

Abstract
The immunosuppressant and anticancer drug rapamycin works by inducing inhibitory protein complexes with the kinase mTOR, an important regulator of growth and proliferation. The obligatory accessory partner of rapamycin is believed to be FK506-binding protein 12 (FKBP12). Here we show that rapamycin complexes of larger FKBP family members can tightly bind to mTOR and potently inhibit its kinase activity. Cocrystal structures with FKBP51 and FKBP52 reveal the modified molecular binding mode of these alternative ternary complexes in detail. In cellular model systems, FKBP12 can be functionally replaced by larger FKBPs. When the rapamycin dosage is limiting, mTOR inhibition of S6K phosphorylation can be enhanced by FKBP51 overexpression in mammalian cells, whereas FKBP12 is dispensable. FKBP51 could also enable the rapamycin-induced hyperphosphorylation of Akt, which depended on higher FKBP levels than rapamycin-induced inhibition of S6K phosphorylation. These insights provide a mechanistic rationale for preferential mTOR inhibition in specific cell or tissue types by engaging specific FKBP homologs.
INTRODUCTION
The kinase mTOR (mammalian or mechanistic target of rapamycin) integrates growth factor-dependent stimuli, amino acid availability, and cellular energy levels to coordinate cell growth and proliferation. This phosphatidylinositide 3-kinase (PI3K)-related kinase is part of at least two distinct multiprotein complexes (mTORC1 and mTORC2) that display different substrate specificities, depending on the presence of the scaffolding protein Raptor or Rictor. While Raptor recruits the mTORC1 substrates p70 S6 kinase and 4E-BP1, two key regulators of protein translation, Rictor mediates the activation of Akt, a key “survival” kinase that is hyperactive in many cancers. Altogether, the PI3K/Akt/mTOR signaling pathway is crucial for the regulation of cell cycle progression and protein metabolism (1, 2).
mTORC1 can be specifically inhibited by rapamycin, a small molecule originally isolated from the bacterium Streptomyces hygroscopicus (3). This compound is known to bind to FKBP12, and the resulting complex specifically interacts with the FRB (FK506-rapamycin binding) domain of mTOR, allosterically inhibiting the kinase activity (4). Importantly, the phosphorylation of 4E-BP1 is only partially inhibited in many cell types, while the critical T389 phosphorylation of p70 S6 kinase is totally abolished (5). The FKBP12-rapamycin complex does not interact with mTORC2, and Akt kinase activation is not affected by acute rapamycin treatment (6). However, mTORC2 assembly is rapamycin sensitive in susceptible cell lines upon long-term treatment (7), and this was shown to be responsible for the metabolic side effects of rapamycin in vivo (8).
Rapamycin is an FDA-approved drug used after organ transplantations that exerts its immunosuppressive action in humans by modulating T, B, or dendritic cell responses (9). Rapamycin and pharmacokinetically improved analogs thereof (called rapalogs) also inhibit the growth of certain cancer cell lines. These compounds are approved or in clinical trials for the treatment of various cancer types and have been shown to be especially effective in advanced renal cell carcinoma (10). Rapamycin also prevents pathological protein aggregation or accumulation in animal models of neurodegenerative disorders which was attributed in part to its autophagy-promoting activity. Finally, rapamycin treatment was shown to prolong the life spans of numerous organisms, including mice (11). Unfortunately, the clinical use of rapamycin is generally restricted in the latter applications, as chronic rapamycin treatment is associated with severe side effects (12).
Rapamycin does not exclusively interact with FKBP12 but rather binds with high affinity to most members of the FK506-binding protein (FKBP) family (13), resulting in the inhibition of their peptidyl-prolyl cis-trans isomerase activity. For FKBP38, Bai and colleagues reported direct inhibitory effects on mTOR (14) but these results were challenged by others (15). Recently, the larger FKBP51 protein was shown to function as a scaffold protein to facilitate the dephosphorylation of Akt by the phosphatase PHLPP (16).
At present, the pharmacological effects of rapamycin are almost exclusively interpreted and discussed in the context of a complex with the prototypical protein FKBP12. Since many of the 14 known human FKBP family members can form tight complexes with rapamycin (13), we set out to investigate whether the pharmacology of rapamycin is indeed restricted to FKBP12.
MATERIALS AND METHODS
Plasmids and other materials.
Plasmids pRK7-HA-S6K1-WT and pRK-5-myc-Raptor were purchased from Addgene, Cambridge, MA (catalog numbers 8984 and 1859) (17, 18). Plasmids for expression of pcDNA3-FLAG-mTOR-WT and pcDNA3-FLAG-mTOR-S2035T were a kind gift of Jie Chen (19).
pcDNA3 constructs for expression in mammalian cells were generated with primer pairs 5′-CGGAATTCATGGACTACAAGGACGATGACGATAAGATGGGAGTGCAGGTGGAAACCATC-3′ and 5′-GGCTCGAGTCATTCCAGTTTTAGAAGCTCCACA-3′ (FLAG_FKBP12), 5′-CGGAATTCATGACTACTGATGAAGGTGC-3′ and 5′-GCAGTCGACTCTCCTTTGAAATCAAGGAGC-3′ (FLAG_FKBP51), and 5′-CCGAATTCATGACAGCCGAGGAGATG-3′ and 5′-GTCGACTCATTCTCCCTTAAACTCAAACAACTC-3′ (FLAG_FKBP52) (underlining indicates restriction sites). For the expression of human FKBP12 in shFKBP12-SH-SY5Y cells, silent mutations destroying the shFKBP12 recognition sequence were generated by using 5′-CTACACCGGAATGCTGGAGGACGGCAAAAAATTTG-3′ and 5′-CAAATTTTTTGCCGTCCTCCAGCATTCCGGTGTAG-3′ as primers. The amplified cDNAs were cloned into the pcDNA3 vector at the EcoRI and Xho or XbaI and KpnI restriction sites.
Plasmids for the expression of enhanced green fluorescent protein (EGFP)-FKBP and glutathione S-transferase (GST)–FRB domain fusion proteins were generated by using an EGFP-specific pair of primers (5′-GGCGCCATGGTGAGCAAGGGCGAGGA-3′ and 5′-CCATGGCCTTGTACAGCTCGTCCATGCCGAG-3′) with the vector pEGFP-N1 (Clontech Laboratories Inc., Mountain View, CA) as the template. All EGFP cDNAs including EGFP-4E-BP1 cDNA were amplified and cloned into pProEx-Hta vectors at the KasI and NcoI or EcoRI and XhoI restriction sites. The resulting modified vector, pProEx-Hta-His-EGFP, was used for the cloning and expression of EGFP fusion proteins in Escherichia coli.
The expression plasmid for GST-FRB was generated by cloning cDNA of the mTOR FRB domain (amino acids [aa] 2025 to 2114) into pGEX-4T2 (Invitrogen, Carlsbad, CA). Plasmids for the expression of His-tagged FKBPs in E. coli were prepared as described before (13). For expression in mammalian cell lines, cDNA of FKBPs was cloned into pcDNA3 (Invitrogen), and for expression in yeast cells, it was cloned into pYX223 (Invitrogen).
All cell culture media (Dulbecco's modified Eagle's medium [DMEM], Dulbecco's phosphate-buffered saline [D-PBS], fetal bovine serum [FBS], penicillin-streptomycin, and human insulin solution) were purchased from Gibco-Invitrogen. All other chemicals were from Sigma-Aldrich (Steinheim, Germany) or from Carl Roth (Karlsruhe, Germany). F1706 was a kind gift from Astellas Pharma Inc. (Tokyo, Japan).
Antibodies against the hemagglutinin (HA) tag, the FLAG tag, and the myc tag were purchased from Sigma-Aldrich, as were the corresponding peptides for immunoprecipitate (IP) elution. Antibodies against FKBP12 were from Abcam (Cambridge, MA). Antibodies against FKBP51 and FKBP52 were from Bethyl Laboratories (Montgomery, TX), while antibodies against S6 kinase, pan-Akt, 4E-BP1, phospho-T389 S6K, phospho-S473 Akt, and phospho-T37/46 4E-BP1 were from Cell Signaling (Beverly, MA).
ON TARGET plus small interfering RNA (siRNA) targeting FKBP12 and a nontargeting control were purchased from Dharmacon (Lafayette, CO). Nickel-nitrilotriacetic acid agarose was from Qiagen (Hilden, Germany), and rapamycin was from Cfm (Marktredwitz, Germany).
For yeast experiments, Saccharomyces cerevisiae strain BY4742 (accession no. Y12941; MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 YNL135c::kanMX4; deletion, YNL135c) was obtained from EUROSCARF (Frankfurt, Germany).
GST pulldown assay.
FKBP51 FK1 or FKBP52 FK1 concentrations of 0.5, 1.0, and 1.5 μM were each combined with 1 μM GST-FRB and 1 μM rapamycin (where indicated) in 100 μl PBS and incubated for 30 min at room temperature. A 50-μl volume of glutathione resin (Miltenyi Biotech, Bergisch Gladbach, Germany) was added, mixed well, and incubated on a shaker for 10 min. The resin was collected, washed three times each with 500 μl PBS, and finally boiled in 20 μl SDS sample buffer. The supernatant was subjected to SDS-gel electrophoresis and analyzed by Coomassie brilliant blue staining.
Cell culture, transfection, and stimulation.
HEK293 and HeLa cells were maintained in DMEM supplemented with 10% FBS and a 1% antibiotic (penicillin and streptomycin) solution. For transfection, cells were plated in six-well plates and subjected to the Lipofectamine 2000 or Lipofectamine LTX transfection procedure as recommended by the manufacturer (Invitrogen). For siRNA transfection, standard protocols provided by Dharmacon (Thermo Fisher Scientific, Lafayette, CO) were used together with the transfection reagent DharmaFECT1.
After 24 h, the medium was changed to starvation medium (DMEM without supplementation). After another 16 h, cells were stimulated by the addition of DMEM supplemented with FBS and 100 nM insulin for 60 min. Rapamycin or other compounds were added where indicated.
Cell lysis and immunoprecipitation.
After stimulation, cells were washed twice with Tris-buffered saline (TBS) and lysed in lysis buffer {40 mM HEPES (pH 7.5), 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 0.5 mM orthovanadate, 50 mM NaF, 0.3% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) or 1% (vol/vol) Triton X-100, 1% (vol/vol) protease inhibitor mix (Sigma-Aldrich)} by shaking at 1,000 rpm at 4°C for 20 min. After 20 min of centrifugation at 14.000 rpm and 4°C, the supernatant was collected and total protein was determined by Bradford assay (Bio-Rad, Munich, Germany). The mTOR activity and phosphorylation status of downstream targets were assessed as described with the appropriate phospho-specific antibodies (20).
Cell lysates were combined with FLAG affinity resin (Sigma-Aldrich) and incubated under mild agitation for 2 h at 4°C. Beads were pelleted and washed four times with lysis buffer. Finally, beads were directly boiled after the addition of an equal amount of SDS sample buffer for 5 min. Extracts were subjected to SDS-PAGE and immunoblotting.
In vitro binding and kinase assays.
The fluorescence resonance energy transfer (FRET)-based FRB domain-binding assay and the mTOR activity assay (LanthaScreen TR-FRET assay; Invitrogen) were performed as recommended by the manufacturer. Kinase assays with mTORC1 were performed as described previously (21). Briefly, myc-Raptor was transiently expressed in HEK293 cells. Transfected cells were starved overnight in DMEM, followed by 40 min in PBS, and stimulated with DMEM–10% FBS for 60 min. After lysis in CHAPS-containing buffer, myc affinity beads were added to the lysate and the IP mixture was incubated for 2 h under agitation. Beads were collected and washed five times with lysis buffer. Beads were extracted with myc-peptide in TBS. IP eluate was directly added to recombinant EGFP–4E-BP1 or immunoprecipitated inactive HA-S6K in kinase buffer (20 mM Tris HCl [pH 7.4], 10 mM MgCl2, 0.2 mM ATP). After 30 min of incubation at 37°C, the reaction was stopped by the addition of SDS sample buffer and boiling for 5 min. Finally, the samples were analyzed by SDS-PAGE and immunoblotting.
Yeast cell culture.
Yeast cells were maintained in selective medium (containing 0.5% [wt/vol] ammonium sulfate, 0.17% [wt/vol] yeast nitrogen base, 2% [wt/vol] glucose, 20 mg/liter uracil, 20 mg/liter l-histidine, 50 mg/liter l-lysine, 60 mg/liter l-leucine) or on selective plates (medium plus 1% [wt/vol] agar). Transfection of pYX242-FKBP expression plasmids was performed in accordance with a standard transformation protocol (22). Positive clones were identified by using selective plates lacking l-histidine. For the yeast dilution assays, yeast cells were grown in liquid selective medium lacking l-histidine and supplemented with 0.2% galactose to induce FKBP expression. Cultures at an optical density at 600 nm (OD600) of 0.5 were serially diluted 1 to 10 in sterile water, and 4 μl of each dilution was spotted onto SGal-His plates (synthetic defined yeast medium with 0.2% galactose lacking l-histidine) supplemented with the indicated final concentration of rapamycin. The plates were incubated for 3 to 4 days at 30°C. For growth curves, yeast was grown in 96-well plates at 30°C in 100 μl of medium. OD600 was measured every 180 min in a Tecan reader.
Crystal structure determination.
The complexes were crystallized by the vapor diffusion method at 291 K. The FKBP51 FK1-rapamycin-FRB domain complex was crystallized with either 25% polyethylene glycol 3350–0.1 M NaCl–0.1 M HEPES-NaOH (pH 7.5) or 0.1 M citric acid (pH 3.5)–2 M (NH4)2SO4 (low pH) as a precipitant. For the FKBP52 FK1-rapamycin-FRB domain complex, 0.1 M Bis-Tris (pH 6.5)–1.95 M (NH4)2SO4 was used. The latter two conditions are from the Hampton Research Index screen. Diffraction data were collected at 100 K and a 0.98-Å wavelength at European Synchrotron Radiation Facility beamline ID29. For data processing and evaluation, the CCP4i software suite (23) was used. Data integration was done with MOSFLM (24), data reduction was carried out by SCALA (25), and molecular replacement was performed with Molrep with the crystal structure of the ternary complex FKBP12–C15-(R)-methylthienyl rapamycin–FRB domain (PDB entry 3FAP) as the initial model (26). Model bias was reduced by autobuilding with ArpWarp (27). The models were completed manually with Coot (28) and refined with Refmac (29). The final models have plausible stereochemistry with more than 92% of the residues in the most favored region of the Ramachandran plot.
Cellular Akt pS473 HTRF and mTOR pS2448 HTRF assays.
The commercially available HTRF phospho-Akt (Ser473) and mTOR phospho-(S2448) cell-based assays (Cisbio, Codolet, France) were used in accordance with the manufacturer's recommendations. Briefly, wild-type SH-SY5Y, FKBP12 knockdown SH-SY5Y (30), or HeLa cells were grown in 96-well plates, starved for 24 h, and stimulated with 10% FBS in the presence of the indicated rapamycin or torin-1 concentrations for 60 min. Cells were lysed in 50 μl of assay lysis buffer, and 16 μl of the lysate was transferred to a white 384-well plate. After the addition of both 2 μl of d2-labeled anti-Akt or d2-labeled anti-mTOR antibody and K-labeled anti-phospho-Akt or K-labeled anti-phospho-mTOR antibody to each well, the mixtures were incubated at room temperature for 4 h or overnight. With a Tecan GENios Pro reader, emission levels at 620 and 665 nm were determined. FRET efficiency was assessed by calculation of emission ratios (665 nm/620 nm). For rapamycin competition experiments, wild-type SH-SY5Y and HeLa cells were treated with FKBP inhibitors for 60 min in addition to the above-described rapamycin treatment, followed by cell lysis and assay readout as described above.
RESULTS
Numerous FKBP-rapamycin complexes bind and inhibit mTOR.
To assess whether other FKBP-rapamycin complexes could bind to mTOR as well, we first performed pulldown assays with the purified FRB domain of mTOR and the FK1 domains of FKBP51 and FKBP52. Both FKBP homologs bound specifically to the FRB domain in the presence but not in the absence of rapamycin (Fig. 1A). To characterize such alternative complexes in more detail, we used a FRET-based protein-protein dimerization assay. All of the EGFP-FKBPs tested displayed tight binding to the GST-FRB domain in a clearly rapamycin-dependent manner (Table 1; see Fig. S1 in the supplemental material). The 50% effective concentrations (EC50s) ranged from 3.8 nM for FKBP12 to 25.5 nM for FKBP52. Identical results were observed for the full-length proteins and for the FK1 domains of the larger FKBP52 protein and FKBP52, confirming that the additional domains do not influence the interaction with the FRB domain. The value obtained for FKBP12 is in good accordance with previously published data (31).
Fig 1.
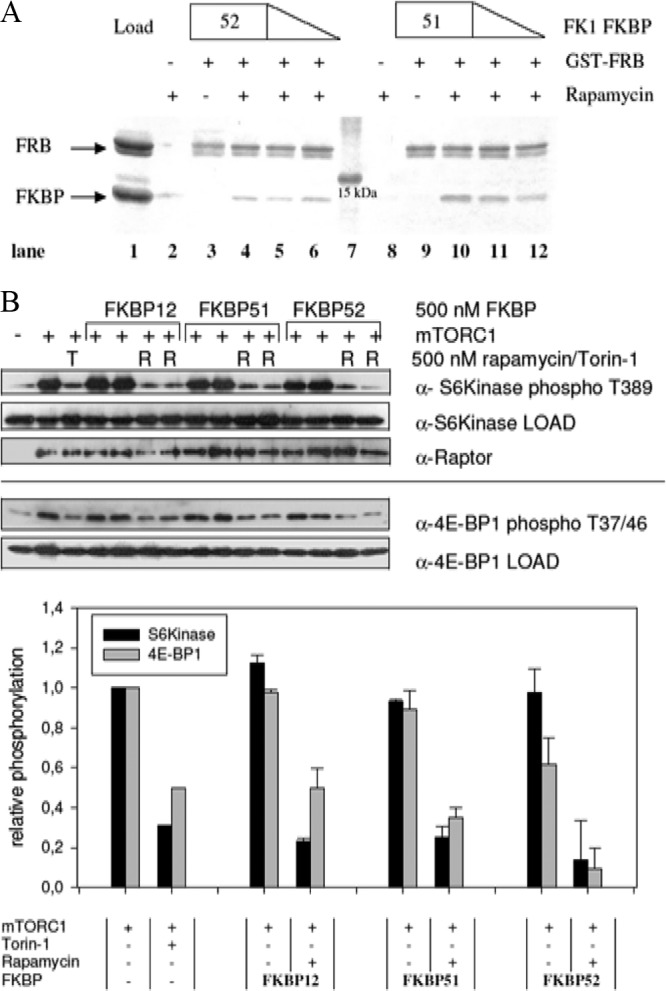
Large FKBPs bind to the FRB domain of mTOR and inhibit its kinase activity in vitro. (A) Rapamycin dependence of the interaction of FKBP51 and FKBP52 with the FRB domain of mTOR. The binding of the isolated FK1 domains of FKBP51 and FKBP52 to the FRB domain was tested in a GST pulldown assay. FKBP51 FK1 and FKBP52 FK1 at 0.5, 1.0, and 1.5 μM were incubated with a 1 μM concentration of the GST-FRB domain on GST beads in the absence (lanes 3 and 9) or presence of 1 μM rapamycin (lanes 4 to 6 and 10 to 12). After washing, matrix-bound proteins were analyzed by SDS-PAGE. Lane 1 is the GST-FRB domain and FKBP52 FK1 at 1.5 μM each. Lanes 2 and 8 are controls for unspecific binding of 1.5 μM FKBP51 FK1 or FKBP52 FK1 in the absence of the GST-FRB domain. Lane 7 is a molecular size standard. (B) mTORC1 kinase activity is inhibited by large FKBPs in vitro. Kinase assays were performed by incubating immunoprecipitated mTORC1 with EGFP–4E-BP1 or HA-S6 kinase at 37°C in the presence or absence of 500 nM FKBP, 500 nM rapamycin (R), or 100 nM torin-1 (T). After 30 min, the samples were boiled and 4E-BP1 phospho-T37/46 and S6 kinase phospho-T389 signal intensities were assessed by Western blotting (top), quantified, and normalized to those of a sample treated with mTORC1 only (bottom). Results for FKBPs are displayed as standard deviations from the mean.
Table 1.
Ternary-complex formation and inhibition of mTOR by FKBP homologs
| Protein | EC50a,b (nM) | IC50a,c (nM) |
|---|---|---|
| FKBP12 | 3.8 ± 0.6 | 0.69 ± 0.17 |
| FKBP12.6 | 5.6 ± 0.9 | 4.79 ± 1.76 |
| FKBP13 | 6.7 ± 1.6 | 2.48 ± 0.82 |
| FKBP25 | 4.3 ± 0.9 | 2.61 ± 0.57 |
| FKBP51 | 25 ± 6.7 | 1.95 ± 0.36 |
| FKBP52 | 25.5 ± 5.8 | 1.68 ± 0.51 |
| FKBP51 FK1 | 10.5 ± 1.8 | |
| FKBP52 FK1 | 26.2 ± 5.6 |
The values shown are means and standard deviations.
EC50s for ternary GST-FRB/rapamycin/EGFP-FKBP complex formation are shown (see Fig. S1 in the supplemental material).
IC50s for inhibition of the kinase activity of purified, truncated mTOR by different FKBP-rapamycin complexes are shown (see Fig. S2 in the supplemental material).
To address the possible inhibitory activity of large FKBPs on the kinase activity of mTOR, an in vitro kinase assay was performed. This assay determines the activity of a heterologously expressed, truncated mTOR protein by assessing the phosphorylation of the mTOR substrate 4E-BP1. The data show that all of the FKBPs examined exert a potent inhibitory effect on mTOR kinase activity at low nanomolar rapamycin concentrations (Table 1; see Fig. S2 in the supplemental material). For FKBP12, FKBP51, and FKBP52, 50% inhibitory concentrations (IC50s) below 2 nM were measured, which is well below the clinically achieved concentrations of rapamycin (around 6 nM [32]).
To mimic cellular conditions more closely, we next performed an in vitro kinase assay with the intact mTOR complex 1. As a readout, we determined the phosphorylation status of 4E-BP1 and S6 kinase at the sites that directly reflect mTOR activity. In the presence of rapamycin FKBP12, FKBP51 and FKBP52 abolished S6 kinase and 4E-BP1 phosphorylation to the same extent as the ATP-competitive mTOR inhibitor torin-1 (33) (Fig. 1B). Thus, the large FKBP homologs can functionally substitute for FKBP12 with regard to mTOR inhibition in vitro.
Crystal structures of alternative ternary complexes.
The available apo structures of the larger FKBPs revealed substantial deviations in the 80s loop, the main direct interface between FKBP12 and the FRB domain. This indicated potential clashes for larger FKBPs in putative ternary complexes that could preclude larger FKB-rapamycin complexes from efficiently engaging the FRB domain (26). To clarify the molecular basis of ternary complexes for larger FKBPs, we solved the cocrystal structures of the FKBP51 and FKBP52 FK1 domains with rapamycin and the FRB domain of mTOR at resolutions of 1.45 and 1.8 Å, respectively (see Table S1 in the supplemental material). The overall architecture of the complexes was very similar to that of the known FKBP12-containing complex (Fig. 2A) (26). The macrocyclic compound rapamycin was located in the center of the complex, interacting with the FK1 and FRB domains through distinct, separate segments.
Fig 2.

Crystal structures of the complexes of FKBP51 and FKBP52 with rapamycin and mTOR. (A) Ribbon representation of the ternary minimal complexes of FKBP12 (2FAP) and of the FK1 domains (aa 1 to 143) of FKBP51 and FKBP52 with rapamycin and the FRB domain of mTOR. The FKBP domains were superimposed and are in red (FKBP12), pale blue (FKBP51 FK1), and yellow (FKBP52 FK1), respectively. The 40s and 80s loops, which directly contact the FRB domain, are indicated. For clarity, only a single rapamycin molecule (dark blue sticks) and a single FRB domain (green ribbon) are shown. (B) Direct protein-protein contacts in the ternary complexes of FKBP12 (left) and FKBP51 (right). The orientation is the same as in panel A. Interacting residues are shown as sticks. van der Waals interactions are gray dashed lines, and hydrogen bonds are black lines. FRB domains and FKBPs are shown as backbone traces. (C) Network of ordered water molecules at the FKBP-FRB domain interface. The proteins are shown as surface representations, and the water molecules are shown as red spheres. Hydrogen bonds are indicated by dashed lines. Water molecules that directly mediate interactions between the FRB domain and FKBPs are circled.
Direct interactions between the larger FKBPs and the FRB domain were surprisingly sparse. Similar to FKBP12, most of the direct contacts were formed by the 40s and 80s loops of the FKBPs with the α4 helix of the FRB domain. Unlike FKBP12, however, only minor contacts were observed between the α1-α2 loop of the FRB domain and FKBP52, and none were observed with FKBP51. In the FKBP12 complex, most contacts with the FRB domain were formed by the 80s loop (T85 to I91) (26). These contact areas were substantially reduced in the FKBP52 complex (A116, P119, and P120 contact R2042 and V2094 of the FRB domain) and almost eliminated in FKBP51 (P120-V2094 and A116-R2042 contacts). In contrast, FKBP51 formed several additional contacts between its 40s loop (R73, E75 to V78) and the C terminus of α4 of the FRB domain (D2102, Y2105, H2106, and R2109), including three novel hydrogen bonds. The S78-Y2105 hydrogen bond was also observed in the complex with FKBP52. In summary, the main protein-protein interface with the FRB domain was shifted from the 80s loop in FKBP12 to the 40s loop in FKBP51, while FKBP52 formed fewer direct contacts through both its 40s and 80s loops (Fig. 2B and C).
Interestingly, shape complementarity (SC) at the contact interface between the FRB domain and FKBP51 and FKBP52 was higher (SC values of 0.675 and 0.697, respectively) than with FKBP12 (SC value of 0.423). These values are, however, lower than those typically seen in constitutive complexes (e.g., hemoglobin SC value of 0.74) (34). In addition, numerous well-ordered water molecules were resolved at the interface between the FRB domain and FKBP51 in the 1.45-Å resolution structure, suggesting that this extended hydrogen bond network might contribute substantially to complex stability (Fig. 2C).
Alternative functional ternary complexes are formed in eukaryotic cells.
To study the pharmacological relevance of alternative FKBP-rapamycin complexes in eukaryotic cells, we first employed a yeast model lacking the endogenous FKBP12 homolog Fpr1p, the only cytoplasmic yeast FKBP (35). This yeast strain is insensitive to rapamycin. In this strain, we overexpressed human FKBPs to evaluate their ability to reconstitute sensitivity (Fig. 3A). The overexpression of both the human FKBP51 and FKBP52 FK1 domains reduced cell growth in the presence of rapamycin, but the sensitivity was less pronounced than that obtained by the overexpression of human FKBP12. However, full-length FKBP52 restored rapamycin sensitivity to the same extent as FKBP12. The ability of FKBP51 FK1 domain to restore the inhibition of yeast proliferation by rapamycin was also observed in growth curve assays (see Fig. S3 in the supplemental material). Full-length FKBP51 could not be assessed in this growth assay since it was toxic alone and substantially repressed yeast proliferation even in the absence of rapamycin.
Fig 3.

TOR-FKBP interactions in eukaryotic cells. (A) Overexpression of FKBP homologs restores rapamycin sensitivity in yeast cells. Yeast strain ΔFPR1p, lacking FKBP12, was transformed with plasmids encoding human FKBP12, FKBP51 FK1, FKBP52 FK1, or full-length FKBP52 under the control of the Gal promoter. A 1:10 dilution series of the resulting strains was spotted onto yeast minimal medium plates lacking histidine with 0.2% galactose as the carbon source with or without supplementation with the indicated concentrations of rapamycin and grown for 3 to 4 days at 30°C. (B) mTOR interacts with FKBP12, FKBP51, and FKBP52 in a rapamycin-dependent manner. HEK293 cells were transfected with a FLAG-mTOR or FLAG-mTOR-S2035T expression plasmid. At 30 min prior to lysis, rapamycin (Rap), FK506 (FK) (final concentration, 25 nM each), or the dimethyl sulfoxide vehicle (veh.) was added. The cell lysates were subjected to immunoprecipitation with anti-FLAG antibody, and the eluates were analyzed by immunoblotting. (C) FKBP12 and FKBP51 interact with the mTORC1 component Raptor. HEK293 cells were transfected with myc-Raptor and FLAG-FKBPs, starved, and stimulated with FBS in the absence or presence of 20 nM rapamycin for 60 min. Cell lysates were subjected to immunoprecipitation with anti-FLAG antibody. Beads were washed four times with CHAPS-containing lysis buffer and eluted by boiling with SDS sample buffer for 5 min. Supernatants were analyzed by SDS-PAGE and immunoblotting. Expression of FKBPs was induced by SG medium.
We next analyzed mammalian cells for a rapamycin-mediated interaction of mTOR and FKBPs. In coimmunoprecipitation experiments with overexpressed mTOR, endogenous FKBP12, FKBP51, and FKBP52 were clearly coprecipitated in the presence of rapamycin, while the related FKBP ligand FK506 had no effect (Fig. 3B). Furthermore, rapamycin failed to induce an interaction of FKBPs with S2035T mutant mTOR, which is compromised in the binding of rapamycin (36). Rapamycin also mediated an interaction of FKBP12 and FKBP51 with the mTORC1-specific component Raptor (Fig. 3C).
FKBP51 can contribute to the effects of rapamycin.
We next investigated the contributions of individual FKBPs to the functional effects of rapamycin in human cells. To test whether the prototypical FKBP12 protein is required for rapamycin function, we knocked down endogenous FKBP12 levels by at least 90% (Fig. 4A). We then treated these cells with increasing rapamycin concentrations and used T389 phosphorylation of S6K as a readout of cellular mTORC1 activity. For both FKBP12 knockdown and mock-treated control cells, a clear dose-dependent reduction of the phospho-T389 signal was observed, with partial inhibition at 1.5 nM and complete inhibition at 3 nM for both cell types, indicating functional redundancy of FKBP12.
Fig 4.

Contributions of FKBPs to the cellular action of rapamycin. (A) Effect of rapamycin on S6K phosphorylation in FKBP12 knockdown HeLa cells. HeLa cells were transfected with siRNA for FKBP12 or nontargeting controls, starved, and stimulated with 10% FBS–100 nM insulin for 60 min in the presence of 0 to 3 nM rapamycin. Cells were lysed and analyzed by immunoblotting. FKBP25 probing served as an additional loading control. (B) Overexpression of FKBP12 or FKBP51 has different effects on S6 kinase phosphorylation. HeLa cells were transfected with an FKBP12 or FKBP51 overexpression vector, starved for 16 h, and stimulated with 10% FBS–100 nM insulin for 60 min in the absence or presence of 0.25 nM rapamycin. Thereafter, cells were lysed and subjected to SDS-PAGE and Western blotting. The membrane was probed with antibodies against the indicated proteins or antigens. Unrelated lanes between mock-transfected controls and FKBP12 and FKBP51 overexpression samples have been removed for clarity. (C) In FKBP12 knockdown cells, rapamycin-induced Akt hyperphosphorylation is blunted. Wild-type (wt) and FKBP12 knockdown neuroblastoma cells were starved for 24 h and stimulated with 10% FCS–100 nM insulin for 60 min in the presence of 0 to 10 nM rapamycin. Cells were lysed and subjected to SDS-PAGE and immunoblotting. S6 kinase and Akt phosphorylation was assessed with the appropriate phospho-specific antibodies. (D) FKBP12 is necessary for rapamycin-induced Akt hyperphosphorylation. Wild-type and FKBP12 knockdown SH-SY5Y neuroblastoma cells were starved for 24 h and stimulated with 10% FCS–100 nM insulin for 60 min in the presence of 0 to 500 nM rapamycin or 100 nM torin-1. Cells were lysed, and Akt S473 phosphorylation was assessed with a phospho-antibody-based FRET assay. Mean values of two independent duplicates are shown. Values were normalized to 1 for 0 nM rapamycin for each cell type. (E) FKBP51 can replace FKBP12 in enabling rapamycin (Rap)-induced Akt hyperphosphorylation. FKBP12 knockdown SH-SY5Y cells were transfected with FKBP12, FKBP51, or a control plasmid and treated 24 h later with 10 nM rapamycin or 100 nM torin-1 for 60 min. Cellular Akt S473 phosphorylation was determined with a homogeneous time-resolved FRET assay. Mean values of three independent data points are shown. Results were normalized to those of dimethyl sulfoxide (DMSO)-treated controls (100%) for each transfection condition. ***, P < 0.001; **, P < 0.01.
In order to probe the role of FKBP51 in rapamycin pharmacology, we overexpressed human FKBP51 and tested its influence at subeffective concentrations of rapamycin. In mock-transfected cells, S6K phosphorylation was unchanged at 0.25 nM rapamycin. While overexpression of FKBP12 only minimally facilitated the inhibition of S6K phosphorylation at this low concentration of rapamycin, similar overexpression of FKBP51 clearly enhanced the inhibitory effect of 0.25 nM rapamycin (Fig. 4B).
To further explore the role of FKBP12 in the pharmacology of rapamycin, we referred to FKBP12-depleted SH-SY5Y neuroblastoma cells (30). In these cells, S6K(T389) phosphorylation was slightly increased under basal conditions (Fig. 4C), similar to that in transiently FKBP12-depleted HeLa cells (Fig. 4A) and consistent with observations in conditional FKBP12 knockout mice (37). Basal Akt phosphorylation was also increased in the FKBP12 knockdown SH-SY5Y cells, suggesting that the whole Akt-mTOR-S6K pathway was activated by FKBP12 reduction. Importantly, rapamycin reduced S6K(T389) phosphorylation in the FKBP12 knockdown SH-SY5Y cells with an IC50 of 2 to 4 nM, similar to that in wild-type SH-SY5Y cells (Fig. 4C).
The antiproliferative efficiency of rapamycin has been modest for many cancer cell types (10), and this had been attributed in part to the counterproductive repression of a negative feedback, which increases signaling via the PI3K-Akt pathway. Surprisingly, we observed a differential influence of rapamycin on Akt phosphorylation in the presence or absence of FKBP12. In wild-type SH-SY5Y cells, rapamycin potently and dose dependently induced Akt hyperphosphorylation whereas this effect was blunted in FKBP12-depleted cells (Fig. 4C; see Fig. S4 in the supplemental material). Even very high rapamycin concentrations did not induce Akt hyperphosphorylation in the FKBP12-depleted cells (Fig. 4D). Reexpression of FKBP12 restored the inducibility of Akt(S473) phosphorylation by rapamycin in these cells (Fig. 4E). Importantly, FKBP51 was almost as efficient as FKBP12 in enabling rapamycin-induced Akt(S473) hyperphosphorylation, suggesting that FKBP51 can functionally replace FKBP12. An enhancement of rapamycin-induced Akt(S473) hyperphosphorylation was also observed upon FKBP51 overexpression in HeLa cells (see Fig. S5 in the supplemental material).
FKBP12 inhibition alone does not block the effects of rapamycin.
To further distinguish whether the effects of rapamycin rely on binding to FKBPs in general or specifically on binding to FKBP12, we used a panel of FKBP inhibitors. The inhibition of mTOR autophosphorylation and rapamycin-induced Akt hyperphosphorylation was blocked or significantly reduced by the panselective FKBP ligand FK506, which potently binds to all cytosolic human FKBPs (Fig. 5) (13). FK1706, a nonimmunosuppressive analog of FK506 (38) that tightly binds to FKBP12, as well as to FKBP51 (Ki = 50 nM) and to FKBP52 (Ki = 20 nM) (13, 38), likewise blocked the effects of rapamycin. Biricodar (39) and compound 44 (40) are two structurally different substances that bind to FKBP12 almost as tightly as FK506 and FK1706 but more strongly discriminate against the larger FKBP51 and FKBP52 proteins (>500-fold selectivity for FKBP12). In contrast to FK506 or FK1706, neither of the FKBP12-specific inhibitors affected the effects of rapamycin on mTOR autophosphorylation or Akt hyperphosphorylation, even at concentrations 3- to 10-fold higher than those of FK506 and FK1706. Almost identical results were obtained with SHSY-5Y neuroblastoma cells (Fig. 5A and B), as well as with HeLa cells (Fig. 5C and D).
Fig 5.

FKBP12-selective ligands do not block the cellular effects of rapamycin. SHSY-5Y cells (A and B) and HeLa cells (C and D) were treated with dimethyl sulfoxide, rapamycin (Rap), or torin-1 (100 nM) in the absence or presence of the FKBP12-selective inhibitor Biricodar (Biri) (40) or compound 44 (cmpd44) (58) or the nonselective FKBP inhibitor FK1706 or FK506 (45) at the indicated concentration. After 60 min, cellular Akt phosphorylation (A and C) and cellular mTOR phosphorylation (B and D) were determined with a homogeneous time-resolved assay. The panselective FKBP inhibitors, but not the FKBP12-selective inhibitors, blocked the effect of rapamycin. For all experiments, mean values from at least three data points are shown. The dimethyl sulfoxide control was set to 100% phosphorylation, while torin-1 was defined as 0% phosphorylation. The values are presented as means ± standard deviations. Two-way analysis of variance and a priori testing were used for statistical analysis. Comparison with dimethyl sulfoxide treatment: ***, P < 0.001; **, P < 0.01; *, P < 0.05. Comparison with treatment with rapamycin alone: +++, P < 0.001; ++, P < 0.01; +, P < 0.05.
DISCUSSION
Taken together, our results suggest an unexpected and substantial contribution of larger FKBP homologs like FKBP51 in mediating the effect of rapamycin. Our biochemical studies unambiguously show that tight, rapamycin-dependent binding of the FRB domain is possible for all of the FKBP family members tested and that these alternative ternary complexes all potently inhibit mTOR kinase activity. In mammalian cells, rapamycin induced mTOR complexes with larger FKBPs. In yeast, FKBP52 could mediate the antiproliferative effect of rapamycin, while FKBP51 enhanced mTOR inhibition in HeLa cells. Inhibition of FKBP12 alone was not sufficient to block the effect of rapamycin. We thus postulate that larger FKBPs contribute to the effects of rapamycin in mammalian cells.
Throughout the literature, only FKBP12 is discussed as a relevant target mediating the inhibitory effects of rapamycin on mTOR, although there is little experimental evidence for this exclusive role in mammals. In several fungi, knockout of the respective homolog of FKBP12 was shown to abolish sensitivity to rapamycin (35, 41–43). However, the FKBP repertoire in these organisms is much smaller than that in mammals and fungal FKBP12 is the only well-established ortholog of human FKBPs (44). Moreover, fungal FKBP12 is usually the only cytoplasmic FKBP expressed, explaining the strict FKBP12 dependence of rapamycin in these organisms.
For mammalian FKBPs, only for FKBP12.6, the closest homolog of FKBP12, had ternary-complex formation with rapamycin and the FRB domain been demonstrated (45). However, experiments with FKBP12.6-deficient T cells clearly showed that FKB12.6 can be redundant for the action of rapamycin (22). Recently, FKBP38 has been proposed as a player in mTOR inhibition, but this effect was largely rapamycin independent and might be indirect (14). The rapamycin sensitivity of glioma cell lines was shown to be affected by FKBP51 expression levels, but this was attributed to the direct role of FKBP51 in NF-κB signaling, which is inhibited by FKBP51 ligands (46). The rapamycin levels used in the present work are very low and unlikely to saturate FKBP-binding sites in cells. The rapamycin effects observed in our studies are thus unlikely to reflect direct effects on FKBPs.
Our results suggest that for the inhibition of S6K phosphorylation by rapamycin, very small amounts of FKBPs seem to be sufficient. In our knockdown experiments a substantial reduction of FKBP12 was achieved. We cannot fully exclude the possibility that the inhibitory effect of rapamycin on S6K phosphorylation was mediated by remaining amounts of FKBP12. However, our results are in striking contrast to studies with FK506, where even slight reductions in FKBP12 dramatically reduced the effect of FK506 on the cellular phosphatase activity of calcineurin (47). The results reported by Weiwad et al. were fully consistent with the much higher efficacy of FKBP12 than other human FKBP homologs in biochemical calcineurin inhibition assays. Our biochemical and cellular results suggest that the human FKBP system is much more versatile in forming inhibitory FKBP-rapamycin-mTOR complexes than in forming FKBP-FK506-calcineurin complexes, for which FKBP12 is strongly preferred.
The model in which larger FKBP homologs can substitute for FKBP12 in mediating the effect of rapamycin is further supported by our FKBP inhibition studies, where the inhibition of all known cytosolic FKBPs was necessary to block the effect of rapamycin, while inhibition of FKBP12 alone was insufficient. The FKBP12-selective inhibitors compound 44 and Biricodar are highly lipophilic substances, and several close analogs easily permeate cells. The inability of these substances to block the effect of rapamycin is therefore unlikely to be due to insufficient cell penetration.
The antiproliferative efficiency of rapamycin has been shown to be disappointingly modest for several cancer cell types. This was attributed in part to a counterproductive repression of a negative feedback on Akt that increases the PI3K-Akt signaling pathway. This feedback effect can be mediated either via Grb10 or S6 kinase and the insulin receptor substrates (48, 49) or via a reduction of PHLPP expression (50). Intriguingly, we show that hyperphosphorylation of Akt by rapamycin—in contrast to the inhibition of S6K phosphorylation—seems to depend on the availability of higher FKBP levels. Our reconstitution experiments clearly show that both FKBP12 and the larger FKBP51 protein can mediate this effect of rapamycin. The mechanisms of the different rapamycin sensitivities of S6K phosphorylation and Akt hyperphosphorylation, depending on different FKBP levels, remain to be determined. In addition to the above-mentioned rapamycin-induced feedback loops, possible alternative mechanisms are an altered FKBP-rapamycin dependence of mTORC1 (phosphorylating S6K) and mTORC2 (phosphorylating Akt) or rapamycin-independent effects of FKBP12 and FKBP51 on the Akt-mTOR-S6K pathway.
Rapamycin inhibits the late phases of neuronal long-term potentiation, which represent adaptive processes thought to underlie learning and memory. This was reduced in FKBP12-deficient neurons and interpreted as indicating that FKBP12 is necessary for the action of rapamycin on mTOR (37). Our results suggest a diminished activating effect of rapamycin on the PI3K-Akt pathway in the absence of FKBP12 as an additional explanation for the functional alterations in neurons lacking FKBP12.
Structural arguments against the involvement of larger FKBPs have been raised on the basis of the comparison of the rapamycin complexes of FKBP13 and FKBP25 with the ternary-complex structures of FKBP12 (26). Notably, the 80s loop conformation, as observed in the apo structures of the larger FKBPs, would indeed clash with the FRB domain in a rigid FKBP12-like binding mode. Our crystal structures show that (i) the contact areas adapt efficiently, avoiding potential clashes and instead forming compensatory novel contacts (e.g., via the 40s loop for FKBP51), and that (ii) many FKBP-FRB domain interactions are indirect, mediated by an dense but presumably dynamic water network at the interface.
The energetics of the FKBP12-FRB domain interactions are poorly understood. Protein-protein interactions must add at least 20 kJ/mol to the binding energy of the ternary-complex formation compared to the interactions of rapamycin alone (rapamycin alone is a very weak FRB domain binder [26 μM]) (31), whereas the rapamycin-FKBP complexes are strong FRB domain binders (about 10 nM [Table 1]) yet they engage in only limited direct protein-protein contacts. The numerous well-ordered bridging water molecules that were resolved in our high-resolution structure likely contribute substantially to the stability of the FKBP-rapamycin-FRB domain complex, thus providing a plausible explanation for this conundrum.
Mechanistically, rapamycin is thought to inhibit mTOR by allosterically restraining the access of substrates (e.g., S6K) to the kinase active site or by interfering with the mTOR complex's integrity (51) or assembly (7). The biochemical details of the mechanism of actions of rapamycin are, however, far from clear. Notably, the activity of mTORC1 toward some, but not all, downstream targets has been shown to be rapamycin resistant, often in a cell context-dependent manner (reviewed in reference 5). Likewise, mTORC2 integrity has been shown to be sensitive to long-term rapamycin treatment only in some cell types (7). Importantly, the substrate bias of rapamycin has emerged recently as a key factor responsible for the limited efficacy of rapamycin in cancer or for its metabolic side effects (52). The cellular factors involved in partial rapamycin resistance have yet to be identified.
The identity of specific FKBP homologs in rapamycin-mTOR complexes will be functionally important when they add novel functionalities to the ternary complexes. For example, the maturation of mTOR complexes has been shown to depend on the Hsp90 machinery (53) and to be sensitive to rapamycin treatment in certain cell types (7). In this context, FKBP51- or FKBP52-rapamycin complexes, which are cochaperones of Hsp90, could be more effective than the smaller single-domain homolog FKBP12. In summary, our results suggest that the customary focus on FKBP12-rapamycin is too narrow and that additional FKBP-rapamycin-mTOR complexes must be considered for a comprehensive understanding of rapamycin function.
Most FKBP family members bind to rapamycin with comparably high affinities (13). Consequently, a mixture of different FKBPs will bind to rapamycin when the compound is administered. Since all of the FKBP-rapamycin complexes tested have the biochemical potential to engage mTOR, the importance of specific FKBP-rapamycin-mTOR complexes might rather depend on the cell type and state. FKBP13 and FKBP25 are probably not involved in mTOR inhibition in vivo, since they are located mainly in the endoplasmic reticulum (54) and the nucleus (55), respectively, and thus are physically separated from mTOR. For the cytosolic FKBPs, the contributions of the particular FKBP homologs might be largely dictated by their relative cellular abundance. Baughman et al. characterized the tissue distribution and cellular concentration of FKBP51 and directly compared it to FKBP12 (56). They found that FKBP51 was more abundant in 11 of 17 tissues, with an up-to-12-fold molar excess in the liver. Furthermore, mRNA quantification suggests that FKBP51 is highly overexpressed, especially in adipocytes and in skeletal muscle cells, compared to median expression (57). These tissues are extremely important for the metabolic (side) effects of rapamycin, and FKBP51 might be the most relevant FKBP subtype for the inhibition of mTOR by rapamycin in these tissues.
Besides immunomodulation, rapamycin treatment has been suggested for several other applications, including type II diabetes, neurodegenerative diseases (2), or even life span extension (58), but the prolonged systemic use of rapamycin is hampered by unacceptable side effects (12). For these applications, tissue-selective mTOR inhibition would be very advantageous. Our results suggest a mechanistic rationale for how tissue- or cell type-biased mTOR inhibition might be realized, namely, by engaging specific FKBP homologs dominating the FKBP pool in the desired cell types. This could be accomplished by blocking undesired FKBPs with nonimmunosuppressive, FKBP-subselective FKBP ligands such as Biricodar or compound 44, which leave other FKBPs like FKBP51 or FKBP52 as the mediators of rapamycin's action. A more direct way would be tailoring of rapalogs for the desired FKBP homologs. Such FKBP subtype-selective rapamycin analogs can be expected to be active primarily in those cells where their cognate FKBP is highly expressed, e.g., liver cells, skeletal muscle cells, or adipocytes in the case of FKBP51 (56, 57). Conversely, rapalogs devoid of FKBP51 binding might be less active in these than in other tissues. While the development of such analogs represents a substantial synthetic challenge, our FKBP51- or FKBP52-rapamycin-FRB domain crystal structures provide a first starting point to support a rational drug design, e.g., by exploiting differential conformations in the 40s or 80s loop (39, 40).
Taken together, our results call for expansion of the customary focus on FKBP12-rapamycin and for consideration of additional FKBP-rapamycin-mTOR complexes to comprehensively understand and optimally exploit the action of rapamycin.
Supplementary Material
ACKNOWLEDGMENTS
Initial crystal screening was performed at the crystallization facility of the Max Planck Institute of Biochemistry. The generous support during data collection by the Joint Structural Biology Group at the European Synchrotron Radiation Facility in Grenoble, France, is gratefully acknowledged. We are indebted to F. Holsboer (Max Planck Institute of Psychiatry, Munich, Germany) for continuous financial support.
We thank D. M. Sabatini and N. S. Gray for a sample of torin-1 and M. Gerard and V. Blaekelandt for a sample of shFKBP12-SH-SY5Y cells. We thank J. Chen (UCLA, Riverside, CA) for providing FLAG-mTOR wt/S2035T expression plasmids; T. Rein (Max Planck Institute of Psychiatry, Munich, Germany) for FKBP12, FKBP51, and FKBP52 cDNAs; and G. Fischer (Max Planck Gesellschaft, Halle, Germany) for FKBP12.6, FKBP13, and FKBP25 cDNAs.
Footnotes
Published ahead of print 28 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00678-12.
REFERENCES
- 1. Proud CG. 2011. mTOR signalling in health and disease. Biochem. Soc. Trans. 39: 431–436 [DOI] [PubMed] [Google Scholar]
- 2. Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vezina C, Kudelski A, Sehgal SN. 1975. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 28: 721–726 [DOI] [PubMed] [Google Scholar]
- 4. Gaali S, Gopalakrishnan R, Wang Y, Kozany C, Hausch F. 2011. The chemical biology of immunophilin ligands. Curr. Med. Chem. 18: 5355–5379 [DOI] [PubMed] [Google Scholar]
- 5. Choo AY, Blenis J. 2009. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 8: 567–572 [DOI] [PubMed] [Google Scholar]
- 6. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- 7. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22: 159–168 [DOI] [PubMed] [Google Scholar]
- 8. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. 2012. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335: 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Araki K, Ellebedy AH, Ahmed R. 2011. TOR in the immune system. Curr. Opin. Cell Biol. 23: 707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benjamin D, Colombi M, Moroni C, Hall MN. 2011. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10: 868–880 [DOI] [PubMed] [Google Scholar]
- 11. Bové J, Martinez-Vicente M, Vila M. 2011. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat. Rev. Neurosci. 12: 437–452 [DOI] [PubMed] [Google Scholar]
- 12. Saunders RN, Metcalfe MS, Nicholson ML. 2001. Rapamycin in transplantation: a review of the evidence. Kidney Int. 59: 3–16 [DOI] [PubMed] [Google Scholar]
- 13. Kozany C, Marz A, Kress C, Hausch F. 2009. Fluorescent probes to characterise FK506-binding proteins. Chembiochem 10: 1402–1410 [DOI] [PubMed] [Google Scholar]
- 14. Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. 2007. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318: 977–980 [DOI] [PubMed] [Google Scholar]
- 15. Uhlenbrock K, Weiwad M, Wetzker R, Fischer G, Wittinghofer A, Rubio I. 2009. Reassessment of the role of FKBP38 in the Rheb/mTORC1 pathway. FEBS Lett. 583: 965–970 [DOI] [PubMed] [Google Scholar]
- 16. Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. 2009. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16: 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14: 1296–1302 [DOI] [PubMed] [Google Scholar]
- 18. Schalm SS, Blenis J. 2002. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12: 632–639 [DOI] [PubMed] [Google Scholar]
- 19. Vilella-Bach M, Nuzzi P, Fang Y, Chen J. 1999. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J. Biol. Chem. 274: 4266–4272 [DOI] [PubMed] [Google Scholar]
- 20. Ikenoue T, Hong S, Inoki K. 2009. Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol. 452: 165–180 [DOI] [PubMed] [Google Scholar]
- 21. Sato T, Umetsu A, Tamanoi F. 2008. Characterization of the Rheb-mTOR signaling pathway in mammalian cells: constitutive active mutants of Rheb and mTOR. Methods Enzymol. 438: 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dubois S, Shou W, Haneline LS, Fleischer S, Waldmann TA, Muller JR. 2003. Distinct pathways involving the FK506-binding proteins 12 and 12.6 underlie IL-2- versus IL-15-mediated proliferation of T cells. Proc. Natl. Acad. Sci. U. S. A. 100: 14169–14174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leslie AGW. 1992. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography, no. 26. Daresbury Laboratory, Warrington, UK [Google Scholar]
- 25. Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62: 72–82 [DOI] [PubMed] [Google Scholar]
- 26. Liang J, Choi J, Clardy J. 1999. Refined structure of the FKBP12-rapamycin-FRB ternary complex at 2.2 A resolution. Acta Crystallogr. D Biol. Crystallogr. 55: 736–744 [DOI] [PubMed] [Google Scholar]
- 27. Perrakis A, Morris R, Lamzin VS. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255 [DOI] [PubMed] [Google Scholar]
- 30. Gerard M, Deleersnijder A, Daniels V, Schreurs S, Munck S, Reumers V, Pottel H, Engelborghs Y, Van den Haute C, Taymans JM, Debyser Z, Baekelandt V. 2010. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson's disease-like pathology. J. Neurosci. 30: 2454–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banaszynski LA, Liu CW, Wandless TJ. 2005. Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 127: 4715–4721 [DOI] [PubMed] [Google Scholar]
- 32. Waksman R, Ajani AE, Pichard AD, Torguson R, Pinnow E, Canos D, Satler LF, Kent KM, Kuchulakanti P, Pappas C, Gambone L, Weissman N, Abbott MC, Lindsay J. 2004. Oral rapamycin to inhibit restenosis after stenting of de novo coronary lesions: the Oral Rapamune to Inhibit Restenosis (ORBIT) study. J. Am. Coll. Cardiol. 44: 1386–1392 [DOI] [PubMed] [Google Scholar]
- 33. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284: 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lawrence MC, Colman PM. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234: 946–950 [DOI] [PubMed] [Google Scholar]
- 35. Heitman J, Movva NR, Hall MN. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253: 905–909 [DOI] [PubMed] [Google Scholar]
- 36. Chen J, Zheng XF, Brown EJ, Schreiber SL. 1995. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. U. S. A. 92: 4947–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. 2008. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron 60: 832–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Price RD, Yamaji T, Yamamoto H, Higashi Y, Hanaoka K, Yamazaki S, Ishiye M, Aramori I, Matsuoka N, Mutoh S, Yanagihara T, Gold BG. 2005. FK1706, a novel non-immunosuppressive immunophilin: neurotrophic activity and mechanism of action. Eur. J. Pharmacol. 509: 11–19 [DOI] [PubMed] [Google Scholar]
- 39. Gopalakrishnan R, Kozany C, Gaali S, Kress C, Hoogeland B, Bracher A, Hausch F. 2012. Evaluation of synthetic FK506 analogues as ligands for the FK506-binding proteins 51 and 52. J. Med. Chem. 55: 4114–4122 [DOI] [PubMed] [Google Scholar]
- 40. Gopalakrishnan R, Kozany C, Wang Y, Schneider S, Hoogeland B, Bracher A, Hausch F. 2012. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J. Med. Chem. 55: 4123–4131 [DOI] [PubMed] [Google Scholar]
- 41. Bastidas RJ, Shertz CA, Lee SC, Heitman J, Cardenas ME. 2012. Rapamycin exerts antifungal activity in vitro and in vivo against Mucor circinelloides via FKBP12-dependent inhibition of Tor. Eukaryot. Cell 11: 270–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cruz MC, Cavallo LM, Gorlach JM, Cox G, Perfect JR, Cardenas ME, Heitman J. 1999. Rapamycin antifungal action is mediated via conserved complexes with FKBP12 and TOR kinase homologs in Cryptococcus neoformans. Mol. Cell. Biol. 19: 4101–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cruz MC, Goldstein AL, Blankenship J, Del Poeta M, Perfect JR, McCusker JH, Bennani YL, Cardenas ME, Heitman J. 2001. Rapamycin and less immunosuppressive analogs are toxic to Candida albicans and Cryptococcus neoformans via FKBP12-dependent inhibition of TOR. Antimicrob. Agents Chemother. 45: 3162–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pemberton TJ. 2006. Identification and comparative analysis of sixteen fungal peptidyl-prolyl cis/trans isomerase repertoires. BMC Genomics 7: 244 doi:10.1186/1471-2164-7-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, Abraham RT, O'Keefe SJ, O'Neill EA, Wiederrecht GJ. 1995. A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J. Biol. Chem. 270: 26511–26522 [DOI] [PubMed] [Google Scholar]
- 46. Jiang W, Cazacu S, Xiang C, Zenklusen JC, Fine HA, Berens M, Armstrong B, Brodie C, Mikkelsen T. 2008. FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-kappaB signaling pathway. Neoplasia 10: 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weiwad M, Edlich F, Kilka S, Erdmann F, Jarczowski F, Dorn M, Moutty MC, Fischer G. 2006. Comparative analysis of calcineurin inhibition by complexes of immunosuppressive drugs with human FK506 binding proteins. Biochemistry 45: 15776–15784 [DOI] [PubMed] [Google Scholar]
- 48. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332: 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. 2011. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332: 1322–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu J, Stevens PD, Gao T. 2011. mTOR-dependent regulation of PHLPP expression controls the rapamycin sensitivity in cancer cells. J. Biol. Chem. 286: 6510–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. 2010. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38: 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takai H, Xie Y, de Lange T, Pavletich NP. 2010. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 24: 2019–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nigam SK, Jin YJ, Jin MJ, Bush KT, Bierer BE, Burakoff SJ. 1993. Localization of the FK506-binding protein, FKBP 13, to the lumen of the endoplasmic reticulum. Biochem. J. 294(Pt 2): 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jin YJ, Burakoff SJ. 1993. The 25-kDa FK506-binding protein is localized in the nucleus and associates with casein kinase II and nucleolin. Proc. Natl. Acad. Sci. U. S. A. 90: 7769–7773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baughman G, Wiederrecht GJ, Chang F, Martin MM, Bourgeois S. 1997. Tissue distribution and abundance of human FKBP51, and FK506-binding protein that can mediate calcineurin inhibition. Biochem. Biophys. Res. Commun. 232: 437–443 [DOI] [PubMed] [Google Scholar]
- 57. Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U. S. A. 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. 2009. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.