

Abstract
Suppression of major histocompatibility complex (MHC) class I-mediated presentation of human cytomegalovirus (HCMV) peptides is an important mechanism to avoid CD8 T lymphocyte recognition and killing of infected cells. Of particular interest is how MHC class I presentation of essential regulatory immediate early (IE) proteins of HCMV can be effectively compromised at times when known viral immunoevasins are not abundantly expressed. The tegument protein pp71 had been suggested to be involved in MHC class I downregulation. Intriguingly, this polypeptide is also critically engaged in the initial derepression of the major IE gene locus, leading to enhanced expression of IE proteins IE1-pp72 and IE2-pp86. Using a set of viral mutants, we addressed the role of pp71 in MHC class I presentation of IE1-pp72-derived peptides. We show that the amount of “incoming” pp71 positively correlates with IE1-pp72 protein levels and with the presentation of IE1-derived peptides. This indicates that the amount of the IE1 protein, induced by pp71, rather than a putative immunoevasive function of the tegument protein, determines MHC class I antigen presentation of IE1-derived peptides. This process proved to be independent of the presence of pp65, which had been reported to interfere with IE1 presentation. It may thus be beneficial for the success of HCMV replication to limit the level of pp71 delivered from infecting particles in order to avoid critical levels of MHC class I presentation of IE protein-derived peptides.
INTRODUCTION
Infection with human cytomegalovirus (HCMV) is tightly controlled by the immune system. Only immature or compromised immune defense functions may allow viral replication to proceed to an extent that results in overt clinical disease. Cytomegaloviruses likely would have been eradicated if it was not for the expression of immunoevasive functions that antagonize intrinsic, innate, and adaptive responses in order to allow the establishment of latency (1–4).
CD8 T cells are particularly powerful in restricting both acute infection and viral reactivation (5–7). Glycoproteins encoded by the US2-11 gene region efficiently disturb the major histocompatibility complex (MHC) class I antigen presentation pathway, thereby antagonizing CD8 T cell control (8–11). Expression of these genes in a rhesus cytomegalovirus model prevented superinfection in seropositive animals (12). Besides gpUS2-gpUS11, the tegument proteins pp65 and pp71 have also been suggested to interfere with MHC class I-mediated antigen presentation (13, 14).
Although considerable knowledge has been gathered about the regulation of the MHC class I presentation pathway by studying HCMV immunoevasive proteins, there is still only limited information available on how single evasins act in the context of infection (8, 9). One question of particular interest is how MHC class I antigen presentation was suppressed at the immediate early (IE) stages of infection. Abundant expression of the IE proteins IE1 (IE1-pp72) and IE2 (IE2-pp86), a crucial event for efficient progression of lytic infection, relies on pp71-mediated derepression of the major IE enhancer-promoter (15–18). On the other hand, IE1 and IE2 are major target antigens of the CD8 T cell response against HCMV (19–21). Presentation of antigenic peptides from the two proteins by MHC class I at IE phases may be detrimental to the success of viral infection. Mechanisms that suppress presentation are likely implemented soon after infection, thereby preventing recognition by cytotoxic CD8 T lymphocytes (CTL) and cell lysis.
The gpUS3 has been reported to be expressed at IE times (22–26). However, it may have only limited impact on IE1 peptide presentation (27). We have recently shown that both gpUS2 and gpUS11 are also expressed at IE times, and both proteins interfere significantly with IE1 peptide presentation by MHC class I (27). All these evasive proteins are, however, synthesized concomitantly with IE1 and IE2. Thus, their kinetics of expression may simply be insufficient to prevent presentation of IE1 and IE2 early in lytic infection. In contrast, tegument proteins, delivered to the cell via incoming viral particles, are directly available for suppression of antigen presentation. Using an HCMV deletion mutant as well as vaccinia virus constructs, a selective effect of pp65 on presentation of peptides from IE1 has been reported (13). The role of incoming pp71 in the suppression of CD8 T cell recognition of HCMV-infected cells has not been fully elucidated. In glioblastoma cells, pp71 appeared to have an MHC class I-suppressive effect only after its de novo synthesis (14). It remained unclear whether pp71 has an impact on MHC class I expression in permissively infected human fibroblasts and whether, in these cells, incoming pp71 would have an effect on the presentation of IE protein-derived peptides.
The pp71 protein is a key regulator of lytic HCMV infection (16, 28). Transcription of IE genes of HCMV critically depends on pp71, originating from infecting viral particles (4, 28). Deleting the gene encoding pp71 from the viral genome results in mutants with severely impaired growth properties (15). This renders investigations of pp71-mediated effects on MHC class I in the course of infection complicated. We thus focused our analysis of the immunoevasive impact of pp71 on HCMV mutants that expressed various levels of pp71. We found that increased amounts of pp71 in viral particles, resulting from enhanced expression by mutant viruses, correlated with increased presentation of IE1-derived peptides by MHC class I. This indicated that the tegument protein was supporting rather than suppressing CD8 T cell recognition.
(Part of this research was conducted by J. Hesse in fulfillment of the requirements for a doctoral degree from Johannes Gutenberg University, Mainz, Germany.)
MATERIALS AND METHODS
Cells and viruses.
Primary human foreskin fibroblasts (HFF) and cytotoxic CD8 T lymphocytes (CTL) were cultured as described previously (8). Viral stocks of the laboratory strain AD169, the wild-type (wt) strains RV-BADwt (RV stands for recombinant virus) (29) and RV-HB15 (AD169-BAC [30]), the pp65 deletion mutants RV-Ad65 (31), RV-Hd65 (8), and RV-KB14, the EYFP-pp71 (EYFP stands for enhanced yellow fluorescent protein) mutant AD169/EYFP-pp71 (32), and the US2-11 deletion mutants AD169_ΔUS2-11 and AD169/EYFP-pp71_ΔUS2-11 were prepared and titrated by counting IE1-positive cells or by quantifying intracellular viral genomes as described previously (9).
For experiments requiring restriction of gene expression to IE genes in infected cells, cycloheximide (CX) (250 μg/ml) was added 1 h prior to infection. After an incubation period of 9 h, the culture medium was replaced and actinomycin D (AcD) (10 μg/ml) was added. Following another 13.5 h of incubation, the cells were fixed using 0.5% paraformaldehyde and subjected to gamma interferon (IFN-γ) enzyme-linked immunosorbent spot assay (ELISpot) analysis.
BAC mutagenesis.
The viral mutants RV-KB14, AD169_ΔUS2-11, and AD169/EYFP-pp71_ΔUS2-11 were generated by bacterial artificial chromosome (BAC) mutagenesis, using Red recombination in Escherichia coli strain EL250 as described by Lee et al. (33). For generation of BAC pKB14, the UL83 (pp65) gene was deleted in BAC pAD/Cre (29) by inserting a tetracycline resistance gene, which was retained at the deletion site. The tetracycline resistance gene was amplified from plasmid pACYC184 (34), using primers KB38 (5′-GCT GCC GCA CGA GAC GCG ACT CCT GCA GAC GGG TAT CCA CGT ACG CGT GAG CGC ATT CAC AGT TCT CC-3′) and KB39 (5′-GGA CGT GGG TTT TTA TAG AGT CGT CCT AAG CGC GTG CGG CGG GTG GCT CAC TGA AGT CAG CCC CAT AC-3′). In the resulting BAC clone, the complete coding sequence of pp65 was deleted except for 150 bp at the 5′ end of UL83 and the stop codon. For generation of AD169_ΔUS2-11 and AD169/EYFP-pp71_ΔUS2-11, the entire region containing the US2-11 genes was deleted in AD169-BAC (30) or in BAC pHB15/EYFP-pp71 (32), respectively, by inserting a kanamycin resistance gene. The kanamycin resistance gene was amplified from a derivative vector of pCP15 (35), using primers with 48- to 51-bp identity to the nucleotide sequence directly adjacent to the deletion site (JH5 fwd [fwd stands for forward] [5′-CTT ACA GCT TTT GAG TCT AGA CAG GGT AAC AGC CTT CCC TTG TAA GAC AGA AGA GCG CTT TTG AAG CTG GG-3′] and JH5 rev [rev stands for reverse] [5′-GGG TAC TCG TGG CTA GAT TTA TTG AAA TAA ACC GCG ATC CCG GGC GTC GGA ATA GGA ACT TCA AGA TCC CCC-3′]). The reconstitution of BAC vector-free recombinant viruses was performed by the method of Yu et al. (29) (RV-KB14) or by the method of Hobom et al. (30) (AD169_ΔUS2-11 and AD169/EYFP-pp71_ΔUS2-11).
Growth kinetics.
To quantify viral DNA replication, HFF were infected at 0.4 or 0.04 human cytomegalovirus (HCMV) genome copies per cell. Infected cells were harvested at different time points. DNA was isolated. Viral genomes were quantified by TaqMan PCR as described previously (9).
Polyacrylamide gel electrophoresis and quantitative Western blot analysis.
Western blot analyses were performed using the Odyssey system (Licor Biosciences, Lincoln, NE, USA) as described previously (9) with a few modifications. HFF were infected either at a multiplicity of infection (MOI) of 1 or with viral inocula normalized for an equivalent uptake of 125 or 300 viral genomes per cell. For sodium dodecyl sulfate (SDS) gel electrophoresis, 8% or 10% polyacrylamide gels containing 0.1% SDS were used. Protein staining was performed by soaking the gel with Roti-Black P-Siberfärbungskit für Proteine (Carl Roth, Karlsruhe, Germany). For Bis-Tris gel electrophoresis, the Bolt Bis-Tris Plus gel system (Life Technologies, Darmstadt, Germany) was used. Electrophoresis was performed with 4 to 12% gels according to the manufacturer's instructions.
For blotting, a methanol-reduced transfer buffer (25 mM Tris–192 mM glycine–10% methanol) was employed. Membranes, which were air dried for 1 h after blotting, were reactivated by rinsing in methanol, H2O, and phosphate-buffered saline (PBS). Membrane blocking was carried out with PBS plus 5% milk powder (PBS–5% milk powder). Primary antibodies were diluted in PBS–5% milk powder–0.1% Tween 20. Rabbit polyclonal antibody directed against human β-actin (Rockland, Gilbertsville, PA, USA; distributed by Biotrend, Köln, Germany) was diluted 1:400 to 1:2,000, the monoclonal antibody directed against IE1 (p63-27 [36]) was diluted 1:125 to 1:250, the monoclonal antibody directed against pp65 (65-33) was diluted 1:250, and the goat polyclonal antibody directed against pp71 (vC-20; Santa Cruz Biotechnology, Heidelberg, Germany) was diluted 1:100 to 1:200. For detection of primary antibody binding, the following secondary antibodies were used: IRDye 800-conjugated donkey anti-mouse IgG (H+L) and IRDye 800-conjugated donkey anti-goat IgG (H+L) (Rockland, Gilbertsville, PA, USA; distributed by Biotrend, Köln, Germany); Alexa Fluor 680-conjugated goat anti-rabbit IgG (H+L), Alexa Fluor 680-conjugated donkey anti-rabbit IgG (H+L), and Alexa Fluor 680-conjugated donkey anti-goat IgG (H+L) (Invitrogen, Karlsruhe, Germany); and IRDye 800-conjugated goat anti-rabbit IgG (H+L) (Licor Biosciences, Lincoln, NE, USA). Secondary goat anti-rabbit antibodies from Invitrogen or Licor and donkey anti-goat antibodies from Invitrogen were diluted 1:10,000, donkey anti-mouse antibodies from Rockland were diluted 1:5,000 to 1:10,000, donkey anti-goat antibodies from Rockland were diluted 1:5,000, and donkey anti-rabbit antibodies from Invitrogen were diluted 1:400 in PBS–0.1% Tween–0.01% SDS. Membranes were incubated with secondary antibodies in the dark for 2 h. For dual detection with donkey anti-goat and goat anti-rabbit antibodies, the membranes were first incubated with donkey anti-goat antibody for 1 h (donkey anti-goat antibody from Invitrogen diluted in PBS–0.1% Tween without SDS) and, after 3 washing steps with PBS–0.2% Tween, then incubated for 1 h with goat anti-rabbit antibody (diluted in PBS–0.1% Tween without SDS). For reprobing after detection, the membranes were washed for 5 min in H2O, for 5 min in 0.2 M NaOH, and again for 5 min in H2O. To calculate relative fluorescence intensities of IE1 and pp71, the fluorescence intensity obtained after staining against each of the proteins was divided by the fluorescence intensity obtained after β-actin staining (9).
Flow cytometry.
A Cytomics FC500 flow cytometer (Beckman Coulter, Brea, CA, USA) was used to analyze surface expression of major histocompatibility complex (MHC) class I and human leukocyte antigen HLA-A2 on HCMV-infected HFF. Viral samples were normalized for an equivalent uptake of 125 viral genomes per cell. The flow cytometry analyses were performed as described earlier (9).
IFN-γ ELISpot assay.
ELISpot assays were performed as described previously (9). HFF as stimulator cells were infected with viral samples normalized for an equivalent uptake of 125 viral genomes per cell or with an MOI of 5. For fixation, CX-and-AcD (CX/AcD)-treated stimulator cells were resuspended in PBS–0.5% paraformaldehyde, incubated for 20 min at room temperature, and washed twice with PBS. CTL clones specific for the HLA-A2-restricted HCMV-derived peptides IE1 from amino acids 297 to 305 (IE1TMY-CTL [8, 37]) and pp65 from amino acids 495 to 503 (pp65NLV-CTL [8, 38, 39]) were generated by peptide immunization of HLA-A2/huCD8 (hu stands for human) double-transgenic mice (a kind gift from L. Sherman, The Scripps Institute, La Jolla, CA, USA). Details of establishment and culturing of these cells were previously described (8). For ELISpot experiments with 5,000 CTL per well, CD8 T cells were enriched using the magnetically activated cell sorting (MACS) system from Miltenyi Biotech (Bergisch Gladbach, Germany). For this, cells were centrifuged at 472 × g for 5 min and resuspended in MACS buffer (PBS–0.5% bovine serum albumin–2 mM EDTA). After that procedure, the cells were incubated with anti-human CD8 Miltenyi MACS MicroBeads for 15 min at 4°C. Subsequently, the cells were washed twice with MACS buffer (centrifugation at 302 × g for 10 min) and loaded on a Miltenyi LS column. The MACS cell separation was performed according to the manufacturer's instructions.
Mass spectrometry. (i) Sample preparation.
Virions of RV-BADwt, RV-KB14, RV-HB15, or RV-Hd65 were purified from late-stage infected HFF using glycerol tartrate gradient ultracentrifugation by the method of Irmiere and Gibson (40). The protein concentration of the purified virions was determined with the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Bonn, Germany). Then, virions (20 μg) were pelleted for protein digestion (100,000 × g, 1 h).
(ii) Protein digestion.
Aliquots (20 μg) of purified virions were dissolved in lysis buffer. Lysis buffer consisted of 7 M urea, 2 M thiourea, and 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS). Subsequently, proteins were digested using a modified filter-aided sample preparation (FASP) method (41). Briefly, redissolved protein was loaded on the filter, and detergents were removed by washing the filter three times with buffer containing 8 M urea. The proteins were then reduced using dithiothreitol and alkylated using iodoacetamide, and the excess reagent was quenched by supplementation of additional dithiothreitol and washed through the filters. Buffer was exchanged by washing with 50 mM NH4HCO3, and proteins were digested overnight by trypsin (Trypsin Gold; Promega, Mannheim, Germany) with an enzyme-to-protein ratio of 1:50. After overnight digestion, peptides were recovered by centrifugation and two additional washes using 50 mM NH4HCO3. The flowthrough fractions were combined, lyophilized, and redissolved in 20 μl of 0.1% formic acid by sonication. The resulting tryptic digest solutions were diluted with aqueous 0.1% (vol/vol) formic acid to a concentration of 200 ng/μl and spiked with 25 fmol/μl of enolase 1 (Saccharomyces cerevisiae) tryptic digest standard (Waters Corporation, Milford, MA, USA).
(iii) Ultraperformance liquid chromatography (UPLC)-mass spectrometry (MS) configuration.
Nanoscale LC separation of tryptic peptides was performed with a nanoAcquity system (Waters Corporation) equipped with an ethylene bridged hybrid (BEH) C18 analytical reversed-phase column (1.7 μm; 75 μm by 150 mm) (Waters Corporation) in direct injection mode as described previously (42). Part (0.2 μl) of the sample (50 ng of total protein) was injected per technical replicate. Mobile phase A was water containing 0.1% (vol/vol) formic acid, while mobile phase B was acetonitrile (ACN) containing 0.1% (vol/vol) formic acid. The peptides were separated with a gradient of 3 to 40% mobile phase B over 60 min at a flow rate of 300 nl/min, followed by a 10-min column rinse with 90% of mobile phase B. The columns were reequilibrated under the initial conditions for 15 min. The analytical column temperature was maintained at 55°C. The lock mass compound, [Glu1]-Fibrinopeptide B (100 fmol/μl), was delivered by the auxiliary pump of the LC system at 300 nl/min to the reference sprayer of the NanoLockSpray source of the mass spectrometer.
Mass spectrometric analysis of tryptic peptides was performed using a Synapt G2-S mass spectrometer (Waters Corporation, Manchester, United Kingdom). For all measurements, the mass spectrometer was operated in v-mode with a typical resolution of at least 25,000 FWHM (full width half maximum). All analyses were performed in positive-mode electrospray ionization (ESI). The time of flight analyzer of the mass spectrometer was externally calibrated with a NaI mixture from m/z 50 to 1,990. The data were acquisition lock mass corrected using the doubly charged monoisotopic ion of [Glu1]-Fibrinopeptide B. The reference sprayer was sampled with a frequency of 30 s. Accurate mass LC-MS data were collected in data-independent modes of analysis (43, 44). The spectral acquisition time in each mode was 0.6 s with a 0.05-s interscan delay. In low-energy MS mode, data were collected at a constant collision energy of 4 eV. In elevated-energy MS mode, the collision energy was ramped from 25 to 55 eV during each 0.6-s integration. One cycle of low- and elevated-energy data was acquired every 1.3 s. The radio frequency amplitude applied to the quadrupole mass analyzer was adjusted so that ions from m/z 350 to 2,000 were efficiently transmitted, ensuring that any ions observed in the LC-MS data less than m/z 350 were known to arise from dissociations in the collision cell. All samples were analyzed in quintuplicate.
Data processing and protein identification.
Continuum LC-MS data were processed and searched using ProteinLynx GlobalSERVER version 2.5.2 (Waters Corporation). Protein identifications were obtained by searching a custom compiled database containing sequences of human and HCMV proteins from the Uniprot database. Sequence information of enolase 1 (S. cerevisiae) and bovine trypsin were added to the databases to normalize the data sets or to conduct absolute quantification as described previously (45). Guideline identification criteria were applied (46) for all searches. The experimental data were typically searched with a 3-ppm precursor and 10-ppm product ion tolerance, respectively, with one missed cleavage allowed and fixed carbamidomethylcysteine and variable methionine oxidation set as the modifications.
RESULTS
IE1 peptide presentation by HCMV-infected cells is strain dependent but pp65 independent.
Both pp65 and pp71 had been reported to interfere with MHC class I antigen presentation (13, 14). To investigate which of the tegument proteins would have an impact on presentation of peptides derived from IE1-pp72 under IE conditions of infection, a set of viral strains was used in initial experiments. In two of these strains, the genes encoding pp65 were intentionally deleted to abolish pp65 expression (RV-KB14 and RV-Hd65) (8) (Fig. 1A). Note that the genetic manipulation used to mutagenize the pp65 gene UL83 differed. For both mutants, the parental, BAC-derived wild-type (wt) strains were used for control.
Fig 1.
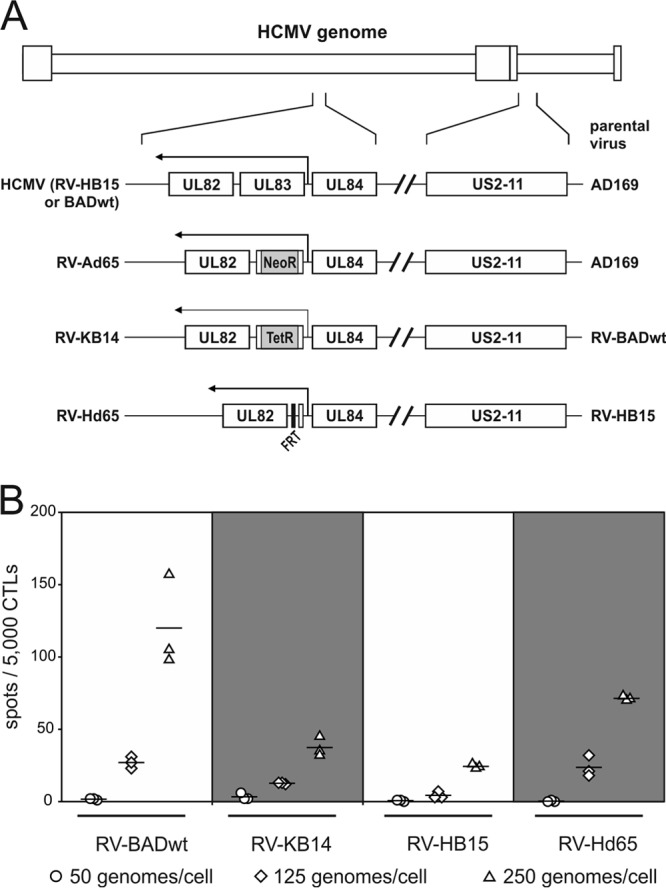
Strain-dependent MHC class I presentation of IE1 peptides at IE times of infection. (A) Schematic representation of the viral mutants used for analysis. The direction of transcription from the genes encoding pp65 (UL83) or pp71 (UL82) in their native or mutated configuration is shown by arrows. NeoR, neomycin resistance gene; TetR, tetracycline resistance gene; FRT, recognition site for the Flp recombinase. (B) IFN-γ ELISpot analysis of infected HFF. Cells were infected with the indicated viruses in the presence of 250 μg/ml cycloheximide (CX) for 9 h. The medium was then replaced by medium containing 10 μg/ml actinomycin D (AcD). Prior to ELISpot analysis with a CTL clone directed against IE1TMY, cells were fixed with 0.5% paraformaldehyde. The results for three wells (each symbol represents the value for one well) and the mean values (horizontal lines) are shown.
Infected cells were incubated in the presence of cycloheximide (CX) to reversibly block protein translation for 9 h. After removal of CX, the cells were incubated for another 13.5 h in the presence of actinomycin D (AcD) to enable translation of IE RNA without allowing further transcription of downstream HCMV genes. This experimental setup was necessary, as the sensitivity of the assay was below the level of detectability in cells infected for 1 to 6 h without CX/AcD enhancement (data not shown). After incubation with CX/AcD, cells were fixed using paraformaldehyde to terminate proteasomal processing and MHC class I loading and transport. The cells were subsequently subjected to IFN-γ ELISpot analysis, using a CTL clone directed against an HLA-A2-presented peptide from the IE1 protein (amino acids [aa] 297 to 305) (8). The detection limit of this clone was monitored before the assays were performed, using different concentrations of peptide loaded on T2 presenter cells. Recognition was still seen at peptide concentrations of 10−7 M (data not shown).
An increase in IE1 presentation depending on the amount of input virus was seen for all four strains investigated (Fig. 1B). The pp65-negative mutant RV-Hd65 showed higher levels of IE1 presentation at all MOIs tested than its parental strain (RV-HB15) did. This would have been in agreement with previous findings, indicating that pp65 was involved in suppression of IE1 peptide presentation (13). However, a reverse correlation was found when comparing the pp65-negative strain RV-KB14 with its parental strain, RV-BADwt. In this case, presentation by the parental strain was higher, arguing against a role of pp65 in this instance.
Various levels of pp71 are expressed when the adjacent pp65 gene is deleted from the HCMV genome.
The experimental results suggested that HCMV strains differed in the extent by which IE1-derived peptides were introduced into MHC class I presentation. As pp71 was known to induce IE1 gene expression, we next tested whether these viruses would express different levels of the regulator protein. Using a quantitative immunoblot analysis of cell lysates, enhanced expression of pp71 was found in HFF infected with the mutant RV-Hd65, compared to its parental strain, RV-HB15 (Fig. 2A and B). Also, cells infected with the pp65 deletion mutant RV-Ad65 (31) (Fig. 1A), which had been used to analyze the immunoevasive effect of pp65 previously (13), showed increased levels of pp71 (data not shown). These results indicated that genetic modification of the UL83 open reading frame, encoding pp65, interfered with the expression levels of pp71 in infected HFF, as also suggested by others (47). The pp65 and pp71 proteins are encoded by a bicistronic 4-kb RNA (48). RV-Hd65 expressed a truncated version of that RNA with large parts of UL83 missing upstream of UL82, encoding pp71. Higher levels of that transcript, compared with the full-length 4-kb RNA, were observed in Northern blot analyses (data not shown). This may explain the higher expression rates of pp71 compared to the parental strain. In cells infected with the pp65-negative mutant RV-KB14, no elevated expression of pp71 was detected (Fig. 2A and B).
Fig 2.
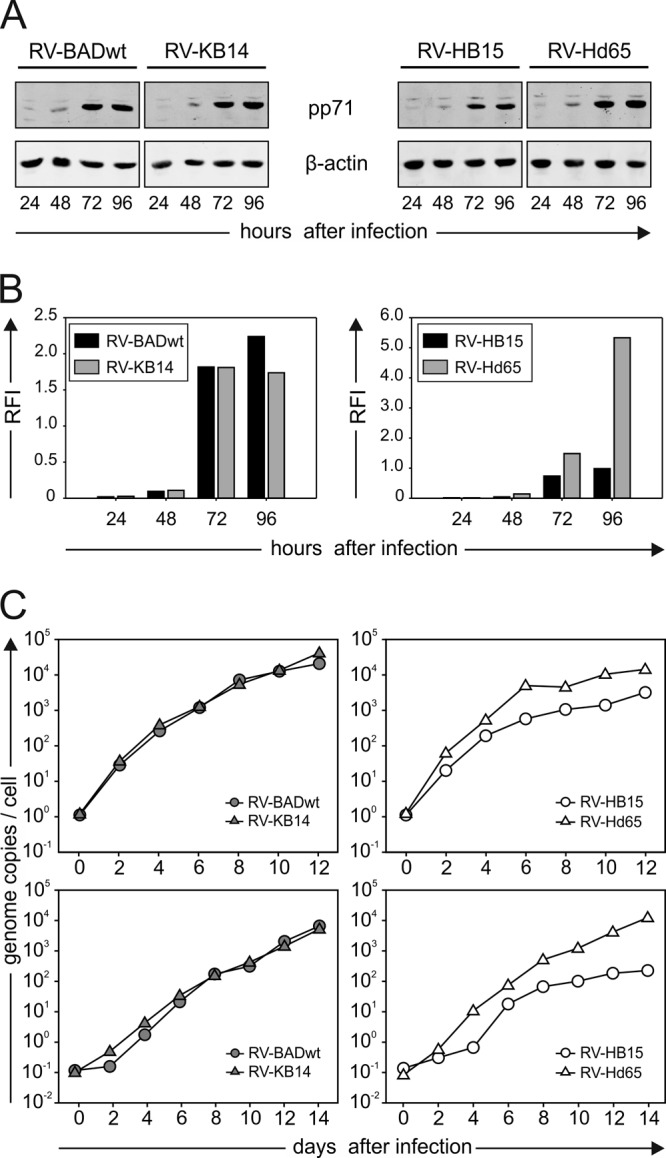
Impact of UL83 mutagenesis on pp71 expression levels and DNA replication kinetics. (A) Immunoblot analyses of lysates from cells infected with the indicated viruses at 125 genomes per cell and collected at the indicated times after infection. Lysates from 1 × 105 cells were applied to each lane on SDS-polyacrylamide gels. Antibodies directed against pp71 or β-actin were used for detection. (B) Quantification of the pp71 bands from panel A, using the Odyssey analysis system. Signals from individual bands were normalized to β-actin in each case (RFI, relative fluorescence intensity). (C) Quantification of viral DNA replication at low infectious doses. Cells were infected with either 0.4 genome copies per cell (top graphs) or 0.04 genome copies per cell (bottom graphs). Infected cells were collected at the indicated time points of infection. Cellular DNA was purified and analyzed by quantitative DNA PCR, using HCMV-specific primers.
To test whether the overexpression of pp71 was sufficient to mediate the well-established effects of pp71 (32), viral DNA replication was monitored (Fig. 2C). RV-Hd65 indeed replicated to higher genome copy levels at a low MOI than its parental strain did. This effect was not detected when RV-KB14 was compared to its parental strain. These results showed that biologically significant differences in the levels of pp71 were imparted by deleting pp65.
Virions of viral mutants contain different amounts of pp71.
The results obtained so far indicated that the differences in IE1 peptide presentation at IE times of infection with pp65-negative mutants correlated with the particular pp71 levels that the mutants expressed. To verify that the expression levels of pp71 translated into different levels of packaging into virions, mass spectrometry was performed. Virions of the four strains were purified from infected-cell culture supernatants, using glycerol-tartrate gradient centrifugation (40). The band corresponding to virions was collected in each case. A fraction of the material was subjected to SDS-polyacrylamide gel electrophoresis, followed by silver staining (Fig. 3A). A pattern of bands typical of virion-associated HCMV proteins was detected in each of the four preparations. This material was then subjected to label-free quantitative proteomic analysis. The results obtained for pp71 were correlated to the amount of the major capsid protein (Fig. 3B). As suggested from the intracellular expression pattern, RV-Hd65 virions contained significantly larger amounts of pp71 than virions of the parental strain RV-HB15 did. In contrast, similar or even reduced levels of pp71 were seen in virions of RV-KB14 compared to its parental strain (RV-BADwt) (see the bands in Fig. 3A). To investigate the amount of pp71 delivered into cells, a quantitative immunoblot analysis was performed. For this analysis, cells were infected at a high MOI in the presence of CX for 9 h and subjected to Bis-Tris-polyacrylamide gel electrophoresis (Fig. 4A). After transfer, membranes were probed with pp71-specific antibodies and, for a control, with pp65-specific antibodies. The levels of pp71, delivered into cells by the different strains were quantified, using β-actin as the standard (Fig. 4B). Concordant with the expression levels of pp71 in infected cells and the level of packaging of the tegument protein in the particles of the different strains, RV-Hd65 delivered higher levels of pp71 than its parental strain (RV-HB15) did. Also in accordance, RV-BADwt introduced slightly more pp71 than RV-KB14 did. These results indicated that different amounts of pp71 were packaged into virions of the different strains, and this translated into delivery of different amounts of the tegument protein into cells, when infection was normalized to genome copy numbers.
Fig 3.
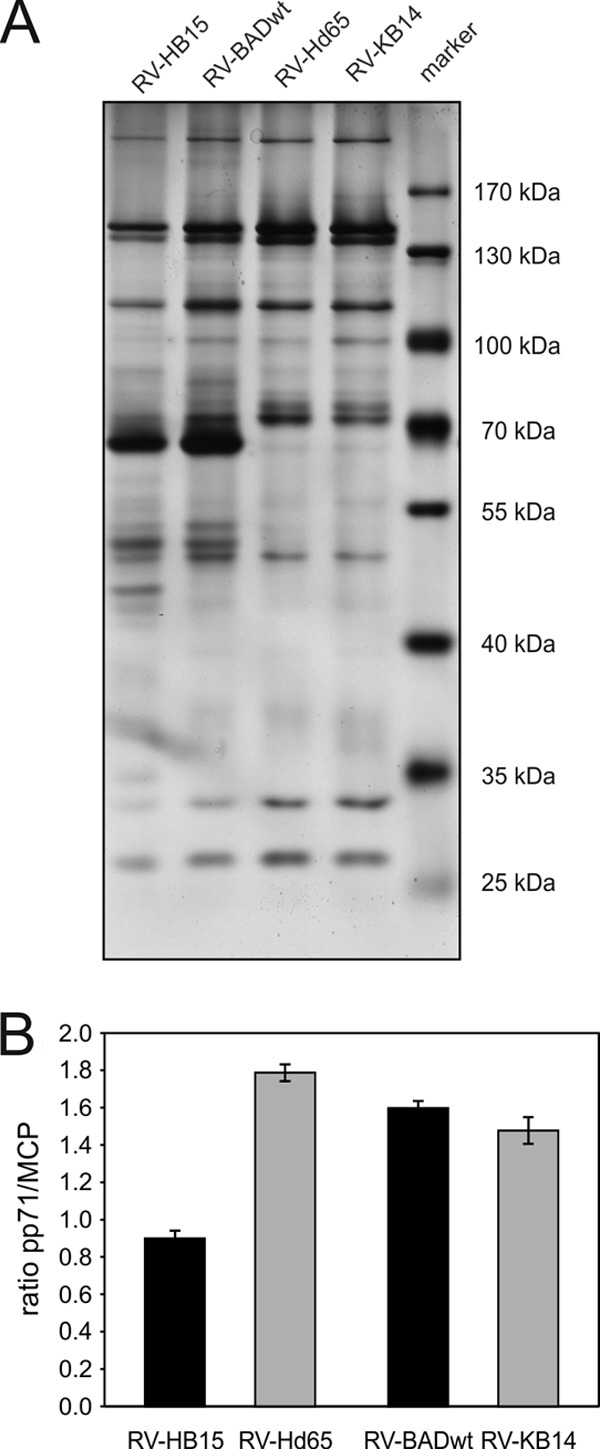
Mass spectrometry of the pp71 content of purified virions from different viral strains. (A) SDS-polyacrylamide gel electrophoreses of purified virions of the indicated strains. After the proteins were separated, the gel was silver stained. The positions of the molecular masses (in kilodaltons) of the standards (markers) are indicated to the right of the gel. Two micrograms of virions were loaded onto each lane. (B) Ratio of pp71 molecules to major capsid protein (MCP) molecules of purified virions, analyzed by LC-MS mass spectrometry. The samples were analyzed in quintuplicate.
Fig 4.
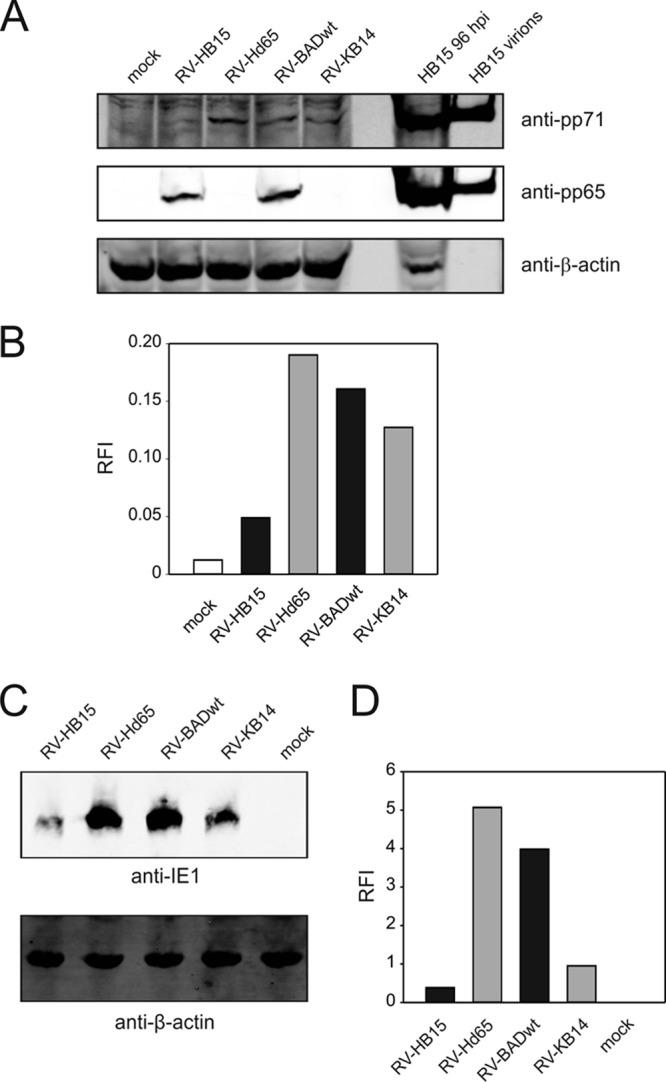
Influence of the amount of incoming pp71 on IE1 expression levels. (A) Immunoblot analysis of the amount of pp71 in HFF, infected with the different virus strains in the presence of CX at 300 genomes per cell. Lysates from 6 × 105 cells were applied to each lane on a Bis-Tris gel. For a positive control, a lysate from 2 × 105 cells, infected for 96 h with RV-HB15, and 2 μg RV-HB15 virions were additionally loaded. After blotting, the membrane was first probed with antibodies directed against pp71 and β-actin and then reprobed with antibodies against pp65. (B) Quantification of the pp71 bands from panel A, using the Odyssey analysis system. Signals from individual bands were normalized to β-actin in each case (RFI, relative fluorescence intensity). (C) Immunoblot analysis of the expression levels of IE1 in HFF, infected with the different virus strains under CX/AcD blocking conditions at 125 genomes per cell. Lysates from 6 × 105 cells were applied to each lane on an SDS-polyacrylamide gel, blotted, and probed with an IE1-specific antibody. (D) Quantification of the IE1 bands from panel C, using the Odyssey analysis system. Signals from individual bands were normalized to β-actin in each case.
The amount of pp71 in mutant virus particles correlates with IE1 levels.
It has been shown that incoming, particle-associated pp71 determines the level of IE1 gene expression (32). It was thus possible that the differences seen in IE1 peptide presentation were based on different IE1 expression levels. To test this, immunoblot analyses were performed on cell lysates from HFF infected with the different mutants. Identical genome copy numbers were applied under CX/AcD blocking conditions (Fig. 4C and D). RV-Hd65 clearly expressed higher IE1 levels than its parental strain, RV-HB15, did. Surprisingly, an inverse correlation was seen for RV-KB14 and RV-BADwt. More IE1 was expressed, in this case, when cells were infected with the parental strain. Note that the experiments shown in Fig. 4C and D have been reproduced with identical results on three different occasions. These results confirmed that the differential packaging of pp71 into particles of the mutants resulted in differential expression of IE1.
High levels of pp71 in virus particles increase recognition of infected cells by CD8 T cells at IE times.
To confirm the results without interrupting pp65 synthesis, an HCMV mutant that expressed an enhanced yellow fluorescent protein (EYFP)-tagged version of pp71 was used (AD169/EYFP-pp71 [Fig. 5A]). As shown in previous experiments (32), this modification leads to a markedly increased expression and packaging of pp71 (Fig. 5B and C). Like cells infected with RV-Hd65, cells infected with the EYFP-tagged virus in the presence of CX and AcD showed enhanced levels of IE1 expression (32) (Fig. 5D and E). Using this virus, an IFN-γ ELISpot analysis with IE1TMY-CTL was performed. Cells infected with AD169/EYFP-pp71 generated roughly 3-fold more spots than cells infected with the parental wt strain, AD169 (RV-HB15). This confirmed the findings with the pp65-negative HCMV mutants, indicating that levels of pp71 expression and packaging positively correlate with IE1 peptide presentation.
Fig 5.
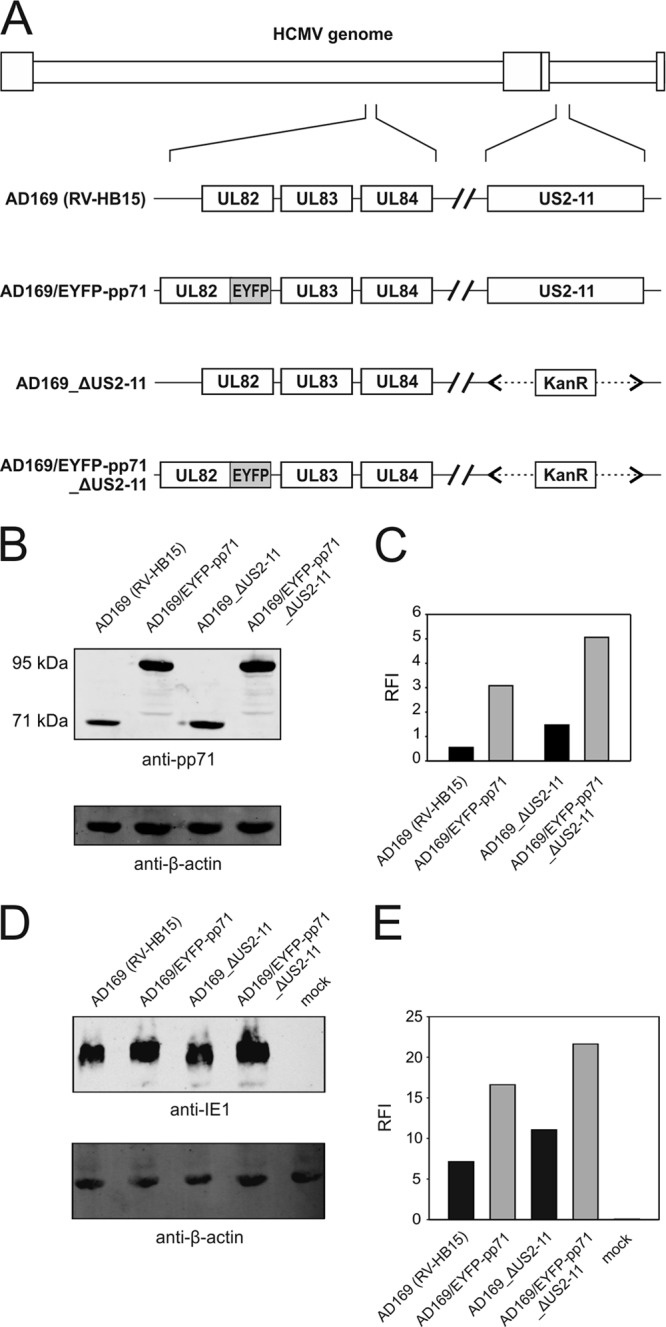
Characterization of US2-11-competent and US2-11-negative HCMV strains, expressing an EYFP-tagged version of pp71. (A) Schematic representation of the viral mutants. The US2-11 gene region in Ad169/EYFP-pp71 (32) or wt HCMV (RV-HB15) was deleted and replaced by a kanamycin resistance gene, using BAC recombineering. The resulting mutants were designated AD169/EYFP-pp71_ΔUS2-11 or HCMV_ΔUS2-11. (B) Immunoblot analysis of cell lysates of HFF infected with the indicated viruses at an MOI of 1 for 4 days. Lysates from 1 × 105 cells were applied to each lane. Antibodies directed against pp71 or β-actin were used for detection. (C) Quantification of the pp71 or EYFP-pp71 bands from panel B, using the Odyssey analysis system. Signals from individual bands were normalized to β-actin in each case (RFI, relative fluorescence intensity). (D) Immunoblot analysis of the expression levels of IE1 in HFF infected with the different virus strains under CX/AcD blocking conditions at 125 genomes per cell. Lysates from 2 × 105 cells were applied to each lane. (E) Quantification of the IE1 bands from panel D, using the Odyssey analysis system. Signals from individual bands were normalized to β-actin in each case.
To exclude any effect of the US2-11-encoded immune evasion proteins on the ELISpot results, the respective genomic region was deleted from AD169/EYFP-pp71, generating AD169/EYFP-pp71_ΔUS2-11 (Fig. 5A). In accordance with AD169/EYFP-pp71, AD169/EYFP-pp71_ΔUS2-11 expressed higher levels of both pp71 and IE1 proteins than the control mutant AD169_ΔUS2-11, which expressed pp71 without modification (Fig. 5B to E). There was significant IE1 presentation by cells infected with the control mutant AD169_ΔUS2-11 (Fig. 6). Cells infected with the wt strain AD169 (RV-HB15) showed only very limited IE1 presentation. We recently showed that gpUS2 and gpUS11 were expressed and effective under these IE conditions (27), but again, enhanced levels of pp71 in cells infected with AD169/EYFP-pp71_ΔUS2-11 also correlated with roughly 3-fold more spots in the absence of US2-11. This suggested that gpUS2-11 had no limiting effect on the relative enhancement of IE gene expression through particle-associated pp71.
Fig 6.
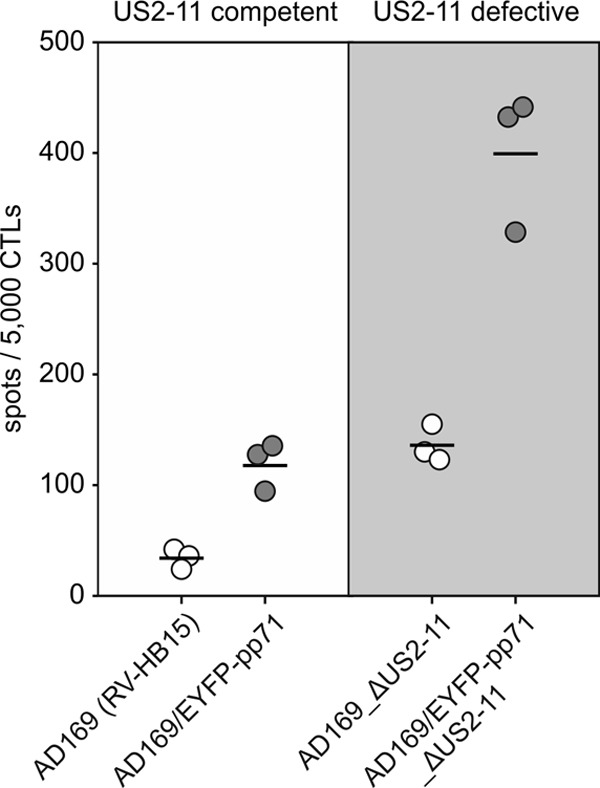
MHC class I presentation of IE1-derived peptides at IE times after infection with mutants expressing EYFP-pp71. HFF were infected with the indicated viruses at 125 genomes per cell in the presence of 250 μg/ml CX. At 9 h after infection, culture medium was replaced by medium containing 10 μg/ml AcD. Incubation of infected cells was continued for another 13.5 h. The cells were then fixed with 0.5% paraformaldehyde and subjected to IFN-γ ELISpot analysis, using IE1TMY-specific CD8 T cells as responders. The results for three wells (white and gray circles) (each symbol represents the value for one well) and the mean values (horizontal lines) are shown.
Overexpression of pp71 by pp65-competent HCMV strains has no impact on MHC class I surface expression or antigen presentation at early or late phases of infection.
The pp71 protein had been shown to influence surface expression of MHC class I on glioblastoma cells at later stages of infection (14). We consequently focused our attention on the question of whether overexpression of pp71 would also have an effect on MHC class I in fibroblasts at early and late times of infection.
HFF were infected with the two US2-11-negative mutants. The US2-11-expressing variant AD169/EYFP-pp71 was excluded at this point, since previous experiments had shown a complete suppression of IE1 presentation at early and late times, mediated by gpUS2-US11 (49). Cells were analyzed by flow cytometry for expression of HLA-ABC (MHC class I) or for expression of HLA-A2 (Fig. 7A). Unexpectedly, enhanced levels of pp71 had no effect on the detection of these molecules on the surfaces of infected HFF. This indicated that, in contrast to glioblastoma cells, pp71 had little effect on MHC class I surface expression in fibroblasts.
Fig 7.

MHC class I surface expression and IE1 peptide presentation on cells infected with AD169/EYFP-pp71_ΔUS2-11 at 24 and 96 h after infection. (A) Cells were infected with the indicated viruses at 125 genomes per cell and were subjected to flow cytometry analysis of HLA-ABC or HLA-A2 surface expression at the indicated time points (in hours) after infection. (B and C) IFN-γ ELISpot analysis of infected HFF, using a CTL clone directed against IE1TMY. HFF were infected either with 125 genomes per cell (B) or at an MOI of 5 (C). The results for three wells (white and gray circles) (each symbol represents the value for one well) and the mean values (horizontal lines) are shown. h.p.i., hours postinfection. (D and E) IFN-γ ELISpot analysis of infected HFF, using a CTL clone directed against pp65NLV. HFF were infected either with 125 genomes per cell (D) or at an MOI of 5 (E). The results for three wells (white and gray circles) (each symbol represents the value for one well) and the mean values (horizontal lines) are shown.
Next we tested the impact of pp71 on antigen presentation at early and late times in the IFN-γ ELISpot assay. Presentation of IE1TMY was first tested. Tavalai and colleagues had shown that the IE1 protein was expressed to similar levels in cells, infected with either wt HCMV or AD169/EYFP-pp71 overexpressing pp71 (32). Different from the IE conditions, IE1 protein levels could thus not influence presentation, and consequently, specific effects of pp71 on MHC class I presentation would become visible. Infection was carried out at 125 genomes per cell (Fig. 7B) or at an MOI of 5 (Fig. 7C). Upon ELISpot analysis, presentation of IE1 was comparable in AD169_ΔUS2-11- and AD169/EYFP-pp71_ΔUS2-11-infected cells at 24 h after infection, indicating that there was little impact of pp71 at early times of HCMV infection. Also, after 96 h of infection, no clear difference in the number of spots was detected after infection with AD169/EYFP-pp71_ΔUS2-11, compared to infection with AD169_ΔUS2-11. To corroborate these findings with a different antigen, presentation of the pp65-derived peptide NLVPMVATV was tested, using a specific CTL clone (8). This clone was shown to detect its cognate peptide in HLA-A2 with a minimal concentration of 10−8 M (data not shown). As with IE1, no influence of pp71 levels on the presentation of pp65NLV by HLA-A2 could be demonstrated, irrespective of whether infection was normalized to the genome copy number (Fig. 7D) or to the MOI (Fig. 7E). These data indicated that pp71 has little impact on MHC class I presentation of viral peptides by fibroblasts at early or late times of infection.
DISCUSSION
Expression of IE1-pp72 and IE2-pp86 is essential for efficient lytic HCMV replication. The former protein is also known as a prominent CD8 T cell target. It had thus been suggested that tegument components of HCMV virions not only antagonize nuclear domain 10 (ND10)-mediated repression mechanisms of IE gene expression during lytic viral infection (16, 17, 50, 51), but also interfere with MHC class I peptide presentation of IE proteins very early in infection (52).
It was shown before that pp65 specifically interfered with cytolysis of infected cells by IE1-specific cytotoxic T cell lines; modification of the phosphorylation status of IE1 as well as interference of pp65 with alpha/beta interferon signal transduction pathways were taken as possible mechanisms to explain this effect (13, 53, 54). Our results could not confirm the effect of pp65 on MHC class I presentation of IE1 peptides. There are several explanations for this apparent discrepancy, including differences in experimental conditions and, in particular, differences in MHC class I alleles, used for analysis. Allelic differences in the effect of particular immunoevasive proteins of HCMV are well established (55–58). Note that, in the course of our experiments, we found that mutant RV-Ad65, used in the original analysis (13), expressed higher levels of pp71 in the absence of pp65. It could thus be that part of the phenotype seen with this virus was due to the enhanced IE1 expression, mediated by pp71.
Infection with mutant viruses that expressed higher levels of pp71 resulted in an increase of pp71 packaging into virions, increased delivery of the tegument protein into cells, and in an increase of IE1 steady-state levels following infection under CX/AcD blocking conditions. In parallel, IE1 peptide presentation was enhanced after infection with these viruses using the same conditions. This suggests that the level of incoming pp71 not only contributes to the efficiency of lytic replication; it also influences the detection of these cells by antiviral CD8 T cells. This effect may be alleviated by US2-11. Viral mutants that expressed US2-11 but comparable levels of pp71 showed markedly reduced levels of IE1 presentation under IE conditions. This indicated that at least one of these immunoevasins was operative at IE times (27). However, the increase in IE1 presentation by pp71 overexpression could not be overcome by US2-11, although presentation remained at a lower level, compared to US2-11-negative viruses. This indicates that, although US2-11 are effectively suppressing MHC class I antigen presentation, higher levels of pp71 could well lead to a labeling of infected cells for CTL destruction early in infection.
The pp71 protein has been shown to delay maturation of MHC class I molecules when expressed in glioblastoma cells (14). Using a viral mutant which significantly overexpressed the EYFP-tagged pp71 in the absence of gpUS2-11, no influence of pp71 on MHC class I and HLA-A2 surface expression or presentation of IE1 was seen at 24 or 96 h of infection. The latter result was corroborated by using a pp65-specific CTL clone. Concordant results were also seen with a US2-11-negative virus that overexpressed an untagged version of pp71 (data not shown). This suggests that pp71 has no significant immunoevasive effect on MHC class I presentation in cells, like HFF, which support high-level viral replication. It can, however, not formally be excluded that wt levels of pp71 display an effect on MHC class I surface expression that cannot be further enhanced by overexpression of the tegument protein. Further analyses are required to elucidate this and the question of whether pp71 imparts an effect on MHC class I on cells like endothelial cells or epithelial cells that differ from fibroblasts in their potential to support viral replication.
In summary, from the viral perspective, particle-associated pp71 may not only lead to a favorable enhancement of IE gene expression. As shown in this study, this may also cause enhanced MHC class I presentation of IE1 peptides and thus to enhanced destruction of infected cells by CD8 T cells. Consequently, it may be hypothesized that viruses, as exemplified by HCMV here, may have to tightly control the packaging of transactivating proteins into particles in order to avoid early labeling of the cells for destruction by the immune system.
ACKNOWLEDGMENTS
This work was supported by grants from Deutsche Forschungsgemeinschaft DFG Clinical Research Unit 183 (KFO 183, B.P.), SFB 490 (J.H., K.B., and B.P.), and SFB796 (N.R. and T.S.). S.T. was supported by the Forschungszentrum Immunologie (FZI) of the Johannes Gutenberg University Mainz.
The donations of BAC clones by Thomas Shenk, Princeton University, and by Ulrich Koszinowski and Gaby Hahn, University of Munich, and the donation of antibodies by William Britt, University of Alabama at Birmingham, are gratefully acknowledged.
Footnotes
Published ahead of print 28 February 2013
REFERENCES
- 1. Powers C, DeFilippis V, Malouli D, Früh K. 2008. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 325:333–360 [DOI] [PubMed] [Google Scholar]
- 2. Reddehase MJ. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2:831–844 [DOI] [PubMed] [Google Scholar]
- 3. Roy CR, Mocarski ES. 2007. Pathogen subversion of cell-intrinsic innate immunity. Nat. Immunol. 8:1179–1187 [DOI] [PubMed] [Google Scholar]
- 4. Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133 [DOI] [PubMed] [Google Scholar]
- 5. Reddehase MJ, Mutter W, Münch K, Bühring HJ, Koszinowski UH. 1987. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J. Virol. 61:3102–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steffens HP, Kurz S, Holtappels R, Reddehase MJ. 1998. Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J. Virol. 72:1797–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. 1995. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 333:1038–1044 [DOI] [PubMed] [Google Scholar]
- 8. Besold K, Frankenberg N, Pepperl-Klindworth S, Kuball J, Theobald M, Hahn G, Plachter B. 2007. Processing and MHC class I presentation of human cytomegalovirus pp65-derived peptides persist despite gpUS2-11-mediated immune evasion. J. Gen. Virol. 88:1429–1439 [DOI] [PubMed] [Google Scholar]
- 9. Besold K, Wills M, Plachter B. 2009. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 391:5–19 [DOI] [PubMed] [Google Scholar]
- 10. Jones TR, Hanson LK, Sun L, Slater JS, Stenberg RM, Campbell AE. 1995. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol. 69:4830–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Z, Winkler M, Biegalke B. 2009. Human cytomegalovirus: host immune modulation by the viral US3 gene. Int. J. Biochem. Cell Biol. 41:503–506 [DOI] [PubMed] [Google Scholar]
- 12. Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, Axthelm MK, Nelson JA, Jarvis MA, Picker LJ, Fruh K. 2010. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328:102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gilbert MJ, Riddell SR, Plachter B, Greenberg PD. 1996. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate-early gene product. Nature 383:720–722 [DOI] [PubMed] [Google Scholar]
- 14. Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD. 2006. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J. Virol. 80:951–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 97:14506–14511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu B, Stinski MF. 1992. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 66:4434–4444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schierling K, Stamminger T, Mertens T, Winkler M. 2004. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 78:9512–9523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borysiewicz LK, Hickling JK, Graham S, Sinclair J, Cranage MP, Smith GL, Sissons JG. 1988. Human cytomegalovirus-specific cytotoxic T cells. Relative frequency of stage-specific CTL recognizing the 72-kD immediate early protein and glycoprotein B expressed by recombinant vaccinia viruses. J. Exp. Med. 168:919–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kern F, Surel IP, Faulhaber N, Frömmel C, Schneider-Mergener J, Schonemann C, Reinke P, Volk HD. 1999. Target structures of the CD8+-T-cell response to human cytomegalovirus: the 72-kilodalton major immediate-early protein revisited. J. Virol. 73:8179–8184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202:673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biegalke BJ. 1995. Regulation of human cytomegalovirus US3 gene transcription by a cis-repressive sequence. J. Virol. 69:5362–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan YJ, Tseng WP, Hayward GS. 1996. Two distinct upstream regulatory domains containing multicopy cellular transcription factor binding sites provide basal repression and inducible enhancer characteristics to the immediate-early IES (US3) promoter from human cytomegalovirus. J. Virol. 70:5312–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL. 1996. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. U. S. A. 93:11327–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thrower AR, Bullock GC, Bissell JE, Stinski MF. 1996. Regulation of a human cytomegalovirus immediate-early gene (US3) by a silencer-enhancer combination. J. Virol. 70:91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weston K. 1988. An enhancer element in the short unique region of human cytomegalovirus regulates the production of a group of abundant immediate early transcripts. Virology 162:406–416 [DOI] [PubMed] [Google Scholar]
- 27. Hesse J, Ameres S, Besold K, Krauter S, Moosmann A, Plachter B. 2013. Suppression of CD8+ T cell recognition in the immediate-early phase of human cytomegalovirus infection. J. Gen. Virol. 94:376–386 [DOI] [PubMed] [Google Scholar]
- 28. Kalejta RF. 2008. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 325:101–115 [DOI] [PubMed] [Google Scholar]
- 29. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmolke S, Kern HF, Drescher P, Jahn G, Plachter B. 1995. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J. Virol. 69:5959–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tavalai N, Kraiger M, Kaiser N, Stamminger T. 2008. Insertion of an EYFP-pp71 (UL82) coding sequence into the human cytomegalovirus genome results in a recombinant virus with enhanced viral growth. J. Virol. 82:10543–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65 [DOI] [PubMed] [Google Scholar]
- 34. Rose RE. 1988. The nucleotide sequence of pACYC184. Nucleic Acids Res. 16:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 36. Andreoni M, Faircloth M, Vugler L, Britt WJ. 1989. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 23:157–167 [DOI] [PubMed] [Google Scholar]
- 37. Gallez-Hawkins G, Villacres MC, Li X, Sanborn MC, Lomeli NA, Zaia JA. 2003. Use of transgenic HLA A*0201/Kb and HHD II mice to evaluate frequency of cytomegalovirus IE1-derived peptide usage in eliciting human CD8 cytokine response. J. Virol. 77:4457–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diamond DJ, York J, Sun JY, Wright CL, Forman SJ. 1997. Development of a candidate HLA A*0201 restricted peptide-based vaccine against human cytomegalovirus infection. Blood 90:1751–1767 [PubMed] [Google Scholar]
- 39. Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, Sissons JG. 1996. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 70:7569–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irmiere A, Gibson W. 1983. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 130:118–133 [DOI] [PubMed] [Google Scholar]
- 41. Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362 [DOI] [PubMed] [Google Scholar]
- 42. Tenzer S, Docter D, Rosfa S, Wlodarski A, Kuharev J, Rekik A, Knauer SK, Bantz C, Nawroth T, Bier C, Sirirattanapan J, Mann W, Treuel L, Zellner R, Maskos M, Schild H, Stauber RH. 2011. Nanoparticle size is a critical physicochemical determinant of the human blood plasma corona: a comprehensive quantitative proteomic analysis. ACS Nano. 5:7155–7167 [DOI] [PubMed] [Google Scholar]
- 43. Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI. 2009. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependent LC-MS/MS. Proteomics 9:1683–1695 [DOI] [PubMed] [Google Scholar]
- 44. Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, Geromanos S. 2005. Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 77:2187–2200 [DOI] [PubMed] [Google Scholar]
- 45. Patzig J, Jahn O, Tenzer S, Wichert SP, de Monasterio-Schrader P, Rosfa S, Kuharev J, Yan K, Bormuth I, Bremer J, Aguzzi A, Orfaniotou F, Hesse D, Schwab MH, Mobius W, Nave KA, Werner HB. 2011. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J. Neurosci. 31:16369–16386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bradshaw RA, Burlingame AL, Carr S, Aebersold R. 2006. Reporting protein identification data: the next generation of guidelines. Mol. Cell. Proteomics 5:787–788 [DOI] [PubMed] [Google Scholar]
- 47. Taylor RT, Bresnahan WA. 2006. Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J. Virol. 80:920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rüger B, Klages S, Walla B, Albrecht J, Fleckenstein B, Tomlinson P, Barrell B. 1987. Primary structure and transcription of the genes coding for the two virion phosphoproteins pp65 and pp71 of human cytomegalovirus. J. Virol. 61:446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Besold K, Plachter B. 2008. Recombinant viruses as tools to study human cytomegalovirus immune modulation. Med. Microbiol. Immunol. 197:215–222 [DOI] [PubMed] [Google Scholar]
- 50. Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tavalai N, Papior P, Rechter S, Stamminger T. 2008. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 82:126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gilbert MJ, Riddell SR, Li CR, Greenberg PD. 1993. Selective interference with class I major histocompatibility complex presentation of the major immediate-early protein following infection with human cytomegalovirus. J. Virol. 67:3461–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 78:10995–11006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. U. S. A. 100:11439–11444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barel MT, Pizzato N, Le Bouteiller P, Wiertz EJ, Lenfant F. 2006. Subtle sequence variation among MHC class I locus products greatly influences sensitivity to HCMV US2- and US11-mediated degradation. Int. Immunol. 18:173–182 [DOI] [PubMed] [Google Scholar]
- 56. Llano M, Guma M, Ortega M, Angulo A, Lopez-Botet M. 2003. Differential effects of US2, US6 and US11 human cytomegalovirus proteins on HLA class Ia and HLA-E expression: impact on target susceptibility to NK cell subsets. Eur. J. Immunol. 33:2744–2754 [DOI] [PubMed] [Google Scholar]
- 57. Park B, Kim Y, Shin J, Lee S, Cho K, Früh K, Lee S, Ahn K. 2004. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 20:71–85 [DOI] [PubMed] [Google Scholar]
- 58. Thilo C, Berglund P, Applequist SE, Yewdell JW, Ljunggren HG, Achour A. 2006. Dissection of the interaction of the human cytomegalovirus-derived US2 protein with major histocompatibility complex class I molecules: prominent role of a single arginine residue in human leukocyte antigen-A2. J. Biol. Chem. 281:8950–8957 [DOI] [PubMed] [Google Scholar]