

Abstract
Novel antivirals are needed to supplement existing control strategies for influenza A virus (IAV). A promising new class of drug, exemplified by the compound nucleozin, has recently been identified that targets the viral nucleoprotein (NP). These inhibitors are thought to act as “molecular staples” that stabilize interactions between NP monomers, promoting the formation of nonfunctional aggregates. Here we detail the inhibitory mechanism of nucleozin, finding that the drug has both early- and late-acting effects on the IAV life cycle. When present at the start of infection, it inhibited viral RNA and protein synthesis. However, when added at later time points, it still potently blocked the production of infectious progeny but without affecting viral macromolecular synthesis. Instead, nucleozin blocked the cytoplasmic trafficking of ribonucleoproteins (RNPs) that had undergone nuclear export, promoting the formation of large perinuclear aggregates of RNPs along with cellular Rab11. This effect led to the production of much reduced amounts of often markedly smaller virus particles. We conclude that the primary target of nucleozin is the viral RNP, not NP, and this work also provides proof of the principle that IAV replication can be effectively inhibited by blocking cytoplasmic trafficking of the viral genome.
INTRODUCTION
The influenza A virus (IAV) genome consists of eight segments of negative-sense, single-stranded RNA (vRNA) that encode 10 to 14 identified proteins (1–3). Each vRNA is separately encapsidated into ribonucleoprotein (RNP) particles by the trimeric viral RNA-dependent RNA polymerase (PB1, PB2, and PA) and single-strand RNA-binding nucleoprotein (NP) (4). At the start of infection, RNPs are released into the cytoplasm and imported into the nucleus by the host importin alpha/beta pathway (5). Once in the nucleus, the vRNAs are transcribed by the viral polymerase to produce capped and polyadenylated mRNAs, as well as replicated via an alternative plus sense transcript (cRNA). Both the cRNA and progeny vRNA molecules are encapsidated into RNPs, necessitating the nuclear import of newly translated NP, again via interactions with importin alpha (5). Once levels of the viral M1 and NS2/NEP polypeptides are sufficient, newly synthesized RNPs are exported to the cytoplasm via the cellular CRM1/exportin 1 pathway (5). We and others have recently reported that RNPs accumulate at the perinuclear recycling endosome (RE) after leaving the nucleus. There, they piggyback onto Rab11-positive vesicles to access the microtubule network for transit through the cytoplasm (6–9). The assembly/budding phase then starts as RNPs join with the three viral membrane proteins (hemagglutinin [HA], neuraminidase [NA], and M2), as well as with the M1 matrix protein and small amounts of NS2 and the RNPs, to form new enveloped virus particles at the apical plasma membrane (PM) (10).
Influenza virus transmission and influenza disease are partially controlled by a global vaccination program and antiviral therapy. The available antivirals target the ion channel protein M2 (amantadine, rimantadine) or NA (oseltamivir, zanamivir), which are responsible for entrance into and exit from the host cell, respectively. However, both therapies are rendered less effective by resistance (11). New antivirals developed against other IAV targets are therefore required. One attractive strategy is to target NP, since this protein plays many essential roles in vRNA synthesis, genome trafficking, and virus assembly (4, 12). Recently, several groups identified NP-interacting molecules able to inhibit virus replication (13–17). These compounds, nucleozin or related derivatives, bind to at least two sites on NP and appear to act as “molecular staples” that promote the formation of higher-order oligomers or aggregates by stabilizing monomer interactions. Evidence for this comes from various in vitro biophysical analyses of purified NP, cocrystallization studies with the inhibitor, and fluorescence microscopy of infected, drug-treated cells (13, 15–17). Further support for this model comes from the identification of several resistance mutations in NP that map to the separate drug-binding pockets (15–17). The drug-induced aggregation evidently interferes with NP function, because the compounds potently inhibit virus replication in tissue culture and infected animals (13, 15–17). However, the precise mechanism(s) of action has not been investigated in detail. While the compounds clearly inhibit viral transcription, either from RNPs reconstituted in cells by plasmid transfection or from RNPs purified from virus (15–17), discrepancies between the 50% inhibitory concentrations for overall virus replication versus transcription raise the possibility of more than one mechanism of action (14).
Here we describe a distinct effect of nucleozin on the later stages of viral infection, finding that it inhibits the cytoplasmic transport of Rab11-RNP complexes. This is a novel mechanism of action for any antiviral that provides proof of principle for new strategies to interfere with viral replication.
MATERIALS AND METHODS
Cells, viruses, antisera, and drug.
Human embryonic kidney 293T, Madin-Darby canine kidney (MDCK), and human alveolar basal epithelial (A549) cells were cultured as previously described (18). Reverse-genetics-derived A/Puerto Rico/8/34 (PR8; H1N1) was used as a model virus (19) and titrated according to reference 20. Mutations of segment 5 (NP Y289H) were made, and viruses were rescued as described in reference 21. Infections were carried out at a multiplicity of infection of 3 in serum-free medium (SFM). After 30 min, infected cells were overlaid with SFM and 0.14% bovine serum albumin. The antibodies used included mouse monoclonal antibodies against green fluorescent protein (GFP) (JL8; Clontech; catalog no. 632380); GM130 (BD Transduction Laboratories; catalog no. 610822); calnexin, EEA1, and clathrin (BD Transduction Laboratories; catalog no. 610524, 610457, and 610499, respectively); LAMP1 (BD Pharmingen; catalog no. 553792); influenza virus M2 (14C2; AbCam; catalog no. ab5416) and NA (7D8; a gift from Susanna Colaco and Phil Stevenson); rat monoclonal anti-α-tubulin (YL1/2 MCA77G; AbD-Serotec); and sheep polyclonal antibody to TGN46 (AbD-Serotec; catalog no. AHP500G). The rabbit polyclonal antisera used were to whole PR8 virions, PR8 M1 (22), PB1 (V19), PA (V35) (23), PB2 (2N580) (24), and NP (2915) (25). Anti-NS2 antibody was a gift from A. Portela. All of the secondary antibodies used were from the Alexa Fluor range (Invitrogen). Nucleozin (16) was dissolved in dimethyl sulfoxide and used at a concentration of 1 μM unless otherwise stated.
Plasmids and transfections.
Plasmids encoding GFP-tagged NP (26) or red fluorescent protein (RFP)-tagged wild-type (WT) Rab11a (6) have been previously described. pCDNA3 plasmids used to synthesize fluorescence in situ hybridization (FISH) probes to detect vRNA from segments 1, 4, and 7 were described in references 22, 27, and 28, and that used to detect mRNA for segment 5 was described in reference 29. To GFP tag influenza virus RNPs, 1 × 105 293T or A549 cells were transfected with GFP-NP (200 ng) by using Lipofectamine Plus and LTX (Invitrogen) according to the manufacturer's instructions, incubated overnight, and superinfected with PR8. Cells were imaged 8 h later (unless stated otherwise).
Microscopy.
The FISH methods used to detect vRNA were described in reference 22, and those used to detect mRNA were described in reference 29. Immunofluorescence assays were carried out as described in reference 30. Samples were imaged with a Leica SP5 confocal microscope and postprocessed with Adobe Photoshop and ImageJ (NIH). Single optical sections are shown.
For live imaging, cells were grown in chambered glass bottom dishes (Lab-Tek) and maintained at 37°C in Leibovitz L-15 CO2-independent medium (Gibco) during analysis. Cells were transfected with GFP-NP and infected with PR8 12 h later. For nucleozin treatment, images were acquired for 5 min and then nucleozin was added to a final concentration of 2 μM before imaging for around 20 to 30 min (as stated). Images were acquired at 0.25 or 0.71 frame/s and processed with ImageJ.
Pull-down assays, Western blotting, and primer extension analysis.
Pull-down assays of GFP-tagged proteins were performed with GFP-Trap beads (Chromotek) as described in reference 6. Confluent six-well dishes of 293T cells were transfected with 500 ng of each plasmid (GFP or GFP-NP) and, where applicable, infected 12 h later with virus. All samples were collected at 8 h postinfection (hpi). At the end of the process, bound proteins were eluted by boiling in SDS-PAGE sample buffer, while bound RNA was extracted by adding 1 ml of TRIzol (Invitrogen) and 200 μl chloroform directly to the Trap beads and recovered by ethanol precipitation. Specific RNA species were detected by reverse transcriptase-radiolabeled primer extension, followed by urea-PAGE and autoradiography as described previously (31). Western blotting was performed according to standard procedures and imaged with a LiCor Biosciences Odyssey near-infrared platform (32).
Transmission electron microscopy (TEM).
A549 cells were seeded onto 24-well plates, transfected, and infected as described above. Cells were washed in 0.1 M HEPES, pH 7.4, and incubated for 2 h at 4°C in 0.1 M HEPES (pH 7.4)–2% glutaraldehyde–0.1% H2O2. The monolayer was washed with HEPES buffer, and cells were scraped into buffer and harvested by centrifugation at 14,000 × g for 10 min. The supernatant was replaced with fresh HEPES buffer and processed by the Cambridge University Multi-Imaging Centre. Images were acquired with an FEI Philips CM100 transmission electron microscope equipped with a Deben AMT digital camera and an EDAX Genesis XM4 EDX system.
RESULTS
Nucleozin has distinct effects on virus replication, depending on the time of addition.
As a first test of how nucleozin inhibits IAV replication, we examined the effects of adding the drug at different times postinfection on the titer of replicated virus, by using the WT PR8 virus and a derivative engineered to contain the nucleozin resistance mutation NP Y289H. In the absence of the drug, WT virus release was detectable by 6 hpi but was around 20-fold higher at 8 hpi. However, addition of the drug at 0.5, 2.5, or 4 hpi decreased titers at 8 hpi by around 1,000-fold and, on average, by 10-fold if added at 6 hpi (Fig. 1A). The NP Y289H mutant reached similar titers in the absence or presence of the drug but had an overall growth defect of about 10-fold compared to the WT, as reported for the A/WSN/33 strain (16). Thus, nucleozin is an effective inhibitor of WT virus replication even when added at relatively late times in the replication cycle.
Fig 1.

Time-of-addition experiments examining the effect of nucleozin on IAV replication and macromolecular synthesis. (A) A549 cells were infected with either the WT or the NP Y289H mutant virus, and 1 μM nucleozin (NCZ) was added at the indicated time points. Supernatants were collected and plaque titrated in MDCK cells. Virus titers (white bars) are expressed as the mean percentage ± the standard deviation (n = 3). (B) Cell lysates were analyzed by Western blotting for the indicated proteins. (C) Total cellular RNA was extracted and analyzed by radioactive primer extension, urea-PAGE and autoradiography for mRNA and vRNA from segments 3 and 5, as well as 5S rRNA. (D) vRNA accumulation was quantified by densitometry of primer extension autoradiographs with ImageJ and plotted as the mean percentage of the amount from untreated cells at 8 hpi ± the standard deviation (n = 3).
We next tested the effect of nucleozin on WT viral macromolecular synthesis over a time course of infection. Expression of viral proteins increased over time in untreated cells, as expected (Fig. 1B, lanes 1 to 4). When nucleozin was added at 0.5 hpi, viral polypeptide synthesis was much reduced but not completely abolished, with small amounts of NP, M1, and PB2 detected at 8 hpi (Fig. 1B, lanes 5 to 8). Inhibition of viral protein synthesis was much less noticeable when the drug was added at 2.5 hpi (lanes 9 to 11) and was not significant when the drug was added at 4 hpi (lanes 12 and 13), where protein expression reached the same levels as in untreated cells. These alterations in polypeptide accumulation were reflected in drug-induced changes in the abundance of viral mRNA when PA-segment 3 and NP-segment 5 transcripts were examined as examples. In untreated cells, viral mRNA peaked at 4 hpi and declined thereafter (Fig. 1C, lanes 1 to 4), as previously observed (28, 31). When the drug was added at the beginning of infection, transcript accumulation was much delayed and reduced (lanes 5 to 8), but this effect was not seen when the compound was added at 2.5 or 4 hpi (lanes 9 to 13). Notably, this failure to inhibit mRNA production when the drug was added later was seen even when the compound was added before substantial amounts of transcription had occurred (compare lanes 1 and 2 with lanes 9 and 10). Synthesis of vRNA followed a kinetic pattern similar to that of the viral proteins, with detectable and increasing synthesis from 4 hpi in normal cells (Fig. 1C, lanes 1 to 4) that was markedly reduced when nucleozin was added at 0.5 hpi (lanes 5 to 8). Limited (∼25% of normal) vRNA synthesis was seen when the drug was added at 2.5 hpi, with a relatively early peak at 4 hpi and little increase thereafter (lanes 9 to 11). With later time points of drug addition, only minimal inhibition of vRNA synthesis was seen; quantification of replicate experiments with the WT virus showed a less-than-2-fold decrease when the drug was added at 4 or 6 hpi (Fig. 1C and D). The NP Y289H mutant produced similar levels of vRNA at 8 hpi, independently of the time of addition of the drug (Fig. 1D).
Therefore, nucleozin showed two distinct effects on virus replication, depending on the time of addition. When added early in infection, it inhibited the synthesis of all classes of viral RNA and (if added early enough to block transcription) viral proteins. However, when the drug was added later in infection, at 4 hpi or later, viral macromolecular synthesis seemed near normal and yet the drug still profoundly inhibited the release of infectious viral particles, a differential effect that could be clearly seen when the relative effects of the time of drug addition on vRNA accumulation and the titer of the released virus were compared (Fig. 1A and D).
Nucleozin aggregates outgoing viral RNPs (vRNPs) and Rab11 in the cytoplasm.
Previous work has suggested two mechanisms of action for nucleozin: direct inhibition of RNP transcription, or a block to NP trafficking, postulated to be prior to nuclear import and RNP assembly (13, 15–17). However, neither mechanism seemed a plausible explanation of the late-acting drug effect, since vRNA synthesis, and therefore, by extrapolation, RNP formation, was near normal. Furthermore, after ∼5 hpi, the major flow of NP is from the nucleus to the cytoplasm (33, 34). Accordingly, to better understand the effect of nucleozin on NP/RNP trafficking, we infected A549 cells with the WT virus and the NP Y289H mutant virus and followed the localization of NP (used as a proxy for vRNPs) by indirect immunofluorescence assay during the later stages of infection. We also double stained cells for Rab11, since vRNPs use Rab11-positive vesicles to reach the PM (6–9). In untreated cells infected with the WT virus, NP localized in the nucleus early in infection and then accumulated in the cytoplasm, where it colocalized with Rab11 at both 6 and 8 hpi (Fig. 2A; see high-magnification images in Fig. S1 in the supplemental material), as previously observed (6–9). When the WT virus was treated with nucleozin at 4 hpi (and fixed at 8 h), NP accumulation in the cytoplasm was retarded and confined largely to the perinuclear region, where it colocalized with a small proportion of Rab11. The majority of Rab11 staining was, however, distributed throughout the cytoplasm. Similar nuclear retention of NP was seen when nucleozin was added at 6 hpi and incubated for a further 2 h prior to fixation of the cells but with the striking addition of large juxtanuclear accumulations of cytoplasmic staining that colocalized strongly with Rab11 at low magnification (Fig. 2A). At higher magnification, NP staining was more pronounced toward the exterior of these structures and Rab11 staining was more pronounced in their interior (see Fig. S1 in the supplemental material). However, the distribution of Rab11 was not altered by the presence of the drug in uninfected cells, even when it was present for 4 h (Fig. 2A; see Fig. S1), nor were any differences in the localization of either NP or Rab11 observed in the NP Y289H mutant in the presence or absence of nucleozin (see Fig. S1).
Fig 2.

Nucleozin induces cytoplasmic aggregates containing RNP and Rab11. A549 cells were infected with the WT virus or mock infected and either left untreated or treated with 1 μM nucleozin as shown. Samples were fixed at the times shown and stained for Rab11 and NP (A) or processed for FISH for the indicated vRNAs (B) before imaging by confocal microscopy. Merged images include a 4′,6-diamidino-2-phenylindole (DAPI) channel shown in blue. Scale bars, 10 μm. Images are representative of three independent experiments.
Nucleozin-induced cytoplasmic aggregates of NP have been noted before but interpreted to be newly synthesized NP on the way into the nucleus (13, 16, 17). However, the timing of the effect we observed, as well as the inclusion of Rab11 in the aggregates (Rab11 interacts with RNPs but not non-RNP NP [6, 9]), suggested that the drug was altering the trafficking of RNPs. To test this, we used FISH to examine the localization of vRNA. As expected, in untreated cells, the three genomic segments examined (1, 4, and 7) progressed from early accumulation in the nucleus at 4 hpi to a later punctate distribution in the cytoplasm with a perinuclear focus (Fig. 2B). However, as observed for NP, when nucleozin was added at either 4 or 6 hpi (and cells were fixed at 8 hpi), prominent cytoplasmic aggregates of vRNA were seen surrounding the nucleus (Fig. 2B). Furthermore, this effect was specific for vRNA, because when similarly treated cells were double stained by FISH for segment 3 vRNA and segment 7 mRNA, no drug-induced alterations of mRNA localization were apparent and there was no significant cytoplasmic colocalization of the two senses of vRNA under any condition (see Fig. S2 in the supplemental material). Thus, nucleozin has a late-acting effect of causing the cytoplasmic aggregation of viral genomic RNPs along with cellular Rab11.
The prominent cytoplasmic aggregates of NP and vRNA clearly reflected the disruption of normal RNP trafficking, so we next tested if this resulted from the drug “dismantling” assembled RNPs. For this, we used a system in which cells are transfected with GFP-NP and superinfected with virus to produce GFP-tagged RNPs that can be affinity purified or followed by live-cell microscopy (6). Cells were infected with PR8 virus 12 h after GFP or GFP-NP transfection, treated (or not) with nucleozin at 6 hpi, and lysed at 8 hpi. Aliquots of the cell lysates were then examined by Western blotting for proteins of interest or by radioactive reverse transcriptase primer extension reactions for RNA before or after affinity fractionation over GFP-Trap beads. Examination of unfractionated cell lysate showed that GFP and GFP-NP were expressed as expected and that all of the viral proteins tested were present in the supernatant of infected cells in equal quantities, as was the cellular protein α-tubulin (Fig. 3, lanes 1 to 6). Primer extension reactions confirmed that segment 3 vRNA and cellular 5S rRNA were also present in approximately equal quantities. When GFP-NP (but not GFP) was affinity purified, the NP and polymerase components of RNPs were coprecipitated equally in the absence and presence of nucleozin (lanes 7 to 9 and 11). Moreover, primer extension analysis detected vRNA but not 5S rRNA when GFP-NP was purified. Thus, the system successfully detected RNP formation and this was not substantially changed by nucleozin treatment. We also tested for the coprecipitation of other viral structural proteins, M1 and NS2, both known to interact with RNPs during nuclear export and virion assembly (5), and M2. A small proportion of M1 was selectively purified by GFP-NP (Fig. 3, lanes 7 and 8), but no detectable amounts of NS2 or M2 were precipitated with or without drug treatment. Nucleozin therefore induces the cytoplasmic aggregation of RNPs but not other viral proteins.
Fig 3.
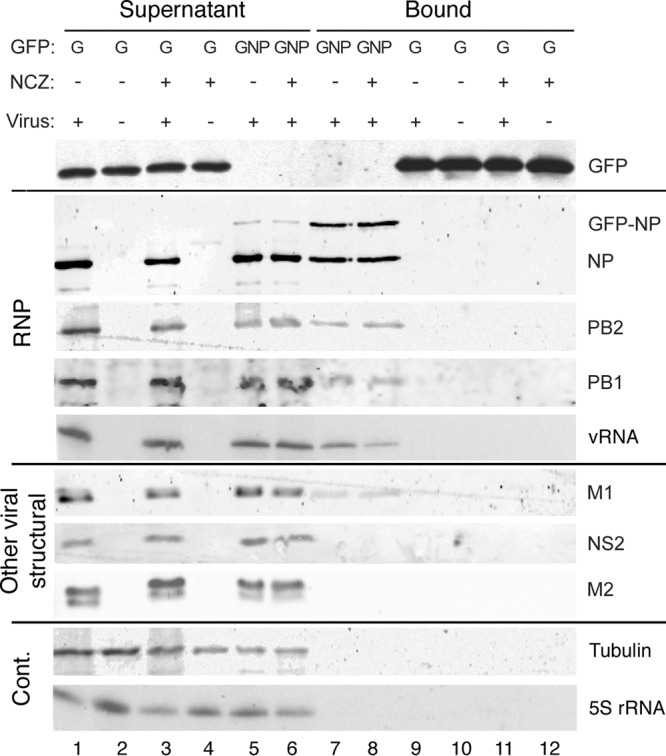
Nucleozin treatment does not disrupt RNPs. 293T cells were transfected with plasmids expressing GFP alone or GFP-NP, incubated for 24 h, and infected with the WT virus or mock infected. At 6 hpi, one set of cells was treated with 1 μM nucleozin (NCZ). Cell lysates prepared at 8 hpi were analyzed by Western blotting for the indicated polypeptides after GFP-Trap affinity selection into supernatant and bound fractions. Sample loading is such that the supernatants are equivalent to 1/10 of the bound fractions. RNA was analyzed by primer extension for segment 3 vRNA and 5S rRNA before or after GFP-Trap selection. The experimental procedure was performed twice. Cont., control.
Nucleozin impairs the trafficking of circulating RNPs in the cytoplasm.
Nucleozin induces aggregation of vRNPs in the perinuclear RE area, a region of the cell hypothesized to represent a “way station” for transport of the virus genome from the nucleus to the apical PM (6, 8, 9). To test if nucleozin blocked onward transport from the RE, we acquired images of living virus-infected cells containing GFP-NP-tagged RNPs, as well as RFP-Rab11. A549 cells were transfected with GFP-NP and RFP-Rab11, infected with PR8 virus or mock infected 12 h later, and imaged at 8 hpi. Inspection of still images of uninfected cells showed that GFP-NP localized mostly in the nucleus, while RFP-Rab11 was dispersed throughout the cytoplasm but with a prominent perinuclear concentration, probably representing the RE (Fig. 4A). Examination of time-lapse movies showed that both nucleoplasmic GFP-NP and cytoplasmic RFP-Rab11 were highly mobile but did not coincide substantially (see the first 4.5 min of Movie S1 in the supplemental material). When cells were infected, a distinct population of GFP-NP was seen as punctate objects in the cytoplasm that colocalized with Rab11 (Fig. 4B). Time-lapse movies showed that these too were highly mobile bodies, often forming filamentous or tubular structures. This pattern of movement was stable under imaging conditions for at least 25 min (Fig. 4B; see Movie S2). On the basis of prior work in our laboratory and others (6, 8, 9), this pattern was likely detecting GFP-NP that had been incorporated into RNPs which, following nuclear export, were undergoing, in part, microtubule-mediated transport courtesy of interactions with Rab11-positive vesicles. In confirmation of this, when cells were fixed after imaging and stained by FISH for segment 4 and 7 vRNAs, the signal colocalized with GFP-NP (data not shown). We then tested the effect of adding nucleozin. In mock-infected cells, drug addition had little apparent effect, in terms of both the overall localization patterns of GFP-NP and RFP-Rab11 (Fig. 4A) and their movement in time-lapse movies (see min 4.5 to 29 of Movie S1). However, when the drug was added to infected cells, the result was a dramatic loss of the small, highly mobile GFP-NP- and RFP-Rab11-positive bodies, which were replaced by increasingly large cytoplasmic aggregates that still contained both proteins but that in many cases collapsed back onto the perinuclear region (Fig. 4C; see Movie S3). High-magnification movies of vRNP-decorated vesicles moving in the cytoplasm of infected cells showed that they were highly mobile and that while there were apparently continual interactions among them, these were transient (see Fig. S3A [white arrowheads] and Movie S4 in the supplemental material). However, in the presence of nucleozin, interacting vesicular structures tended to remain attached over time, thus forming large agglomerations. (see Fig. S3B [white arrowheads] and Movie S5). Thus, nucleozin exerts a general block to cytoplasmic transport of RNP-Rab11 complexes by sticking vRNPs and their associated transport machinery together, most likely via NP-NP interactions.
Fig 4.

Live cell trafficking of GFP-NP and RFP-Rab11. A549 cells were transfected with GFP-NP and RFP-Rab11 and 12 h later infected with WT PR8 or mock infected before imaging under time-lapse conditions (approximately every 4 s) at 8 hpi. Selected still images are shown. Arrows indicate the time of drug addition (1 μM). (A) Mock-infected cell. (B) Infected cell without drug treatment. (C) Infected cell with nucleozin treatment. Scale bars, 10 μm. Images were acquired with an SPE confocal microscope, and images were processed with LAS AF Lite. See Movies S1 to S3 in the supplemental material.
Nucleozin does not widely disrupt the exocytic pathway in infected cells.
The components of the secretory pathway are interconnected in an intricate network whereby disruption of one pathway can have secondary effects on other vesicular pathways (35–37). Since nucleozin disrupted Rab11 localization in infected cells, we tested whether it has a generalized effect on the exocytic pathway and, as a consequence, induces disruption of the trafficking of viral membrane proteins. First, we tested whether the localization of standard cellular marker proteins for the secretory pathway was altered by the addition of nucleozin at late times postinfection. Neither endosomes (highlighted with EEA1, clathrin, and LAMP1), nor the endoplasmic reticulum (as assessed by calnexin), nor the Golgi compartment (GM130 marker), nor the trans-Golgi network (marked with TGN 46; data not shown) aggregated in treated cells (Fig. 5A to C) or, indeed, noticeably changed localization at all after drug addition (Fig. 5A and B; data not shown for panel C). In addition, double staining for NP confirmed that the drug affected RNP localization as expected, but in contrast to the result seen with Rab11 (Fig. 2A), little to no colocalization of the viral protein and the cellular markers was seen (Fig. 5A to C).
Fig 5.

Nucleozin does not disrupt the main exocytic pathway. (A to C, E, F) A549 cells were infected with the WT virus, and at 6 hpi, 1 μM nucleozin (NCZ) was added to one set. Samples were fixed at 8 hpi and stained for NP and calnexin (A); NP and GM130 (B); NP and clathrin, EEA1, or LAMP1 in the presence of nucleozin (C); M2 (E); HA (PR8) and NA (F); or NP and α-tubulin (α-tub) (G). Merged images include a DAPI channel shown in blue. Scale bars, 10 μm unless indicated otherwise. In panel D, lysates from cells treated and harvested as indicated were analyzed by Western blotting for HA, M2, and (as a loading control) α-tubulin (α-tub). Images are representative of three independent experiments.
Since M1 and NS2/NEP are involved in RNP nuclear export (5) and therefore might accompany the genome while it crosses the cytoplasm, we also examined whether nucleozin affects their localization. NS2 localization did not dramatically change in the presence of the drug (data not shown), nor did that of M1 (data not shown).
Next, we tested the effect of nucleozin on the synthesis and accumulation at the PM of viral transmembrane proteins M2, NA, and HA. By Western blotting, expression of the M2 and HA proteins increased up to 8 hpi in a typical untreated time course (Fig. 5D, lanes 1 to 3). The addition of nucleozin at either 4 or 6 hpi had little effect on the accumulation of either protein at 8 hpi (lanes 4 and 5), consistent with previous data showing the general insensitivity of viral protein expression to drug addition after early times postinfection (Fig. 1C). NA accumulation could not be similarly assessed because of the lack of an appropriate antibody. We then tested HA, M2, and NA localization at the PM by immunofluorescence confocal microscopy. Staining of nonpermeabilized cells for the three transmembrane proteins showed readily detectable levels of cell surface protein at 8 hpi (Fig. 5E and F). However, no diminution of staining intensity or obvious differences in localization were seen after treatment with nucleozin from 6 hpi onward. No effect on cell surface HA or M2 amounts was seen when cells were examined by fluorescence-activated cell sorting (data not shown). Thus, the effects of nucleozin on viral and cellular trafficking pathways are apparently specific to the conjunction of RNPs and Rab11 in infected cells.
The normal trafficking of cellular and additional viral proteins suggests that nucleozin did not affect the function of the microtubule network. Consistent with this, α-tubulin staining in treated cells was identical to that in untreated cells (Fig. 5G).
Virions budding from PM in the presence of nucleozin exhibit marked defects.
Nucleozin impedes the viral genome from reaching the PM in the later stages of viral infection. To understand the implications of this for virion formation, we analyzed the WT and NP Y289H mutant viruses by TEM with and without drug treatment. Budding virions of either strain were not detected at 4 hpi (Fig. 6A). By 6 hpi, cells infected with both viruses displayed the characteristic features of budding virus, i.e., ellipsoidal structures surrounded by a glycoprotein fringe, generally attached to the PM by the tip of their shorter axis. When the budding events in WT virus-infected cells were counted at 8 hpi, the numbers varied around a mean of 4 to 5 particles/μm of PM (Fig. 6B), while measurement of the lengths of budding virus showed a mean long-axis dimension of 132 ± 2.9 nm (Fig. 6C; a gallery of representative images is shown in Fig. 6D). When nucleozin was added at either 4 or 6 hpi to cells infected with the nucleozin-resistant virus NP Y289H, little effect was apparent on the budding (Fig. 6A, right hand panels) or size of the virus particles formed at 8 hpi (Fig. 6C and D), with sizes of 113.9 ± 2.1, 124.9 ± 2.5, and 122.1 ± 2.8 nm for no drug and nucleozin addition at 4 and 6 hpi, respectively. The WT virus, however, showed marked and distinct defects. When the drug was added at 4 hpi, very few virions were detected and in the rare cases where they were observed, they had an altered morphology, appearing much rounder, with an average height of around 71.5 ± 3.8 nm (Fig. 6C and D). In addition, they were often still attached to the PM by wide necks in a manner similar to that seen when virus budding was inhibited through small interfering RNA (siRNA)-mediated depletion of Rab11 (38). When the drug was added at 6 hpi and the cells were fixed at 8 hpi, the number of budding events was still very low compared to that in untreated cells (Fig. 6B) and although some normal particles were seen, the morphology of many others was abnormal; many virions were shorter than in untreated cells (105.4 ± 5.9 nm), while others lacked an electron-dense content (Fig. 6A and D). Thus, the late-acting effects of nucleozin on vRNP trafficking and titer output can be correlated with matching effects on virus particle formation, suggesting a direct link between RNP localization and virus budding.
Fig 6.

TEM visualization of budding virions in nucleozin-treated cells. A549 cells were infected with WT or NP Y289H mutant virus, treated with the drug, and fixed at the time points shown before imaging by TEM. Arrowheads indicate defective budding events (A) Representative images. Scale bars, 100 nm. (B) Numbers of WT budding virions were calculated over the whole surface of one side of the cell. (C) Sizes of budding virions from cells infected with WT PR8 were determined (as shown) for at least 30 particles from three independent experiments and plotted as the average ± the standard error of the mean. Nonparametric statistical analysis values obtained by Kruskal-Wallis analysis of variance with a significance of 95% were calculated with GraphPad Prism. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, no statistically significant difference. (D) Gallery of representative budding events with or without nucleozin (NCZ) treatment (1 μM, added at the indicted times postinfection). Arrows indicate defective particles. Images were compiled from three independent experiments.
DISCUSSION
Independent work in several laboratories has identified nucleozin and related compounds as drugs that promote the formation of cytoplasmic NP aggregates, inhibit vRNA synthesis, and effectively interfere with virus replication (13, 15–17). Structural analyses suggest that the drugs act as “molecular staples” that stabilize NP-NP interactions, presumably either by inducing functionally inappropriate binding modes that do not normally occur or by locking ordinarily transient interactions in place so that they become inhibitory. The 50% effective concentration of nucleozin for overall virus replication is around 15-fold lower than the corresponding concentration required to inhibit transcription by isolated RNPs (15), suggesting that the direct inhibition of vRNA synthesis is not the primary block to virus replication. Instead, initial studies hypothesized that the drug caused the cytoplasmic aggregation of newly translated NP, thereby blocking its nuclear import and “starving” viral genome replication of the building blocks required for encapsidation (13, 15–17). However, on the basis of our time-of-addition experiments and detailed analyses of individual steps within viral replication, we propose that RNPs, not NP, are the primary target of the drug and that the sensitive step is their cytoplasmic transport.
We show that nucleozin shows distinct effects on virus replication, depending on the time of addition. When added early in infection, it inhibited the synthesis of viral proteins and all classes of RNA (Fig. 1). While failure to produce vRNA could result from a depleted pool of intranuclear NP, viral transcription and translation are independent of this (4) and a distinct inhibitory mechanism must operate here. Furthermore, the inhibitory effect of nucleozin on overall virus replication could be decoupled from any reduction of the synthesis of viral mRNA, proteins, or vRNA by adding the drug later in infection. Tellingly, adding the drug at 4 hpi had no effect on viral protein synthesis, caused an only 2-fold drop in vRNA accumulation, and yet reduced the output titer by around 100-fold compared to that in untreated cells (Fig. 1). Instead, the drug caused a striking and rapid aggregation of cytoplasmic RNPs (Fig. 2, 4, and 5; see Fig. S1 and S2 in the supplemental material). This blockade of RNP transport also correlated with a marked reduction in the amount of virus budding, as well as the formation of abnormally small virus particles (Fig. 6). Notably, however, we saw little effect of drug treatment on intranuclear NP localization, either on GFP-NP with or without RNP formation or on authentic NP (Fig. 2A). We therefore conclude that the late-acting effect of nucleozin on virus replication is directly attributable to effects on RNP localization.
Regarding the undoubted ability of nucleozin to inhibit earlier steps in the virus life cycle, given that the drug did not inhibit mRNA synthesis when added at 2.5 hpi (before large amounts of mRNA have accumulated) and that we were working at drug concentrations substantially below those required to inhibit in vitro transcription by isolated RNPs, we think it plausible that the transcriptional inhibition observed when the drug was added at 30 min postinfection can also be explained by inhibition of cytoplasmic RNP transport, but on the way into the nucleus rather than away from it. Thus, although NP is regarded primarily as part of the vRNA synthesis machinery, this study provides proof of the principle that targeting its role in intracellular trafficking of the virus genome can lead to effective inhibition of virus replication. This mechanism has parallels with the block to RNP nuclear export caused by mitogen-activated protein kinase kinase inhibitors (39, 40), and it would be interesting to test these compounds for synergy with nucleozin.
A further striking outcome of the late-acting effect of nucleozin was the inclusion of Rab11 in the cytoplasmic aggregates of RNPs (Fig. 2 and 4; see Fig. S1 in the supplemental material). Since Rab11 trafficking was not affected by nucleozin in the absence of vRNPs, it seems likely that the cellular protein is secondarily recruited to the aggregates via its interaction with RNPs. This argues against the simple hypothesis that nucleozin disrupts RNP trafficking by breaking interactions with Rab11. Taking all of this into account suggests a model in which Rab11-vRNP-decorated vesicles are “stapled” together via persistent RNP-RNP interactions that override ordinarily transient interactions between vesicular cargos (see Fig. S3 and Movies S4 and S5 in the supplemental material). Transfection of dominant negative GFP-Rab11 or depletion of endogenous Rab11 siRNA did not prevent these accumulations in cells infected with the WT viruses (data not shown), suggesting that Rab11 may not be essential for aggregate formation. While this is consistent with data suggesting that Rab11 interacts with the viral polymerase rather than NP (6, 9), it leaves open the question of why the aggregates cluster near the nucleus. The speed with which this occurs suggests that the RNP aggregates remain attached to microtubules (Fig. 5G) but that the net flow becomes biased toward the inward (minus-end) direction. Why the increase in the size of RNP-Rab11 cargo complexes should favor this direction of travel is not obvious, but it may become clearer when the motor proteins involved in RNP transport are identified. Dynein has, however, been implicated in the transport of “aggresomes,” created by misfolded proteins, so it is possible that the same mechanism operates here (41).
Nucleozin treatment also inhibited virus budding, leading to the production of markedly fewer and smaller virus particles (with the latter potentially because of reduced RNP content) (Fig. 6). For normal virus budding, all virion components need to meet at the PM from where a membrane curvature protrudes, followed by the formation of a neck and finally membrane scission. All three viral integral membrane proteins have been implicated in this process (42–44). Despite the major effect nucleozin had on Rab11 localization (and thus presumably traffic throughout the recycling endosomal pathway), we did not detect any drug-induced alterations of the standard exocytic pathway or of the localization of HA, NA, and M2 (Fig. 5). We therefore favor the hypothesis that correct trafficking of the viral genome is specifically required to allow efficient virus assembly and/or budding. This is consistent with our previous findings of defective budding and RNP trafficking in the absence of normal levels of Rab11 (6, 38). Although the effect of nucleozin does not help distinguish between a budding defect resulting primarily from the disruption of Rab11 function and a secondary effect on RNP localization, observations that viruses lacking an individual segment or containing disrupted segment-specific packaging signals produce reduced numbers of particles (21, 45–47) favor a role for RNP organization in normal virion formation.
The Rab11 pathway has been implicated in the transport of progeny RNA from other negative-strand viruses, notably, Sendai virus and human respiratory syncytial virus, members of the Paramyxoviridae family (48, 49); hantavirus (50); and hepatitis C virus, a positive-sense RNA virus in the Flaviviridae family (51). The common exploitation of a pathway by several circulating virus families able to infect mammals and on occasion cause severe disease in humans suggests that the inhibitory strategy described herein could be applied to other viruses.
Supplementary Material
ACKNOWLEDGMENTS
We thank Susanna Colaco and Phil Stevenson for the gift of reagents. We also thank the multi-imaging center of the University of Cambridge for processing images for TEM.
This work was supported by a grant from the MRC (G0700815) to P.D. M.J.A. was supported by a Newton Trust grant from Trinity College, University of Cambridge, Cambridge, United Kingdom. R.Y.K. was supported by GRF 768010 M from RGC Hong Kong.
Footnotes
Published ahead of print 13 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03123-12.
REFERENCES
- 1. Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P. 2009. A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 83:8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. 2012. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 8:e1002998 doi:10.1371/journal.ppat.1002998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83:723–734 [DOI] [PubMed] [Google Scholar]
- 5. Boulo S, Akarsu H, Ruigrok RW, Baudin F. 2007. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 124:12–21 [DOI] [PubMed] [Google Scholar]
- 6. Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 85:4143–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Avilov SV, Moisy D, Munier S, Schraidt O, Naffakh N, Cusack S. 2012. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J. Virol. 86:1433–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. 2011. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 85:6117–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Momose F, Sekimoto T, Ohkura T, Jo S, Kawaguchi A, Nagata K, Morikawa Y. 2011. Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS One 6:e21123 doi:10.1371/journal.pone.0021123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das K, Aramini JM, Ma LC, Krug RM, Arnold E. 2010. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 17:530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang TS, Palese P, Krystal M. 1990. Determination of influenza virus proteins required for genome replication. J. Virol. 64:5669–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H, Wan J, Lin MI, Liu Y, Lu X, Liu J, Xu Y, Chen J, Tu Z, Cheng YS, Ding K. 2012. Design, synthesis, and in vitro biological evaluation of 1H-1,2,3-triazole-4-carboxamide derivatives as new anti-influenza A agents targeting virus nucleoprotein. J. Med. Chem. 55:2144–2153 [DOI] [PubMed] [Google Scholar]
- 14. Cianci C, Gerritz SW, Deminie C, Krystal M. 26 July 2012. Influenza nucleoprotein: promising target for antiviral chemotherapy. Antivir. Chem. Chemother. (Epub ahead of print.) doi:10.3851/IMP2235 [DOI] [PubMed] [Google Scholar]
- 15. Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva-Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. 2011. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. U. S. A. 108:15366–15371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 28:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin-Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. 2010. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc. Natl. Acad. Sci. U. S. A. 107:19151–19156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrasco M, Amorim MJ, Digard P. 2004. Lipid raft-dependent targeting of the influenza A virus nucleoprotein to the apical plasma membrane. Traffic 5:979–992 [DOI] [PubMed] [Google Scholar]
- 19. de Wit E, Spronken MI, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2004. Efficient generation and growth of influenza virus A/PR/8/34 from eight cDNA fragments. Virus Res. 103:155–161 [DOI] [PubMed] [Google Scholar]
- 20. Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63 doi:10.1186/1743-422X-3-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. 2008. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J. Virol. 82:11869–11879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amorim MJ, Read EK, Dalton RM, Medcalf L, Digard P. 2007. Nuclear export of influenza A virus mRNAs requires ongoing RNA polymerase II activity. Traffic 8:1–11 [DOI] [PubMed] [Google Scholar]
- 23. Digard P, Blok VC, Inglis SC. 1989. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology 171:162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poole E, Elton D, Medcalf L, Digard P. 2004. Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology 321:120–133 [DOI] [PubMed] [Google Scholar]
- 25. Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. 2007. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J. Gen. Virol. 88:2280–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loucaides EM, von Kirchbach JC, Foeglein A, Sharps J, Fodor E, Digard P. 2009. Nuclear dynamics of influenza A virus ribonucleoproteins revealed by live-cell imaging studies. Virology 394:154–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harman A, Browne H, Minson T. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 76:10708–10716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mullin AE, Dalton RM, Amorim MJ, Elton D, Digard P. 2004. Increased amounts of the influenza virus nucleoprotein do not promote higher levels of viral genome replication. J. Gen. Virol. 85:3689–3698 [DOI] [PubMed] [Google Scholar]
- 29. Read EK, Digard P. 2010. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J. Gen. Virol. 91:1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simpson-Holley M, Ellis D, Fisher D, Elton D, McCauley J, Digard P. 2002. A functional link between the actin cytoskeleton and lipid rafts during budding of filamentous influenza virions. Virology 301:212–225 [DOI] [PubMed] [Google Scholar]
- 31. Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 90:1398–1407 [DOI] [PubMed] [Google Scholar]
- 32. Bruce EA, Medcalf L, Crump CM, Noton SL, Stuart AD, Wise HM, Elton D, Bowers K, Digard P. 2009. Budding of filamentous and non-filamentous influenza A virus occurs via a VPS4 and VPS28-independent pathway. Virology 390:268–278 [DOI] [PubMed] [Google Scholar]
- 33. Breitenfeld PM, Schafer W. 1957. The formation of fowl plague virus antigens in infected cells, as studied with fluorescent antibodies. Virology 4:328–345 [DOI] [PubMed] [Google Scholar]
- 34. Elton D, Amorim MJ, Medcalf L, Digard P. 2005. ‘Genome gating’; polarized intranuclear trafficking of influenza virus RNPs. Biol. Lett. 1:113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bard F, Malhotra V. 2006. The formation of TGN-to-plasma-membrane transport carriers. Annu. Rev. Cell Dev. Biol. 22:439–455 [DOI] [PubMed] [Google Scholar]
- 36. Gonnord P, Blouin CM, Lamaze C. 2012. Membrane trafficking and signaling: two sides of the same coin. Semin. Cell Dev. Biol. 23:154–164 [DOI] [PubMed] [Google Scholar]
- 37. Hsu VW, Prekeris R. 2010. Transport at the recycling endosome. Curr. Opin. Cell Biol. 22:528–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bruce EA, Digard P, Stuart AD. 2010. The Rab11 pathway is required for influenza A virus budding and filament formation. J. Virol. 84:5848–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Droebner K, Pleschka S, Ludwig S, Planz O. 2011. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. 92:195–203 [DOI] [PubMed] [Google Scholar]
- 40. Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301–305 [DOI] [PubMed] [Google Scholar]
- 41. Wan Y, Yang Z, Guo J, Zhang Q, Zeng L, Song W, Xiao Y, Zhu X. 2012. Misfolded Gbeta is recruited to cytoplasmic dynein by Nudel for efficient clearance. Cell Res. 22:1140–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen BJ, Leser GP, Morita E, Lamb RA. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 81:7111–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yondola MA, Fernandes F, Belicha-Villanueva A, Uccelini M, Gao Q, Carter C, Palese P. 2011. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J. Virol. 85:2480–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Wit E, Spronken MI, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2006. Evidence for specific packaging of the influenza A virus genome from conditionally defective virus particles lacking a polymerase gene. Vaccine 24:6647–6650 [DOI] [PubMed] [Google Scholar]
- 46. Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. 2003. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. U. S. A. 100:2002–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muramoto Y, Takada A, Fujii K, Noda T, Iwatsuki-Horimoto K, Watanabe S, Horimoto T, Kida H, Kawaoka Y. 2006. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 80:2318–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chambers R, Takimoto T. 2010. Trafficking of Sendai virus nucleocapsids is mediated by intracellular vesicles. PLoS One 5:e10994 doi:10.1371/journal.pone.0010994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE., Jr 2008. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc. Natl. Acad. Sci. U. S. A. 105:10209–10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rowe RK, Suszko JW, Pekosz A. 2008. Roles for the recycling endosome, Rab8, and Rab11 in hantavirus release from epithelial cells. Virology 382:239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8:e1002466 doi:10.1371/journal.ppat.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.