. Fluorescence profiles from the sections indicated by the arrows are shown in the lower panels. (A) Localization of AP-LDLR with IDOL C387A-GFP. (B) Localization of AP-LDLR with IDOL C387A-V5. (C) Localization of AP-K6R K20R C29A LDLR (K6/K20R C29A LDLR) with IDOL-V5. (D) Localization of AP-LDLR with ΔFERM IDOL-GFP. (E) Localization of AP-LDLR with FERM-GFP. (F) Localization of AP-Δ792 LDLR with IDOL-GFP.
We also analyzed the localization of wild-type IDOL in the presence of LDLR. To circumvent the problem of IDOL degrading the LDLR, we employed a mutant LDLR (K6R K20R C29A LDLR) that is insensitive to ubiquitination (5). Both IDOL-GFP and V5-IDOL colocalized with the mutant LDLR at the plasma membrane (Fig. 2C and data not shown). Thus, the cellular localization of IDOL is dependent on the availability of its target protein, and the IDOL-LDLR interaction is independent of the ability of the LDLR to be ubiquitinated.
Previous work showed that the IDOL FERM domain binds directly to lipoprotein receptor tails (17). We confirmed that IDOL utilizes the FERM domain to interact with LDLR in cells using confocal microscopy. The IDOL FERM domain alone (residues 1 to 344) colocalized with AP-LDLR at the plasma membrane, whereas an IDOL protein lacking the FERM domain (ΔFERM; residues 345 to 445) did not (Fig. 2D and E). The interaction of the FERM domain is weaker than that of intact IDOL likely because IDOL is a functional dimer, whereas the FERM domain protein is monomeric (16). Furthermore, IDOL was unable to interact with AP-LDLR that lacks the cytoplasmic tail (AP-Δ792 LDLR), confirming that the LDLR tail mediates the interaction with IDOL (Fig. 2F). The results of the localization studies of Fig. 2 are quantitated in Fig. S3 in the supplemental material.
Changes in diffusion of single LDLR molecules upon interaction with the Idol submembrane scaffold.
We employed single-particle tracking in combination with live-cell imaging to visualize the behavior of LDLR at the plasma membrane in the presence or absence of IDOL. T-Rex CHO A7 cells were transfected with AP-LDLR and IDOL-GFP, and biotinylated AP-LDLR was then visualized with streptavidin-quantum dots (QDs) emitting at 655 nm. We recorded real-time changes in the behavior of LDLR in regions of the plasma membrane in association with IDOL-GFP clusters (Fig. 3A; see also Video S1 in the supplemental material). Most of the LDLR molecules on the plasma membrane colocalized with submembrane IDOL, and their mobility was limited. However, in a limited number of cases, we detected single LDLR molecules exhibiting faster diffusion while drifting away from IDOL clusters (Fig. 3B to D). The diffusion coefficient (D) extrapolated from the mean square displacement (MSD)-against-time plot was found to increase up to three orders of magnitude. This sudden change of mobility depending on protein-protein interactions is reminiscent of results reported for gephyrin and the glycine receptor (28, 29).
Fig 3.

IDOL induces clustering and restricts the mobility of LDLR in the plasma membrane. Single-particle tracking using QDs was used to study the interaction between LDLR and IDOL. (A) Living T-Rex cells cotransfected for 36 h with AP-LDLR, BirA-ER, and IDOL-GFP, pretreated with tetracycline and biotin, and surface-labeled with streptavidin-QDs emitting at 655 nm. Scale bar, 5 μm. (B) Magnification of the white region in panel A shows real-time changes in the trajectory of an LDLR molecule in relation to IDOL submembrane clusters. Scale bar, 1 μm. (C to D) Position vector and MSD analysis of the LDLR molecule indicated by colored arrows in panel B. Colors correspond to changes in LDLR mobility, which are dependent on the interaction with IDOL. The MSD was calculated over the time indicated according to the colors in panel C. (E) Examples of cells overexpressing AP-LDLR alone, AP-LDLR and IDOL-GFP, or AP-Δ792 LDLR and IDOL-GFP. Scale bar, 2 μm. (F and G) QD-labeled LDLR trajectories corresponding to images in panel E. Scale bar, 1 μm. (H) The cumulative probability distributions of diffusion coefficients showing decreased mobility of LDLR in cells cotransfected with IDOL-GFP. No significant differences were observed between cells expressing LDLR alone and AP-Δ792 LDLR together with IDOL.
Simultaneous dual-view recordings allowed us to monitor the diffusion of single IDOL clusters colocalizing with LDLR molecules (see Video S2 in the supplemental material). The trajectories of the IDOL cluster and the LDLR molecule were almost identical and overlapping (30). To quantify the extent to which IDOL affects the lateral diffusion of LDLR molecules, we compared the D distributions of LDLR in the absence and presence of cotransfected IDOL-GFP (Fig. 3E to I; Table 1). The median D value of LDLR alone was found to be one order of magnitude higher than in cells overexpressing IDOL-GFP (Table 1). The D distributions of AP-Δ792 LDLR did not differ significantly in the presence or absence of IDOL (Fig. 3H and I and Table 1).
Table 1.
Reported median D values of LDLR alone and in the absence and presence of cotransfected IDOL-GFP
| Condition | nb | Lateral diffusion of LDLRa |
||
|---|---|---|---|---|
| Dmedian (μm2 s−1) | Δmax | P | ||
| LDLR | 481 | 1.99 ×10−3 | ||
| LDLR + IDOL-GFP | 331 | 6.42 ×10−4 | 3.5 × 10−1 | ≤10−4 |
| Δ792 LDLR + IDOL-GFP | 138 | 2.02 ×10−3 | 9.0 × 10−2 | 3.4 × 10−1 |
On the plasma membrane of T-rex cells. Dmedian, median diffusion value; Δmax, maximum difference in cumulative fraction; P, Kolmogorov-Smirnov test P value calculated using Δmax as the statistic.
Number of trajectories analyzed.
A clathrin-independent pathway for lysosomal degradation.
Multiple studies have documented that PCSK9 induces lysosome-dependent LDLR degradation (8, 10, 15). We compared the effects of proteasome and lysosome inhibitors on LDLR degradation induced by PCSK9 or IDOL in parallel experiments. Protein expression of ATP binding cassette transporter A1 (ABCA1), a known target of LXR, was used as a control for LXR activation. As shown in Fig. 4A and B, the lysosome inhibitor bafilomycin A1 prevented both PCSK9- and IDOL-mediated LDLR degradation. We also visualized trafficking of the LDLR after LXR stimulation using confocal microscopy. After 4 h of LXR agonist treatment, LDLR began to accumulate in a lysosome-associated membrane protein 1 (LAMP-1)-positive compartment (Fig. 4C). After 24 h, the small amounts of LDLR remaining in the cells colocalized entirely with LAMP-1.
Fig 4.

IDOL induces late-endosome/lysosome localization of LDLR coincident with degradation. (A) HepG2 cells were treated with GW3965 (GW; 1 μM) or PCSK9 (5 μg/ml) for 1 h, and then proteasome inhibitor (25 μM MG132 [MG]) or lysosome inhibitor (50 nM bafilomycin [Baf]) was added for an additional 5 h. The levels of ABCA1 and LDLR were determined by immunoblotting. Similar results were obtained in three independent experiments. Numbers to the right of the blots are molecular weight markers. (B) HeLa cells were processed and analyzed as described for panel A. (C) HeLa cells were cultured in 10% LPDS medium for 8 h and then treated with GW3965 (1 μM) for the indicated times. Cells were immunostained with LDLR and LAMP-1 antibodies. Nuclei were counterstained with DAPI (blue). Representative confocal images are shown. T, time; O/N, overnight.
Clathrin-mediated endocytosis is a major route of entry for a broad array of cargo, including LDL (31, 32). Clathrin-independent pathways for internalization include those mediated by caveolae and macropinocytosis (33). As a first step to determine the involvement of these pathways in IDOL action, we tested the effects of dynasore (an inhibitor of dynamin GTPase activity essential for the formation of clathrin-coated pits and caveolae) (34, 35), filipin (an inhibitor of the lipid raft/caveola pathway) (36–39), and dimethyl amiloride (which inhibits macropinocytosis) (40). Unexpectedly, none of these treatments had a prominent effect on IDOL-mediated LDLR degradation in ES cells, macrophages, or HeLa cells (Fig. 5A and data not shown). Neither the LXR agonist nor the other inhibitors affected LDLR levels in IDOL-null ES cells (Fig. 5B).
Fig 5.

IDOL induces LDLR internalization independent of clathrin, caveolae, or macropinocytosis. (A) Wild-type (WT) ES cells were grown in sterol depletion medium (10% LPDS with 5 μM simvastatin and 100 μM mevalonic acid) for 16 h. Cells were then pretreated with GW3965 (1 μM) for 1 h, and subsequently dynasore (Dyn; 80 μM), filipin (Fil; 1 μg/ml), or 5-(N,N-dimethyl) amiloride hydrochloride (DMA; 100 μM) was added for the indicated times. Surface proteins were collected and analyzed as described in Materials and Methods. Results from replicate experiments are quantitated below each blot. (B) IDOL-null (IKO) ES cells were analyzed as described for panel A.
We also used siRNA-mediated knockdown to inhibit clathrin-dependent endocytosis. siRNA against clathrin heavy chain (CHC) reduced CHC protein levels and inhibited the ability of cells to internalize transferrin (Fig. 6A; see also Fig. S4 in the supplemental material). Consistent with published work (7, 41, 42), CHC knockdown also inhibited PCSK9-mediated LDLR degradation. However, CHC had no effect on IDOL-dependent LDLR degradation (Fig. 6A). Additional studies indicated that caveolae were also dispensable for IDOL-mediated LDLR degradation. Cavin-1 is an essential component of caveolae (43, 44). Treatment with LXR agonist decreased membrane LDLR levels to similar extents in both wild-type and Cav-1-null MEFs (Fig. 6B). Thus, IDOL-mediated LDLR degradation, in contrast to PCSK9, does not depend on endocytic pathways involving clathrin or caveolae.
Fig 6.

IDOL stimulates LDLR internalization through a clathrin- and caveola-independent pathway. (A) HeLa cells were transfected with siRNA targeting CHC (clathrin heavy chain) or a universal control siRNA or the transfection reagent only (mock) for 96 h as described in Materials and Methods. Then the cells were cultured in 10% LPDS medium for 8 h and treated with GW3965 (1 μM) or PCSK9 (5 μg/ml) overnight. Expression of ABCA1, LDLR, and CHC was assessed by immunoblotting. Endogenous transferrin receptor (TfR) and actin levels were also determined as loading controls for membrane and intracellular lysates, respectively. (B) Wild-type and cavin-1 KO MEFs were cultured in 10% LPDS medium for 16 h and then treated with GW3965 (1 μM) for the indicated times. Membrane lysates were analyzed by immunoblotting. Similar results were obtained in two independent experiments. (C) Visualization of IDOL-dependent LDLR trafficking to MVBs and lysosomes. HepG2 cells were transduced with an AP-Y807C LDLR expression vector along with control or tetracycline-inducible IDOL expression vectors. After 12 h of serum, AP-Y807C LDLR was labeled with biotin. Cells were then treated with doxycycline to induce IDOL and labeled with streptavidin gold beads for 2 to 6 h. Representative electron micrographs are shown.
Visualizing the IDOL-LDLR degradation pathway by electron microscopy.
To visualize the fate of LDLR receptors ubiquitinated by IDOL at the plasma membrane, we employed electron microscopy. HepG2 cells were transduced with a vector expressing AP-Y807C LDLR, which cannot undergo clathrin-dependent endocytosis in coated pits but can be still be degraded by IDOL (see Fig. S2C in the supplemental material). Cells were simultaneously transduced with control or doxycycline-inducible IDOL expression vectors. AP-Y807C LDLR was labeled with biotin, and cells were then treated with doxycycline to induce IDOL and labeled with streptavidin-gold beads for 2 to 6 h. In the absence of doxycycline or IDOL expression vector, all gold particles were present at the cell surface and were not observed intracellularly (data not shown). In contrast, the presence of IDOL stimulated the uptake of gold-labeled LDLR into the cell and its localization in multivesicular bodies (MVBs) (Fig. 6C) (45, 46). By 6 h, some gold particles could be found in lysosomes in cells expressing IDOL. In the absence of IDOL, we did not observe gold in either MVBs or lysosomes at any time point. These data strongly suggest that IDOL employs the MVB pathway to sort ubiquitinated LDLR to the lysosome for degradation.
The ESCRT pathway is required for IDOL-mediated LDLR degradation.
The ESCRT (endosomal-sorting complex required for transport) machinery mediates the sorting of ubiquitinated membrane cargo destined for the lysosome (47, 48). To determine whether the ESCRT pathway is involved in IDOL action, we used siRNA to silence key components. HGS, part of the ESCRT-0 complex, plays a role in the selection of ubiquitinated cargo at the endosomal membrane (49–51). TSG101, a subunit of ESCRT-I, acts downstream and is involved in interactions with HGS and ubiquitinated cargo (50, 52). We also analyzed two deubiquitinases, AMSH and USP8, previously reported to function in the multivesicular body (MVB) pathway. siRNA knockdown of IDOL blocked LXR-induced LDLR degradation, serving as a positive control (Fig. 7A). Remarkably, silencing of HGS completely blocked the effect of LXR agonist on LDLR protein levels in both the plasma membrane and intracellular pools (Fig. 7B). Knockdown of TSG101 did not block LDLR removal from the membrane but led to the accumulation of LDLR in the intracellular pool (Fig. 7C), consistent with its point of action downstream of HGS. Knockdown of USP8 also blocked IDOL-dependent degradation, but depletion of AMSH had no effect (Fig. 7C and D). We also confirmed that knockdown of the USP8 inhibited IDOL-dependent degradation of the very-low-density lipoprotein receptor (VLDLR), suggesting that the MVB pathway is likely to be the common route of degradation for all IDOL targets.
Fig 7.

IDOL facilitates LDLR degradation by shuttling it into the MVB protein-sorting pathway. (A to D) HeLa cells were transfected with indicated siRNAs for 48 or 96 h as indicated in Materials and Methods, followed by GW3965 (1 μM) treatment in lipoprotein-deficient medium for the indicated times. Membrane and intracellular cell lysates were prepared and analyzed by immunoblotting. Efficiency of knockdown was confirmed by immunoblotting using polyclonal anti-HGS (B), anti-TSG101 (C), anti-AMSH (C), and anti-USP8 (D) antibodies. Endogenous transferrin receptor (TfR) and actin levels were also measured as controls for membrane and intracellular lysates, respectively. Blots are representative of three independent experiments. *, specific band.
We also analyzed LDLR distribution in siRNA-treated cells by confocal microscopy. Depletion of HGS, TSG101, or USP8 led to a marked accumulation of intracellular LDLR and prevented its trafficking to the lysosome, as indicated by the lack of colocalization with LAMP-1 (Fig. 8).
Fig 8.

Silencing of the MVB pathway blocks IDOL-mediated LDLR degradation and localization of the LDLR in late endosomes/lysosomes. HeLa cells were transfected with the indicated siRNAs for 48 or 96 h, followed by GW3965 (GW; 1 μM) treatment in lipoprotein-deficient medium for 6 h. Cells were then fixed and stained with LDLR and LAMP-1 antibodies. Nuclei were counterstained with DAPI (blue). Representative confocal images are shown. Scr, scrambled siRNA; siHGS, siRNA targeting HGS; siTSG101, siRNA targeting TSG101; siUSP8, siRNA targeting USP8.
In parallel, we determined the effect of knockdown of ESCRT proteins on PCSK9-mediated LDLR intracellular sorting. Consistent with recently reported results (41), the silencing of HGS or TSG101 did not affect PCSK9-mediated LDLR degradation (Fig. 9). Furthermore, we found that, in contrast to IDOL, PCSK9 does not require USP8 (Fig. 9). These results implicate the MVB pathway in IDOL-dependent LDLR ubiquitination and trafficking from the plasma membrane to the lysosome.
Fig 9.
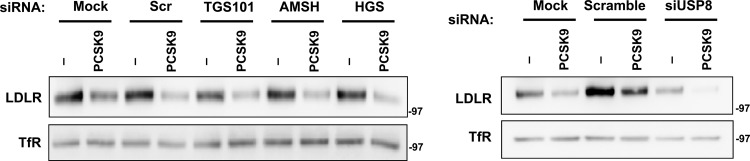
Inactivation of MVB pathway does not affect PCSK9-driven LDLR degradation. HeLa cells were transfected with the indicated siRNA for 48 or 96 h as described in the legend of Fig. 7, followed by PCSK9 (5 μg/ml) or BSA (Ctrl) treatment in lipoprotein-deficient medium for 12 h. Membrane cell lysates were prepared, and proteins were analyzed by immunoblotting.
Target deubiquitination by USP8 is critical for IDOL-dependent degradation.
Since USP8 is a ubiquitin-specific protease, we hypothesized that it might function in the IDOL pathway to deubiquitinate the LDLR and thereby allow it to proceed into the MVB pathway for lysosomal degradation. To test this idea, we first analyzed the IDOL-dependent ubiquitination status of the LDLR upon depletion of USP8. Cells were treated with the lysosomal inhibitor bafilomycin A1 in order to prevent LDLR degradation. We found that USP8 knockdown led to the accumulation of higher-molecular-weight LDLR species, specifically in the presence of IDOL (Fig. 10A). Immunoprecipitation of ubiquitinated proteins followed by immunoblotting for LDLR confirmed that inhibition of USP8 increases IDOL-dependent LDLR ubiquitination (Fig. 10B). These data implicate USP8 as a key mechanistic component of the IDOL degradation pathway.
Fig 10.

USP8 is required for deubiquitination of LDLR downstream of IDOL action in the MVB pathway. (A) 293T cells were transfected with a control siRNA (scramble) or siRNA targeting USP8. After 24 h, the cells were transfected with V5-tagged LDLR, HA-tagged ubiquitin (HA-Ub), and human IDOL expression vectors. The siRNA transfection was then repeated after 24 h, and bafilomycin was added to the medium for 6 h. Total lysates were analyzed by immunoblotting. *, specific band. (B) V5-tagged LDLR in the cell lysates was immunoprecipitated (IP) using anti-V5 antibody followed by immunoblotting for HA-ubiquitin. Blots are representative of two independent experiments.
DISCUSSION
LDLR protein levels are regulated by both PCSK9 and IDOL. PCSK9 binds to the extracellular domain of LDLR and induces its internalization in clathrin-coated pits. In contrast, IDOL triggers degradation of the LDLR through ubiquitination of its cytoplasmic tail. In this study, we showed that IDOL and PCSK9 employ distinct mechanisms to degrade the LDLR in the lysosome. We defined interactions between IDOL and LDLR at the plasma membrane that lead to LDLR internalization via a clathrin- and caveola-independent mechanism. We further showed that LDLR ubiquitination by IDOL leads to recognition by ESCRT complexes, deubiquitination by USP8, and subsequent MVB-dependent lysosomal degradation. These results provide important new insights into an evolutionarily conserved pathway for the control of lipoprotein receptor expression and lipid uptake.
IDOL contains two functional domains. The RING domain supports target ubiquitination in conjunction with UBE2D, and the FERM domain is responsible for target recognition (16–18). The physical interactions between IDOL and LDLR in living cells have not been characterized in detail previously. We showed here that IDOL is actively recruited to the plasma membrane by its target. This interaction is dependent on the IDOL FERM domain and the LDLR cytoplasmic tail but independent of ubiquitination. Real-time single-particle tracking revealed that IDOL promotes clustering of LDLR on the plasma membrane and dramatically reduces its mobility. The isolated IDOL FERM domain also localizes to the plasma membrane; however, this interaction is weaker than that observed with the full-length protein. IDOL dimerization is likely important for optimal binding and receptor clustering, and the dimerization interface lies within the RING domain (16).
Our study has clarified the pathway for IDOL-mediated target internalization. The classical mode of receptor-mediated endocytosis of macromolecules involves uptake through clathrin-coated pits, as exemplified by endocytosis of LDL or transferrin (32, 53). PCSK9 has been shown to induce LDLR internalization in clathrin-coated pits, similar to the binding of lipoprotein ligands (7, 41, 54–56), and we initially suspected that IDOL would also use this pathway. Surprisingly, however, we found that IDOL induces LDLR internalization through a pathway independent of a clathrin, caveolae, or macropinocytosis. Thus, the two major regulators of LDLR protein abundance rely on distinct mechanisms to accomplish LDLR removal from the plasma membrane. There is precedence for membrane receptors using different routes of internalization in different contexts. For example, at high doses of ligand, the epidermal growth factor receptor (EGFR) is ubiquitinated and endocytosed through a clathrin-independent, lipid raft-dependent route (57–59). We speculate that IDOL-mediated LDLR ubiquitination may serve as a signal for the formation of plasma membrane invaginations instead of clathrin-coated pits, possibly using a different set of adaptor proteins.
Once internalized, certain membrane proteins are sorted from early endosomes to MVBs, specialized structures that traffic proteins to lysosomes for degradation (60). The addition of ubiquitin to target proteins is postulated to be a critical signal for their entry into the MVB pathway (61). ESCRT complexes recognize this ubiquitin tag and sort the protein into luminal vesicles in MVBs (48, 62–64). Our data indicate that the IDOL degradation pathway employs the ESCRT machinery. Depletion of HGS or TGS101 (components of the ESCRT-0 and ESCRT-I complexes, respectively) blocks IDOL-dependent LDLR degradation. We also directly visualized IDOL-dependent localization of LDLR to MVBs by electron microscopy. In contrast to IDOL, it was recently reported that PCSK9-dependent LDLR degradation does not require the ESCRT pathway (41), and our data confirm those results. These findings are not surprising given that PCSK9 does not promote LDLR ubiquitination.
Studies of other receptor degradation pathways have shown that ubiquitin must be removed from the cargo by ESCRT-III-associated deubiquitinating enzymes prior to cargo internalization into luminal vesicles (59, 62, 65–67). The deubiquitinases USP8 and AMSH have been implicated in degradation of the EGFR (67–70). Our studies have identified USP8 but not AMSH as a critical factor that deubiquitinates LDLR downstream of IDOL. Knockdown of USP8 in the presence of IDOL blocks LDLR degradation and leads to an accumulation of ubiquitinated LDLR.
In conclusion, we have elucidated the cellular pathway by which IDOL redirects LDLR trafficking and accomplishes its degradation. Given the importance of LDLR in human lipid metabolism and cardiovascular disease risk, a better understanding of pathways for LDLR degradation could lead to new therapeutic opportunities. Based on our findings that the mechanisms for IDOL- and PCSK9-induced degradation are distinct, IDOL and PCSK9 inhibitors might have additive effects in a therapeutic setting.
Supplementary Material
ACKNOWLEDGMENTS
P.T. is an Investigator of the Howard Hughes Medical Institute. This work was supported by NIH grants HL-066088 and HL-090553, American Heart Association Postdoctoral Fellowship 11POST6600001, the Italian Ministry for Education, Flagship Project NANOMAX, the EU Seventh Framework Programme (FP7/2007-2013, 284464), and the Ente Cassa di Risparmio di Firenze. Confocal laser scanning microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA, supported with funding from NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and NSF Major Research Instrumentation grant (CHE-0722519).
We thank Douglas Black assistance with confocal microscopy and Jay Horton (UTSW) for the gift of PCSK9 protein.
We have no conflicts of interest related to this work.
Footnotes
Published ahead of print 4 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01716-12.
REFERENCES
- 1. Russell DW, Schneider WJ, Yamamoto T, Luskey KL, Brown MS, Goldstein JL. 1984. Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell 37:577–585 [DOI] [PubMed] [Google Scholar]
- 2. Brown MS, Goldstein JL. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232:34–47 [DOI] [PubMed] [Google Scholar]
- 3. Tolleshaug H, Hobgood KK, Brown MS, Goldstein JL. 1983. The LDL receptor locus in familial hypercholesterolemia: multiple mutations disrupt transport and processing of a membrane receptor. Cell 32:941–951 [DOI] [PubMed] [Google Scholar]
- 4. Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. 1993. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. U. S. A. 90:11603–11607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zelcer N, Hong C, Boyadjian R, Tontonoz P. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scotti E, Hong C, Yoshinaga Y, Tu Y, Hu Y, Zelcer N, Boyadjian R, de Jong PJ, Young SG, Fong LG, Tontonoz P. 2011. Targeted disruption of the Idol gene alters cellular regulation of the low-density lipoprotein receptor by sterols and liver X receptor agonists. Mol. Cell. Biol. 31:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nassoury N, Blasiole DA, Tebon Oler A, Benjannet S, Hamelin J, Poupon V, McPherson PS, Attie AD, Prat A, Seidah NG. 2007. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8:718–732 [DOI] [PubMed] [Google Scholar]
- 8. Maxwell KN, Fisher EA, Breslow JL. 2005. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc. Natl. Acad. Sci. U. S. A. 102:2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG. 2004. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279:48865–48875 [DOI] [PubMed] [Google Scholar]
- 10. Qian YW, Schmidt RJ, Zhang Y, Chu S, Lin A, Wang H, Wang X, Beyer TP, Bensch WR, Li W, Ehsani ME, Lu D, Konrad RJ, Eacho PI, Moller DE, Karathanasis SK, Cao G. 2007. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J. Lipid Res. 48:1488–1498 [DOI] [PubMed] [Google Scholar]
- 11. Horton JD, Cohen JC, Hobbs HH. 2009. PCSK9: a convertase that coordinates LDL catabolism. J. Lipid Res. 50(Suppl):S172–S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lalanne F, Lambert G, Amar MJ, Chetiveaux M, Zair Y, Jarnoux AL, Ouguerram K, Friburg J, Seidah NG, Brewer HB, Jr, Krempf M, Costet P. 2005. Wild-type PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J. Lipid Res. 46:1312–1319 [DOI] [PubMed] [Google Scholar]
- 13. Hong C, Duit S, Jalonen P, Out R, Scheer L, Sorrentino V, Boyadjian R, Rodenburg KW, Foley E, Korhonen L, Lindholm D, Nimpf J, van Berkel TJ, Tontonoz P, Zelcer N. 2010. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J. Biol. Chem. 285:19720–19726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. 2008. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem. Biophys. Res. Commun. 375:69–73 [DOI] [PubMed] [Google Scholar]
- 15. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. 2008. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283:2363–2372 [DOI] [PubMed] [Google Scholar]
- 16. Zhang L, Fairall L, Goult BT, Calkin AC, Hong C, Millard CJ, Tontonoz P, Schwabe JW. 2011. The IDOL-UBE2D complex mediates sterol-dependent degradation of the LDL receptor. Genes Dev. 25:1262–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Calkin AC, Goult BT, Zhang L, Fairall L, Hong C, Schwabe JW, Tontonoz P. 2011. FERM-dependent E3 ligase recognition is a conserved mechanism for targeted degradation of lipoprotein receptors. Proc. Natl. Acad. Sci. U. S. A. 108:20107–20112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorrentino V, Scheer L, Santos A, Reits E, Bleijlevens B, Zelcer N. 2011. Distinct functional domains contribute to degradation of the low density lipoprotein receptor (LDLR) by the E3 ubiquitin ligase inducible Degrader of the LDLR (IDOL). J. Biol. Chem. 286:30190–30199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zou P, Ting AY. 2011. Imaging LDL receptor oligomerization during endocytosis using a co-internalization assay. ACS Chem. Biol. 6:308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM. 2006. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol. Biol. Cell 17:4300–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bannai H, Levi S, Schweizer C, Dahan M, Triller A. 2006. Imaging the lateral diffusion of membrane molecules with quantum dots. Nat. Protoc. 1:2628–2634 [DOI] [PubMed] [Google Scholar]
- 22. Bonneau S, Dahan M, Cohen LD. 2005. Single quantum dot tracking based on perceptual grouping using minimal paths in a spatiotemporal volume. IEEE Trans. Image Process 14:1384–1395 [DOI] [PubMed] [Google Scholar]
- 23. Ehrensperger MV, Hanus C, Vannier C, Triller A, Dahan M. 2007. Multiple association states between glycine receptors and gephyrin identified by SPT analysis. Biophys. J. 92:3706–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sbalzarini IF, Koumoutsakos P. 2005. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 151:182–195 [DOI] [PubMed] [Google Scholar]
- 25. Saxton MJ, Jacobson K. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399 [DOI] [PubMed] [Google Scholar]
- 26. Ishibashi M, Masson D, Westerterp M, Wang N, Sayers S, Li R, Welch CL, Tall AR. 2010. Reduced VLDL clearance in Apoe−/− Npc1−/− mice is associated with increased Pcsk9 and Idol expression and decreased hepatic LDL-receptor levels. J. Lipid Res. 51:2655–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Do HT, Tselykh TV, Makela J, Ho TH, Olkkonen VM, Bornhauser BC, Korhonen L, Zelcer N, Lindholm D. 2012. Fibroblast growth factor-21 (FGF21) regulates low-density lipoprotein receptor (LDLR) levels in cells via the E3-ubiquitin ligase Mylip/Idol and the Canopy2 (Cnpy2)/Mylip-interacting saposin-like protein (Msap). J. Biol. Chem. 287:12602–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meier J, Vannier C, Serge A, Triller A, Choquet D. 2001. Fast and reversible trapping of surface glycine receptors by gephyrin. Nat. Neurosci. 4:253–260 [DOI] [PubMed] [Google Scholar]
- 29. Calamai M, Specht CG, Heller J, Alcor D, Machado P, Vannier C, Triller A. 2009. Gephyrin oligomerization controls GlyR mobility and synaptic clustering. J. Neurosci. 29:7639–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brown A. 2000. Slow axonal transport: stop and go traffic in the axon. Nat. Rev. Mol. Cell Biol. 1:153–156 [DOI] [PubMed] [Google Scholar]
- 31. McNiven MA, Thompson HM. 2006. Vesicle formation at the plasma membrane and trans-Golgi network: the same but different. Science 313:1591–1594 [DOI] [PubMed] [Google Scholar]
- 32. Motley A, Bright NA, Seaman MN, Robinson MS. 2003. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162:909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansen CG, Nichols BJ. 2009. Molecular mechanisms of clathrin-independent endocytosis. J. Cell Sci. 122:1713–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10:839–850 [DOI] [PubMed] [Google Scholar]
- 35. Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. 2009. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J. Biol. Chem. 284:28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh RD, Puri V, Valiyaveettil JT, Marks DL, Bittman R, Pagano RE. 2003. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol. Biol. Cell 14:3254–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orlandi PA, Fishman PH. 1998. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 141:905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rothberg KG, Ying YS, Kamen BA, Anderson RG. 1990. Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J. Cell Biol. 111:2931–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ros-Baro A, Lopez-Iglesias C, Peiro S, Bellido D, Palacin M, Zorzano A, Camps M. 2001. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc. Natl. Acad. Sci. U. S. A. 98:12050–12055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O. 2001. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 75:11166–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Huang Y, Hobbs HH, Cohen JC. 2012. Molecular characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9)-mediated degradation of the LDLR. J. Lipid Res. 53:1932–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. 2006. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Invest. 116:2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. 2008. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 8:310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu L, Pilch PF. 2008. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J. Biol. Chem. 283:4314–4322 [DOI] [PubMed] [Google Scholar]
- 45. Murk JL, Humbel BM, Ziese U, Griffith JM, Posthuma G, Slot JW, Koster AJ, Verkleij AJ, Geuze HJ, Kleijmeer MJ. 2003. Endosomal compartmentalization in three dimensions: implications for membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 100:13332–13337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, Bellen HJ. 2005. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 170:127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wollert T, Yang D, Ren X, Lee HH, Im YJ, Hurley JH. 2009. The ESCRT machinery at a glance. J. Cell Sci. 122:2163–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raiborg C, Stenmark H. 2009. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458:445–452 [DOI] [PubMed] [Google Scholar]
- 49. Bache KG, Brech A, Mehlum A, Stenmark H. 2003. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 162:435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bache KG, Raiborg C, Mehlum A, Stenmark H. 2003. STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J. Biol. Chem. 278:12513–12521 [DOI] [PubMed] [Google Scholar]
- 51. Raiborg C, Malerod L, Pedersen NM, Stenmark H. 2008. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 314:801–813 [DOI] [PubMed] [Google Scholar]
- 52. Rusten TE, Vaccari T, Stenmark H. 2012. Shaping development with ESCRTs. Nat. Cell Biol. 14:38–45 [DOI] [PubMed] [Google Scholar]
- 53. Liu AP, Aguet F, Danuser G, Schmid SL. 2010. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol. 191:1381–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown MS, Herz J, Goldstein JL. 1997. LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature 388:629–630 [DOI] [PubMed] [Google Scholar]
- 55. Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340 [DOI] [PubMed] [Google Scholar]
- 56. Brown MS, Goldstein JL. 1976. Receptor-mediated control of cholesterol metabolism. Science 191:150–154 [DOI] [PubMed] [Google Scholar]
- 57. Chen H, De Camilli P. 2005. The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proc. Natl. Acad. Sci. U. S. A. 102:2766–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. 2005. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. U. S. A. 102:2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haglund K, Dikic I. 2012. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 125:265–275 [DOI] [PubMed] [Google Scholar]
- 60. Miller K, Beardmore J, Kanety H, Schlessinger J, Hopkins CR. 1986. Localization of the epidermal growth factor (EGF) receptor within the endosome of EGF-stimulated epidermoid carcinoma (A431) cells. J. Cell Biol. 102:500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hicke L, Dunn R. 2003. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 19:141–172 [DOI] [PubMed] [Google Scholar]
- 62. Hurley JH. 2008. ESCRT complexes and the biogenesis of multivesicular bodies. Curr. Opin. Cell Biol. 20:4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Williams RL, Urbe S. 2007. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 8:355–368 [DOI] [PubMed] [Google Scholar]
- 64. Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. 2007. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449:735–739 [DOI] [PubMed] [Google Scholar]
- 65. McCullough J, Row PE, Lorenzo O, Doherty M, Beynon R, Clague MJ, Urbe S. 2006. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 16:160–165 [DOI] [PubMed] [Google Scholar]
- 66. Kyuuma M, Kikuchi K, Kojima K, Sugawara Y, Sato M, Mano N, Goto J, Takeshita T, Yamamoto A, Sugamura K, Tanaka N. 2007. AMSH, an ESCRT-III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct. Funct. 31:159–172 [DOI] [PubMed] [Google Scholar]
- 67. Row PE, Prior IA, McCullough J, Clague MJ, Urbe S. 2006. The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J. Biol. Chem. 281:12618–12624 [DOI] [PubMed] [Google Scholar]
- 68. Bowers K, Piper SC, Edeling MA, Gray SR, Owen DJ, Lehner PJ, Luzio JP. 2006. Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J. Biol. Chem. 281:5094–5105 [DOI] [PubMed] [Google Scholar]
- 69. Row PE, Liu H, Hayes S, Welchman R, Charalabous P, Hofmann K, Clague MJ, Sanderson CM, Urbe S. 2007. The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J. Biol. Chem. 282:30929–30937 [DOI] [PubMed] [Google Scholar]
- 70. Ma YM, Boucrot E, Villen J, Affar EB, Gygi SP, Gottlinger HG, Kirchhausen T. 2007. Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J. Biol. Chem. 282:9805–9812 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.