

Abstract
Many viruses use the host trafficking system at a variety of their replication steps. Measles virus (MV) possesses a nonsegmented negative-strand RNA genome that encodes three components of the ribonucleoprotein (RNP) complex (N, P, and L), two surface glycoproteins, a matrix protein, and two nonstructural proteins. A subset of immune cells and polarized epithelial cells are in vivo targets of MV, and MV is selectively released from the apical membrane of polarized epithelial cells. However, the molecular mechanisms for the apical release of MV remain largely unknown. In the present study, the localization and trafficking mechanisms of the RNP complex of MV were analyzed in detail using recombinant MVs expressing fluorescent protein-tagged L proteins. Live cell imaging analyses demonstrated that the MV RNP complex was transported in a manner dependent on the microtubule network and together with Rab11A-containing recycling endosomes. The RNP complex was accumulated at the apical membrane and the apical recycling compartment. The accumulation and shedding of infectious virions were severely impaired by expression of a dominant negative form of Rab11A. On the other hand, recycling endosome-mediated RNP transport was totally dispensable for virus production in nonpolarized cells. These data provide the first demonstration of the regulated intracellular trafficking events of the MV RNP complex that define the directional viral release from polarized epithelial cells.
INTRODUCTION
For airborne viruses, efficient shedding of progeny viruses is critical for transmission. Measles virus (MV) is the causative agent of measles, which is an acute and highly contagious disease characterized by high fever and a maculopapular rash. MV is an enveloped virus that belongs to the genus Morbillivirus in the family Paramyxoviridae. The MV genome is a nonsegmented negative-sense RNA encoding six tandemly linked genes, N, P/V/C, M, F, H, and L (1). Each virion contains the RNA genome encapsidated by the nucleocapsid (N) protein. The encapsidated genome is associated with a viral RNA-dependent RNA polymerase composed of the phospho- (P) and large (L) proteins and forms the ribonucleoprotein (RNP) complex. The virus particle possesses two types of glycoprotein spikes, the hemagglutinin (H) and fusion (F) proteins, on the envelope. The H protein is responsible for binding to cellular receptors, while the F protein mediates membrane fusion between the virus envelope and the host cell plasma membrane. The matrix (M) protein is also a major component of the virus particle and plays crucial roles in virus assembly by associating with the cytoplasmic tails of the F and H proteins as well as the RNP complex. In addition to these structural components, the MV genome encodes two nonstructural proteins, V and C, which are synthesized in infected cells and counteract host interferon responses. Recent studies showed that polarized epithelial cells are infected with MV via nectin4 as a receptor, a process that is critical for virus shedding in vivo (2–4). Progeny MV particles are selectively released from the apical plasma membrane of polarized epithelial cells (5, 6). It is well known that MV replicates entirely within the cytoplasm, but the detailed location for each event, such as viral RNA synthesis, is poorly elucidated. Moreover, little is known about the molecular mechanisms underlying the directional virus release from polarized epithelial cells. Previous studies demonstrated that the viral RNP complexes of influenza A virus (IAV) in the family Orthomyxoviridae and Sendai virus (SeV) in the family Paramyxoviridae are transported along microtubules (MTs) using Rab11-positive recycling endosomes (REs) (7–10). The Rab11 GTPase subfamily consists of Rab11a, Rab11b, and Rab25/Rab11c, which play key roles in protein traffic by REs. Similarly, vesicular stomatitis virus (VSV) in the family Rhabdoviridae also uses MTs for its protein transport (11). In the present study, the intracellular location and trafficking of the RNP complex of MV were analyzed.
MATERIALS AND METHODS
Plasmids.
The full-length genome plasmid p(+)MV323-EGFPtagL encoding the MV genome with an enhanced green fluorescent protein (EGFP)-tagged L gene was described previously (12, 13). The full-length genome plasmid p(+)MV323-mCherrytagL was generated by replacing the EGFP cDNA region of p(+)MV323-EGFPtagL with an mCherry cDNA. The full-length genome plasmid p(+)MV323-AddmCherry was generated by introducing the mCherry gene into an additional transcriptional unit between the H and L genes, as reported previously (14). pTagRFP-Tubulin and pAcGFP1-Tubulin were purchased from Evrogen (Moscow, Russia) and Clontech (Mountain View, CA), respectively. These plasmids encode red fluorescent protein (RFP)- and green fluorescent protein (GFP)-tagged α-tubulin, which are reported to behave similarly to untagged α-tubulin (10, 15, 16). The expression plasmids pMXsIP-EGFP, -EGFP-Rab5, -EGFP-Rab7, -EGFP-Rab11A, and -EGFP-Rab11ADN (encoding a dominant-negative form of Rab11A, Rab11A-S25N) were generated by inserting the cDNAs of the respective Rab genes with N-terminally fused EGFP into the multicloning site of pMXsIP (17). A retroviral vector encoding a short-hairpin RNA (shRNA) against Rab11A mRNA (pSUPERretro-shRab11A) was constructed by inserting the oligonucleotide fragment (target sequence, 5′-AAGAGCACCATTGGAGTAGAG-3′) into pSUPER.retro.puro (Oligoengine, Seattle, WA). As a negative control, a retroviral vector with an oligonucleotide fragment (target sequence, 5′-AAGCGCGCTTTGTAGGATTCG-3′) (pSUPERretro-shNC) was also prepared. Retroviral preparations were performed according to the manufacturer's instructions.
Cells and virus.
Vero/hSLAM cells (18), BHK/T7-9 cells (kindly provided by N. Ito) (19), and MDCK II cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma, St. Louis, MO) containing 7% fetal bovine serum (FBS). PLAT-gp cells, a 293T-derived Moloney murine leukemia virus (MMLV)-based retroviral vector packaging cell line (kindly provided by M. Shimojima and T. Kitamura) (20), were maintained in DMEM containing 10% FBS. MMLV-based retroviral vectors expressing EGFP or EGFP-tagged Rab proteins were produced by introducing the corresponding pMXsIP vector (pMXsIP-EGFP, -EGFP-Rab5, -EGFP-Rab7, -EGFP-Rab11A, or -EGFP-Rab11ADN) together with a VSV G protein-expressing plasmid, pCVSV-G, into PLAT-gp cells (17). Vero/hSLAM or MDCK cells constitutively expressing EGFP, EGFP-Rab5, EGFP-Rab7, EGFP-Rab11A, or EGFP-Rab11ADN were then generated by transduction of the respective genes using the MMLV-based retroviral vector and selection with puromycin (17). Vero/hSLAM cells constitutively expressing negative-control shRNA (Vero/hSLAM/shNC) or shRNA against Rab11A (Vero/hSLAM/shRab11A) were generated by transduction of the respective shRNA using the retroviral vector and selection with puromycin. IC323 (21) and IC323-EGFPtagL (13) were reported previously. IC323-mCherrytagL and IC323-AddmCherry were generated from p(+)MV323-mCherrytagL and p(+)MV323-AddmCherry, respectively, using an efficient MV reverse genetics system (22). All of the recombinant MVs (rMVs) used in this study were propagated in Vero/hSLAM cells, and the infectious virus titers were determined by plaque assays.
Growth kinetics analysis of MV.
Vero/hSLAM cells were infected with MV at a multiplicity of infection (MOI) of 0.01. At various time points, the culture medium or the cells were harvested and the infectious virus titers (in plaque-forming units [PFU]) were determined by standard plaque assays on Vero/hSLAM cells. MDCK cells on 24-well plates or 12-mm filter units of tissue culture-treated 0.4-μm-pore-size transwell polycarbonate filters (Costar Corp., Cambridge, MA) were infected with MV at an MOI of 0.2 immediately after cell seeding, since confluent monolayers of MDCK cells are poorly susceptible to MV infection (6). The formation of an electrically tight monolayer was checked daily by measuring the transepithelial resistance using an ERS-2 apparatus (Millipore, Bedford, MA). At various time points, the culture medium or the cells were harvested and the infectious virus titers were determined by plaque assays on Vero/hSLAM cells.
Reagents and antibodies.
Nocodazole and paclitaxel were purchased from Sigma. A fusion-blocking peptide (FBP) (Z-D-Phe-Phe-Gly) was purchased from Peptide Institute Inc. (Osaka, Japan). Mouse monoclonal antibodies (MAbs) against the N, C, M, F, and H proteins of MV and a rabbit polyclonal antibody raised against the common N terminus of the P and V proteins were described previously (23, 24). Mouse MAbs against α- and γ-tubulin were purchased from Invitrogen (Carlsbad, CA) and Sigma, respectively. Mouse MAbs raised against EGFP was purchased from Clontech. Rabbit MAbs against Rab11 were purchased from Cell Signaling Technology (Danvers, MA). Alexa Fluor 405-, 488-, 594-, and 647-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Invitrogen. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was purchased from Nacalai Tesque (Kyoto, Japan). Vybrant CM-DiI cell-labeling solution was purchased from Invitrogen and used according to the manufacturer's instructions.
Immunofluorescence and confocal microscopy.
MV-infected cells were cultured for appropriate durations, fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde, and then permeabilized with PBS containing 0.5% Triton X-100. Vero/hSLAM cells were inoculated and incubated for 2 days in the presence of the FBP to prevent cell-to-cell fusion before fixation. MDCK cells were fixed at 4 days postinfection (p.i.) in all experiments. The cells were then washed with PBS and incubated with a primary antibody, followed by incubation with a secondary antibody conjugated with Alexa Fluor 405, 488, 594, or 647. For counterstaining of the nuclei, the cells were incubated with DAPI. After the fluorescence staining, the cells were observed using an FV1000D Spectral Type confocal laser-scanning microscope (inverted microscope IX81) (Olympus, Tokyo, Japan). Data analysis was performed using FV10-ASW (Olympus) and ImageJ (National Institutes of Health, Bethesda, MD) software.
Time-lapse fluorescence microscopy.
Cells were seeded on a 35-mm glass-based dish (Iwaki Glass, Tokyo, Japan), infected with an rMV, and cultured with the FBP for appropriate durations. The dish was placed in an incubation chamber installed in the FV1000D confocal laser-scanning microscope, in which the temperature and CO2 concentration were maintained at 37°C and 5%, respectively. A total of 50 to 180 still images of the cells were taken at appropriate intervals using a 60×/1.35 numerical aperture (NA) oil objective (Olympus). Time intervals for acquisition of images were 10 min for Movie S1 and 1.1 s for Movies S2 to S5 in the supplemental material. Data analysis was performed using FV10-ASW and ImageJ software.
Fluorescence in situ hybridization assay.
MV negative-strand genomic RNA was detected using the QuantiGene ViewRNA ISH cell assay kit (Affymetrix, Santa Clara, CA) and measles virus probe set (Affymetrix) that can hybridize to nucleotides 1489 to 2745 of the MV genome. Cells were applied to glass coverslips, infected with IC323-mCherrytagL, and cultured for 2 days in the presence of the FBP. Cells were fixed in 4% formaldehyde solution, permeabilized, treated with protease, and hybridized according to the manufacturer's instructions. The cells were observed using an FV1000D Spectral Type confocal laser-scanning microscope (inverted microscope IX81). Data analysis was performed using FV10-ASW and ImageJ software.
RESULTS
Generation of rMVs encoding FLP-tagged L proteins.
Previously, Duprex et al. (12) generated rMV based on the Edmonston vaccine strain encoding the L protein fused with EGFP in the variable hinge (H2) region. In the present study, based on the wild-type IC-B strain, rMVs encoding fluorescent protein (FLP)-tagged L proteins were generated. IC323-EGFPtagL and IC323-mCherrytagL encode the L protein fused with EGFP and mCherry, respectively, in the H2 region (12) (Fig. 1A). These rMVs were viable, and their replication kinetics was only slightly delayed compared with that of non-FLP-tagged IC323 in Vero/hSLAM cells (Fig. 1B and C). The growth of the Edmonston strain was more severely affected by the EGFP insertion in the H2 region (12). This difference may be attributed to the fact that the L protein of the IC323 strain exhibits higher activity than that of the Edmonston strain (25), and it may be more tolerant for the insertion than the Edmonston strain. The peak cell-associated and cell-free virus titers of IC323-EGFPtagL and IC323-mCherrytagL were comparable to those of non-FLP-tagged IC323 (Fig. 1B and C). These viruses efficiently produced syncytia expressing the FLP in monolayers of Vero/hSLAM cells (Fig. 1D) and were used for subsequent analyses of the transport and localization of the L protein during virus replication.
Fig 1.
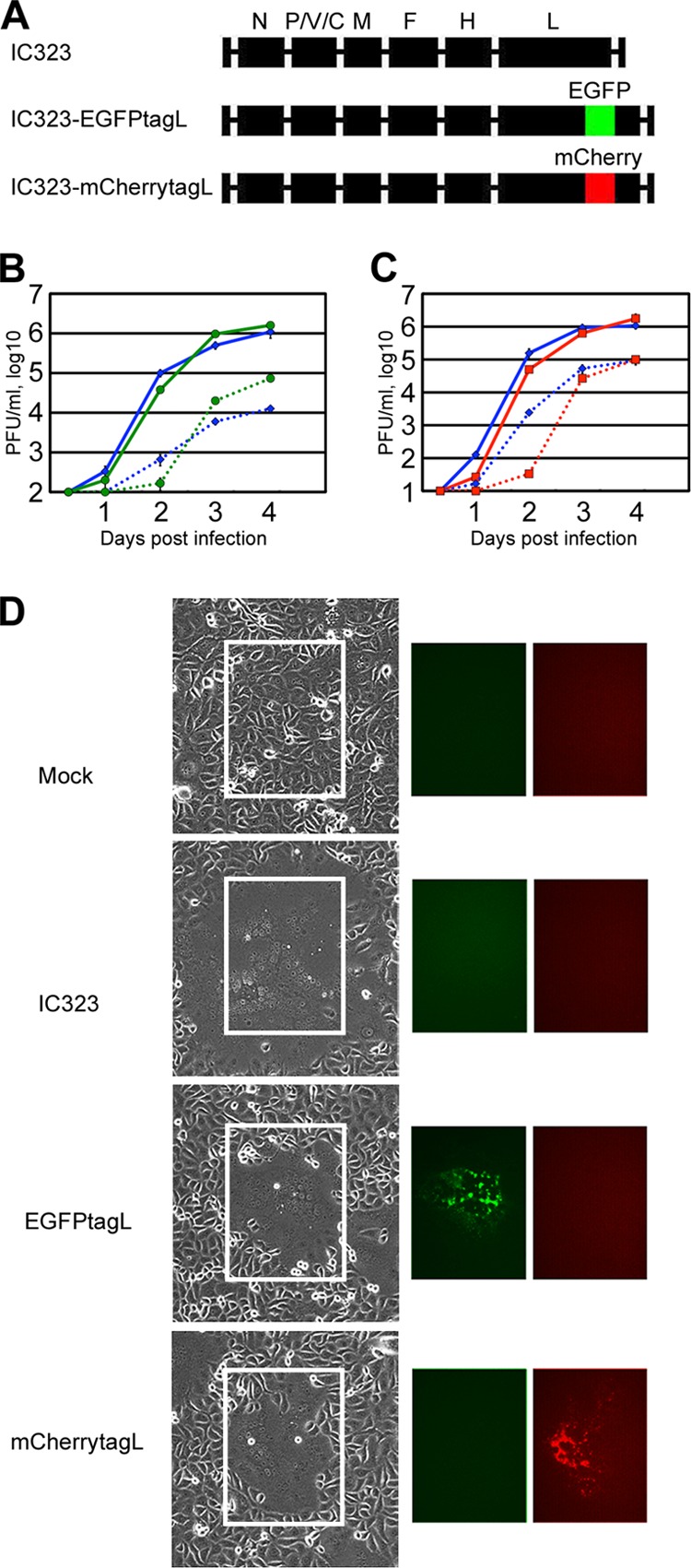
Construction of rMVs expressing FLP-tagged L proteins. (A) Genome structures of the rMVs. The six internal boxes indicate the N, P, M, F, H, and L genes of MV. The portions colored in green and red indicate the coding regions of the green FLP (EGFP) and red FLP (mCherry), respectively. (B and C) Growth kinetics of the rMVs in Vero/hSLAM cells. Vero/hSLAM cells were infected with the rMVs at an MOI of 0.01. At various time points, the cells and culture medium were harvested separately, and the PFU in both samples were determined. The data represent the means ± standard deviations of results from triplicate samples. The solid and dashed lines indicate the data for the cell-associated and cell-free titers, respectively. (B) Blue and green symbols indicate the data for IC323 and IC323-EGFPtagL, respectively. (C) Blue and red symbols indicate the data for IC323 and IC323-mCherrytagL, respectively. (D) Syncytium morphology and FLP-tagged L protein expression in rMV-infected Vero/hSLAM cells. Vero/hSLAM cells were infected with the rMVs at an MOI of 0.01. The cells were observed daily under light and fluorescence microscopes. Data at 2 days p.i. are shown.
The RNP complex of MV accumulated at the perinuclear region in MV-infected Vero/hSLAM cells.
To show the intracellular location and relationship of the L protein with other MV proteins, immunofluorescence assays were performed using a series of antibodies specific to each MV protein (Fig. 2). Previous studies indicated that the components of the RNP complex (N, P, and L proteins) are mostly colocalized and form dots of various sizes in the cytoplasm (13, 26). These observations were reexamined using IC323-EGFPtagL, IC323-mCherrytagL, and non-FLP-tagged IC323 to assess whether the FLP tagging had any untoward effects on the location of the RNP complex proteins. The EGFPtagL and mCherrytagL proteins were typically colocalized with the N and P/V proteins, with punctate fluorescence in the cytoplasm (Fig. 2A and B and data not shown), as reported previously (13). In these experiments, specific MAb against the N protein was used, while a rabbit polyclonal antibody raised against the common N terminus of the P and V proteins was used, since no antibody specific for the P protein was available. Thus, P/V indicates the P and/or V proteins. However, cells infected with the V protein-deficient rMV (MVΔV) (27) also showed a strong colocalization pattern of the N and P proteins (data not shown), indicating that the P protein colocalizes with the N and L proteins.
Fig 2.

Intracellular distributions of mCherry-tagged L protein and other viral proteins. (A) Vero/hSLAM cells were infected with IC323 or IC323-mCherrytagL, cultured for 2 days, and then analyzed by immunofluorescence and confocal microscopy. Red fluorescence indicates the mCherry-tagged L protein. The N and P/V proteins were detected with viral protein-specific primary antibodies and secondary antibodies conjugated with Alexa Fluor 488 (green) and Alexa Fluor 647 (blue, pseudocolor), respectively. The upper and lower panels show the results for IC323 and IC323-mCherrytagL, respectively. (B) RGB line profiles along the lines selected in panel A. The left and right panels show the data for IC323 and IC323-mCherrytagL, respectively. (C) Vero/hSLAM cells were infected with IC323-mCherrytagL, cultured for 2 days, and analyzed by fluorescence in situ hybridization assay. Lower panels show the result of MV-infected cells with a probe for genomic RNA. Green and red fluorescence indicates genomic RNA and mCherry-tagged L protein, respectively. Assays were also performed with mock-infected cells (upper panels). Middle panels show the result of MV-infected cells subjected to the fluorescence in situ hybridization assay without a probe for genomic RNA. Nuclei were counterstained with DAPI (blue). The scale bars indicate 10 μm. (D to G) Vero/hSLAM cells were infected with IC323-mCherrytagL, cultured for 2 days, and then analyzed by immunofluorescence and confocal microscopy. Red fluorescence indicates the mCherry-tagged L protein. The C (D), M (E), F (F), and H (G) proteins were detected with viral protein-specific primary antibodies and Alexa Fluor 488-conjugated secondary antibodies (green). Nuclei were counterstained with DAPI (blue). Scale bars indicate 10 μm.
The localization patterns of the N and P/V proteins were similar among IC323-EGFPtagL-, IC323-mCherrytagL-, and IC323-infected cells (Fig. 2A and B and data not shown). These findings indicate that the FLP tags on the L protein hardly affected the distribution of the RNP complex proteins. Although punctate fluorescent areas indicating the N, P/V, and mCherrytagL (or EGFPtagL) proteins were distributed throughout the cytoplasm, they tended to concentrate at a perinuclear region (Fig. 2A and B and data not shown). In contrast to the N protein in the cytoplasm, the N protein located in the nucleus was not associated with the L and P/V proteins (Fig. 2A and B). The virus genome was also detected by fluorescence in situ hybridization assay (Fig. 2C). Consistent with the knowledge that MV undergoes its replication cycles entirely in the cytoplasm, the genome was detected exclusively in the cytoplasm that was concentrated at a perinuclear region, where the RNP complex proteins were accumulated (Fig. 2C). Additionally, the punctate signals of the L protein were colocalized with fluorescence signals for the virus genome (Fig. 2C). These data confirmed that the L protein signals represent the viral RNP complex.
Subsequently, the distributions of other viral proteins (C, M, F, and H) were analyzed using specific MAbs against these proteins, and representative single optical slice images are shown in Fig. 2D to G. The intracellular distribution patterns of these proteins were compared among IC323-EGFPtagL-, IC323-mCherrytagL-, and non-FLP-tagged IC323-infected cells and were found to be similar (data not shown). The C protein, which modulates viral RNA synthesis by interacting with the RNP complex, was almost perfectly colocalized with EGFPtagL and mCherrytagL (Fig. 2D and data not shown), as reported previously (13). The M protein was detected in both the cytoplasm and the nucleus in small dots and was partly associated with the plasma membrane (Fig. 2E). A portion of the M protein in the cytoplasm was colocalized with the L protein (Fig. 2E) that may represent the RNP-coating M protein destined to be incorporated into virions (28). Large parts of the F and H proteins were localized at the plasma membrane, and only fractions of these proteins in the cytoplasm were colocalized with the L protein in small dots (Fig. 2F and G).
The MV L protein is transported along MTs and concentrates at the microtubule-organizing center (MTOC) in MV-infected Vero/hSLAM cells.
To clarify the overall trend of the L protein (EGFPtagL) movement, IC323-EGFPtagL-infected Vero/hSLAM cells were analyzed by time-lapse fluorescence microscopy. The data confirmed that, overall, the L protein moved to allow concentration at the perinuclear region (Fig. 3A; see Movie S1 in the supplemental material; data at 24 to 40 h p.i. are shown). The MTOC is known to be located at a perinuclear region in the center of cells. The minus ends of MTs face toward the MTOC, and the metronomic array of MTs is important for the directional transport of cellular proteins. MTs were detected using MAbs specific for α-tubulin (a major component of MTs) and γ-tubulin (a constituent of the minus ends of MTs anchored to the MTOC). The data revealed that the perinuclear region where the EGFPtagL protein was concentrated was the MTOC (Fig. 3B and C). To clarify the role of MTs in the transport, the movement of the EGFPtagL protein in cells expressing TagRFP (red FLP)-tagged α-tubulin was analyzed by time-lapse fluorescence microscopy. Each dot moved intermittently in various directions, showing a stop-start or zigzag movement along the MTs (Fig. 3D; see Movie S2 in the supplemental material). The EGFPtagL-containing dots moved in both directions along the MTs (Fig. 3D; see Movie S2), as observed for other viral and cellular proteins (29, 30). To confirm the dependency on MTs, IC323-mCherrytagL-infected cells were treated with 5 μM nocodazole, an MT-disrupting agent, or left untreated as a control, and the movement of the mCherrytagL-containing dots was analyzed by time-lapse fluorescence microscopy. In the control cells, the mCherrytagL-containing dots were transported efficiently throughout the cytoplasm (see Movie S3 in the supplemental material). On the other hand, the mCherrytagL-containing dots no longer moved actively in the nocodazole-treated cells (see Movie S4 in the supplemental material). It was also found that the accumulation of mCherrytagL at the perinuclear region was disrupted by treatment with 5 μM nocodazole (Fig. 3E). These findings indicate that the MV RNP complexes are transported to and concentrated at the MTOC in an MT-dependent manner.
Fig 3.
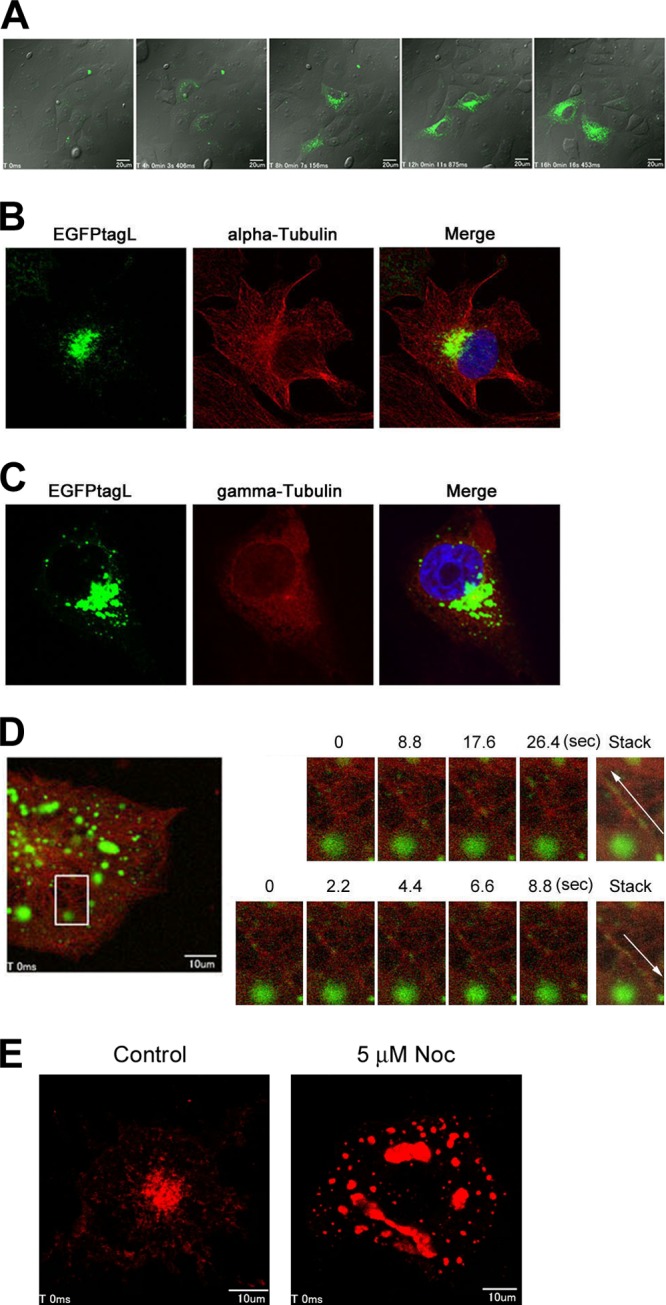
Accumulation of the L protein at the MTOC in an MT-dependent manner. (A) Vero/hSLAM cells were infected with IC323-EGFPtagL, cultured for 24 h, and analyzed by time-lapse florescence microscopy. Five images taken at 4-h intervals are shown. Bars indicate 20 μm. (B and C) Vero/hSLAM cells were infected with IC323-EGFPtagL, cultured for 2 days, and analyzed by immunofluorescence and confocal microscopy. Green fluorescence indicates the EGFPtagL protein. α-Tubulin (B) and γ-tubulin (C) were detected with specific primary antibodies and Alexa Fluor 594-conjugated secondary antibodies (red). Nuclei were counterstained with DAPI (blue). (D) Vero/hSLAM cells were infected with IC323-EGFPtagL and transfected with pTagRFP-Tubulin. At 2 days p.i., the intracellular movement of EGFPtagL was analyzed by time-lapse fluorescence microscopy. A total of 150 still images were taken at 1.1-s intervals. The left panel shows the overall confocal image of the cells at the start of the time-lapse analysis. Green and red fluorescence indicates the EGFPtagL protein and TagRFP-tubulin, respectively. The scale bar indicates 10 μm. The right panels show enlarged time-lapse images and time-stacked images of the area indicated by the box in the left panel. The upper and lower panels show obliquely upward and downward movements of the EGFPtagL-containing dots, respectively, at different time points after starting the time-lapse analysis. Movements of the EGFPtagL fluorescence are indicated with white arrows in the time-stacked images. (E) Vero/hSLAM cells were infected with IC323-mCherrytagL. At 2 days p.i., the cells were treated with 5 μM nocodazole for 15 h (5 μM Noc) or left untreated (control). A total of 50 still images were taken at 1.1-s intervals. The panels show the overall confocal images of the cells at the start of the time-lapse analysis. Red fluorescence indicates the mCherrytagL protein. Scale bars indicate 10 μm.
The L protein moves along with Rab11A-containing REs.
To determine the roles of transport vehicles in the trafficking of the RNP complex of MV, the intracellular distributions of mCherrytagL protein and EGFP-tagged Rab (EGFP-Rab) proteins were analyzed in the same cells. Rab proteins contribute to the synthesis, transport, and function of specific endosomes and are used as endosome markers (31). Recent studies showed that specific endosomes are associated with viral protein transport along MTs (7, 9, 10). Vero/hSLAM cells constitutively expressing EGFP-Rab5, -Rab7, or -Rab11A (Vero/hSLAM/EGFP-Rab cells) were generated and infected with IC323-mCherrytagL. Rab5, Rab7, and Rab11A are markers for early endosomes, late endosomes, and REs, respectively (31). In cells infected with IC323-mCherrytagL, the mCherrytagL-containing dots were mostly colocalized with EGFP-Rab11A (Fig. 4A). On the other hand, the signals were poorly associated with the EGFP signals of Rab5 and Rab7 (Fig. 4B and C). These findings suggest that the MV RNP complex is associated with REs. The intracellular transport of the L protein in association with Rab11A was analyzed by time-lapse fluorescence microscopy. The data clearly showed that mCherrytagL was transported along with Rab11A (Fig. 4D; see Movie S5 in the supplemental material). Therefore, our findings suggest that REs function as vehicles for transport of the MV RNP complex.
Fig 4.
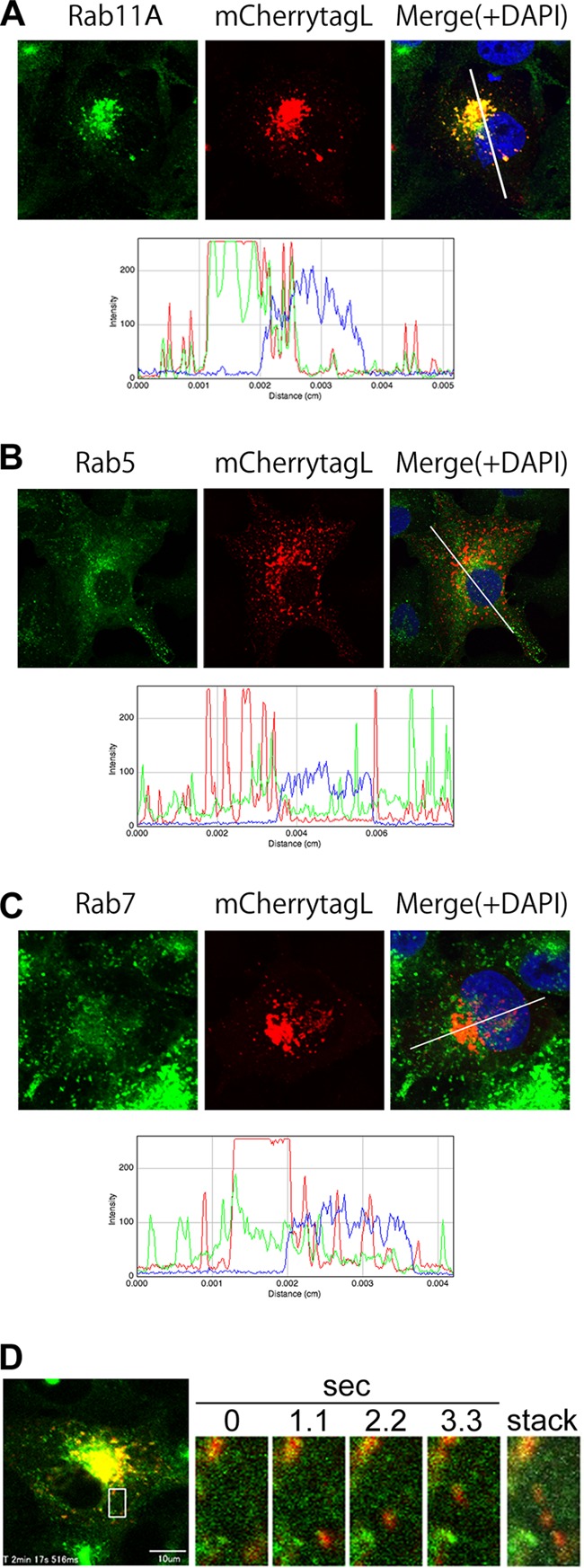
Colocalization and cotransport of mCherry-tagged L protein with EGFP-tagged Rab11A protein. (A–C) Vero/hSLAM cells constitutively expressing EGFP-Rab11A (A), -Rab5 (B), or -Rab7 (C) (Vero/hSLAM/EGFP-Rab cells) were infected with IC323-mCherrytagL. At 2 days p.i., the cells were fixed and analyzed by confocal microscopy. Green and red fluorescence indicates the EGFP-tagged Rab proteins and mCherrytagL protein, respectively. Nuclei were counterstained with DAPI (blue). The lower panels show the RGB line profiles along the selected lines in the merged images (upper right panels). (D) Vero/hSLAM/EGFP-Rab11A cells were infected with IC323-mCherrytagL. At 2 days p.i., the intracellular movements of EGFP-Rab11A and mCherrytagL were analyzed by time-lapse fluorescence microscopy. The left panel shows the overall confocal image of the cells at the start of the time-lapse analysis. Green and red fluorescence indicates the EGFP-Rab11A and mCherrytagL proteins, respectively. The right panels show enlarged time-lapse images of the area indicated by the box in the left panel. A representative series of images taken at 1.1-s intervals and a time-stacked image are shown. The movement of the mCherrytagL dots with EGFP-Rab11A is indicated by the white arrow in the time-stacked image.
Rab11A plays a role for efficient MV release from polarized epithelial cells.
A dominant negative form of Rab11A (Rab11ADN) having a S25N mutation was used to perturb the functions of endogenous Rab11A. The distribution pattern of viral RNP complex in Vero/hSLAM/EGFP-Rab11ADN cells was similar to those in Vero/hSLAM/EGFP and Vero/hSLAM/EGFP-Rab11A cells (Fig. 5A). In subsequent experiments, IC323-AddmCherry encoding a red FLP (mCherry) in the additional independent transcription unit between the H and L genes was used to visualize MV-infected cells. At 2 days p.i., both the cell-associated and cell-free virus titers in Vero/hSLAM/EGFP-Rab11ADN cells were unaffected or were slightly higher than those in other cells (Fig. 5B and C). Vero/hSLAM/EGFP-Rab11ADN cells showed a stronger cytopathic effect (CPE) than the other cells, and at 100 h p.i., the cell-associated virus titer in Vero/hSLAM/EGFP-Rab11ADN cells was markedly decreased (Fig. 5B and D). Knockdown of Rab11A expression also hardly affected MV growth in Vero/hSLAM cells (Fig. 5E to H). These data show that, although the transport of MV RNP complex is mediated by Rab11A-positive REs, it is not critical for MV growth in Vero/hSLAM cells.
Fig 5.

Growth kinetics of the IC323-AddmCherry virus in Rab11ADN-expressing Vero/hSLAM or MDCK cells. (A) Vero/hSLAM cells constitutively expressing EGFP, EGFP-Rab11A, or EGFP-Rab11ADN were infected with IC323, cultured for 1 day, and analyzed by immunofluorescence and confocal microscopy. The N protein was detected by indirect immunofluorescence assay. (B to D) Vero/hSLAM cells constitutively expressing EGFP, EGFP-Rab11A, or EGFP-Rab11ADN were infected with IC323-AddmCherry at an MOI of 0.01. At various time points, the cells and culture medium were harvested separately, and the PFU in both samples were determined. The data for the cell-associated and cell-free titers are plotted in panels B and C, respectively. The data represent the means ± standard deviations of results from triplicate samples. The blue, red, yellow, and green lines indicate the data for Vero/hSLAM, Vero/hSLAM/EGFP, Vero/hSLAM/EGFP-Rab11A, and Vero/hSLAM/EGFP-Rab11ADN, respectively. In panel D, representative CPEs at 48 and 100 h p.i. are shown. Red fluorescence indicates the virus-derived mCherry expression. (E) Vero/hSLAM cells constitutively expressing negative-control shRNA (shNC) or shRNA against Rab11A mRNA (shRab11A) were transfected with pMXsIP-EGFP-Rab11A. At 2 days posttransfection, EGFP-Rab11A expression levels were analyzed by Western blotting. (F) Vero/hSLAM cells constitutively expressing shNC or shRab11A were lysed and endogenous Rab11 expression was analyzed by Western blotting. (G and H) Vero/hSLAM cells constitutively expressing shNC or shRab11A were infected with IC323-AddmCherry at an MOI of 0.01. At various time points, PFU in cells (G) and culture medium (H) were determined. The data represent the means ± standard deviations of results from triplicate samples. The blue, red, and green lines indicate the data for Vero/hSLAM, Vero/hSLAM/shNC, and Vero/hSLAM/shRab11A, respectively. (I to K) MDCK cells constitutively expressing EGFP, EGFP-Rab11A, or EGFP-Rab11ADN were plated on 24-well plates and immediately infected with IC323-AddmCherry at an MOI of 0.2. At various time points, the cells and culture medium were harvested separately, and the PFU in both samples were determined. The data for the cell-associated and cell-free titer are plotted in panels I and J, respectively. The data represent the means ± standard deviations of results from triplicate samples. The blue, red, yellow, and green lines indicate the data for MDCK, MDCK/EGFP, MDCK/EGFP-Rab11A, and MDCK/EGFP-Rab11ADN, respectively. In panel K, representative CPEs at 4 days p.i. are shown. Red fluorescence indicates the virus-derived mCherry expression. (L and M) MDCK cells constitutively expressing EGFP, EGFP-Rab11A, or EGFP-Rab11ADN were seeded on transwell filters with 0.4-μm pores and immediately infected with IC323-AddmCherry at an MOI of 0.2. At various time points, the apical and basolateral media were harvested separately, and the PFU in both samples were determined. The data for the apical and basolateral titers are plotted in panels L and M, respectively. The data represent the means ± standard deviations of results from triplicate samples. The blue, red, yellow, and green lines indicate the data for MDCK, MDCK/EGFP, MDCK/EGFP-Rab11A, and MDCK/EGFP-Rab11ADN, respectively. (N and O) MDCK (N) and Vero/hSLAM (O) cells were infected with IC323-AddmCherry at MOIs of 0.2 and 0.01, respectively. At 4 and 2 days p.i., MDCK cells and Vero cells, respectively, were treated with reagents (5 μM nocodazole [5 μM Noc], 5 μM paclitaxel [5 μM Pac], or dimethyl sulfoxide [DMSO] [as vehicle control]) for 1 day. PFU counts in cells and culture medium were determined. The ratios of cell-free virus titer to cell-associated virus titer are plotted. The data represent the means ± standard deviations of results from triplicate samples. (P and Q) MDCK (P) and Vero/hSLAM (Q) cells, which were transfected with pAcGFP1-Tubulin, were infected with IC323-mCherrytagL at MOIs of 0.2 and 0.1, respectively. At 4 and 2 days p.i., MDCK cells and Vero/hSLAM cells, respectively, were treated with reagents for 1 day. Then, cells were fixed and nuclei were stained with DAPI (blue). Z-stacks of images were acquired by confocal microscopy. Green (Q) and red (P and Q) fluorescence indicates the AcGFP-tagged α-tubulin and mCherrytagL protein, respectively. The Z-projection images of the maximum intensities are shown.
Similar virus growth assays were conducted in polarized epithelial cells, since directional budding of MV was previously reported (6, 32) and the locations and functions of REs differ between nonpolarized and polarized epithelial cells (31). MDCK cells are well-studied polarized epithelial cells. We found that MDCK cells were to some extent susceptible to the IC323 strain infection, as observed from the infection with the laboratory-adapted Edmonston strain (6). Using Vero cells expressing canine and human nectin4 (33, 34), we found that canine nectin4 functions as an MV receptor as well as human nectin4 (data not shown). It was recently reported that MDCK cells express canine nectin4 (35). Thus, MV likely uses this epithelial cell molecule (canine nectin4) to enter MDCK cells. As shown in Fig. 5I and J, the cell-free virus titer of IC323-AddmCherry in MDCK/EGFP-Rab11ADN cells was ∼20-fold lower than those in MDCK/EGFP, MDCK/EGFP-Rab11A, and parental MDCK cells, while the cell-associated virus titer in MDCK/EGFP-Rab11ADN cells was comparable to those in the other cells. The CPE was weak in the MDCK-derived cell lines and similar among the cells (Fig. 5K). These findings suggest that Rab11A is necessary for efficient MV release into the culture medium from MDCK cells. The kinetics of the MV release from these MDCK-derived cell lines was reexamined on transwell filters, since selective MV release from the apical surface was previously described under this culture condition (6). The formation of an electrically tight monolayer was confirmed daily by measuring the transepithelial resistance (36). As expected, MV was released exclusively from the apical side (Fig. 5L and M). In the MDCK/EGFP-Rab11ADN cell culture, the MV titer in the apical culture medium was 10- to 100-fold lower than those in the other three cell cultures (Fig. 5L and M).
We also analyzed effects of MT disrupting (nocodazole) and stabilizing (paclitaxel) drugs on MV release from Vero/hSLAM and MDCK cells. Nocodazole treatment partially affected MV release from MDCK cells but not from Vero/hSLAM cells (Fig. 5N to Q). Although there was only a partial effect, it should be considered that after a 4-day-incubation period, MV-infected MDCK cells were incubated with nocodazole for only 1 day prior to virus harvest, since a long incubation with nocodazole was highly toxic to cells. Paclitaxel treatment showed little, if any, effect on MV release from these cells (Fig. 5N to Q). These data suggest that Rab11A-positive REs specifically function to guide the MV RNP complex to the apical direction (toward the minus end of MTs) of polarized epithelial cells for efficient virus release from the apical membrane. In Vero/hSLAM cells, the MV RNP complex was concentrated at the MTOC (the minus end of MTs). Consistent with these data, the ratio of cell-free virus titers (released virus particles) to cell-associated virus titers in Vero/hSLAM cells was much lower than that in MDCK cells, even in the absence of nocodazole (Fig. 5N and O).
The MV RNP complex accumulates at the apical membrane and the apical recycling compartment (ARC) via functions of Rab11A.
The orientation of MTs and vesicular transport system have been shown to differ between polarized epithelial cells and nonpolarized cells (31, 37). To determine the location of the MV RNP complex in MDCK cells and examine the effects of Rab11ADN, imaging analyses were performed using a confocal laser-scanning microscope. In MDCK/EGFP cells, mCherrytagL was accumulated at or beneath the apical plasma membrane and at a specific intracellular region (Fig. 6A). The data for MDCK/EGFP-Rab11A cells demonstrated that the mCherrytagL located at the apical membrane and the intracellular region was mostly colocalized with EGFP-Rab11A, a marker of the ARC in polarized epithelial cells (31) (Fig. 6B). Therefore, the intracellular region was likely to be the ARC. On the other hand, in MDCK/EGFP-Rab11ADN cells, mCherrytagL was poorly accumulated at the apical membrane, and a region corresponding to the ARC was not detected (Fig. 6C). Instead, mCherrytagL was concentrated in other regions at the bottom of the cytoplasm (Fig. 6C). Similar imaging analyses were performed using a non-FLP-tagged IC323 virus and a MAb against the N protein. For this assay, CM-Dil, a highly lipophilic carbocyanine dye, was used to visualize the overall cell morphology. In MDCK/EGFP-Rab11A cells, the N protein was accumulated beneath the apical membrane and at the ARC and was colocalized with EGFP-Rab11A (Fig. 6D). In MDCK/EGFP-Rab11ADN cells, the N protein was detected throughout the cell and was not concentrated at the apical membrane or the ARC (Fig. 6E). Finally, the colocalization of the N protein with endogenous Rab11 was analyzed in IC323-infected MDCK cells; that is, only unmodified (natural) proteins were evaluated in this experiment. The data demonstrated that the N protein was accumulated beneath the apical plasma membrane and at the ARC and was colocalized with endogenous Rab11 proteins (Fig. 6F). Taken together, these findings indicate that the RNP complex of MV is accumulated at the apical membrane and the ARC in polarized epithelial cells by a mechanism dependent on Rab11A-positive REs.
Fig 6.
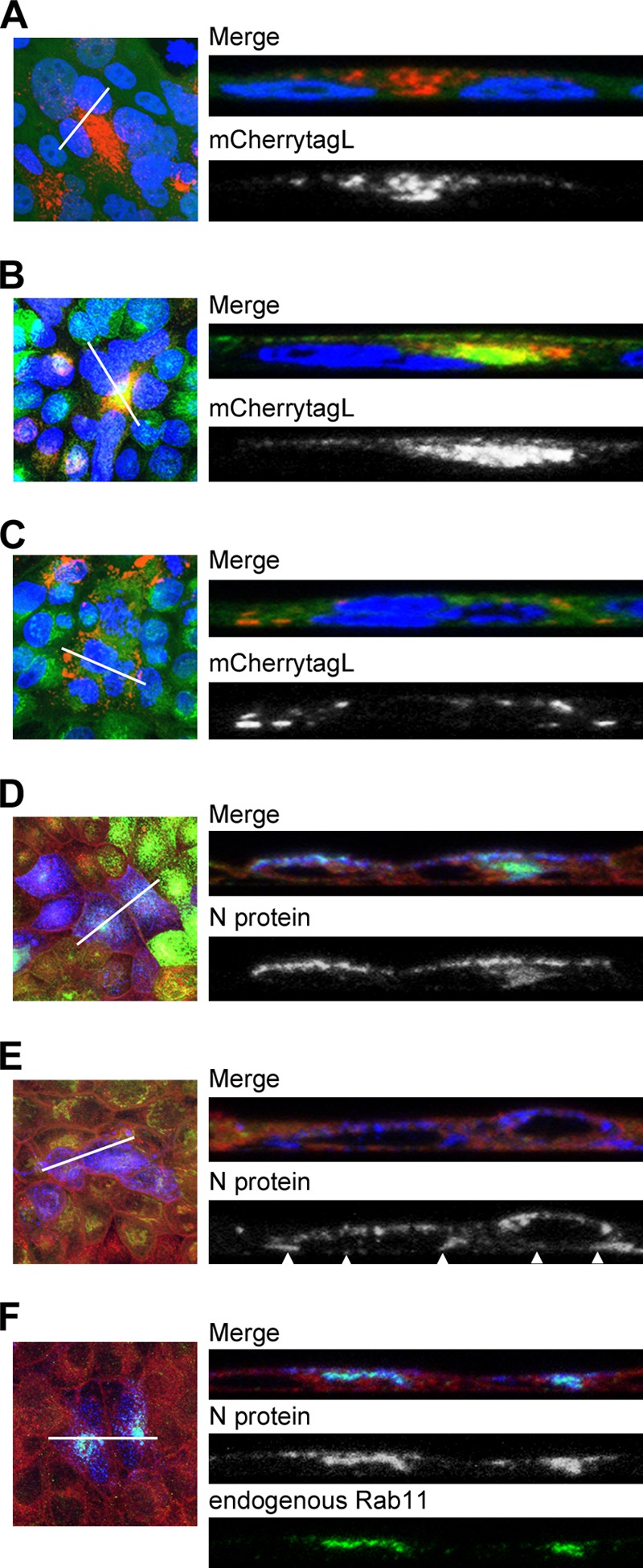
Intracellular localizations of the MV RNP complex and Rab11 in MDCK cells. (A to C) MDCK/EGFP (A), MDCK/EGFP-Rab11A (B), and MDCK/EGFP-Rab11ADN (C) cells were infected with IC323-mCherrytagL at an MOI of 0.2. At 4 days p.i., the cells were fixed and their nuclei were stained with DAPI (blue). Z-stacks of images were acquired by confocal microscopy. Green and red fluorescence in the merged images indicates the EGFP-tagged proteins and mCherrytagL protein, respectively. The left panels show Z-projection images of the maximum intensities, and the right panels show reconstituted images of the XZ plane along the lines in the left panels. (D and E) MDCK/EGFP-Rab11A (D) and MDCK/EGFP-Rab11ADN (E) cells were infected with IC323 at an MOI of 0.2. At 4 days p.i., the cell membrane was stained with CM-Dil (red in the merged images) and the cells were fixed for analyses by immunofluorescence and confocal microscopy. Green fluorescence in the merged images indicates the EGFP-tagged proteins. The N protein was detected with a specific primary antibody and an Alexa Fluor 405-conjugated secondary antibody (blue in the merged images). The left and right panels show Z-projection images and reconstituted images of the XZ plane, respectively, as shown in panels A to C. Arrowheads in panel E indicate the accumulation of the N proteins at the bottom of the cytoplasm. (F) MDCK cells were infected with IC323 at an MOI of 0.2. At 4 days p.i., the cell membrane was stained with CM-Dil (red in the merged images) and the cells were fixed for analyses by immunofluorescence and confocal microscopy. The N protein and endogenous Rab11 were detected with specific primary antibodies and secondary antibodies conjugated with Alexa Fluor 405 (blue in the merged images) and Alexa Fluor 488 (green), respectively. The left and right panels show Z-projection images and reconstituted images of the XZ plane, respectively, as shown in panels A to C.
DISCUSSION
Measles is an airborne disease, and MV is evidently one of the most transmissible pathogens. MV must have mechanisms to shed progeny viruses efficiently. Recent findings for MV infection of polarized epithelial cells and selective budding from the apical membrane can, in part, nicely explain the efficient virus shedding in vivo. However, the intracellular events for trafficking of MV proteins that lead to virus shedding from the apical membrane remain largely unknown.
Much information about the intracellular trafficking of cellular proteins and viral proteins has been obtained using FLP technology (38). Although an rMV expressing the L protein fused with EGFP was generated previously (12, 13), detailed analyses of the trafficking of the MV L protein have not been conducted. The present data obtained using FLP-tagged L proteins clearly demonstrated that, similarly to SeV and IAV (7, 9, 10), the MV RNP complex was transported in a MT-dependent manner and associated with Rab11A-positive REs. MTs have a polar nature, and the minus ends of most MTs are anchored in the MTOC in nonpolarized cells and directed toward the apical membrane in polarized epithelial cells (37, 39). Previous papers showed that the polar nature of MTs is important for the directional transport of some viral proteins (7, 9, 11, 37, 40) and that transport using REs is critical for particle production of IAV at the cell surface (8, 10). It is also noteworthy that respiratory syncytial virus requires an RE-mediated protein sorting system for efficient budding from the apical membrane in polarized epithelial cells (41, 42). Importantly, our data revealed that the functions of Rab11A are critical only for apical release of MV from polarized epithelial cells and not for virus production in nonpolarized Vero/hSLAM cells. Even destruction of the MTs by nocodazole did not significantly affect virus production in Vero/hSLAM cells. Therefore, Rab11A- and RE-dependent RNP transport is not a general requirement for MV replication and particle production but is specifically required for the apical release of MV particles from polarized epithelial cells. The work will raise questions in the field regarding differences in sorting mechanisms between MV, respiratory syncytial virus, SeV, and IAV.
These observations may, in part, be inconsistent with the previous finding by Berghall et al. (43) that nocodazole treatment of Vero cells causes a reduction in MV progeny production. However, nocodazole has a broad-spectrum effect, because the MTs are important for almost all cellular activities, including cell division, intracellular signal transduction, and cell metabolism. Therefore, the effects of nocodazole on MV replication might be affected by subtle differences in the cell conditions. Similarly, the importance of accumulation of the MV RNP complex at the MTOC in Vero/hSLAM cells remains unclear, because nonpolarized epithelial cells are unnatural host cells for MV, and it is also largely unclear whether the accumulation of viral proteins, called viral inclusion, at the perinuclear site close to the MTOC is beneficial to the virus or the host cell (44). The MV RNP was accumulated beneath the apical membrane and at the ARC in polarized epithelial cells, a natural target in vivo.
Our data show that the selective progeny virus release from the apical membrane (5, 6, 32) is at least partly driven by the apical transport of the RNP complex. However, other molecular mechanisms and the contributions of other viral proteins remain to be investigated. The two glycoproteins, H and F, were selectively transported to the basolateral surface in MDCK cells, and the M protein redirected the H and F proteins to the apical surface in MV-infected cells (6, 45). However, the M protein expressed alone using a plasmid was homogeneously distributed throughout the cell and did not accumulate as large clusters at the cell surface in MDCK cells (46). Thus, the roles of the M protein in the apical virus release are still uncertain. It is necessary to address which virus components or cellular proteins are necessary for MV release from the apical membrane. Consistent with our imaging analysis data that the L protein was often associated with the M protein, a recent study showed that the MV M protein mainly functions to coat the RNP complex and not for association with the viral envelope (28). For IAV, the viral glycoproteins and matrix proteins are transported by mechanisms distinct from that of the viral RNP complex (47) and the process of the RNP complex transport is mediated by direct or indirect interactions between the viral polymerase complex (PB1, PB2, and PA complex) and Rab11 (8, 10, 48). The impacts of the M protein and H and F glycoproteins on the transport of the MV RNP complex are now under investigation in our laboratory.
It is of interest to analyze the intracellular transport of MV proteins in different cell types, including lymphocytes and neurons, because they are also in vivo target cells of MV, and MV infection of these cells is associated with immunosuppression and encephalitis. Duprex and colleagues demonstrated cell-to-cell spread of MV in astrocytoma cells, and MV infection caused disruption of the glial-fibrillary-acidic protein filament but not MTs (49, 50). The implementation of these studies in various cell types is expected to elucidate the molecular mechanisms of MV pathogenicity. Interestingly, cells in the central nervous system possess differently orientated MTs and distinct cell polarity (37).
In conclusion, the data obtained in the present study demonstrate that the MV RNP complex is transported in an MT-dependent manner together with Rab11A-containing REs to become accumulated at the apical membrane and the ARC. This RE-mediated RNP transport is dispensable for virus production in nonpolarized cells but critical for virus shedding from the apical membrane of polarized epithelial cells. These data provide evidence for the regulated intracellular trafficking events of the MV RNP complex that drive the viral shedding from polarized epithelial cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank T.A. Sato and N. Ito for providing the MAbs against the N, M, F, and H proteins of MV and the BHK/T7-9 cells, respectively. We also thank M. Shimojima and T. Kitamura for providing PLAT-gp cells, pMXs-IP, and pCVSV-G.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, the Ministry of Health, Labor and Welfare of Japan, and the Takeda Science Foundation.
Footnotes
Published ahead of print 13 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02189-12.
REFERENCES
- 1. Griffin DE. 2007. Measles virus, p 1551–1585 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB, Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J. Clin. Invest. 118:2448–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB, Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7:e1002240 doi:10.1371/journal.ppat.1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blau DM, Compans RW. 1995. Entry and release of measles virus are polarized in epithelial cells. Virology 210:91–99 [DOI] [PubMed] [Google Scholar]
- 6. Maisner A, Klenk H, Herrler G. 1998. Polarized budding of measles virus is not determined by viral surface glycoproteins. J. Virol. 72:5276–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chambers R, Takimoto T. 2010. Trafficking of Sendai virus nucleocapsids is mediated by intracellular vesicles. PLoS One 5:e10994 doi:10.1371/journal.pone.0010994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. 2011. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 85:6117–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Momose F, Kikuchi Y, Komase K, Morikawa Y. 2007. Visualization of microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes Infect. 9:1422–1433 [DOI] [PubMed] [Google Scholar]
- 10. Momose F, Sekimoto T, Ohkura T, Jo S, Kawaguchi A, Nagata K, Morikawa Y. 2011. Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS One 6:e21123 doi:10.1371/journal.pone.0021123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das SC, Nayak D, Zhou Y, Pattnaik AK. 2006. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 80:6368–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duprex WP, Collins FM, Rima BK. 2002. Modulating the function of the measles virus RNA-dependent RNA polymerase by insertion of green fluorescent protein into the open reading frame. J. Virol. 76:7322–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. 2008. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 82:8296–8306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakatsu Y, Takeda M, Iwasaki M, Yanagi Y. 2009. A highly attenuated measles virus vaccine strain encodes a fully functional C protein. J. Virol. 83:11996–12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prasain N, Alexeyev M, Balczon R, Stevens T. 2009. Soluble adenylyl cyclase-dependent microtubule disassembly reveals a novel mechanism of endothelial cell retraction. Am. J. Physiol. Lung Cell. Mol. Physiol. 297:L73–L83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serio G, Margaria V, Jensen S, Oldani A, Bartek J, Bussolino F, Lanzetti L. 2011. Small GTPase Rab5 participates in chromosome congression and regulates localization of the centromere-associated protein CENP-F to kinetochores. Proc. Natl. Acad. Sci. U. S. A. 108:17337–17342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. 2003. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31:1007–1014 [PubMed] [Google Scholar]
- 18. Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75:4399–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto N. 2003. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol. Immunol. 47:613–617 [DOI] [PubMed] [Google Scholar]
- 20. Morita S, Kojima T, Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066 [DOI] [PubMed] [Google Scholar]
- 21. Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J. Virol. 74:6643–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seki F, Yamada K, Nakatsu Y, Okamura K, Yanagi Y, Nakayama T, Komase K, Takeda M. 2011. The SI strain of measles virus derived from a patient with subacute sclerosing panencephalitis possesses typical genome alterations and unique amino acid changes that modulate receptor specificity and reduce membrane fusion activity. J. Virol. 85:11871–11882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakatsu Y, Takeda M, Ohno S, Koga R, Yanagi Y. 2006. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J. Virol. 80:11861–11867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tahara M, Takeda M, Yanagi Y. 2007. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J. Virol. 81:6827–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takeda M, Ohno S, Tahara M, Takeuchi H, Shirogane Y, Ohmura H, Nakamura T, Yanagi Y. 2008. Measles viruses possessing the polymerase protein genes of the Edmonston vaccine strain exhibit attenuated gene expression and growth in cultured cells and SLAM knock-in mice. J. Virol. 82:11979–11984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y. 2009. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 83:10374–10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikegame S, Takeda M, Ohno S, Nakatsu Y, Nakanishi Y, Yanagi Y. 2010. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 84:372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liljeroos L, Huiskonen JT, Ora A, Susi P, Butcher SJ. 2011. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc. Natl. Acad. Sci. U. S. A. 108:18085–18090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mallik R, Gross SP. 2004. Molecular motors: strategies to get along. Curr. Biol. 14:R971–R982 [DOI] [PubMed] [Google Scholar]
- 30. Welte MA. 2004. Bidirectional transport along microtubules. Curr. Biol. 14:R525–R537 [DOI] [PubMed] [Google Scholar]
- 31. Somsel Rodman J, Wandinger-Ness A. 2000. Rab GTPases coordinate endocytosis. J. Cell Sci. 113(Part 2):183–192 [DOI] [PubMed] [Google Scholar]
- 32. Tahara M, Takeda M, Shirogane Y, Hashiguchi T, Ohno S, Yanagi Y. 2008. Measles virus infects both polarized epithelial and immune cells by using distinctive receptor-binding sites on its hemagglutinin. J. Virol. 82:4630–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otsuki N, Sekizuka T, Seki F, Sakai K, Kubota T, Nakatsu Y, Chen S, Fukuhara H, Maenaka K, Yamaguchi R, Kuroda M, Takeda M. 2013. Canine distemper virus with the intact C protein has the potential to replicate in human epithelial cells by using human nectin4 as a receptor. Virology 435:485–492 [DOI] [PubMed] [Google Scholar]
- 34. Pratakpiriya W, Seki F, Otsuki N, Sakai K, Fukuhara H, Katamoto H, Hirai T, Maenaka K, Techangamsuwan S, Lan NT, Takeda M, Yamaguchi R. 2012. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J. Virol. 86:10207–10210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Noyce RS, Delpeut S, Richardson CD. 2013. Dog nectin-4 is an epithelial cell receptor for canine distemper virus that facilitates virus entry and syncytia formation. Virology 436:210–220 [DOI] [PubMed] [Google Scholar]
- 36. Yu AS, Enck AH, Lencer WI, Schneeberger EE. 2003. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J. Biol. Chem. 278:17350–17359 [DOI] [PubMed] [Google Scholar]
- 37. Dohner K, Nagel CH, Sodeik B. 2005. Viral stop-and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol. 13:320–327 [DOI] [PubMed] [Google Scholar]
- 38. Campbell EM, Hope TJ. 2008. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 16:580–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Q, Joshi HC. 1995. Gamma-tubulin is a minus end-specific microtubule binding protein. J. Cell Biol. 131:207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Greber UF, Way M. 2006. A superhighway to virus infection. Cell 124:741–754 [DOI] [PubMed] [Google Scholar]
- 41. Brock SC, Goldenring JR, Crowe JE., Jr 2003. Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 100:15143–15148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE., Jr 2008. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc. Natl. Acad. Sci. U. S. A. 105:10209–10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berghall H, Wallen C, Hyypia T, Vainionpaa R. 2004. Role of cytoskeleton components in measles virus replication. Arch. Virol. 149:891–901 [DOI] [PubMed] [Google Scholar]
- 44. Wileman T. 2007. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu. Rev. Microbiol. 61:149–167 [DOI] [PubMed] [Google Scholar]
- 45. Naim HY, Ehler E, Billeter MA. 2000. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 19:3576–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Riedl P, Moll M, Klenk HD, Maisner A. 2002. Measles virus matrix protein is not cotransported with the viral glycoproteins but requires virus infection for efficient surface targeting. Virus Res. 83:1–12 [DOI] [PubMed] [Google Scholar]
- 47. Jo S, Kawaguchi A, Takizawa N, Morikawa Y, Momose F, Nagata K. 2010. Involvement of vesicular trafficking system in membrane targeting of the progeny influenza virus genome. Microbes Infect. 12:1079–1084 [DOI] [PubMed] [Google Scholar]
- 48. Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 85:4143–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. 1999. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 73:9568–9575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duprex WP, McQuaid S, Rima BK. 2000. Measles virus-induced disruption of the glial-fibrillary-acidic protein cytoskeleton in an astrocytoma cell line (U-251). J. Virol. 74:3874–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.