

Abstract
To begin to investigate the mechanism by which the human adenovirus type 5 E1B 55-kDa protein protects against the antiviral effects of type 1 interferon (IFN) (J. S. Chahal, J. Qi, and S. J. Flint, PLoS Pathog. 8:e1002853, 2012 [doi:10.1371/journal.ppat.1002853]), we examined the effects of precise amino acid substitution in this protein on resistance of viral replication to the cytokine. Only substitution of residues 443 to 448 of E1B for alanine (E1B Sub19) specifically impaired production of progeny virus and resulted in a large defect in viral DNA synthesis in IFN-treated normal human fibroblasts. Untreated or IFN-treated cells infected by this mutant virus (AdEasyE1Sub19) contained much higher steady-state concentrations of IFN-inducible GBP1 and IFIT2 mRNAs than did wild-type-infected cells and of the corresponding newly transcribed pre-mRNAs, isolated exploiting 5′-ethynyluridine labeling and click chemistry. These results indicated that the mutations created by substitution of residues 443 to 448 for alanine (Sub19) impair repression of transcription of IFN-inducible genes, by the E1B, 55-kDa protein, consistent with their location in a segment required for repression of p53-dependent transcription. However, when synthesized alone, the E1B 55-kDa protein inhibited expression of the p53-regulated genes BAX and MDM2 but had no impact whatsoever on induction of IFIT2 and GBP1 expression by IFN. These observations correlate repression of transcription of IFN-inducible genes by the E1B 55-kDa protein with protection against inhibition of viral genome replication and indicate that the E1B 55-kDa protein is not sufficient to establish such transcriptional repression.
INTRODUCTION
The E1B gene of species C human adenoviruses such as adenovirus type 5 (Ad5) encodes major, unrelated proteins of 19 and 55 kDa, each of which can cooperate with viral E1A gene products to transform rodent cells and counter host cell responses detrimental to viral replication (1, 2). The E1B 19-kDa protein is a viral homolog of cellular antiapoptotic proteins such as Bcl2 and blocks induction of apoptosis by the E1A proteins in transformed and infected cells (2, 3). The known protective functions of the E1B 55-kDa protein are fulfilled by a virus-specific E3 ubiquitin (Ub) ligase assembled from the E1B and the viral E4 Orf6 proteins and the cellular proteins cullin5, elongins B and C, and Rbx1 (4, 5), which ubiquitinylates multiple cellular substrates to target them for subsequent proteasomal degradation. These substrates include the cellular tumor suppressor p53 (4–7) and the Mre11, Rad50, and Nbs1 proteins, which comprise the MRN complex (8). As the viral immediate-early E1A 243R protein can induce apoptosis via stabilization of p53 (9–12), removal of the latter protein as a result of the action of the E1B 55-kDa protein-containing E3 Ub ligase is thought to prevent induction of G1 arrest or apoptosis in infected cells (1, 2, 13). The proteins of the MRN complex recognize double-strand breaks in DNA to activate signaling pathways that result in repair by recombination or nonhomologous end joining (NHEJ) (14–17). It is well established that when MRN components are not targeted for degradation by the virus-specific E3 Ub ligase or relocalized by the viral E4 Orf3 protein (8, 18), viral DNA synthesis is impaired in infected cells (19–21). Furthermore, late in the infectious cycle, concatemers of randomly oriented copies of the viral genome are formed in NHEJ-dependent reactions (8, 22, 23). Such concatemerization also requires the cellular enzyme DNA ligase IV (8), another substrate that is targeted for proteasomal degradation by the virus-specific E3 Ub ligase (24). Other cellular proteins marked for degradation by this enzyme include Bloom helicase (25) and integrin α3, which may be removed from infected cells to facilitate release of progeny virus particles (26). The assembly of the E1B 55-kDa protein- and E4 Orf6 protein-containing E3 Ub ligase is also necessary for induction of selective export of viral late mRNAs from the nucleus (27, 28), one of the first functions in the infectious cycle to be ascribed to the E1B 55-kDa protein (29, 30).
In addition to its important functions as a component of the virus-specific Ub ligase, in which it is thought to serve as a substrate recognition subunit (6, 7, 31, 32), the E1B 55-kDa protein exhibits additional, E4 Orf6-independent activities. For example, it is also a Sumol E3 ligase (33, 34) that modifies p53 to induce association of this cellular protein with nuclear Pml bodies and its subsequent export from the nucleus (34). This mechanism of blocking regulation of transcription by p53 is thought to contribute to the ability of the E1B 55-kDa protein to cooperate with viral E1A proteins to transform rodent cells in culture (33, 34), as does a second E4 Orf6 protein-independent activity, inhibition of p53-dependent transcription. Early studies using transient expression assays established that the E1B 55-kDa protein is sufficient to repress expression of p53-dependent reporter genes (35). Mutations that result in impaired interaction of the E1B 55-kDa protein with p53 (36), impaired function of the repression domain (37, 38), or inhibition of sumoylation and nuclear entry of the E1B protein (39) inhibit E1B 55-kDa protein-dependent transformation. Conversely, a greater degree of repression of p53-dependent transcription and more efficient transformation were observed when the intranuclear concentration of the E1B protein was increased by substitutions of critical residues within its nuclear export signal (40).
In principle, inhibition of p53-dependent transcription by the E1B 55-kDa protein could also represent a mechanism to ensure prevention of induction of G1 arrest or apoptosis during the infectious cycle. However, this viral protein is not required for inhibition of expression of classic p53-dependent genes, such as p21 (CDKN1) and MDM2, in infected primary human epithelial cells or established cell lines (41, 42), nor is apoptosis induced when the p53 protein accumulates to high concentrations in cells infected by E1B 55-kDa protein null mutants (42, 43). Furthermore, analysis of alterations in cellular gene expression by microarray hybridization demonstrated that infection of normal human cells with a mutant that cannot direct production of the E1B 55-kDa protein (Hr6) blocked the p53 transcriptional program as effectively as Ad5 infection did, even though over 600 cellular genes were observed to be differentially expressed in Hr6- and Ad5-infected cells (44). Subsequently, the E4 Orf3 protein has been reported to be responsible for blocking the transcriptional activity of p53 in infected cells (45).
The set of genes repressed by the E1B 55-kDa protein following infection of normal human cells is highly enriched for those associated with innate immune responses and antiviral defense (44), notably, 130 genes previously identified to be inducible by alpha or beta interferon (IFN-α or IFN-β; type I interferons here designated IFN) (46). Indeed, replication of Hr6 in normal human fibroblasts or epithelial cells was observed to be several orders of magnitude more sensitive to exposure of cells to IFN than replication of Ad5 (46). The identical phenotype was exhibited by an E1B 55-kDa protein null mutant, AdEasyE1Δ2347, engineered to contain the Hr6 mutation (deletion of bp 2347 [48]), which prevents production of the E1B 55-kDa protein, but none of the other mutations recently identified in the Hr6 genome (49). Furthermore, the concentrations of pre-mRNAs synthesized from several IFN-inducible genes were increased substantially in cells infected by Hr6 and AdEasyE1Δ2347 compared to those in cells infected by the wild-type parental viruses in the absence or presence of exogenous IFN (46). These observations indicate that the E1B 55-kDa protein makes an important contribution to the resistance of adenoviral replication to IFN. The experiments reported here were undertaken to investigate further the mechanism by which this E1B protein blocks the action of IFN, in particular, the relationship of this to the transcriptional repression function of the protein.
MATERIALS AND METHODS
Cells and viruses.
Human 293 and A549 cells were maintained in monolayer culture in Dulbecco's modified Eagle medium (DMEM; Gibco-BRL) containing 5% calf serum (Gibco-BRL) and 5% HyClone bovine growth serum (Thermo-Fisher Scientific). Normal human foreskin fibroblasts (HFFs) were maintained in the same medium supplemented with 7.5% HyClone bovine growth serum.
Isolation of the E1B 55-kDa protein null mutant viruses AdEasyE1Δ2347 and AdEasyE1GΔ2347 has been described previously (47, 48). Mutations designed to result in precise substitutions of specific amino acids with a gain or loss of a restriction endonuclease cleavage site were introduced into the coding sequence of the E1B 55-kDa protein present in the shuttle plasmid pShuttleE1 or pShuttle E1-G by using a QuikChange II site-directed mutagenesis kit (Stratagene-Agilent Technologies) (48). The presence of an expression cassette for enhanced green fluorescent protein (eGFP) upstream of the viral E1A transcription unit in the latter plasmid (47) facilitated subsequent identification of mutant virus plaques. Following initial screening by cleavage of the products of mutagenesis with the appropriate restriction endonuclease, the presence of the desired mutation(s) and the absence of other changes in the E1B gene were confirmed by sequencing (Genewiz). The altered E1 regions were recovered into the viral genome by homologous recombination between the shuttle plasmids and pAdEasy (50) in Escherichia coli, and mutant viruses were isolated from these genomes and validated as described previously (44, 48). The mutants and their wild-type parent, AdEasyE1 or AdEasyE1G, were propagated in 293 cells (51), and the concentrations of infectious particles were determined by plaque assay on these same cells.
Type I IFN (peripheral blood leukocyte interferon source) was diluted in phosphate-buffered saline containing 0.1% (wt/vol) bovine serum albumen (BSA; Sigma-Aldrich). To assess the sensitivity of viral replication to IFN, HFFs were incubated with medium containing 250 units/ml IFN or only the BSA vehicle for 24 h prior to infection with the multiplicities of wild-type or mutant viruses indicated. Following adsorption, incubation was continued in medium containing IFN or only BSA for various periods. Viral yields were determined by using plaque assays in 293 cells.
Isolation of HFFs stably expressing the Ad5 E1B 55-kDa protein-coding sequence.
The segment of the Ad5 genome from bp 2019 to 3601, which encompasses the complete E1B 55-kDa protein-coding sequence, was amplified by PCR using FideliTaq polymerase (Affymetrix) according to the manufacturer's instructions and 0.2 μM forward primer GCAGTTACCGGTGGAGCAGGAGCAATGGAGCGAAGAAAC and reverse primer GCAGTCGAATTCTGCACCTGCACCTGCACCGCGGCTGCTGCTGCAAAA, which carry AgeI and EcoRI restriction endonuclease sites, respectively. This segment was ligated into the multiple-cloning site of the pLJM1-eGFP plasmid (Addgene) (52) after removal of the eGFP-coding sequence via AgeI and EcoRI digestion. The sequence of the E1B 55-kDa insert was verified by sequencing (Genewiz). The parental pLJM1-eGFP plasmid was used in parallel experiments as a control for exposure to lentivirus and transduction. Transducing lentivirus particles were generated by triple transfection of 293FT cells with either the pLJM1-eGFP control or the pLJM1-E1B 55-kDa protein construct with packaging (pCMV-dR8.91) and envelope (vesicular stomatitis virus glycoprotein) vectors (53). HFFs were transduced with these lentivirus preparations, and stable transductants were selected by maintenance in medium containing 0.5 μg/ml puromycin (Sigma-Aldrich).
Immunoblotting.
To examine accumulation of viral proteins, HFFs were infected with AdEasyE1, AdEasyE1G, or the mutants indicated for increasing periods, and cell extracts were prepared as described previously (54). Steady-state concentrations of the viral E1A and E1B 55-kDa proteins and late protein V were examined by immunoblotting with monoclonal antibodies M73 (55), 2A6 (56), and F58#1 (57), respectively, as described previously (54). Cellular β-actin was examined in parallel using a horseradish peroxidase-conjugated monoclonal antibody (Abcam) to provide an internal control. The cellular proteins p53 and Stat1 phosphorylated at Y701 were detected by using the mouse monoclonal antibodies DO1 conjugated to horseradish peroxidase (Santa Cruz) and 5806 (Cell Signaling, Inc.), respectively.
Immunofluorescence.
The localization of the E1B 55-kDa protein in wild-type- and mutant-infected HFFs was examined using mouse monoclonal antibody 2A6 and Alexa Fluor 488 anti-mouse IgG as described previously (54).
Measurement of viral DNA concentrations.
HFFs at 80 to 90% confluence were infected with 3 PFU/cell AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Sub19 for 2 or 72 h. Total cell DNA was isolated as described previously (47), and concentrations of viral DNA were determined by using real-time PCR and SBYR green (Applied Biosystems) detection of a 90-bp amplicon in the major late transcription unit with a cellular β-actin amplicon as an internal control exactly as described previously (46). All samples were assayed in triplicate.
Measurement of relative mRNA concentrations.
HFFs were infected with 200 PFU/cell AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Sub19 for 48 h. Cells were harvested using the TRIzol reagent (Invitrogen), and whole-cell RNA was prepared according to the manufacturer's instructions. Following resuspension in DNase I digestion buffer (Roche), the nucleic acid samples were incubated with 0.4 units/μl DNase I prior to phenol-CHCl3 extraction and ethanol precipitation. RNA samples were resuspended in 10 mM Tris, pH 7.5, containing 5 mM NaCl and 0.5 U/μl RNasin (Promega). RNA concentrations were determined from A260 readings made using a NanoDrop ND-1000 spectrophotometer. cDNA was synthesized from RNA by priming with 200-ng random hexamers (Roche) per 1 μg of RNA and extension with SuperScript II reverse transcriptase (Invitrogen) using the conditions recommended by the manufacturer. Quantitative real-time PCR was carried out using an ABI Prism 7900HT sequence detection system with SYBR green master mix and SDS, version 2.1, software (Applied Biosystems). For IFIT2 mRNA, the PCR primers were forward primer AATTGAGGTGGCAACATAGTTTGA and reverse primer CCCGTCGCTTCTAGCTATGTATC; for GBPl mRNA, they were forward primer GTCAACGGGCCTCGTCTAGA and reverse primer CCCACTGCTGATGGCAATG; for BAX mRNA, they were forward primer TTTCTGACGGCAACTTCAACTG and reverse primer GGTGCACAGGGCCTTGAG; for hMDM2 mRNA, they were forward primer TCCTCTCAAGCTCCGTGTTTG and reverse primer TCATGATGTGGTCAGGGTAGATG; and for the β-actin internal control, they were forward primer TCCTCCTGAGCGCAAGTACTC and reverse primer ACTCGTCATACTCCTGCTT. Relative amplicon concentrations were determined by the standard curve method using a 10-fold dilution series of the IFN-treated, AdEasyE1Δ2347-infected HFF cDNA as the standards for GBP1 and IFIT2 and DNA of a recombinant human cytomegalovirus bacterial artificial chromosome containing the genomic human β-actin sequence (a kind gift of Thomas Shenk) as the standard for β-actin.
Isolation of newly synthesized RNA.
HFFs at 80 to 90% confluence were infected with 50 PFU/cell AdEasyE1 or AdEasyE1Sub19 or mock infected. The medium was replaced with DMEM containing 0.5 mM 5-ethynyluridine (5-EU) (Invitrogen), and incubation at 37°C continued for an additional 40 min. The cells were then harvested, and total RNA was purified as described in the previous paragraph. The RNA newly labeled with 5-EU was biotinylated using click chemistry and isolated by binding to streptavidin attached to magnetic beads using a click-iT nascent RNA capture kit (Invitrogen) following the protocol recommended by the manufacturer. cDNA was synthesized from the newly synthesized RNA populations isolated from equal numbers of cells infected by the different viruses as described above, and IFIT2 and interleukin-6 (IL-6) pre-mRNAs were detected by PCR with the primers spanning exon-intron junctions described previously (47). Viral E1A pre-mRNA was examined using such pre-mRNA-specific PCR primers with the sequences GACCTGTGGCATGTTTGTCTACA (forward primer) and CACCAAACCCACCACTCTATCA (reverse primer), while β-actin mRNA was detected with the primers described in the previous section. All PCR products were examined by electrophoresis in 8% polyacrylamide gels cast and run in 40 mM Tris-acetate, pH 8.3, containing 1 mM EDTA, followed by staining with 2.5 μg/ml ethidium bromide.
RESULTS
Identification of substitutions within the E1B 55-kDa protein that render viral replication sensitive to IFN.
Previous studies have suggested that the Ad5 E1B 55-kDa protein does not comprise discrete structural and functional domains, as the stability, localization, and interactions of the protein are sensitive to even relatively small insertions at multiple sites throughout the coding sequence (54, 58, 59). To identify sequences required for this protein to confer resistance of viral replication to IFN (46), we therefore introduced mutations that result in precise amino acid substitutions. These mutations were designed to alter previously described sequence motifs, residues reported to be required for specific functions of the protein, and/or sequences predicted to be exposed on the surface of the protein (Table 1). Mutations were introduced into the E1B 55-kDa protein-coding sequence in shuttle plasmids that contain both the E1A and E1B genes (47, 48) and recovered in viral genomes prior to isolation, validation, and propagation of mutant viruses as described in Materials and Methods. Four of the mutations were introduced into a viral genome that contains an expression cassette for eGFP upstream of the E1A transcription unit (47). To assess the effects of the mutations on the sensitivity of viral replication to IFN, HFFs were incubated with 250 units/ml IFN or a BSA-only control for 24 h prior to and following infection. Cells were infected in parallel with 3 PFU/cell of the substitution mutants and the corresponding phenotypically wild-type parent and E1B 55-kDa protein null mutant viruses, as summarized in Table 1. Viral yields were determined 72 h after infection, as described in Materials and Methods. The results of the initial screen based on two independent infections with each mutant are summarized in Table 1.
Table 1.
Summary of effects of E1B 55-kDa protein substitution mutations on sensitivity of replication to IFN
| Name | Substitution | Motif/sequence target | Protein stabilityg | IFN sensitivity |
|---|---|---|---|---|
| AdEasyE1Sub1a | C25G, E26A, T27Aa | Surface exposedb | +c | −e |
| AdEasyE1Sub2a | R37G, P38A, P39Aa | Surface exposedb | +c | −e |
| AdEasyE1Sub3a | E67A, S68A, R69A, P70Aa | Surface exposed | +c | −e |
| AdEasyE1GSub4 | L83A, L87A, L91A | Nuclear export signal | +d | −e |
| AdEasyE1GSub6 | T211A, E212A, R214A, V215A | Surface exposed | ND | −e |
| AdEasyE1GSub7 | V239E, R240A, F241A | p53 binding | − | +f |
| AdEasyE1Sub12 | A284S, F285L | RNPI motif | +d | −e |
| AdEasyE1Sub13 | R295A, P296A, K297A, S298A, R299A | RNPI motif | +d | −e |
| AdEasyE1GSub14 | R240A | p53 binding | +d | −e |
| AdEasyE1Sub17 | S490A, S491A | C-terminal phosphorylation sites, repression domain | +/−d | +/− |
| AdEasyE1Sub18 | H373A | Surface-exposedb putative C2-H2 zinc finger | ND | −e |
| AdEasyE1Sub19 | R443A, Y444A, D445A, E446A, T447A, R448A | Surface exposedb; repression domain | ++d | +f |
The mutations that alter the E1B 55-kDa protein-coding sequence did not alter the overlapping coding sequence for the E1B 19-kDa protein.
Predicted from hydropathy plots (54).
Based on comparison of steady-state concentrations in cells transfected with pAdEasyE1 shuttle vectors carrying wild type or mutated E1B genes.
Based on comparison to wild type in infected cells. + and ++, more stable than the wild type; −, decreased stability.
Ratios of yields in the presence and absence of 250 units/ml IFN were within a factor of 2 of wild-type values.
Ratios of yields in the presence and absence of 250 units/ml IFN were ≤10-fold lower than that of wild type.
ND, not determined.
It is well established that replication of wild-type Ad5 is quite refractory to inhibition by IFN (46, 60, 61). The majority of the E1B 55-kDa substitution mutations did not reduce such resistance significantly: the ratios of viral yields in the presence compared to those in the absence of IFN were within a factor of 2 of the values observed for the corresponding wild-type virus (Table 1). However, the replication of three mutants in the presence of IFN was observed to be decreased to a greater degree than that of the wild type (Table 1). Replication of AdEasyE1GSub7 and AdEasyE1Sub17 was 25-fold more sensitive to IFN than that of their wild-type parents, whereas AdEasyE1Sub19 replication was some 10-fold more sensitive. To determine whether any of these defects was a trivial consequence of alterations in protein concentration, the steady-state concentrations of the viral E1B 55-kDa proteins were compared by immunoblotting 24 h after infection with 50 PFU/cell mutant or wild-type virus. Viral E1A proteins were examined in parallel, to provide an internal control. As summarized in Table 1, the substitutions introduced into AdEasyE1GSub7 reduced substantially the steady-state concentration of the E1B 55-kDa protein in infected cells, with no effect on E1A protein accumulation (data not shown). Two of the three substitutions present in this mutant replaced large hydrophobic residues with Ala (Table 1), consistent with the conclusion that the stability of this altered E1B protein was reduced. Regardless, the very small quantities of the E1B 55-kDa protein produced in AdEasyE1GSub7-infected cells account for the sensitivity of the replication of this mutant to IFN. The accumulation of the E1B 55-kDa protein was also observed to be reduced in cells infected for 24 h with AdEasyE1Sub17 compared to cells infected with its wild-type parent, but by 36 h after infection, the concentration of this altered E1B protein increased significantly (Table 1; data not shown). These observations suggested that the increased sensitivity to IFN of replication of this mutant observed in the low-multiplicity-of-infection screen (Table 1) was at least in part the result of delayed accumulation of the E1B 55-kDa protein. Consistent with this view, replication of this mutant was considerably less sensitive to IFN when HFFs were infected at a high multiplicity (50 PFU/cell) (data not shown).
As summarized in Table 1, replication of AdEasyE1Sub19 exhibited an increase in sensitivity compared to that of the wild-type parent of some 10-fold, without a defect in the accumulation of the E1B 55-kDa protein. Indeed, increased steady-state concentrations of both this and the E1A proteins were observed in mutant virus-infected cells (see, e.g., Fig. 2A). As these initial observations indicated that the enhanced sensitivity of replication of this mutant to IFN is not a trivial consequence of failure to synthesize stable E1B protein, we examined the properties of this mutant in more detail. We first compared its sensitivity to IFN to that of the corresponding E1B 55-kDa protein null mutant, AdEasyE1Δ2347 (48), following infection of HFFs at a high multiplicity (50 PFU/cell) for 36 h or at a low multiplicity (3 PFU/cell) for 88 h. The former condition corresponds to a single infectious cycle with the great majority of cells initially infected (62). In contrast, the low-multiplicity protocol allowed spread of infection from the small number of cells initially infected and is therefore likely to be more representative of infection and the impact of host antiviral defenses in vivo. In agreement with our previous studies (46), replication of AdEasyE1Δ2347 was much more sensitive to inhibition by IFN than that of its phenotypically wild-type parent, particularly following low-multiplicity infection (Fig. 1). The 2- to 3-order-of-magnitude decreases in yield of this null mutant observed when cells were not exposed to IFN are also consistent with the requirement for the E1B 55-kDa protein to allow maximally efficient viral DNA synthesis in HFFs (47, 62). In the absence of IFN, no significant impact of the E1B Sub19 substitutions on virus yield was observed (Fig. 1). However, replication of this mutant was more sensitive to IFN-induced inhibition than that of the wild type following infection at either a high or a low multiplicity, although it was less sensitive than replication of AdEasyE1Δ2347 (Fig. 1). For example, following low-multiplicity infection, the ratio of the yields of AdEasyE1Sub19 in the presence and absence of IFN was some 13-fold lower than that of the wild type. We therefore next assessed the effects of the E1B Sub19 substitutions on expression of viral genes and genome replication.
Fig 2.

Viral DNA synthesis is inhibited in AdEasyE1Sub19-infected cells treated with IFN. (A) HFFs were infected with 30 PFU/cell AdEasyE1 (wild type [WT]), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Sub19 (S19) for the periods indicated or mock infected (M). The proteins listed at the right were examined by immunoblotting of total cell lysates as described in Materials and Methods. (B) HFFs were infected with 30 PFU/cell AdEasyE1 (wild type [WT]) or AdEasyE1Sub19 (S19) for 36 h or mock infected (mock), and the E1B 55-kDa protein was visualized by immunofluorescence as described in Materials and Methods. DAPI, 4′,6-diamidino-2-phenylindole. (C) HFFs were treated with IFN or untreated (BSA) and infected with 3 PFU/cell of the virus indicated for 2 or 72 h. Viral DNA concentrations were measured by quantitative PCR, with cellular β-actin DNA used as an internal control, as described in Materials and Methods, and are expressed relative to the input (2 h) concentrations. The values represent the averages of two independent experiments, with cumulative standard deviations indicated by the error bars.
Fig 1.
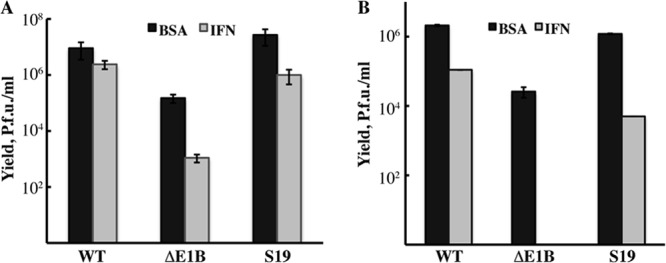
Inhibition of AdEasyE1Sub19 replication by exogenous IFN. HFFs were treated with IFN as described in Materials and Methods or with the vehicle-only control (BSA) and infected with 50 PFU/cell AdEasyE1 (wild type [WT]), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Sub19 (S19) for 36 h (A) or with 3 PFU each virus for 88 h (B). In both cases, virus yields were determined by plaque assay on complementing 293 cells. The results represent those of two independent experiments, and error bars indicate cumulative standard deviations.
The Sub19 substitutions impair viral genome replication only in IFN-treated HFFs.
To examine expression of viral early and late genes, HFFs were infected with 30 PFU/cell AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Sub19 for increasing periods or mock infected, and the accumulation of immediate-early E1A proteins, the early E1B 55-kDa protein, and late protein V was monitored by using immunoblotting, as described in Materials and Methods. As expected (48), the E1B 55-kDa protein could not be detected at any time after infection with AdEasyE1Δ2347, and production of late protein V was severely impaired (Fig. 2A). Higher concentrations of the E1A and E1B 55-kDa proteins were observed in cells at 20 and 40 h after infection with AdEasyE1Sub19 than in cells infected in parallel by the wild type, and higher concentrations of late protein V were observed by 40 h postinfection (p.i.) (Fig. 2A). Quantification of the E1B 55-kDa protein signal at 20 h. p.i., using β-actin as the internal control, indicated that the E1B protein accumulated to a 10-fold higher concentration in AdEasyE1Sub19-infected cells than in wild-type-infected cells. This value was in excellent agreement with the 11-fold increase measured in an independent experiment in which dilutions of extracts of AdEasyE1Sub19-infected cells harvested 24 h after infection were compared to those of AdEasyE1-infected cell extracts, again using β-actin as an internal control (data not shown). We attribute the increased concentrations of all viral proteins examined in AdEasyE1Sub19- compared to those in wild-type-infected cells (Fig. 2A) to underestimation of the titer of the mutant virus, which formed exceptionally small plaques on complementing 293 cells. Regardless, it is clear that the increased sensitivity of replication of AdEasyE1Sub19 to IFN-induced inhibition is not merely an uninteresting consequence of destabilization of the E1B 55-kDa protein. Furthermore, like the protein synthesized in the wild type, the altered E1B 55-kDa protein synthesized in AdEasyE1Sub19-infected cells was concentrated in nuclei (Fig. 2B).
We have reported previously that when the E1B 55-kDa protein is not present during infection of normal human cells, exposure to exogenous IFN induces inhibition of viral DNA synthesis (46). The effect of the Sub19 substitutions on this reaction in the infectious cycle was therefore examined by using quantitative PCR, as described in Materials and Methods. Consistent with our previous observations (46), replication of the E1B 55-kDa protein null mutant AdEasyE1Δ2347 genome was impaired in HFFs that were not treated with IFN (Fig. 2C). However, replication of this mutant genome was inhibited far more severely than that of the wild type when HFFs were exposed to IFN (Fig. 2C), as was previously observed in normal human bronchial/tracheal epithelial cells (NHBECs) (46). In the latter experiments, the majority of cells were infected using a relatively high multiplicity for a period (24 h) insufficient to allow significant release of progeny virus particles. In contrast, HFFs, which are poorly infected by Ad5 (62), were infected with a multiplicity (3 PFU/cell) at which only some 5% of cells became infected for a period longer than the infectious cycle. This protocol, which permits secondary infection and hence amplification of any inhibitory effects of IFN, seems likely to account for the degree of IFN-induced inhibition of viral DNA synthesis observed in both the E1B 55-kDa protein null mutant- and wild-type-infected HFFs (Fig. 2C) greater than that observed previously in NHBECs (47).
No significant difference in the efficiencies of viral DNA synthesis was detected in AdEasyE1Sub19-infected and AdEasyE1-infected cells that were not treated with IFN (Fig. 2C). However, replication of this mutant genome was considerably more sensitive to inhibition in IFN-treated cells, exhibiting a decrease some 300-fold greater than that of the wild type (Fig. 2C). In these experiments, increases in viral DNA concentration in infected cells were calculated relative to the input value measured 2 h after infection, an approach that accounts for any differences in the quantities of viral DNA entering wild-type- and mutant-infected cells. This value was, in fact, some 5-fold higher in cells infected by AdEasyE1Sub19 than those infected by AdEasyE1, consistent with the results of analysis of viral early proteins described previously. Replication of this mutant was also observed to be some 300-fold more sensitive to exogenous IFN than that of AdEasyE1 when cells were infected with equal numbers of genomes, and mutant-infected cells did not accumulate higher concentrations of viral E1A proteins than wild-type-infected cells (data not shown). We therefore conclude that replacement of residues R443 to R448 with Ala in the E1B Sub19 55-kDa protein results in impaired viral genome replication specifically in cells exposed to IFN.
The Sub19 substitutions impair repression of IFN-inducible genes by the E1B 55-kDa protein.
When fused to the DNA-binding domain of the Saccharomyces cerevisiae transcriptional activator Gal4, the E1B 55-kDa protein can act as a strong repressor of transcription from promoters that contain Gal4 binding sites (38). Such repression is severely impaired by the insertion of 4 amino acids at R443 in the E1B protein (38), the first of the 6 charged or hydrophilic residues replaced by Ala by the Sub19 mutations (Table 1). Our previous studies have established that the E1B 55-kDa protein inhibits expression of over 100 IFN-inducible genes and synthesis of pre-mRNA from such genes (44, 46). As it was therefore of considerable interest to investigate whether the Sub19 substitutions had any impact on this function of the E1B protein, we compared expression of IFN-inducible genes in cells infected by the wild-type and mutant viruses.
HFFs exposed to IFN or the vehicle-only control were infected with AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Sub19 for 30 h or mock infected. To allow a quantitative comparison, cDNAs were synthesized from total cell RNA using random priming, and their relative concentrations were measured by quantitative PCR as described in Materials and Methods with primer pairs specific for mRNAs encoded by the IFN-inducible GBP1 and IFIT2 genes or, as an internal control, for β-actin mRNA. Typical results of these experiments are shown in Fig. 3. Exposure of mock-infected cells to IFN induced substantial increases in the concentrations of both GBP1 and IFIT2 mRNAs of some 30- and 100-fold, respectively (Fig. 3). The low levels of IFIT2 mRNA observed in mock-infected cells in the absence of IFN treatment (Fig. 3B) and the greater induction of synthesis of this than of GBP1 mRNA by IFN (Fig. 3A and B) are in excellent agreement with the results of our previous qualitative analyses of the responses of HFFs and NHBECs to IFN (46). The expression of the IFN-inducible genes was not decreased in wild-type- compared to mock-infected cells (Fig. 3), as we observed previously in normal human cells both untreated and exposed to IFN (46). This difference can be attributed to infection until significantly later in the late phase of infection in these experiments (47 h p.i.) compared to previous experiments (24 h p.i.): at such very late times of infection, expression of IFN-inducible genes increases significantly in wild-type-infected cells (D. Miller, B. Rickards, and S. J. Flint, unpublished observations). Nevertheless, consistent with our previous observations summarized above, both these IFN-inducible mRNAs accumulated to higher concentrations in both untreated and IFN-treated cells infected with the E1B 55-kDa protein null mutant than in cells infected by its wild-type parent (Fig. 3). The same pattern was observed in AdEasyE1Sub19-infected cells exposed to IFN (Fig. 3), indicating that substitution of residues 443 to 448 impairs the ability of the E1B 55-kDa protein to block expression of genes in response to IFN. However, synthesis of the E1B Sub19 55-kDa protein in cells that were not treated with IFN led to accumulation of concentrations of GBP1 and IFIT2 mRNAs significantly higher than those observed in null mutant-infected cells (Fig. 3).
Fig 3.
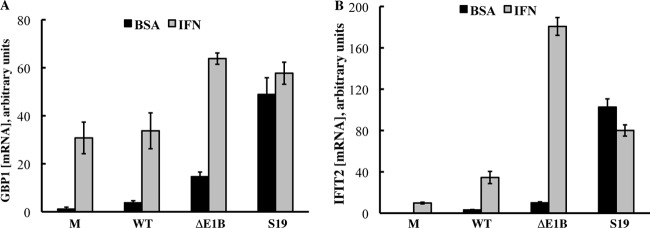
The Sub19 substitutions impair repression of expression of IFN-inducible genes by the E1B 55-kDa protein. HFFs untreated (BSA) or treated with IFN (IFN) were infected with 200 PFU/cell AdEasyE1 (wild type [WT]), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Sub19 (S19), and total cell RNA was isolated 47 h after infection. The concentrations of GBP1 (A) and IFIT2 (B) mRNAs were determined by quantitative PCR, after synthesis of cDNA by reverse transcription from random primers, as described in Materials and Methods. The values shown represent the averages and cumulative standard deviations (bars) of two independent experiments.
This disparity between the phenotypes exhibited by the null and substitution mutants might appear to be paradoxical. However, it can be explained by differences in another important function of the E1B 55-kDa protein during the adenoviral infectious cycle, induction of selective export of viral late mRNAs from the nucleus during the late phase of infection and, hence, maximally efficient expression of viral late genes (29, 30, 58): cells infected by the AdEasyE1Δ2347 mutant lack the E1B 55-kDa protein and, hence, selective regulation of mRNA export, whereas the E1B Sub19 substitutions do not impair this function, as indicated by the absence of defects in viral late gene expression (Fig. 2C). Furthermore, the virus-specific E3 ubiquitin ligase, which is required for selective export of viral late mRNAs (27, 28), assembles efficiently in AdEasyE1Sub19-infected cells (see Fig. 5). The set of mRNAs selectively exported in an E1B 55-kDa protein-dependent manner during the late phase of infection includes not only viral late mRNAs but also mRNAs encoded by cellular genes transcriptionally activated during the late phase of infection (63–65). In HFFs not exposed to IFN, the latter class includes the IFN-inducible genes repressed by the E1B 55-kDa protein (44; J. S. Chahal and S. J. Flint, unpublished observations). As such mRNAs that are not exported selectively do not accumulate in the nucleus (65), their concentration would be expected to be lower when the E1B 55-kDa protein does not induce selective export than when it does, as indeed was observed for the representative IFN-inducible mRNAs in AdEasyE1Δ2347-infected cells compared to AdEasyE1Sub19-infected cells (Fig. 3).
Fig 5.
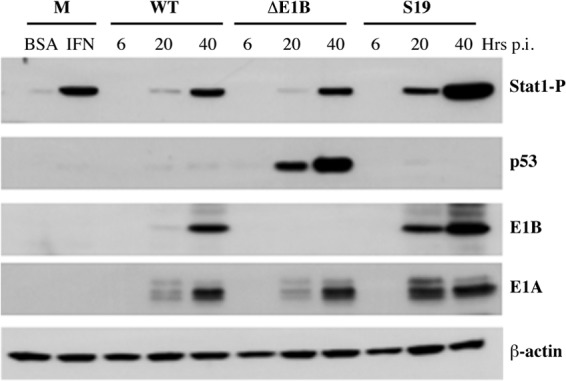
Effects of the E1B 55-kDa Sub19 alterations on activation of Stat1. Mock-infected HFFs were treated with 250 units/ml IFN or vehicle-only control (BSA) for 24 h. Cells were infected with 200 PFU/cell AdEasyE1 (wild type [WT]), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Sub19 (S19) for the periods indicated, and the proteins listed at the right were examined by immunoblotting of whole-cell lysates as described in Materials and Methods. Stat1 phosphorylated on Tyr701 is designated Stat1-P.
We have previously reported that the concentrations of the pre-mRNAs synthesized from several IFN-inducible genes are increased in infected HFFs (and NHBECs) when the E1B 55-kDa protein is not present (46). This observation, in conjunction with the significant stimulation of expression of such genes induced by the E1B Sub19 mutations that alter the previously described transcriptional repression domain, suggested that the E1B 55-kDa protein acts as a repressor of transcription of IFN-inducible genes. To test this inference directly, we exploited pulse-labeling of RNA with 5-ethynyluridine and subsequent click chemistry (66, 67) for attachment of biotin to isolate newly transcribed RNA, as described in Materials and Methods. HFFs infected with 50 PFU/cell AdEasyE1 or AdEasyE1Sub19 for 24 h or mock infected were exposed to the uridine analogue for 40 min, a period sufficient to complete synthesis and processing of pre-mRNAs but short enough to minimize the impact of any differences in turnover. The concentrations of pre-mRNAs as well as of control β-actin mRNA were compared by PCR, following reverse transcription using random priming of newly synthesized RNA isolated from the same number of cells. The concentrations of β-actin mRNA in these populations were similar in mock-, wild-type-, and mutant-infected cells (Fig. 4). Consistent with the levels of viral proteins described previously (Fig. 2A), somewhat higher concentrations of E1A pre-mRNA were observed in cells infected by the mutant virus (Fig. 4). Consistent with previous observations (46), significantly more IFIT2 and IL-6 pre-mRNAs was detected in AdEasyE1Sub19-infected cells than AdEasyE1-infected cells (Fig. 4).
Fig 4.
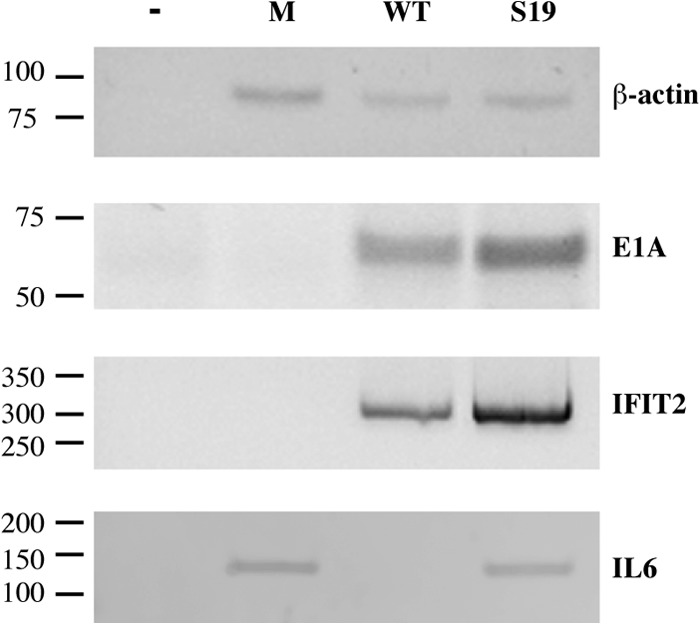
Stimulation of synthesis of primary transcripts of IFN-inducible genes in AdEasyE1Sub19-infected cells. Newly synthesized RNA was isolated from HFFs infected with 50 PFU/cell AdEasyE1 (wild type [WT]) or AdEasyE1Sub19 (S19) for 29 h or mock infected (M), and the concentrations of E1A, IFIT2, and IL-6 pre-mRNAs and β-actin mRNA were examined as described in Materials and Methods. The positions of DNA molecular mass markers (in bp) are indicated at the left.
The E1B 55-kDa protein does not prevent activation of Stat1.
Binding of exogenous IFN to its heterodimeric receptor initiates a signal transduction cascade to result in phosphorylation of Stat1 and Stat2 at specific tyrosine residues, their entry into the nucleus, and formation of the sequence-specific activator of transcription of IFN-inducible genes, Isgf3, by association with Irf9 (68–70). As the E1B 55-kDa protein acts upstream of transcription of such genes, we assessed the impact of this protein on activation of Stat1. HFFs were infected with AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Sub19 for increasing periods, and the accumulation of Stat1 phosphorylated on Tyr701 was examined by immunoblotting. Extracts of mock-infected cells treated with 250 units/ml IFN for 40 h or untreated were examined in parallel to provide a positive control for induction of Stat1 phosphorylation. As described previously, somewhat higher concentrations of the E1A and E1B 55-kDa proteins were observed in cells infected by AdEasyE1Sub19 than in cells infected by its wild-type parent (Fig. 5). In wild-type-infected cells, Tyr701-phosphorylated Stat1 attained a level only slightly lower than that observed in IFN-treated, uninfected cells, but not until between 20 and 40 h after infection (Fig. 5). Neither the absence of the E1B 55-kDa protein nor the presence of higher-than-wild-type concentrations of the Sub19 derivative, which cannot block expression of IFN-inducible genes (Fig. 3 and 4), induced reduced phosphorylation of Stat1 (Fig. 5). In fact, this modification was accelerated in AdEasyE1Sub19-infected cells, in parallel with the alterations in expression of viral immediate-early and early genes (Fig. 5). No differences in the largely nuclear localization of Stat1 phosphorylated on Tyr701 could be discerned in cells infected by AdEasyE1Sub19 and AdEasyE1 (data not shown).
We also wished to investigate the impact, if any, of the E1B Sub19 alterations on the assembly of the Ad E3 Ub ligase and therefore also examined the concentrations of a classic substrate of this enzyme, p53 (see Introduction), by immunoblotting. As expected, p53 accumulated to high concentrations in cells infected by the E1B 55-kDa protein null mutant but was barely detectable in cells infected with either the wild-type virus or AdEasyE1Sub19 (Fig. 5). As different sequences of the E1B 55-kDa protein mediate recognition of different substrates of the Ad E3 Ub ligase (32, 48), it remains possible that replacement of residues 443 to 448 by Ala impairs the effects on one or more other substrates. Nevertheless, this observation indicates that the E1B Sub19 mutations do not impair assembly or the catalytic activity of this enzyme.
The E1B 55-kDa protein is not sufficient to repress expression of IFN-inducible genes.
We have previously reported that mutations that prevent assembly of the virus-specific E3 ubiquitin ligase that contains the E1B 55-kDa and E4 Orf6 proteins do not increase the sensitivity of viral replication to inhibition by exogenous IFN (46). These observations establish that the ability of the E1B 55-kDa protein to block the inhibitory effects of IFN, in contrast to the majority of its functions during the infectious cycle (see Introduction), does not depend on interaction with the E4 Orf6 protein but provide no information about whether other viral proteins might be required. As the E1B 55-kDa protein alone can repress transcription of appropriate reporter genes in in vitro and transient expression assays (37, 71), it was of particular interest to determine whether it was also sufficient to inhibit expression of IFN-inducible genes in normal human cells.
To address this question, derivatives of HFFs that stably express the E1B 55-kDa protein or, as a control, the eGFP-coding sequence under the control of the human cytomegalovirus immediate-early enhancer/promoter were isolated by puromycin selection following transduction with lentiviral vectors, as described in Materials and Methods. Expression of the exogenous genes was then examined by immunoblotting of total cell extracts. The E1B 55-kDa protein was readily detected in HFFs transduced with vector carrying the expression cassette for this protein (HFF-E1B cells), as was eGFP in the control, HFF-GFP cells (Fig. 6A). To determine whether the E1B protein made in HFFs in the absence of other viral proteins was functional, its ability to complement the defect in genome replication in IFN-treated cells infected by the E1B 55-kDa protein null mutant was assessed. HFF-E1B or control HFF-GFP cells were exposed to exogenous IFN or the BSA-only control, as described above, and infected with 3 PFU/cell AdEasyE1 or AdEasyE1Δ2347, and the accumulation of viral DNA at 40 h after infection was measured by quantitative PCR. In HFF-GFP cells infected by the null mutant, synthesis of viral DNA was reduced somewhat in the absence of IFN treatment and was reduced very severely when the cells were exposed to this cytokine (Fig. 6B), exactly as observed in the parental HFFs (Fig. 2B). In contrast, no significant differences in the efficiencies of replication of mutant and wild-type genomes were observed in infected HFF-E1B cells (Fig. 6B). Such efficient complementation of the defects in viral genome replication characteristic of AdEasyE1Δ2347-infected, IFN-treated HFFs established unequivocally that the E1B 55-kDa protein made in HFF-E1B cells is functional and present at sufficient concentrations.
Fig 6.

The E1B 55-kDa protein cannot repress expression of IFN-inducible genes in uninfected cells. HFFs that stably express the sequences encoding the E1B 55-kDa protein (E1B) or eGFP (GFP) were isolated as described in Materials and Methods. (A) The proteins listed at the right were examined by immunoblotting of total cell lysates. (B) Cells were infected with 3 PFU/cell AdEasyE1 (wild type [WT]) or AdEasyE1Δ2347 (ΔE1B), and the concentrations of viral DNA attained by 44 h after infection relative to those at 2 h p.i. were determined by quantitative PCR as described in Materials and Methods. The values shown represent the results of two independent experiments, and bars represent the cumulative standard deviations. (C) Cells were incubated with medium containing 250 units/ml IFN or the vehicle-only control (BSA) for 24 h, and the concentrations of GBP1 mRNA were measured as described in the legend to Fig. 3. The values shown indicate the averages of two independent experiments, and bars represent the cumulative standard deviations. (D) As for panel C, except that the concentrations of IFIT2 mRNA were measured. (E) The concentrations of BAX and MDM2 mRNAs were compared in HFF-GFP and HFF-E1B cells as described for panel C and in Materials and Methods.
We therefore next compared the effect of exogenous IFN on expression of IFN-inducible genes in HFF-E1B and HFF-GFP cells. Cells were incubated with 250 units/ml IFN or the BSA-only control for 8.5 h, and the concentrations of GBP1 and IFIT2 mRNAs relative to the concentration of β-actin mRNA were measured as described in a previous section. In HFF-GFP cells, IFN induced large increases in expression of both the GBP1 and IFIT2 genes, and closely similar increases were observed in HFF-E1B cells (Fig. 6C and D). For example, the concentrations of GBP1 mRNA were increased 11.6- and 14.5-fold following IFN treatment of HFF-GFP and HFF-E1B cells, respectively. We therefore conclude that, when synthesized in the absence of other viral proteins, the E1B 55-kDa protein cannot repress expression of IFN-inducible genes.
In parallel, we compared expression of the p53-regulated BAX and MDM2 genes in HFFs that produce the E1B 55-kDa protein or eGFP. Even though untreated, uninfected HFFs contain low concentrations of p53 (43), the concentrations of BAX and MDM2 mRNAs were some 2-fold lower in HFF-E1B cells than in HFF-GFP cells (Fig. 6E).
Efficient viral DNA synthesis in A549 cells exposed to interferon does not require the E1B 55-kDa protein.
The phenotypes exhibited by AdEasyE1Sub19 correlate with the ability of the E1B 55-kDa protein to protect against IFN-induced inhibition of viral genome replication with repression of expression of IFN-inducible genes. The set of 300 genes repressed by the E1B 55-kDa protein in HFFs (44) contains 130 identified to be IFN inducible, including several associated with induction of apoptosis (46). However, as IFN-induced inhibition of viral replication in the absence of the E1B protein cannot be attributed to programmed cell death (46), the relevant gene(s) remains unknown. To facilitate identification of these genes or their products, we wished to identify lines of established human cells in which replication of the E1B 55-kDa protein null mutant virus is inhibited by exogenous IFN. We therefore examined the effects of IFN treatment on replication of the AdEasyE1, AdEasyE1Δ2347, and AdEasyE1Sub19 genomes in human cell lines commonly used in studies of adenovirus replication and known to be sensitive to IFN, namely, HeLa (72) and A549 (60) cells. In the former, replication of the mutant virus genomes was 70-fold lower than that of the wild type following infection of cells exposed to IFN at a multiplicity of 1 PFU/cell (data not shown).
Treatment of A549 cells with 1,000 units/ml IFN using the protocol described above for HFFs had no significant effect on accumulation of wild-type genomes (Fig. 7). Remarkably, IFN treatment induced only 5- and 2-fold reductions in the quantities of viral DNA molecules synthesized in AdEasyE1Δ2347- and AdEasyE1Sub19-infected A549 cells, respectively (Fig. 7). As these differences are insubstantial in comparison to those of several orders of magnitude resulting from these E1B mutations in HFFs (Fig. 2B), we conclude that the E1B 55-kDa protein is not necessary to prevent IFN-induced inhibition of viral genome replication in A549 cells. These cells are not simply refractory to type I IFN, as exposure to this cytokine activates expression of numerous IFN-inducible genes (73). It seems likely that these tumor-derived cells physically or functionally lack one or more IFN-inducible gene products that inhibit viral genome replication and that are targeted by the E1B 55-kDa protein.
Fig 7.
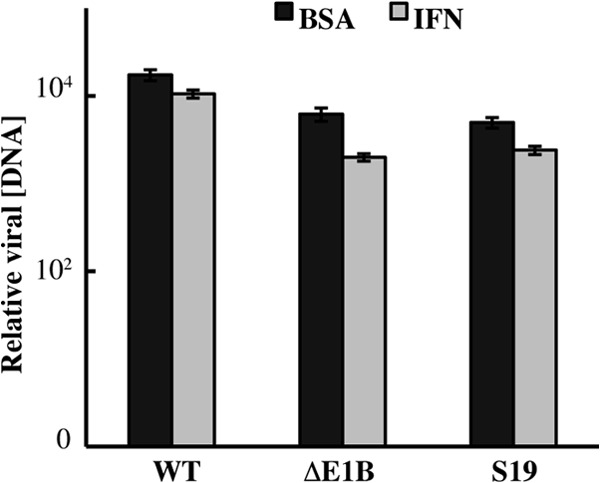
Replication of E1B 55-kDa protein null mutant genomes is not sensitive to IFN in A549 cells. A549 cells were treated with 1,000 units/ml IFN or the vehicle-only control (BSA) for 24 h prior to and during infection with 0.5 PFU/cell AdEasyE1 (wild type [WT]), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Sub19 (S19). Relative viral DNA concentrations at 46 h after infection were determined as described in the legend to Fig. 2. These values represent the average of two independent experiments, and the error bars represent the cumulative standard deviations.
DISCUSSION
Although no structural information about the multifunctional species C adenovirus E1B 55-kDa protein is available, a considerable body of information about sequence motifs, sites of posttranslational modification, and sequences required for interaction with various viral or cellular proteins has been collected (reviewed in reference 58). None of the substitution mutations in the E1B protein-coding sequence that altered such previously characterized features, including the nuclear export signal, RNP motif, and C-terminal sites of phosphorylation, examined in these experiments specifically impaired viral replication in normal human cells exposed to IFN (Table 1). In conjunction with our previous observation that mutations that prevent assembly of the virus-specific E3 ubiquitin ligase in infected cells also fail to render viral replication sensitive to exogenous IFN (46), these data suggest that the E1B 55-kDa protein blocks the antiviral action of IFN by a mechanism that does not depend on previously described properties of the protein. Several Ala substitutions in segments of the E1B 55-kDa protein predicted to be surface exposed and not previously studied, such as the alterations in the N-terminal segment that also encodes the C terminus of the E1B 19-kDa protein in a different reading frame introduced by the AdEasyE1Sub1, AdEasyE1Sub2, and AdEasyE1Sub3 mutations, were also without effect (Table 1). In fact, only the replacement with Ala of residues 443 to 448, a run of 6 charged or hydrophilic residues, in the E1B Sub19 mutations increased the sensitivity of viral replication to IFN (Fig. 1) independently of any reductions in accumulation of the altered protein (Fig. 2A). As mentioned previously, estimation of the impact of the Sub19 mutations on viral yield in IFN-treated cells was complicated by the very small plaque phenotype exhibited by AdEasyE1Sub19 (see Results), which suggests that this altered E1B 55-kDa protein acts as a dominant negative. This interpretation is consistent with both the larger quantities of the E1B Sub19 protein than of the endogenous wild-type protein likely produced in AdEasyE1Sub19-infected 293 cells (Fig. 2A) and the ability of the E1B 55-kDa protein to self-associate when synthesized in the absence of other viral proteins (71, 74). Regardless, measurement of the increase in the concentration of viral genomes over the value determined soon after entry (2 h p.i.), a parameter that is independent of any differences in the numbers of infecting particles or genomes, established that the E1B Sub19 mutations result in a severe defect in viral DNA synthesis specifically in IFN-treated cells (Fig. 2C).
We have previously reported that expression of some 130 IFN-inducible genes is increased significantly when the E1B 55-kDa protein is not present in infected cells, as are the concentrations of several pre-mRNAs synthesized from such genes (44, 46). The large increases in accumulation of IFN-inducible mRNAs in untreated or IFN-treated cells infected with AdEasyE1Sub19 over the levels measured in AdEasyE1-infected cells (Fig. 3) suggested that protection of viral replication and DNA synthesis against IFN-induced inhibition depends on transcriptional repression by the E1B protein: insertion of 4 amino acids at R443, the first residue replaced by Ala in the E1B Sub19 protein (Table 1), impairs both repression of p53-dependent transcription and the ability of an E1B 55-kDa protein–Gal4 DNA-binding domain fusion protein to act as a direct repressor of transcription in transient expression assays (35, 38). The increases in concentrations of newly synthesized, IFN-inducible pre-mRNAs observed in AdEasyE1Sub19-infected cells compared to AdEasyE1-infected cells (Fig. 4) provide direct experimental support for the conclusion that the E1B 55-kDa protein represses transcription of specific genes in infected normal human cells.
Although the mechanism of such repression is not yet known (see below), it can be distinguished in several respects from that proposed for inhibition of transcription of p53-regulated genes (2, 38), which has been implicated in the ability of the E1B 55-kDa protein to cooperate with viral E1A proteins in transformation of rodent cells (see Introduction). Substitutions of the C-terminal sites of phosphorylation of the Ad5 or Ad12 E1B protein reduced its ability to inhibit p53-dependent transcription or to act as a repressor when fused to a heterologous DNA-binding domain (37, 75, 76). However, such mutations neither induced increased expression of E1B 55-kDa protein-repressed genes in infected HFFs (44) nor increased the sensitivity of viral replication (Table 1) or DNA synthesis (data not shown) to IFN. The E1B protein alone is sufficient to repress p53-dependent transcription in cells transiently or stably synthesizing the viral protein (35, 37, 38, 75–78). The E1B 55-kDa protein fully competent to rescue the defects in genome replication of the null mutant AdEasyE1Δ2347 when stably produced in HFFs (Fig. 6B) also inhibited accumulation of mRNAs encoded by the p53-regulated BAX and MDM2 genes (Fig. 6E). In contrast, it had no effect whatsoever on activation of expression of IFN-inducible genes by exogenous IFN (Fig. 6C and D). It therefore appears that the E1B 55-kDa protein can inhibit transcription of cellular genes by multiple mechanisms, depending on whether it is synthesized in the absence of other viral proteins or in the context of infected cells.
The impaired repression of transcription of IFN-inducible genes in cells infected by AdEasyE1Δ2347 or AdEasyE1Sub19 could not be attributed to alterations in the kinetics or degree of activation of Stat1 (Fig. 5) or its nuclear localization (data not shown), suggesting that the E1B 55-kDa protein does not target the signal transduction pathway responsible for activation of transcription of IFN-inducible genes. However, it is not clear whether this protein acts directly to repress transcription of IFN-inducible genes as it can in in vitro or transient expression assays. The proximity of the E1B Sub19 substitutions to the R443 insertion in the previously described repression domain is consistent with such a mechanism, and the well-characterized E1B protein-containing E3 ubiquitin ligase is dispensable for protection against IFN-induced inhibition of viral replication (47). On the other hand, mutations that prevent binding by the only motif in the protein implicated in interaction with nucleic acids, the RNP motif (79), did not reduce the resistance of viral replication to IFN (Table 1), nor did analysis of the clusters of genes differentially expressed in cells infected by Ad5 and an E1B 55-kDa protein null mutant (44) by using FIRE (Finding Informative Regulatory Elements) (80) identify any sequence motif(s) common to or overrepresented among the promoters of E1B-repressed genes (data not shown). Furthermore, the E1B 55-kDa protein has more recently been demonstrated to function as a Sumol E3 ligase (33, 34) and could therefore regulate transcription indirectly via this activity.
The failure of the E1B protein to block activation of expression of IFN-inducible genes in response to exogenous IFN when synthesized in uninfected HFFs (Fig. 6) implies that either one or more additional viral gene products or, perhaps less likely, alterations in the host cell environment induced by infection are also required. Analysis of proteins associated with the E1B 55-kDa protein by mass spectrometry identified the viral IVa2 and L4 100-kDa proteins (4). The E1B-IVa2 protein interaction was confirmed by coimmunoprecipitation (4) but seems unlikely to contribute to inhibition of expression of IFN-inducible genes: although this protein possesses sequence-specific DNA-binding activity, it contributes to activation of transcription from the viral major late promoter (81, 82). Furthermore, only 15 cellular genes were found to carry sequences corresponding to the sequence recognized by the IVa2 protein between positions −1000 and +500, and none were repressed by the E1B 55-kDa protein (44, 83). The small RNA VA-RNAI was the first of several adenovirus-encoded inhibitors of the antiviral effects of IFN to be identified (84). However, this RNA acts downstream of expression of IFN-inducible genes by blocking activation of a specific effector of the IFN response, interferon-induced double-stranded RNA activated protein kinase E1 (E1F2AK2, or Pkr) (85). The viral E1A proteins prevent inhibition of replication of vesicular stomatitis virus by exogenous IFN (60) and inhibit the activation of transcription of IFN-inducible genes (86–88). Such inhibition is the result of suppression of the Jak-Stat signaling pathway that leads to assembly in the nucleus of the crucial activator Isgf3 (89–92) and also sequestration of the coactivator p300 (93). Although the E1A proteins are potent repressors of expression of IFN-inducible genes when synthesized in the absence of any other viral proteins (86–88, 90), they are incapable of maintaining suppression of expression of such cellular genes (Fig. 3), even when produced in large quantities in AdEasyE1Sub19-infected cells (Fig. 2A and 5).
Like the E1B 55-kDa protein (Fig. 2) (46), the viral E4 Orf3 protein prevents inhibition of viral genome replication in cells exposed to exogenous IFN (61). This protective function of the E4 Orf3 protein, which has long been known to reorganize components of Pml nuclear bodies, including the Pml protein, into track-like structures (94–96), becomes dispensable when production of Pml or Daxx is impaired by RNA interference (97). Although the relocalization of Pml and association with the E4 Orf3 protein are indistinguishable in IFN-treated (or untreated) HFFs infected by wild-type and E1B 55-kDa protein null mutant viruses (46), it is possible that the E1B protein functions downstream of E4 Orf3-dependent Pml body disruption. Indeed, IFN-treated cells infected by E1B 55-kDa protein null or E4 Orf3 protein null mutants display a strikingly similar failure in the formation of viral replication centers (46, 61). Furthermore, the E1B protein has been reported to interact with both E4 Orf3 (96) and Daxx (98) and to induce proteasomal degradation of the latter by a mechanism that does not require the Ad E3 ubiquitin ligase (99). The species C human adenovirus E4 orf3 protein also sequesters components of the Mre11-Rad50-Nbs1 (MRN) double-strand break repair complex into the track-like structures (8, 18, 22), a function that is necessary for efficient viral DNA synthesis when these cellular proteins cannot be targeted for proteasomal degradation by the virus-specific E3 ubiquitin ligase (19). Replication of the genome of a double mutant virus null for production of both the E1B 55-kDa and E4 Orf3 proteins proved to be so defective in HFFs that it was not possible to compare the sensitivity of its replication to exogenous IFN to that of the single mutant parents (data not shown). It will therefore be of considerable interest to determine whether the E1A or E4 Orf3 proteins allow repression of IFN-inducible genes when also made in HFFs stably producing the E1B 55-kDa protein.
ACKNOWLEDGMENTS
We thank Ellen Brindle-Clark for assistance with preparation of the manuscript.
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (R56AI1091785), to S.J.F.
Footnotes
Published ahead of print 6 February 2013
REFERENCES
- 1. Berk AJ. 2007. Adenoviridae: the viruses and their replication, p 2355–2394 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Berk AJ. 2005. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24:7673–7685 [DOI] [PubMed] [Google Scholar]
- 3. Cuconati A, White E. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 16:2465–2478 [DOI] [PubMed] [Google Scholar]
- 4. Harada JN, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76:9194–9206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15:3104–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng CY, Blanchette P, Branton PE. 2007. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology 364:36–44 [DOI] [PubMed] [Google Scholar]
- 7. Luo K, Ehrlich E, Xiao Z, Zhang W, Ketner G, Yu XF. 2007. Adenovirus E4orf6 assembles with Cullin5-ElonginB-ElonginC E3 ubiquitin ligase through an HIV/SIV Vif-like BC-box to regulate p53. FASEB J. 21:1742–1750 [DOI] [PubMed] [Google Scholar]
- 8. Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348–352 [DOI] [PubMed] [Google Scholar]
- 9. Debbas M, White E. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 7:546–554 [DOI] [PubMed] [Google Scholar]
- 10. Lowe SW, Ruley HE. 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 7:535–545 [DOI] [PubMed] [Google Scholar]
- 11. Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. 1997. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71:3788–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teodoro JG, Shore GC, Branton PE. 1995. Adenovirus E1A proteins induce apoptosis by both p53-dependent and p53-independent mechanisms. Oncogene 11:467–474 [PubMed] [Google Scholar]
- 13. Levine AJ. 2009. The common mechanisms of transformation by the small DNA tumor viruses: the inactivation of tumor suppressor gene products: p53. Virology 384:285–293 [DOI] [PubMed] [Google Scholar]
- 14. D'Amours D, Jackson SP. 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 3:317–327 [DOI] [PubMed] [Google Scholar]
- 15. Lavin MF. 2007. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26:7749–7758 [DOI] [PubMed] [Google Scholar]
- 16. Stracker TH, Petrini JH. 2011. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12:90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van den Bosch M, Bree RT, Lowndes NF. 2003. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 4:844–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 79:6664–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evans JD, Hearing P. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79:6207–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lakdawala SS, Schwartz RA, Ferenchak K, Carson CT, McSharry BP, Wilkinson GW, Weitzman MD. 2008. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 82:8362–8372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathew SS, Bridge E. 2007. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365:346–355 [DOI] [PubMed] [Google Scholar]
- 22. Evans JD, Hearing P. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77:5295–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weiden MD, Ginsberg HS. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. U. S. A. 91:153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker A, Rohleder KJ, Hanakahi LA, Ketner G. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 81:7034–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orazio NI, Naeger CM, Karlseder J, Weitzman MD. 2011. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 85:1887–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dallaire F, Blanchette P, Groitl P, Dobner T, Branton PE. 2009. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 83:5329–5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blanchette P, Kindsmuller K, Groitl P, Dallaire F, Speiseder T, Branton PE, Dobner T. 2008. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 82:2642–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woo JL, Berk AJ. 2007. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 81:575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pilder S, Moore M, Logan J, Shenk T. 1986. The adenovirus E1B-55kd transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 6:470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Williams J, Karger BD, Ho YS, Castiglia CL, Mann T, Flint SJ. 1986. The adenovirus E1B 495R protein plays a role in regulating the transport and stability of the viral late messages. Cancer Cells 4:275–284 [Google Scholar]
- 31. Blanchette P, Cheng CY, Yan Q, Ketner G, Ornelles DA, Dobner T, Conaway RC, Conaway JW, Branton PE. 2004. Both BC-box motifs of adenovirus protein E4orf6 are required to efficiently assemble an E3 ligase complex that degrades p53. Mol. Cell. Biol. 24:9619–9629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwartz RA, Lakdawala SS, Eshleman HD, Russell MR, Carson CT, Weitzman MD. 2008. Distinct requirements of adenovirus E1b55K protein for degradation of cellular substrates. J. Virol. 82:9043–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muller S, Dobner T. 2008. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 7:754–758 [DOI] [PubMed] [Google Scholar]
- 34. Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. 2010. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 84:12210–12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82–85 [DOI] [PubMed] [Google Scholar]
- 36. Kao CC, Yew PR, Berk AJ. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806–814 [DOI] [PubMed] [Google Scholar]
- 37. Teodoro JG, Branton PE. 1997. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J. Virol. 71:3620–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yew PR, Liu X, Berk AJ. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 8:190–202 [DOI] [PubMed] [Google Scholar]
- 39. Endter C, Kzhyshkowska J, Stauber R, Dobner T. 2001. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc. Natl. Acad. Sci. U. S. A. 98:11312–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Endter C, Hartl B, Spruss T, Hauber J, Dobner T. 2005. Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24:55–64 [DOI] [PubMed] [Google Scholar]
- 41. Hobom U, Dobbelstein M. 2004. E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J. Virol. 78:7685–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, Shen Y, Habets G, Ginzinger D, McCormick F. 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6:611–623 [DOI] [PubMed] [Google Scholar]
- 43. Cardoso FM, Kato SE, Huang W, Flint SJ, Gonzalez RA. 2008. An early function of the adenoviral E1B 55 kDa protein is required for the nuclear relocalization of the cellular p53 protein in adenovirus-infected normal human cells. Virology 378:339–346 [DOI] [PubMed] [Google Scholar]
- 44. Miller DL, Rickards B, Mashiba M, Huang W, Flint SJ. 2009. The adenoviral E1B 55-kilodalton protein controls expression of immune response genes but not p53-dependent transcription. J. Virol. 83:3591–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chahal JS, Qi J, Flint SJ. 2012. The human adenovirus type 5 E1B 55 kDa protein obstructs inhibition of viral replication by type I interferon in normal human cells. PLoS Pathog. 8:e1002853 doi:10.1371/journal.ppat.1002853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chahal JS, Flint SJ. 2012. Timely synthesis of the adenovirus type 5 E1B 55-kilodalton protein is required for efficient genome replication in normal human cells. J. Virol. 86:3064–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kato SE, Huang W, Flint SJ. 2011. Role of the RNA recognition motif of the E1B 55 kDa protein in the adenovirus type 5 infectious cycle. Virology 417:9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kato SE, Chahal JS, Flint SJ. 2012. Reduced infectivity of adenovirus type 5 particles and degradation of entering viral genomes associated with incomplete processing of the preterminal protein. J. Virol. 86:13554–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U. S. A. 95:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–72 [DOI] [PubMed] [Google Scholar]
- 52. Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gonzalez RA, Flint SJ. 2002. Effects of mutations in the adenoviral E1B 55 kDa protein coding sequence on viral late mRNA metabolism. J. Virol. 76:4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harlow E, Franza B, Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 55:533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sarnow P, Sullivan CA, Levine AJ. 1982. A monoclonal antibody detecting the Ad5 E1B-58K tumor antigen in adenovirus-infected and transformed cells. Virology 120:387–394 [DOI] [PubMed] [Google Scholar]
- 57. Lunt R, Vayda ME, Young M, Flint SJ. 1988. Isolation and characterization of monoclonal antibodies against the adenovirus core proteins. Virology 164:275–279 [DOI] [PubMed] [Google Scholar]
- 58. Blackford AN, Grand RJ. 2009. Adenovirus E1B 55-kilodalton protein: multiple roles in viral infection and cell transformation. J. Virol. 83:4000–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yew PR, Kao CC, Berk AJ. 1990. Dissection of functional domains in the adenovirus 2 early 1B 55k polypeptide by suppressor-linker-insertional mutagenesis. Virology 179:795–805 [DOI] [PubMed] [Google Scholar]
- 60. Anderson KP, Fennie EH. 1987. Adenovirus early region 1A modulation of interferon antiviral activity. J. Virol. 61:787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J. Virol. 81:4744–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gonzalez R, Huang W, Finnen R, Bragg C, Flint SJ. 2006. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J. Virol. 80:964–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Flint SJ, Gonzalez RA. 2003. Regulation of mRNA production by the adenoviral E1B 55-kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 272:287–330 [DOI] [PubMed] [Google Scholar]
- 64. Moore M, Schaack J, Baim SR, Morimoto RI, Shenk T. 1987. Induced heat shock mRNAs escape the nucleocytoplasmic transport block in adenovirus-infected HeLa cells. Mol. Cell. Biol. 7:4505–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang U-C, Huang W, Flint SJ. 1996. mRNA export correlates with activation of transcription in human subgroup C adenovirus-infected cells. J. Virol. 70:4071–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jao CY, Salic A. 2008. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U. S. A. 105:15779–15784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tani H, Akimitsu N. 2012. Genome-wide technology for determining RNA stability in mammalian cells: historical perspective and recent advantages based on modified nucleotide labeling. RNA Biol. 9:1233–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 69. Samuel CE. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282:20059–20063 [DOI] [PubMed] [Google Scholar]
- 71. Martin ME, Berk AJ. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72:3146–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Larner AC, Jonak G, Cheng YS, Korant B, Knight E, Darnell JE., Jr 1984. Transcriptional induction of two genes in human cells by beta interferon. Proc. Natl. Acad. Sci. U. S. A. 81:6733–6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanda C, Weitzel P, Tsukahara T, Schaley J, Edenberg HJ, Stephens MA, McClintick JN, Blatt LM, Li L, Brodsky L, Taylor MW. 2006. Differential gene induction by type I and type II interferons and their combination. J. Interferon Cytokine Res. 26:462–472 [DOI] [PubMed] [Google Scholar]
- 74. Morawska-Onyszczuk M, Bienkowska-Szewczyk K, Dobbelstein M. 2010. Self-association of adenovirus type 5 E1B-55 kDa as well as p53 is essential for their mutual interaction. Oncogene 29:1773–1786 [DOI] [PubMed] [Google Scholar]
- 75. Teodoro JG, Halliday T, Whalen SG, Takayesu D, Graham FL, Branton PE. 1994. Phosphorylation at the carboxy terminus of the 55-kilodalton adenovirus type 5 E1B protein regulates transforming activity. J. Virol. 68:776–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhao LY, Santiago A, Liu J, Liao D. 2007. Repression of p53-mediated transcription by adenovirus E1B 55-kDa does not require corepressor mSin3A and histone deacetylases. J. Biol. Chem. 282:7001–7010 [DOI] [PubMed] [Google Scholar]
- 77. Hartl B, Zeller T, Blanchette P, Kremmer E, Dobner T. 2008. Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene 27:3673–3684 [DOI] [PubMed] [Google Scholar]
- 78. Steegenga WT, Shvarts A, Riteco N, Bos JL, Jochemsen AG. 1999. Distinct regulation of p53 and p73 activity by adenovirus E1A, E1B, and E4orf6 proteins. Mol. Cell. Biol. 19:3885–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Horridge JJ, Leppard KN. 1998. RNA-binding activity of the E1B 55-kilodalton protein from human adenovirus type 5. J. Virol. 72:9374–9379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Elemento O, Slonim N, Tavazoie S. 2007. A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell 28:337–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pardo-Mateos A, Young CS. 2004. Adenovirus IVa2 protein plays an important role in transcription from the major late promoter in vivo. Virology 327:50–59 [DOI] [PubMed] [Google Scholar]
- 82. Tribouley C, Lutz P, Staub A, Kedinger C. 1994. The product of the adenovirus intermediate gene IVa2 is a transcription activator of the major late promoter. J. Virol. 68:4450–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Miller DL. 2006. Control of cellular gene expression by adenovirus and myc. Ph.D. thesis Princeton University, Princeton, NJ [Google Scholar]
- 84. Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195–200 [DOI] [PubMed] [Google Scholar]
- 85. Mathews MB, Shenk TE. 1991. Adenovirus virus associated RNA and translation control. J. Virol. 65:5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ackrill AM, Foster GR, Laxton CD, Flavell DM, Stark GR, Kerr IM. 1991. Inhibition of the cellular response to interferons by products of the adenovirus type 5 E1A oncogene. Nucleic Acids Res. 19:4387–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gutch MJ, Reich NC. 1991. Repression of the interferon signal transduction pathway by the adenovirus E1A oncogene. Proc. Natl. Acad. Sci. U. S. A. 88:7913–7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Reich N, Pine R, Levy D, Darnell JE., Jr 1988. Transcription of interferon-stimulated genes is induced by adenovirus particles but is suppressed by E1A gene products. J. Virol. 62:114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Joseph TD, Look DC. 2001. Specific inhibition of interferon signal transduction pathways by adenoviral infection. J. Biol. Chem. 276:47136–47142 [DOI] [PubMed] [Google Scholar]
- 90. Kalvakolanu DV, Bandyopadhyay SK, Harter ML, Sen GC. 1991. Inhibition of interferon-inducible gene expression by adenovirus E1A proteins: block in transcriptional complex formation. Proc. Natl. Acad. Sci. U. S. A. 88:7459–7463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Look DC, Roswit WT, Frick AG, Gris-Alevy Y, Dickhaus DM, Walter MJ, Holtzman MJ. 1998. Direct suppression of Stat1 function during adenoviral infection. Immunity 9:871–880 [DOI] [PubMed] [Google Scholar]
- 92. Shi L, Ramaswamy M, Manzel LJ, Look DC. 2007. Inhibition of Jak1-dependent signal transduction in airway epithelial cells infected with adenovirus. Am. J. Respir. Cell Mol. Biol. 37:720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Routes JM, Li H, Bayley ST, Ryan S, Klemm DJ. 1996. Inhibition of IFN-stimulated gene expression and IFN induction of cytolytic resistance to natural killer cell lysis correlate with E1A-p300 binding. J. Immunol. 156:1055–1061 [PubMed] [Google Scholar]
- 94. Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjärvi G, Carmo-Fonseca M, Dejean A. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10:196–207 [DOI] [PubMed] [Google Scholar]
- 96. Leppard KN, Everett RD. 1999. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 80(Pt 4):997–1008 [DOI] [PubMed] [Google Scholar]
- 97. Ullman AJ, Hearing P. 2008. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 82:7325–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao LY, Colosimo AL, Liu Y, Wan Y, Liao D. 2003. Adenovirus E1B 55-kilodalton oncoprotein binds to Daxx and eliminates enhancement of p53-dependent transcription by Daxx. J. Virol. 77:11809–11821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schreiner S, Wimmer P, Sirma H, Everett RD, Blanchette P, Groitl P, Dobner T. 2010. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 84:7029–7038 [DOI] [PMC free article] [PubMed] [Google Scholar]