

Abstract
Skeletal muscle development is orchestrated by the myogenic regulatory factor MyoD, whose activity is blocked in myoblasts by proteins preventing its nuclear translocation and/or binding to G/C-centered E-boxes in target genes. Recent evidence indicates that muscle gene expression is also regulated at the cis level by differential affinity for DNA between MyoD and other E-box binding proteins during myogenesis. MyoD binds to G/C-centered E-boxes, enriched in muscle differentiation genes, in myotubes but not in myoblasts. Here, we used cell-based and in vivo Drosophila, Xenopus laevis, and mouse models to show that ZEB1, a G/C-centered E-box binding transcriptional repressor, imposes a temporary stage-dependent inhibition of muscle gene expression and differentiation via CtBP-mediated transcriptional repression. We found that, contrary to MyoD, ZEB1 binds to G/C-centered E-boxes in muscle differentiation genes at the myoblast stage but not in myotubes. Its knockdown results in precocious expression of muscle differentiation genes and acceleration of myotube formation. Inhibition of muscle genes by ZEB1 occurs via transcriptional repression and involves recruitment of the CtBP corepressor. Lastly, we show that the pattern of gene expression associated with muscle differentiation is accelerated in ZEB1−/− mouse embryos. These results set ZEB1 as an important regulator of the temporal pattern of gene expression controlling muscle differentiation.
INTRODUCTION
Muscle gene expression is orchestrated by a small set of myogenic regulatory factors (MRFs), namely, Myf-5, MyoD, myogenin, and MRF4, that bind to E-box sequences (CANNTG) in the regulatory regions of target genes by forming homodimers with themselves or heterodimers with E proteins (reviewed in references 1 and 2). During myogenesis, there is a division of labor among MRFs, with Myf-5 and MyoD determining lineage commitment and myogenin driving terminal differentiation of myoblasts into myotubes (1). In addition, there is also a regulatory interplay among MRFs, as Myf5 and MyoD induce myogenin, which in turn regulates MRF4, while MyoD and myogenin can activate their own expression. Overexpression of MRFs in nonmuscle cells (e.g., fibroblasts) is sufficient to induce a number of muscle markers and, to different degrees, drive a myogenic differentiation program (3–5).
However, MRFs cannot explain by themselves the sophisticated pattern of temporal and spatial gene expression during myogenesis. Determination and terminal differentiation of skeletal muscle precursors require the concerted action of MRFs with many other proteins, which modulate the expression, intracellular localization, and/or transcriptional activity of MRFs (1, 6). For instance, in myoblasts, MyoD does not induce downstream muscle differentiation target genes, as its function is temporarily blocked through multiple mechanisms. Inter alia, the interaction of MyoD (and/or its E protein partners) with a number of factors prevents MyoD from entering the nucleus (e.g., interaction of MyoD with I-mfa/MDFI), from binding to DNA (e.g., with Twist, Id, and Mist-1), and/or from activating its targets (e.g., with Mist-1 and MyoR) (7–11). However, these negative regulatory factors are still not sufficient to account for the differential gene expression signature in myoblasts and myotubes (1, 2, 6).
Recent works have revealed that, independently of their squelching by proteins like Twist or Id, binding of MRFs to DNA during muscle differentiation is also controlled at the cis level. Differential affinity of MyoD vis-à-vis other E-box binding proteins for E-box sequences dictates their occupancy in myoblasts and myotubes (12, 13). Binding of MyoD to MyoD-specific E-boxes promotes transcriptional activation of muscle differentiation genes much more strongly than does MyoD binding to E-box sequences that are also shared with NeuroD2, another basic helix-loop-helix (bHLH) protein that regulates neuronal differentiation (12). In addition, E-box sequence specificity, e.g., G/C versus A/T as central nucleotides, determines DNA occupancy during muscle differentiation (13). In myoblasts, MyoD binds to A/T-centered E-boxes in genes associated with cell proliferation but does not interact with G/C-centered E-boxes that are enriched in the regulatory regions of muscle differentiation genes (13). The latter E-boxes are occupied during the myoblast stage by transcriptional repressors of the Snail family (13). As differentiation progresses, Snail factors are displaced from these E-boxes by MyoD, thus inducing the expression of a muscle differentiation gene signature (13).
Another transcriptional repressor that specifically recognizes G/C-centered E-boxes is ZEB1 (also known as δEF1) (reviewed in references 14 and 15). Like Snail factors, ZEB1 is best known for its role in cancer progression, where it triggers an epithelial-to-mesenchymal transition (EMT), endowing cancer cells with a proinvasive phenotype (14, 15). ZEB1 is expressed in the epithelium of the undifferentiated somite and in the dermomyotome during mouse and chick embryogenesis (16, 17). Mutation and overexpression of its ortholog in Drosophila melanogaster, zfh-1, leads to altered somatic musculature formation (18, 19). Interestingly, ZEB1 is a downstream target of both MyoD and Snail factors (20–23). In in vitro overexpression experiments, ZEB1 competes with bHLH and Snail factors for binding to E-boxes in different genes (e.g., those for immunoglobulin heavy chain enhancer, α4 integrin promoter, CD4 proximal enhancer/promoter, and p73 promoter/intron 1) (24–28).
This pattern of ZEB1 expression during development and its exclusive affinity for G/C-centered E-boxes prompted us to question whether endogenous ZEB1 is regulating muscle gene expression. Using a number of cell-based and in vivo models, we show here that ZEB1 imposes a temporary stage-dependent inhibition of muscle gene expression and differentiation via CtBP-mediated transcriptional repression. ZEB1 knockdown triggers early protein and mRNA expression of muscle determination and differentiation genes and, accordingly, precocious myotube conversion. In a reverse pattern with respect to MyoD, ZEB1 binds to G/C-centered E-boxes in muscle differentiation genes at the myoblast stage but not in myotubes. Negative regulation of muscle genes by ZEB1 in myoblasts occurs via transcriptional repression. In both cell-based systems and Xenopus laevis and Drosophila embryos, we show that this repression involves an interaction of ZEB1 with the CtBP corepressor. Finally, we found that the temporal pattern of muscle differentiation gene expression is accelerated in ZEB1−/− mouse embryos. Together, these results indicate that endogenous ZEB1 plays an important role regulating the temporal pattern of gene expression during muscle differentiation.
MATERIALS AND METHODS
Cells, cell culture, and transfections.
C2C12 and C3H-10T1/2 cells were obtained from the American Tissue Culture Collection and maintained in Dulbecco's modified Eagle's medium (DMEM) (Lonza) supplemented with 12% fetal bovine serum (FBS) (Sigma). Cells were transiently transfected with expression or reporter vectors by using Lipofectamine 2000 (Life Technologies) and/or with small interfering RNA (siRNA) oligonucleotides by using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions.
Antibodies.
The following commercial antibodies were used in this work: ZEB1 (H-102; Santa Cruz Biotechnology [SCBT]), sarcomeric pan-myosin (MF-20; Development Studies Hybridoma Bank [DSHB]), myosin heavy chain IIa (MyH2) (SC-71; DSHB), myosin heavy chain IIb (MyH4) (BF-F3; DSHB), MyoD (C-20; SCBT), myogenin (F5D; BD Pharmingen), CtBP1/2 (E-12; SCBT), and α-tubulin (B5-1-2; Sigma) antibodies. Antibodies against Drosophila muscle markers were obtained as follows: mouse anti-myosin heavy chain (anti-MHC) antibody was a gift from D. Kiehart (Duke University), and rabbit anti-MEF2 antibody was a gift from B. M. Patterson (National Cancer Institute, NIH). Secondary horseradish peroxidase (HRP)-conjugated donkey anti-mouse IgG and goat anti-rabbit IgG antibodies were purchased from Jackson ImmunoResearch (JIR). For blocking in immunostaining assays, normal serum from the host species of the secondary antibody was purchased from JIR. As an IgG control for chromatin immunoprecipitation (ChIP) assays, normal rabbit IgG and normal goat IgG-containing serum were purchased from SCBT and JIR, respectively.
RNA interference.
The three set of siRNAs used in this study to target mouse ZEB1 originated as follows. The siRNA duplex referred to as si1ZEB1, with the sense sequence 5′-GACCAGAACAGUGUUCCAUGUUUAA-3′, was purchased from Life Technologies as a Select RNAi siRNA (MSS210696) guaranteed to have no off-target effects. si2ZEB1 (sense sequence 5′-AACUGAACCUGUGGAUUAU-3′) was described previously (29). Finally, the siRNA referred to as si3ZEB1 was purchased from SCBT (catalog number sc-38644) and consisted of a pool of three different siRNA duplexes of the following sense sequences: 5′-GAAGAACCCUUGAACUUGU-3′, 5′-GAACAGUGUUCCAUGUUUA-3′, and 5′-CAACCAUGAAGGAUCUAUA-3′. As negative controls in interference experiments, the following siRNAs were used: 5′-UAUAGCUUAGUUCGUAACC-3′ and Select RNAi siRNA LO GC (catalog no. 12935-200; Life Technologies). In Western blot and quantitative real-time PCR (qRT-PCR) studies, an additional control siRNA targeting firefly luciferase (5′-GAUUAUGUCCGGUUAUGUA-3′) (30) was also used. The siRNA duplex against CtBP1/2 with the sense sequence 5′-GAACUGTGUCAACAAGGAC-3′ was previously described (31).
Plasmids.
Expression vectors used in the study were obtained from the following researchers: full-length mouse ZEB1 was obtained from M. Saito (Tokyo University, Japan) (32), full-length mouse ZEB1 with mutated CtBP binding sites was obtained from Y. Higashi (Institute for Developmental Research, Kasugai, Japan) (33), and pEMSV-MyoD was from the late H. Weintraub (Fred Hutchinson Cancer Research Center, Seattle, WA) (3). The Xenopus MyoD construct, containing the full-length cDNA plus the 5′ and 3′ untranslated regions and cloned into pSP64T, was obtained from J. Gurdon (Gurdon Institute, Cambridge, United Kingdom). Simian virus 40 (SV40)–β-galactosidase (β-gal) was purchased from Promega, pcDNA3 was purchased from Invitrogen, and the pBluescript SK vector was purchased from Stratagene-Agilent. Firefly luciferase reporters for the promoters used in this article were provided by the following researchers: 5.5 kb of rat α-MHC promoter was obtained from E. N. Olson (University of Texas Southwestern, Dallas, TX), (34), 2.3 kb of quail troponin I was obtained from S. Konieczny (Purdue University, West Lafayette, IN) (35), a bp −1256 mouse muscle creatine kinase (MCK) promoter was obtained from S. A. Leibovitch (INRA, Montpellier, France) (36), and a 4-kb enhancer plus a 2.7-kb promoter of the human MyoD gene were obtained from J. P. Capone (McMaster University, Hamilton, ON, Canada) (37).
Western blots.
Western blot assays were performed as described previously (38). Briefly, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.5% SDS, 50 mM Tris [pH 8], 2 mM EDTA, plus protease inhibitors) and loaded onto polyacrylamide gels. Gels were then transferred onto a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore). Following blocking for nonspecific antibody binding with 5% nonfat milk, membranes were incubated with the corresponding primary and HRP-conjugated secondary antibodies before the reaction was developed by using the Pierce ECL Western blotting substrate or SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Western blots shown in this study are representative of at least four independent experiments.
Myogenic conversion assays.
For differentiation of C2C12 myoblasts into myotubes, cells were seeded into 6-well plates and grown in growth medium (GM) (DMEM plus 20% FBS) until they reached 80 to 90% confluence, at which time the GM was switched to differentiation medium (DM) (DMEM supplemented with 2% horse serum; Sigma) and maintained in DM for different periods. In selected experiments as specifically indicated, cells were maintained in GM even after reaching full confluence. Cells were fixed in −20°C-chilled methanol and blocked with 2% gelatin (from cold-water fish skin; Sigma) before being stained with pan-myosin antibody MF-20 for 2 h, washed with phosphate-buffered saline (PBS), incubated with HRP-conjugated anti-mouse IgG antibody (JIR), and developed with a 3,3′-diaminobenzidine (DAB) substrate kit (Vector Laboratories). Finally, cells were incubated for 30 s with hematoxylin for nucleus counterstaining and washed with PBS.
RNA extraction and quantitative real-time PCR.
Total RNA from C2C12 cells and Xenopus ectodermal explants (animal caps) (see below) was extracted with the SV Total RNA isolation system kit (Promega) and TRIzol (Life Technologies), respectively. RNA was then used to synthesize cDNA by using a reverse transcription kit [random hexamers and GoScript (Promega) for C2C12 cell experiments and oligo(dT) and SuperScript II reverse transcriptase (Invitrogen) for Xenopus experiments]. mRNA levels were determined by qRT-PCR using either SYBR green/ROX (GoTaq; Promega) (for C2C12 cells) or iQSYBRGreen Supermix (Bio-Rad) (for Xenopus animal caps). Primers used to determine gene expression in C2C12 cells by qRT-PCR were as follows: mouse ZEB1 forward primer 5′-ACCCCTTCAAGAACCGCTTT-3′ and reverse primer 5′-CAATTGGCCACCACTGCTAA-3′, mouse myosin heavy chain IIa (MyH2) forward primer 5′-CGATGATCTTGCCAGTAATG-3′ and reverse primer 5′-ATAACTGAGATACCAGCG-3′ (39), mouse myosin heavy chain IIb (MyH4) forward primer 5′-TAAGCACGAGCGCAGAGTGAAGGAACT-3′ and reverse primer 5′-GCTGGATCTTACGGAACTTGGCCAGGT-3′ (40), mouse troponin T1 (TnnT1) forward primer 5′-GATTCTGTATGAGAGGAAAAAG-3′ and reverse primer 5′-TCATATTTCTGTTGCTTCAACTT-3′ (40), mouse myogenin forward primer 5′-CACTGGAGTTCGGTCCCAA-3′ and reverse primer 5′-TGTGGGCGTCTGTAGGGTC-3′ (41), mouse MyoD forward primer 5′-TGGGATATGGAGCTTCTATCGC-3′ and reverse primer 5′-GGTGAGTCGAAACACGGATCAT-3′ (42), mouse Myf-5 forward primer 5′-TCTGGTCCCGAAAGAACAGC-3′ and reverse primer 5′-CTTTTATCTGCAGCACATGCATT-3′ (43), and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward primer 5′-AACGACCCCTTCATTGAC-3′ and reverse primer 5′-TCCACGACATACTCAGCAC-3′ (44). Primers used for qRT-PCR of Xenopus animal cap cells were as follows: EF1α forward primer 5′-CACCATGAAGCCCTTACTGA-3′ and reverse primer 5′-ACCTGTGCGGTAAAAGAACC-3′, TnnI1 (slow skeletal troponin I type 1) forward primer 5′-CAGTAGCATTCCAGGGCAGT-3′ and reverse primer 5′-TATGTAGCCCCAATGGGAAA-3′, TnnI2 (fast skeletal troponin I type 2) forward primer 5′-CTCTTCAGCGGGGATATTGA-3′ and reverse primer 5′-ATTTGAGCCCCTCCTTGAGT-3′, MCK forward primer 5′-ACAAACCAGTGTCCCCTCTG-3′ and reverse primer 5′-CCACACCAGGAAGGTCTTGT-3′, mActin (muscle actin) forward primer 5′-GCTGACAGAATGCAGAAG-3′ and reverse primer 5′-TTGCTTGGAGGAGTGTGT-3′, and MyoD forward primer 5′-GACCTGCCAATGTTGTGTTG-3′ and reverse primer 5′-CAAAAAGTGGTCCGCAAGTT-3′. Relative gene expression was calculated by the ΔΔCT method, normalizing values relative to either GAPDH (C2C12 cells) or EF1α (Xenopus animal caps) as a reference gene. qRT-PCR data shown are representative of at least three independent experiments, with each point performed in triplicate, and are represented as fold change plus/minus standard deviation.
Chromatin immunoprecipitation assays.
ChIP assays were performed as described previously (38), using an EpiQuick ChIP kit (Epigentek) according to the manufacturer's instructions. Briefly, C2C12 cells as either nonconfluent cycling myoblasts or terminally differentiated myotubes were incubated during 20 min with a 1% formaldehyde solution (Electron Microscopy) at room temperature, followed by incubation with 1.25 M glycine. Upon sonication of cell lysates, chromatin was immunoprecipitated with the corresponding specific or control antibodies, and amplification of DNA fragments was assessed by qRT-PCR. Identification of potential DNA binding sequences for ZEB1/MyoD and design of primers for qRT-PCR were conducted by using MacVector 12.5 software (MacVector Inc.). For the ZEB1/MyoD CACCTG site at position −1068 of the mouse MyH4 promoter, the primers used to amplify the region between bp −1107 and −1014 of this promoter were as follows: forward primer 5′-TATAAAAGATTTTACCTGCCA-3′ and reverse primer 5′-ATATTTTCAACCACTGTTCT-3′. For the ZEB/MyoD CAGGTG site at position −1046 of the mouse skeletal slow troponin T1 (TnnT1) promoter, the primers used to amplify the region between bp −1105 and −1020 of this promoter were as follows: forward primer 5′-TCTCAAACCAAAGCAAAACCAA-3′ and reverse primer 5′-AGTTCCCCGTACCTCATACTCT-3′. A 191-bp region of the mouse GAPDH promoter, lacking consensus binding sites for ZEB1/MyoD, was amplified by using forward primer 5′-AGCTACTCGCGGCTTTACG-3′ and reverse primer 5′-AAGAAGATGCGGCCGTCTCT-3′, modified from those described previously (45). In all qRT-PCRs, values shown represent relative binding in relation to input and are the averages of data from two independent ChIP assays, each performed in triplicate, and representative of at least three experiments.
Transcriptional assays.
Transcriptional assays were carried out as described previously (38). Briefly, cells were transfected with firefly luciferase reporter vectors and equal molar amounts of either expression plasmids encoding ZEB1 or MyoD or, as controls, the corresponding empty expression vector. As an internal control for transfection efficiency, 1 μg of SV40–β-gal was cotransfected at each point. Total DNA used in each cotransfection was equalized by adding the promoterless pBluescript SK vector as required. Firefly luciferase activity was assessed with a Luciferase Assay System kit (Promega Corporation), whereas β-galactosidase activity was determined with Luminescent β-Galactosidase Detection Kit II (Clontech). Relative luciferase units (RLU) were assessed with a Modulus II Glomax microplate detection system (Promega). RLU values were expressed as the means of duplicates and are representative of at least four independent experiments.
Xenopus embryo microinjection and in situ hybridization.
Xenopus laevis embryos were obtained by in vitro fertilization as described previously (46) and were staged according to methods described previously (47). Linearized pcDNA3-ZEB1wt, pcDNA3-ZEB1CIDmut, and pSP64T-MyoD expression vectors were used to produce capped RNA by using the SP6-Message Machine kit (Ambion). A morpholino oligonucleotide (MO) against Xenopus ZEB1 (48) (5′-AGATCTGCCAAAGTTGAGCGTTT-3′) was purchased from Gene Tools LLC. Where indicated, 200 pg of ZEB1 RNA or 20 ng of ZEB1 MO was injected into one blastomere at the 2-cell stage, along with 20 pg of β-galactosidase RNA. Embryos were further cultured in 0.2× Marc's modified Ringer's (MMR) solution containing 4% Ficoll and 100 μg/ml gentamicin. For animal cap isolations, both cells of pigmented embryos at the 2-cell stage were injected with 100 pg of MyoD plus either 100 pg ZEB1wt or ZEB1CIDmut mRNA, and embryos were raised up to the blastula stage (stages 8 to 9). Twenty ectodermal explants per sample were then isolated and incubated at 25°C in 0.7× MMR supplemented with 1 mg/ml bovine serum albumin (BSA) and 100 μg/ml gentamicin. For in situ hybridization assays, embryos were grown to stages 15 to 16, fixed in minimum essential medium with formaldehyde, stained with Red-Gal (Research Organics), and processed as described previously (49).
Drosophila stocks, crosses, and immunostaining.
Full-length cDNA for zfh-1, obtained from Z. C. Lai (The Pennsylvania State University, Philadelphia, PA), was inserted into the NotI/XbaI sites of pUAST to generate independent stocks of upstream activation sequence (UAS)-zfh-1 flies (genotype, w118; P{w+mC = UAS-zfh-1.P}2B). UAS-zfh-1-CIDmut stocks are identical to UAS-zfh-1 stocks, except that the CtBP-interacting domain (CID) at 790PLDLS796 is mutated to a nonbinding sequence, 790ASASA796 (genotype, P{w+mC = UAS-zfh-1-CIDm}2B). UAS-zfh-1 and UAS-zfh-1-CIDmut stocks are homozygous, viable, and fertile second-chromosome insertions deposited at the Bloomington Drosophila Stock Center (Bloomington, IN) under identification numbers 6879 and 6880, respectively. zfh-1 expression in UAS-zfh-1 and UAS-zfh-1-CIDmut was induced by crossing them with 24B-Gal4 or MEF2-Gal4 stocks obtained as a kind gift from M. Bates (University of Cambridge, United Kingdom). Embryos were allowed to develop at 25°C before being examined for expression of different proteins. After standard fixation, embryos were blocked with 50% normal goat serum in PBS and incubated with primary and secondary antibodies before the reaction was developed with a DAB substrate kit (Vector Laboratories), as described previously (19). Following color development, embryos were mounted in 80% glycerol and examined on a Zeiss Axioplan-2 microscope.
Mouse tissue immunohistochemistry.
Mouse tissue samples corresponded to 4-μm sections from embryonic day 18.5 (E18.5) C57BL/6J wild-type (+/+) and δEF1 (ZEB1) null (−/−) embryos (17). Slides were subjected to deparaffination and hydration by using standard protocols, followed by heat-induced antigen retrieval in 10 mM citrate buffer (pH 6.0) for 15 min. Slides were then treated with 0.3% H2O2 in methanol to block endogenous peroxidase before being incubated with a nonspecific binding blocking solution (5% normal goat or donkey serum depending on the host of the secondary antibody, 4% BSA, and 0.5% Tween 20 in PBS). Next, slides were incubated with the corresponding primary (overnight at 4°C) and HRP-conjugated (1 h at 37°C) antibodies diluted in blocking solution. The immunohistochemistry reaction was developed with the DAB substrate kit (Vector Laboratories), followed by counterstaining with hematoxylin, before being mounted in di-N-butylphthalate in xylene solution (DPX; Sigma) for microscopic examination.
RESULTS
Knockdown of ZEB1 induces precocious expression of muscle differentiation genes.
The existence of proteins that bind and titrate out MyoD, thus preventing MyoD from prematurely activating its target genes in myoblasts, is well established (reviewed in reference 6). Recent evidence indicates that MyoD binding to DNA is also temporarily delayed through cis-level mechanisms to control the timing of muscle differentiation (13). In myoblasts, MyoD is excluded from G/C-centered E-boxes at muscle differentiation genes. MyoD only begins to occupy G/C-centered E-boxes and activate these genes as muscle differentiation progresses (13). Since ZEB1 is expressed early in development and binds exclusively to G/C-centered E-boxes, we questioned whether its knockdown in myoblasts would allow MyoD to bind these E-boxes and trigger a precocious expression of muscle differentiation genes.
To test this hypothesis, we made use of the C2C12 cell myogenic conversion model, widely employed in muscle gene expression studies (20, 50, 51). When grown in high serum (commonly referred as growth medium [GM]), C2C12 cells maintain a proliferating myoblast-like phenotype, and despite expression of MyoD, levels of proteins like the intermediate differentiation gene myogenin or the terminal differentiation markers myosin heavy chain (MHC) and troponin remain low or absent. Only when C2C12 cells exit the cell cycle upon reaching confluence and/or are switched into a low-serum medium (differentiation medium [DM]) do they fuse and terminally differentiate to form multinucleated MHC-positive myotubes.
We found here that knockdown of endogenous ZEB1 in C2C12 cells resulted in precocious expression of differentiation markers. Upon switching to DM, myogenin protein became detectable earlier in C2C12 cells knocked down for ZEB1 with a specific siRNA against ZEB1 (si1ZEB1) than in cells transfected with a siRNA control (siCtl) (Fig. 1A). Upregulation of MHC also occurred earlier and reached higher levels in C2C12 cells knocked down for ZEB1 with si1ZEB1 than in siCtl cells (Fig. 1B). A similar premature expression of MHC occurred when using two additional and independent siRNAs against ZEB1, here referred to as si2ZEB1 and si3ZEB1 (Fig. 1C and D).
Fig 1.

Knockdown of ZEB1 induces earlier protein expression of muscle differentiation genes. C2C12 cells, transfected with either 200 nM specific siRNAs against ZEB1 (si1ZEB1, si2ZEB1, or si3ZEB1) or a siRNA control (siCtl), were allowed to differentiate for up to 48 h after being switched to differentiation medium. At the indicated time points, cells were lysed and assessed by Western blotting for ZEB1 (H-102) and either myogenin (F5D) (A) or MHC (MF-20) (B to D) along with α-tubulin (B5-1-2) as a loading control.
The earlier induction of muscle genes following ZEB1 knockdown was also examined at the mRNA level (Fig. 2). Consistent with the above-described results, mRNA for MHC isoforms IIa (MyH2) and IIb (MyH4) accumulated more rapidly and to higher levels in ZEB1 knockdown C2C12 cells than in the counterpart siCtl control cells (Fig. 2A and B). In addition, mRNA expression levels of the differentiation genes for troponin and myogenin and the determination genes for MyoD and Myf-5 also displayed an earlier induction profile in ZEB1-knocked-down cells than in controls (Fig. 2C to G). Together, these results indicate that ZEB1 knockdown induces premature induction of muscle genes and suggest that endogenous ZEB1 may impose a delay on the ability of MRFs to drive muscle differentiation.
Fig 2.

ZEB1 knockdown induces precocious mRNA expression of several muscle genes. As in Fig. 1, C2C12 cells knocked down for ZEB1 (si1ZEB1) or transfected with a siRNA control (siCtl) were allowed to differentiate for up to 48 h after being switched to differentiation medium. At the indicated time points, mRNA levels for MHC type IIa (MyH2) (A), MHC type IIb (MyH4) (B), troponin T1 (C), myogenin (D), MyoD (E), Myf5 (F), and ZEB1 (G) were determined by quantitative real-time PCR (qRT-PCR) relative to levels of GAPDH as a reference gene. qRT-PCR was conducted as detailed in Materials and Methods. Data shown are a representative case of four independent experiments.
ZEB1 knockdown accelerates myotube formation.
Next, we examined whether this induction of muscle differentiation genes in C2C12 cells upon ZEB1 knockdown translated into their earlier terminal differentiation into multinucleated myotubes. Compared to control cells, the formation of myotubes in cells knocked down for ZEB1 was significantly accelerated, as evidenced morphologically and by quantification of the number of nuclei in MHC-positive cells with respect to the total number of nuclei (Fig. 3A to C; also data not shown for si3ZEB1). At late time points, the difference in the number of nuclei in MHC-positive cells between both experimental conditions was reduced. However, myotube size (number of nuclei per myotube) was still considerably larger in ZEB1 knockdown cells than in control cells (Fig. 3D displays the number of MHC-positive myotubes with more than 4 nuclei). Likewise, although by 72 h, the number of MHC-positive myotubes with more than 4 nuclei was similar under both conditions (Fig. 3D), ZEB1 knockdown cells still had more nuclei per myotube than control cells (e.g., more myotubes in siZEB1 than in siCtl exceeded 6 nuclei) (Fig. 3A).
Fig 3.

ZEB1 knockdown accelerates myotube conversion. (A) C2C12 cells transfected with 200 nM of a specific siRNA against ZEB1 (si1ZEB1) or a siRNA control (siCtl) were grown to confluence in growth medium (GM) (denoted time zero) before being switched to differentiation medium (DM) and allowed to differentiate for up to 72 h in DM. At the indicated time points, cells were fixed and immunostained for MHC (MF-20). Nuclei were counterstained with hematoxylin. Magnification, ×4. Captures shown are representative of five independent experiments with si1ZEB1 and siCtl. For all panels, the number of C2C12 cells throughout the myogenic conversion assay was the same under the ZEB1 knockdown (si1ZEB1, si2ZEB1, and si3ZEB1) and control (siCtl) conditions, as assessed by trypan blue staining (at the beginning of the assays) and by scoring the number of nuclei counterstained with hematoxylin (at the different endpoints) (total cell counts are not shown). (B) Quantification of the number of nuclei in MHC-positive cells with respect to the total number of nuclei in the myogenic conversion experiments shown in panel A. A value of 100 was arbitrarily set for comparison. Counts in panels B to D and F are the averages of four representative fields. (C) Same as panel B but with si2ZEB1. Similar results were obtained with si3ZEB1 (not shown). (D) Quantification of the number of MHC-positive myotubes with more than 4 nuclei with respect to the total number of nuclei. Similar results were obtained for si2ZEB1 and si3ZEB1 (data not shown). (E) Knockdown of ZEB1 also accelerates myotube formation in C2C12 cells maintained in GM. As for panel A, C2C12 cells were transfected with siRNAs against ZEB1 or the siRNA control, but upon reaching confluence, cells were maintained for the rest of the experiment in GM. Time zero refers to the time point when cells reached confluence and corresponds to the time point in panels A to D when cells were switched from GM to DM. (F) Quantification of the number of nuclei in MHC-positive cells with respect to the total number of nuclei in myogenic conversion assays where cells were maintained in GM for the entire experiment, as described above for panel E.
Of note, ZEB1 knockdown also induced accelerated myotube formation in C2C12 cells that were continuously maintained in GM, even after they reached confluence (Fig. 3E and F). This suggests that ZEB1 knockdown can trigger differentiation once cells have undergone contact-inhibition-mediated cell cycle exit. Together, these results suggest that under normal conditions, endogenous ZEB1 delays myoblast-to-myotube conversion, while ZEB1 knockdown is sufficient to activate (by relieving repression) a gene signature associated with terminal muscle differentiation.
Stage-dependent differential binding of ZEB1 and MyoD to muscle differentiation genes.
For most of its known target genes, ZEB1 inhibits their expression by binding to G/C-centered E-boxes at their regulatory regions and actively repressing transcription (14, 15, 24–26, 52, 53). We therefore questioned whether the observed changes in muscle gene expression upon ZEB1 knockdown involve binding of ZEB1 to G/C-centered E-boxes in muscle differentiation genes. As MyoD binds G/C-centered E-boxes at muscle differentiation genes in myotubes but not in myoblasts (13), we also wondered whether ZEB1 could be occupying these E-boxes at the myoblast stage.
The regulatory regions of muscle terminal differentiation genes are particularly enriched for G/C-centered E-boxes (12, 13). We therefore decided to test the ability of ZEB1 and MyoD to bind to these E-boxes in the promoters of mouse MyH4 and troponin through chromatin immunoprecipitation (ChIP) assays of C2C12 myoblasts and myotubes (Fig. 4A). In myoblasts, antibodies against ZEB1, but not its respective control IgG, immunoprecipitated regions of the MyH4 and troponin promoters containing G/C-centered E-boxes (Fig. 4A). Interestingly, MyoD did not bind to the same region of either promoter in myoblasts. Conversely, when these experiments were performed with myotubes, the reverse was observed: MyoD, but not ZEB1, was found to bind to G/C-centered E-box-containing regions in both promoters (Fig. 4A). Lastly, both ZEB1 and MyoD antibodies failed to immunoprecipitate a fragment of the mouse GAPDH promoter lacking G/C-centered E-boxes (Fig. 4A). These results indicate that, in line with our original hypothesis and in an opposite pattern from MyoD, endogenous ZEB1 binds directly to muscle differentiation gene promoters in myoblasts but not in myotubes.
Fig 4.


ZEB1 binds to G/C-centered E-boxes in muscle differentiation genes whose transcription is repressed largely through a CtBP-dependent mechanism. (A) ZEB1 and MyoD display differential binding to G/C-centered E-boxes in muscle differentiation genes in myoblasts and myotubes. Shown are data for qRT-PCR of fragments of the mouse Myh4, troponin T1, and GAPDH promoters immunoprecipitated in ChIP assays from C2C12 myoblasts and myotubes with ZEB1 antibody (E-20X), MyoD antibody (C-20), or their respective IgG controls. ChIP assays were performed as described in Materials and Methods. Values represent binding relative to the input averaged from two experiments, each performed in triplicate, and are representative of at least three essays. (B) The basal transcriptional activity of the MHC promoter is under negative regulation by ZEB1 and CtBP in myoblasts but not in myotubes. Nonconfluent cycling C2C12 myoblasts or confluent myotubes allowed to terminally differentiate for 48 h were cotransfected with 100 nM control siRNA (siCtl) or siRNAs against ZEB1 (si1ZEB1 and si2ZEB1) or CtBP (siCtBP) or equal molar amounts of their different combinations along with 0.4 μg of a luciferase reporter for the MHC promoter. Transcriptional assays and assessment of relative luciferase units (RLU) in panels B to E, G, and H were performed as described in Materials and Methods and are representative of at least four independent experiments. The knockdown efficiencies of si1ZEB1, si2ZEB1, and siCtBP in C2C12 cells are shown in panel F and in Fig. 1A to C. (C) Same as panel B but with 0.2 μg of the troponin I promoter as a luciferase reporter. (D) Same as panel B but with 0.2 μg of the MCK promoter as a luciferase reporter. (E) Same as panel B but with 0.2 μg of the MyoD enhancer/promoter. (F) Knockdown efficiency of siRNAs against CtBP and ZEB1 in C2C12 and C3H-10T1/2 cells. (Left) C2C12 cells were transfected with 100 nM either siCtl or siCtBP. CtBP protein levels were assessed by Western blotting using an antibody against CtBP1/2 (E-12) along with α-tubulin (B5-1-2) as a loading control. (Middle) C3H-10T1/2 cells were transfected with 200 nM either siCtl or si1ZEB1. ZEB1 protein levels were assessed by Western blotting using an antibody against ZEB1 (H-102) along with α-tubulin (B5-1-2) as a loading control. (Right) Same as the left panel but with C3H-10T1/2 cells. (G) Muscle genes are also under negative regulation by ZEB1 and CtBP in the C3H-10T1/2 myogenic conversion model. C3H-10T1/2 fibroblasts were cotransfected with luciferase reporters for muscle gene promoters, as in panels B to E, plus either 100 nM siRNA control (siCtl) or specific siRNAs against ZEB1 (siZEB1) or CtBP1/2 (siCtBP). The knockdown efficiencies of si1ZEB1 and siCtBP in C3H-10T1/2 fibroblasts are shown in panel F. (H) Transcriptional repression of muscle genes by ZEB1 depends on CtBP. C3H-10T1/2 fibroblasts were cotransfected with luciferase reporters for muscle gene promoters as in panel G, 0.6 μg of an expression vector for MyoD, and combinations of either 0.4 μg of an expression vector for ZEB1 or equal molar amounts of its corresponding empty expression vectors plus either 100 nM siRNA control (siCtl) or a specific siRNA against CtBP1/2 (siCtBP).
Inhibition of muscle differentiation by ZEB1 at the myoblast stage involves CtBP-mediated transcriptional repression.
Next, we explored whether negative regulation of muscle differentiation genes by ZEB1 occurs via transcriptional repression. C2C12 cells were cotransfected at the myoblast or myotube stage with either siCtl or specific siRNAs against ZEB1 and luciferase reporters containing the promoter regions of selected muscle genes. Knockdown of ZEB1 in nonconfluent cycling C2C12 myoblasts upregulated the basal transcriptional activity—that is, relieving ZEB1-mediated repression—of the MHC, troponin, muscle creatine kinase (MCK), and MyoD gene promoters, the latter to a much lesser extent (Fig. 4B to E). Interestingly, when the experiment was carried out with C2C12 cells that had been maintained in DM and differentiated into myotubes, interference of ZEB1 had no or a very limited effect (Fig. 4B to E).
ZEB1 represses transcription through recruitment of different corepressors, some still undetermined, that seem to act in a promoter- and tissue-specific manner (52, 53). A known ZEB1 corepressor is CtBP (CtBP1 and CtBP2), a non-DNA-binding protein that in turn tethers histone-modifying enzymes (33, 54, 55). CtBP mediates repression of targets of the myogenic MEF2 factor via recruitment of HDAC9/MITR (56). Here, we used a specific siRNA against CtBP (siCtBP) (Fig. 4F, left) to investigate a potential contribution of CtBP to ZEB1-mediated transcriptional inhibition of muscle genes. We found that CtBP knockdown in C2C12 myoblasts upregulated, albeit to different degrees, the basal activity of MHC, MCK, and MyoD promoters (Fig. 4B, D, and E). For these three promoters, the effect of concomitant knockdown of CtBP and ZEB1 on C2C12 myoblasts was similar to the effect of ZEB1 (or CtBP) single knockdown (Fig. 4B, D, and E), suggesting that CtBP is the main cofactor in the repression of these muscle genes by ZEB1 at the myoblast stage. Interestingly, relief of repression of the troponin promoter by CtBP knockdown in C2C12 myoblasts was below the extent of upregulation following knockdown of ZEB1 (Fig. 4C), indicating that ZEB1-mediated regulation of this gene also involves (and with a greater contribution) other corepressors (also see below). In contrast, as occurred for ZEB1 knockdown, interference of CtBP in myotubes had no or only limited effects on the transcription of all four promoters (Fig. 4B to E).
Active transcriptional repression of muscle genes by ZEB1 and its dependence on CtBP were then confirmed in C3H-10T1/2 fibroblasts, a well-established cell-based model to study transcriptional regulation by MRFs (3–5, 8). Constitutively, C3H-10T1/2 fibroblasts do not express muscle genes, but their transfection with MyoD is sufficient to induce the formation of myotubes, although C3H-10T1/2 cells never reached the extent of myogenic conversion observed for C2C12 cells (3, 4). Here, C3H-10T1/2 cells were cotransfected with MyoD and luciferase reporters for the four promoters described above, and the expression of ZEB1 was modulated by its overexpression or knockdown. We found that ZEB1 knockdown in C3H-10T1/2 cells (Fig. 4F, middle) upregulated the transcription of all four promoters (Fig. 4G), while overexpression of ZEB1 inhibited their activity (Fig. 4H).
Knockdown of CtBP in C3H-10T1/2 cells (Fig. 4F, right) augmented the basal activity of these four promoters, including that of troponin (Fig. 4G). The relief of repression obtained with siCtBP was similar to or below that observed with interference of ZEB1 alone. Likewise, elimination of CtBP partially reversed the repressor effect of ZEB1 on all four promoters albeit to different levels (Fig. 4H). The differential degree of dependence on CtBP for the basal endogenous repression of the troponin promoter by ZEB1 in C2C12 and C3H-10T1/2 cells supports the above-mentioned evidence that the identity of ZEB1 corepressors varies in a promoter- and tissue-specific manner (52). In any case, altogether, these results with cell-based systems indicate that CtBP mediates a significant share of ZEB1-mediated repression of muscle gene expression.
ZEB1 dependence on CtBP for the regulation of muscle gene expression was next examined in vivo in Xenopus and Drosophila embryos. Xenopus embryos were microinjected with MyoD and either wild-type ZEB1 (ZEB1wt) or a version of ZEB1 where its CtBP-interacting domain (CID) had been mutated (ZEB1CIDmut). At blastula stages, ectodermal explants were isolated from these embryos, and the effect of both ZEB1 variants on MyoD-mediated induction of muscle gene expression was examined (Fig. 5A). ZEB1 inhibited mRNA expression of muscle actin, MCK, slow skeletal troponin I type 1 (TnnI1), fast skeletal troponin I type 2, tropomyosin 1 α chain, and MyoD. Mutation of the CID region in ZEB1 partially alleviated—and to different degrees—repression of all the genes examined except for TnnI1, where ZEB1 inhibition seemed to take place independently of CtBP (Fig. 5A). These results demonstrate that repression of muscle differentiation genes by ZEB1, both in cell-based systems and in vivo, occurs to a large extent in a CtBP-dependent manner.
Fig 5.
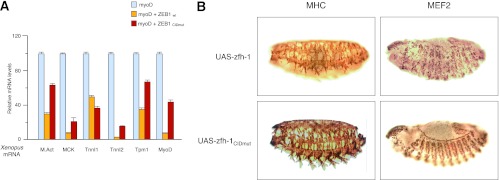
In vivo regulation of muscle gene expression by ZEB1/zfh-1 depends on CtBP. (A) Differential requirement for CtBP in ZEB1-mediated repression of muscle genes in Xenopus ectodermal explants. Xenopus embryos microinjected with mRNA encoding MyoD and either ZEB1wt or ZEB1CIDmut were used to generate ectodermal explants, and qRT-PCR was used to examine MyoD-induced expression of muscle actin (M.Act), MCK, slow skeletal troponin I type 1 (Tnni1), fast skeletal troponin I type 2 (Tnni2), tropomyosin 1 α chain (Tpm1), and MyoD. Isolation of mRNA from ectodermal explants and assessment of relative mRNA levels with respect to the reference gene EF1α were performed as described in Materials and Methods. Data shown are a representative case of three independent experiments. (B) Muscle phenotypes of Drosophila embryos expressing two copies of UAS-zfh-1 or UAS-zfh-1CIDmut under the control of the 24B-Gal4 mesodermal gene as a driver. Embryos were fixed and stained for MEF2 (stage 14) and MHC (stage 16). Note that some myofibers were missing from muscle groups in UAS-zfh-1 embryos compared to UAS-zfh-1CIDmut embryos.
The ZEB1 ortholog in Drosophila, zfh-1, is also a transcriptional repressor that binds to G/C-centered E-boxes and interacts with CtBP (18, 19, 54). Overexpression of zfh-1 in Drosophila embryos under the control of the heat shock protein 70 promoter results in a muscle phenotype that includes, but is not limited to, a downregulation of MHC and MEF2 expression in the somatic musculature (19). As in any overexpression experiment, these results cannot rule out that overexpression of zfh-1 is titrating out other proteins, including CtBP. On the other hand, loss of zfh-1 does not trigger accelerated myogenic differentiation but rather a complex phenotype with simultaneous loss, gain, and mispositioning of muscle precursors (18), thus suggesting that the role of zfh-1 during Drosophila myogenesis is not fully conserved in vertebrate ZEB1. Despite these caveats, we decided to examine whether the muscle phenotype observed upon zfh-1 overexpression was dependent on CtBP. zfh-1 is normally downregulated after gastrulation in most mesodermal derivatives, including muscle (57). We used a Gal4-UAS system to maintain the expression of wild-type zfh-1 (UAS-zfh-1) or a zfh-1 version unable to interact with CtBP (UAS-zfh-1CIDmut) in the developing muscle under the control of the 24B-Gal4 or MEF2-Gal4 mesoderm-specific driver lines (Fig. 5B and data not shown for MEF2-Gal4). As described previously (19), maintaining zfh-1 expression yielded alterations in somatic muscle development and MHC and MEF2 expression that are more complex than just a block in muscle differentiation (Fig. 5B for 24B-Gal4; also data not shown for MEF2-Gal4). Nevertheless, the muscle phenotype observed was dependent on CtBP, as no apparent effect on the embryos crossed with UAS-zfh-1CIDmut was observed (Fig. 5B for 24B-Gal4; also data not shown for MEF2-Gal4), even though the zfh-1CIDmut protein is stably expressed (58). Together, these results demonstrate that repression of muscle differentiation genes by ZEB1 (and likely zfh-1), both in cell-based systems and in in vivo models, occurs to a large extent in a CtBP-dependent manner.
Loss of ZEB1 induces muscle differentiation gene expression in vivo.
We next sought to confirm whether the induction of muscle genes upon ZEB1 knockdown observed by cell-based approaches was recapitulated in vivo. To this end, we examined the effect of overexpressing and knocking down ZEB1 during Xenopus embryonic development. ZEB1 mRNA was coinjected along with β-galactosidase mRNA as a lineage tracer into one of two blastomeres at the 2-cell stage. This introduced the injected mRNA into one bilateral half of the embryo, marked in pink (by staining for the Red-Gal β-galactosidase substrate) and oriented to the right in Fig. 6A. The contralateral half of the embryo served as a control. Embryos were then raised to neurula stages, and in situ hybridization was used to examine the expression of collagen type II (Col II), a marker of differentiated mesoderm, and muscle actin, a muscle marker (Fig. 6A, purple stains). We found that overexpression of ZEB1 strongly repressed the expression of both markers (Fig. 6A). Conversely, ZEB1 knockdown by microinjection of a specific morpholino oligonucleotide (ZEB1 MO) expanded the expression domains of both markers (Fig. 6A). These results parallel our findings in cell-based systems and support a role for ZEB1 in restraining the expression of muscle differentiation genes in vivo.
Fig 6.


ZEB1 represses muscle gene expression in vivo. (A) ZEB1 regulates muscle gene expression in Xenopus embryos. In situ hybridization for collagen II (Col II) and muscle actin (M. Actin) was performed in Xenopus embryos injected with mRNA for ZEB1 (ZEB1 mRNA) or a specific morpholino oligonucleotide against ZEB1 (ZEB1 MO), along with mRNA encoding β-galactosidase to mark the injected side (oriented rightward), as described in Materials and Methods. Embryos were then allowed to develop up to stages 15 to 16. Percentages of embryos with each phenotype are indicated, with the number of embryos analyzed shown in parentheses. The uninjected contralateral half of the embryo serves as an internal control (oriented leftward). (B) Muscle differentiation gene expression is accelerated in embryos from mice with targeted deletion of ZEB1. Shown is immunohistochemistry for MHC IIb (MyH4; BF-F3) in the developing muscle of ZEB1+/+ and ZEB1−/− E18.5 sibling mouse embryos. MyH4 displayed slightly higher expression levels in ZEB1−/− embryos than in the ZEB1+/+ counterparts. Magnifications of ×20 and ×10 are shown. Tissue immunostaining was performed as described in Materials and Methods. (C) Same as panel B but with immunostaining for MHC isoform IIa (MyH2; SC-71). MyH2 was expressed earlier in ZEB1−/− embryos than in the ZEB1+/+ sibling counterparts, where it was barely detectable. (D) Schematic model of the role of endogenous ZEB1 regulating the temporal pattern of gene expression during muscle differentiation. In myoblasts, ZEB1 binds to G/C-centered E-boxes in the regulatory regions of muscle differentiation genes whose transcription would be repressed at least in part via recruitment of CtBP. In myotubes, MyoD would displace ZEB1 from these G/C-centered E-boxes, thus activating the expression of muscle genes as differentiation progresses.
Mice carrying a homozygous targeted deletion of ZEB1 die close to birth with cleft palate and a number of bone and cartilage abnormalities (17). Although at embryonic days 10 to 11, levels of MyoD, myogenin, and MCK are not significantly affected in these mice (17), a molecular analysis of muscle differentiation at later stages has not been carried out. From the results shown up to here, one might expect that the temporal pattern of expression of muscle differentiation gene expression would be accelerated in ZEB1−/− mouse embryos.
Of all adult MHC isoforms, IIb/MyH4 is induced not only the earliest (around E14.5) but also at much higher levels than the rest, while mRNA for isoform IIa/MyH2 starts to be detected only after E17.5 (59). We decided to examine by immunohistochemistry the expression of both adult MHC isoforms during late development in sibling mouse embryos that were either wild type (+/+) or null (−/−) for ZEB1. We found that by E18.5, expression of MyH4 was only slightly higher in ZEB1−/− embryos than in the normal ZEB1+/+ sibling counterparts (Fig. 6B). Meanwhile, MyH2, which was barely detectable in ZEB1+/+ mouse embryos, displayed earlier (higher) expression in the ZEB1−/− sibling counterpart embryos (Fig. 6C). These results, consistent with the cell-based and in vivo data described above, indicate that loss of ZEB1 results in an acceleration of the temporal pattern of the muscle differentiation gene signature.
DISCUSSION
MRFs drive skeletal myogenesis, but the precise temporal and spatial pattern of muscle development involves regulation of the expression and activity of MRFs by a wide range of myogenic agonists and inhibitors. Using cell-based systems and several in vivo models, this study demonstrates that the transcriptional repressor ZEB1, which binds only to G/C-centered E-boxes, imposes a temporary delay in muscle gene expression and differentiation. ZEB1 knockdown resulted in premature expression of muscle differentiation genes and an acceleration of myotube formation. Contrary to MyoD, ZEB1 binds to G/C-centered E-boxes in muscle differentiation genes at the myoblast stage but not in myotubes. Inhibition of muscle differentiation by ZEB1 involves transcriptional repression of its targets at the myoblast stage, largely via recruitment of the CtBP corepressor. Finally, we show that the pattern of gene expression associated with muscle differentiation is accelerated in ZEB1−/− mouse embryos.
Binding of MyoD to the regulatory regions of muscle genes and triggering of myogenesis are temporarily blocked in myoblasts by the action of factors that sequester E-proteins and/or MyoD itself or that bind to the same E-box sequences (1, 6). Accordingly, exogenous overexpression of either type of factor (squelching or E-box binding factors) can inhibit MyoD-induced myotube formation in fibroblasts (7–9, 11, 13, 26, 60). However, in addition to the caveat of overexpression experiments mentioned above, displacement of MyoD from E-boxes when these inhibitory factors are overexpressed occurs independently of their DNA binding affinity relative to MyoD. Recently, it was shown that induction of myogenesis by MyoD is also regulated by its affinity for G/C-centered E-boxes with respect to other endogenous E-box binding proteins like Snail factors (13).
In myoblasts, MyoD binds to A/T-centered E-boxes but not to G/C-centered E-boxes, which are predominant in muscle differentiation genes and can be bound by Snail factors. It is only as differentiation progresses that MyoD occupies these E-boxes and activates muscle differentiation (13). ZEB1 binds G/C-centered E-boxes (mostly of the CACCTG/CAGGTG type also preferred by MyoD) in the promoters of a wide range of genes, where it can act in competition with other bHLH and zinc finger factors (24–28). Upon binding to these E-boxes, ZEB1 regulates gene expression mostly by active transcriptional repression (26, 52–54). Here, we found that knockdown of endogenous ZEB1 in nonconfluent cycling myoblasts, but not in differentiated myotubes, upregulates the basal transcriptional activity of several muscle gene promoters. This result suggests that these genes are normally under negative regulation by ZEB1 at the myoblast stage but not in myotubes (Fig. 6D).
Interestingly, ZEB1 is a downstream target of both Snail1 and Snail2 (21–23). Snail1/2 and ZEB1 have been extensively characterized for their role as inducers of an epithelial-to-mesenchymal transition (EMT) in the context of cancer cell invasion and tumor metastasis (14, 15). In triggering an EMT at the invasive front of carcinomas, both Snail1 and ZEB1 repress an overlapping set of epithelial specification markers, but they participate at different stages, with Snail1 triggering the EMT process and ZEB1 maintaining it (21, 22, 32). Cells in the dermomyotome also undergo an EMT to delaminate and migrate into the primary myotome (1). Conceivably, a similar temporal division of labor between Snail1 and ZEB1 could occur during muscle differentiation, as evidence already exists that Snail1 triggers an EMT in the dermomyotome and regulates its timing (61).
Transcriptional repression by ZEB1 is mediated by the tethering of different corepressors whose identity seems to vary in a tissue- and promoter-specific manner (reviewed in reference 52). Thus, repression of CD4 in lymphocytes is dependent on recruitment of the Tip60 histone acetyltransferase, while CtBP mediates ZEB1 repression of the growth hormone gene in the pituitary gland or interleukin-2 and Bcl-6 in lymphocytes. In heterologous Gal4/LexA reporter systems, ZEB1 antagonizes transcriptional activation by most transcription factors tested, including that induced by the myogenic factor MEF2C (53). ZEB1 repression of MEF2-mediated transcription involves the central region of ZEB1, encompassing the interacting domain for CtBP (33, 53, 54). In the context of muscle gene expression, CtBP is known to mediate repression of MEF2 targets by recruitment of HDAC9/MITR (56). Our results for cell-based systems and in vivo models demonstrated that CtBP participates in ZEB1 repression of muscle genes although to different degrees, being dispensable for the regulation of TnnI1 in Xenopus but largely required in the case of MyoD in C3H-10T1/2 fibroblasts. Similarly, the small dependence on CtBP for the repression of troponin in C2C12 cells relative to C3H-10T1/2 fibroblasts corroborated the promoter and tissue specificity of the cofactors involved in ZEB1-mediated repression. Nevertheless, while CtBP accounts for a large share of ZEB1 repression of muscle differentiation genes, our data also indicate that their inhibition by ZEB1 implicates additional CtBP-independent mechanisms that remain to be elucidated. In line with this, repression of E-cadherin by ZEB1 requires the summative action of several cofactors (62).
MyoD function, rather than its expression, is temporarily blocked in myoblasts (6). Likewise, ZEB1's antimyogenic activity seems to also be regulated during muscle differentiation. Thus, ZEB1 is expressed in the epithelium of the undifferentiated somite and the dermomyotome and continues to be expressed as muscle starts to form (16, 17). The required functional inactivation of ZEB1 during muscle differentiation may take place through several mechanisms, but our results here indicate that, at least in part, this might occur via displacement of ZEB1 from G/C-centered E-boxes by MyoD as muscle differentiation progresses. In keeping with this model, ZEB1 is also in equilibrium with the bHLH E2A protein for binding to the immunoglobulin heavy chain enhancer during B-cell differentiation (24).
In summary, this work has shown that muscle differentiation genes are under transcriptional repression by ZEB1/CtBP in myoblasts. ZEB1 imposes a temporal delay in muscle differentiation, and its regulatory and functional interplay with MyoD appears to play an essential role in controlling the temporal pattern of gene expression during muscle differentiation.
ACKNOWLEDGMENTS
We are indebted to researchers who generously provided us with reagents (see Materials and Methods) and especially to Y. Higashi (Institute for Development Research, Kasugai, Japan) for the δEF1+/− (ZEB1+/−) mice. We are also grateful to J. Skeath (Washington University School of Medicine, St. Louis, MO) for help in Drosophila experiments. We apologize to researchers whose work was cited indirectly through reviews due to space limitations.
The different parts of this study were independently funded by grants from the Spanish Ministry of Economy and Competitiveness (formerly MICINN) (grants BFU2007-60302 and BFU2010-15163), the La Caixa Foundation (LCF), the AVON-SAU Breast Cancer Research Campaign, the Olga Torres Foundation (FOT), the Spanish Association against Cancer (AECC), and the European Commission to A.P. L.S. is the recipient of a Ph.D. scholarship from the Ministry of Education, Culture and Sports (FPU Program, scholarship AP2010-4495), and her salary was previously partly funded by the AECC and FOT. E.S.-T.'s salary was partly funded by the LCF and CIBERehd. We declare that we have no competing financial interests.
Footnotes
Published ahead of print 22 January 2013
REFERENCES
- 1. Braun T, Gautel MM. 2011. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 12: 349– 361 [DOI] [PubMed] [Google Scholar]
- 2. Bentzinger CF, Wang YX, Rudnicki MA. 1 February 2012. Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. [Epub ahead of print.] doi:10.1101/cshperspect.a008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davis RL, Weintraub H, Lassar AB. 1987. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987– 1000 [DOI] [PubMed] [Google Scholar]
- 4. Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD. 1989. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. U. S. A. 86: 5434– 5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Braun T, Buschhausen-Denker G, Bober E, Tannich E, Arnold HH. 1989. A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J. 8: 701– 709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berkes CA, Tapscott SJ. 2005. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16: 585– 595 [DOI] [PubMed] [Google Scholar]
- 7. Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. 1990. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61: 49– 59 [DOI] [PubMed] [Google Scholar]
- 8. Spicer DB, Rhee J, Cheung WL, Lassar AB. 1996. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science 272: 1476– 1480 [DOI] [PubMed] [Google Scholar]
- 9. Chen CMA, Kraut N, Groudine M, Weintraub H. 1996. I-mf, a novel myogenic repressor, interacts with members of the MyoD family. Cell 86: 731– 741 [DOI] [PubMed] [Google Scholar]
- 10. Lu J, Webb R, Richardson JA, Olson EN. 1999. MyoR: a muscle-restricted basic helix-loop-helix transcription factor that antagonizes the actions of MyoD. Proc. Natl. Acad. Sci. U. S. A. 96: 552– 557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lemercier C, To RQ, Carrasco RA, Konieczny SF. 1998. The basic helix-loop-helix transcription factor Mist1 functions as a transcriptional repressor of MyoD. EMBO J. 17: 1412– 1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fong AP, Yao Z, Zhong JW, Cao Y, Ruzzo WL, Gentleman RC, Tapscott SJ. 2012. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell 22: 721– 735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CE, van Ijcken WF, Grosveld F, Rudnicki MA. 2012. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell 47: 457– 468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brabletz S, Brabletz T. 2010. The ZEB/miR-200 feedback loop: a motor of cellular plasticity in development and cancer? EMBO Rep. 11: 670– 677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. 2012. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 69: 3429– 3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Funahashi J, Sekido R, Murai K, Kamachi Y, Kondoh H. 1993. δ-Crystallin enhancer binding protein δEF1 is a zinc finger-homeodomain protein implicated in postgastrulation embryogenesis. Development 119: 433– 446 [DOI] [PubMed] [Google Scholar]
- 17. Takagi T, Moribe H, Kondoh H, Higashi Y. 1998. δEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 125: 21– 31 [DOI] [PubMed] [Google Scholar]
- 18. Lai ZC, Rushton E, Bate M, Rubin GM. 1993. Loss of function of the Drosophila zfh-1 gene results in abnormal development of mesodermally derived tissues. Proc. Natl. Acad. Sci. U. S. A. 90: 4122– 4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Postigo A, Ward E, Skeath JB, Dean DC. 1999. zfh-1, the Drosophila homologue of ZEB, is a transcriptional repressor that regulates somatic myogenesis. Mol. Cell. Biol. 19: 7255– 7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bergstrom DA, Penn BH, Strand A, Perry RL, Rudnicki MA, Tapscott SJ. 2002. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell 9: 587– 600 [DOI] [PubMed] [Google Scholar]
- 21. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA. 2010. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. U. S. A. 107: 15449– 15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG. 2011. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 286: 12024– 12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wels C, Joshi S, Koefinger P, Bergler H, Schaider H. 2011. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Invest. Dermatol. 131: 1877– 1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Genetta T, Ruezinsky D, Kadesch T. 1994. Displacement of an E-box-binding repressor by basic helix-loop-helix proteins: implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol. Cell. Biol. 14: 6153– 6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sekido R, Murai K, Funahashi J, Kamachi Y, Fujisawa-Sehara A, Nabeshima Y, Kondoh H. 1994. The δ-crystallin enhancer-binding protein δEF1 is a repressor of E2-box-mediated gene activation. Mol. Cell. Biol. 14: 5692– 5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Postigo A, Dean DC. 1997. ZEB, a vertebrate homolog of Drosophila Zfh-1, is a negative regulator of muscle differentiation. EMBO J. 16: 3935– 3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brabletz T, Jung A, Hlubek F, Löhberg C, Meiler J, Suchy U, Kirchner T. 1999. Negative regulation of CD4 expression in T cells by the transcriptional repressor ZEB. Int. Immunol. 11: 1701– 1708 [DOI] [PubMed] [Google Scholar]
- 28. Fontemaggi G, Gurtner A, Damalas A, Costanzo A, Higashi Y, Sacchi A, Strano S, Piaggio G, Blandino G. 2005. δEF1 repressor controls selectively p53 family members during differentiation. Oncogene 24: 7273– 7280 [DOI] [PubMed] [Google Scholar]
- 29. Lacher MD, Shiina M, Chang P, Keller D, Tiirikainen MI, Korn WM. 2011. ZEB1 limits adenoviral infectability by transcriptionally repressing the coxsackie virus and adenovirus receptor. Mol. Cancer 10: 91 doi:10.1186/1476-4598-10-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. 2005. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23:457– 462 [DOI] [PubMed] [Google Scholar]
- 31. Wu X, Chang MS, Mitsialis SA, Kourembanas S. 2006. Hypoxia regulates bone morphogenetic protein signaling through C-terminal-binding protein-1. Circ. Res. 99: 240– 247 [DOI] [PubMed] [Google Scholar]
- 32. Shirakihara T, Saitoh M, Miyazono K. 2007. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol. Biol. Cell 18: 3533– 3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furusawa T, Moribe H, Kondoh H, Higashi Y. 1999. Identification of CtBP1 and CtBP2 as corepressors of zinc finger-homeodomain factor δEF1. Mol. Cell. Biol. 19: 8581– 8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu JR, McKinsey TA, Xu H, Wang DZ, Richardson JA, Olson EN. 1999. FOG-2, a heart- and brain-enriched cofactor for GATA transcription factors. Mol. Cell. Biol. 19: 4495– 4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson SE, Wang X, Hardy S, Taparowsky EJ, Konieczny SF. 1996. Casein kinase II increases the transcriptional activities of MRF4 and MyoD independently of their direct phosphorylation. Mol. Cell. Biol. 16: 1604– 1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reynaud EG, Leibovitch MP, Tintignac LA, Pelpel K, Guillier M, Leibovitch SA. 2000. Stabilization of MyoD by direct binding to p57(Kip2). J. Biol. Chem. 275: 18767– 18776 [DOI] [PubMed] [Google Scholar]
- 37. Hunter JG, van Delft MF, Rachubinski RA, Capone JP. 2001. Peroxisome proliferator-activated receptor gamma ligands differentially modulate muscle cell differentiation and MyoD gene expression via peroxisome proliferator-activated receptor γ-dependent and -independent pathways. J. Biol. Chem. 276: 38297– 38306 [DOI] [PubMed] [Google Scholar]
- 38. Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. 2011. β-Catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. U. S. A. 108: 19204– 19209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abe S, Hiroki E, Iwanuma O, Sakiyama K, Shirakura Y, Hirose D, Shimoo Y, Suzuki M, Ikari Y, Kikuchi R, Ide Y, Yoshinari M. 2008. Relationship between function of masticatory muscle in mouse and properties of muscle fibers. Bull. Tokyo Dent. Coll. 49: 53– 58 [DOI] [PubMed] [Google Scholar]
- 40. Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, Fuller-Pace FV, Hoffman EP, Tapscott SJ, Sartorelli V. 2006. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev. Cell 11: 547– 560 [DOI] [PubMed] [Google Scholar]
- 41. Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, Noguchi CT. 2000. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J. Biol. Chem. 275: 39754– 39761 [DOI] [PubMed] [Google Scholar]
- 42. Dogra C, Changotra H, Mohan S, Kumar A. 2006. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-κB and degradation of MyoD protein. J. Biol. Chem. 281: 10327– 10336 [DOI] [PubMed] [Google Scholar]
- 43. Tanaka K, Kitagawa Y, Kadowaki T. 2003. Misexpression of mouse porcupine isoforms modulates the differentiation of P19 embryonic carcinoma cells. Cell Biol. Int. 27: 549– 557 [DOI] [PubMed] [Google Scholar]
- 44. Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. 2008. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 135: 579– 588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Noh OJ, Park YH, Chung YW, Kim IY. 2010. Transcriptional regulation of selenoprotein W by MyoD during early skeletal muscle differentiation. J. Biol. Chem. 285: 40496– 40507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kroll KL, Salic AN, Evans LM, Kirschner MW. 1998. Geminin, a neuralizing molecule that demarcates the future neural plate at the onset of gastrulation. Development 125: 3247– 3258 [DOI] [PubMed] [Google Scholar]
- 47. Nieuwkoop PD, Faber JJ. 1967. Normal table of Xenopus laevis (Daudin). North Holland Publishing Co, Amsterdam, The Netherlands [Google Scholar]
- 48. van Grunsven LA, Taelman V, Michiels C, Opdecamp K, Huylebroeck D, Bellefroid EJ. 2006. δEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev. Dyn. 235: 1491– 1500 [DOI] [PubMed] [Google Scholar]
- 49. Seo S, Herr A, Lim JW, Richardson GA, Richardson H, Kroll KL. 2005. Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev. 19: 1723– 1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blau HM, Chiu CP, Webster C. 1983. Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell 32: 1171– 1180 [DOI] [PubMed] [Google Scholar]
- 51. Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD. 2005. An initial blueprint for myogenic differentiation. Genes Dev. 19: 553– 569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanchez-Tillo E, Siles L, de Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A. 2011. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am. J. Cancer Res. 1: 897– 912 [PMC free article] [PubMed] [Google Scholar]
- 53. Postigo AA, Dean DC. 1999. Independent repressor domains in ZEB regulate muscle and T-cell differentiation. Mol. Cell. Biol. 19: 7961– 7971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Postigo A, Dean DC. 1999. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. U. S. A. 96: 6683– 6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chinnadurai G. 2009. The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res. 69: 731– 734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang CL, McKinsey TA, Lu JR, Olson EN. 2001. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J. Biol. Chem. 276: 35– 39 [DOI] [PubMed] [Google Scholar]
- 57. Lai Z, Fortini ME, Rubin GM. 1991. The embryonic expression patterns of zfh-1 and zfh-2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech. Dev. 34: 123– 134 [DOI] [PubMed] [Google Scholar]
- 58. Leatherman JL, Dinardo S. 2008. zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell 3: 44– 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu BD, Allen DL, Leinwand LA, Lyons GE. 1999. Spatial and temporal changes in myosin heavy chain gene expression in skeletal muscle development. Dev. Biol. 216: 312– 326 [DOI] [PubMed] [Google Scholar]
- 60. Kataoka H, Murayama T, Yokode M, Mori S, Sano H, Ozaki H, Yokota Y, Nishikawa S, Kita T. 2000. A novel snail-related transcription factor Smuc regulates basic helix-loop-helix transcription factor activities via specific E-box motifs. Nucleic Acids Res. 28: 626– 633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delfini MC, De La Celle M, Gros J, Serralbo O, Marics I, Seux M, Scaal M, Marcelle C. 2009. The timing of emergence of muscle progenitors is controlled by an FGF/ERK/SNAIL1 pathway. Dev. Biol. 333: 229– 237 [DOI] [PubMed] [Google Scholar]
- 62. Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A. 2010. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 29: 3490– 3500 [DOI] [PubMed] [Google Scholar]