

Abstract
Proteasomes generally degrade substrates tagged with polyubiquitin chains. In rare cases, however, proteasomes can degrade proteins without prior ubiquitination. For example, the human cytomegalovirus (HCMV) pp71 protein induces the proteasome-dependent, ubiquitin-independent degradation of the retinoblastoma (Rb) and Daxx proteins. These transcriptional corepressors and tumor suppressors inhibit the expression of cellular or viral genes that are required for efficient viral replication. Proteasomes are composed of a 20S catalytic core with or without one or two activator complexes, of which there are four different types. Here, we show that only one of these activators, the 19S regulatory particle that normally participates in ubiquitin-dependent protein degradation, is required for pp71-mediated degradation of Rb and Daxx. We report the unique use of a well-established route of substrate delivery to the proteasome by a viral protein to promote infection.
INTRODUCTION
The proteasome is responsible for the bulk of protein turnover within cells. Most substrates arrive at the proteasome polyubiquitinated, where the ubiquitin chains mediate proteasome binding but are then removed prior to substrate degradation by the proteasomal 20S catalytic core. The core particle can associate with activator complexes that modulate proteasome function (1). For example, the 19S regulatory particle (RP) associates with one or both ends of the 20S core to form the 26S proteasome species responsible for ubiquitin-mediated degradation events. Proteasomal activity is critical for cellular homeostasis, cell cycle progression, transcription, DNA repair, and dichotomously, both the success of viral infections and essential defenses against viral pathogens (2–6).
Proteasomal degradation of viral antigens to generate peptides displayed by major histocompatibility group (MHC) molecules is a well-documented part of adaptive immunity (4). Less well appreciated is the number of cellular intrinsic defense proteins targeted for proteasomal degradation by viral factors (7, 8). For example, human cytomegalovirus (HCMV) infections, which cause severe disease in immunocompromised, -suppressed, or -naive individuals, induce the degradation of several cellular transcriptional corepressors to create an environment conducive to productive, lytic infection. Within the tegument layer of its virion, HCMV packages the viral pp71 protein, which is introduced into cells immediately upon infection, traffics to the nucleus, and induces the degradation of BclAF1, Daxx, and the retinoblastoma (Rb) family members Rb, p107, and p130 (9–11). BclAF1 and Daxx degradation promotes viral immediate early (IE) gene expression. Rb family inactivation, which also occurs through phosphorylation by the virally encoded kinase UL97 (12), likely increases the efficiency of viral DNA replication. These pp71-dependent degradation events are prevented by pharmacologic inhibition of the 20S catalytic core, indicating that they are proteasomal processes. Other experimental evidence, however, indicates that these proteasomal degradation events occur without the usual requirement for substrate polyubiquitination (13, 14).
Ubiquitin-independent protein degradation has recently been associated with the PA28γ proteasomal activator (15–17). To define the proteasomal requirements for pp71-mediated protein degradation, we surveyed all known proteasome activators for potential roles during pp71-mediated Daxx degradation. Interestingly, we found that only the 19S RP was required for the pp71-mediated degradation of both Daxx and Rb. Our results place the 19S RP on a novel virally directed route of nonubiquitinated proteins to the proteasome for degradation that may represent a potential point for therapeutic inhibition of HCMV infection.
MATERIALS AND METHODS
Cells, viruses, and assays.
Human foreskin fibroblasts (HFs), mouse embryonic fibroblasts (MEFS) (kind gifts from Martin Rechsteiner and Lance Barton), and mouse B cells (kind gifts from Barry Sleckman) were cultured as previously described (11, 18–20). Virus strains AD169 and AdsubUL82 (pp71 null) were propagated, UV inactivated, and used to infect cells as previously described (11, 21). Infections with recombinant adenoviruses were performed as previously described (9). Proteins visualized on film were quantified with ImageQuant 5.2 software. Proteins visualized with the LI-COR Odyssey Fc imaging system and IRDyes (926-68170 and 827-08365) were quantified with LI-COR Image Studio software. In both cases, bands were normalized to those of loading controls and are reported as percentages of the results for their respective mock-infected samples. Statistical analyses utilized two-tailed paired t tests.
Inhibitors and antibodies.
Leptomycin B (40 nM) (Calbiochem) was added 2 h prior to infection. Lactacystin (20 μM) (Calbiochem) and Gö6976 (250 nM) (Calbiochem) were added at the time of infection. Primary antibodies are listed in Table S1 in the supplemental material (9, 22). Secondary antibodies conjugated with horseradish peroxidase were purchased from Chemicon (anti-mouse and -rabbit antibodies) or Santa Cruz (anti-goat antibody); those conjugated with Alexa Fluor 488 were from Molecular Probes. Immunoblots, immunofluorescence, and immunoprecipitations were completed as previously described (11, 23).
Nuclear and cytoplasmic fractionation.
Cells were resuspended in a hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 1.0 mM dithiothreitol [DTT], 0.2 mM phenylmethylsulfonyl fluoride [PMSF], and protease inhibitors) and then lysed with 0.25% NP-40. Nuclear (pellet) and cytoplasmic (supernatant) fractions were separated by centrifugation. Cytoplasmic fractions were subjected to five freeze-thaw cycles and a lysate-clearing centrifugation. Nuclear fractions were resuspended in extraction buffer (20 mM HEPES [pH 7.9], 0.45 M NaCl, 1.5 mM MgCl2, 10 mM DTT, 0.2 mM PMSF, and 0.2 mM EDTA) and then treated as described above for the cytoplasmic fractions.
RNA interference.
Reagents for RNA interference were purchased from Dharmacon. Sequences are listed in Table S1 in the supplemental material. For transient knockdowns, an equal number of HFs were transfected with 40 pmol (ubiquitin), 80 pmol (Rpn1 and PA28 isoforms) or 160 pmol (Rpt2 and Rpn11) small interfering RNA (siRNA)/106 cells using Lonza nucleofection kits (VPI-1002; Lonza) following the manufacturer's protocol and equally distributed among culture dishes. (Scrambled control siRNAs were added at concentrations that mimicked ubiquitin or proteasome subunit siRNA conditions in each experiment.) Fresh medium was added 24 h after nucleofection, and infections occurred 48 or 72 h posttransfection at the multiplicities of infection (MOIs) (calculated using scrambled control cell numbers) indicated in the figure legends. Stable PA200 knockdown 293 cells were generated by retroviral transduction as previously described (24) and transfected with a previously described mutant of pp71 (25).
RESULTS
Cytoplasmic proteasome activators are dispensable for the pp71-mediated ubiquitin-independent degradation of Daxx.
pp71 degrades Daxx (Fig. 1A and B) even when ubiquitin levels are decreased by RNA interference to a sufficient level to stabilize p53 (Fig. 1A), a well-documented ubiquitin-dependent proteasomal substrate. Furthermore, pp71 degrades Daxx when cells are treated with leptomycin B (Fig. 1C), which inhibits the CRM1-mediated nuclear export of proteins like NF-κB (Fig. 1D). From this and previous data (13, 14), we conclude that pp71 degrades Daxx in a ubiquitin-independent manner within the nucleus, suggesting that exclusively cytoplasmic proteasome activators would not participate in this process. The known mammalian proteasome activators include the 19S RP (ubiquitin-dependent proteolysis), PA28αβ (the immunoproteasome), PA28γ (ubiquitin-independent proteolysis), and PA200 (spermatogenesis and DNA repair) (reviewed in reference 1). PA200 and PA28β are exclusively cytoplasmic in primary human fibroblast cells fully permissive for HCMV infection, whereas the other activators, as well as 20S cores, are found in both the nucleus and the cytoplasm (Fig. 2A). These localizations generally agree with those described in the published literature, where all subunits have been found in both the cytoplasm and the nucleus (26–30). The only real differences we found include the apparently exclusively cytoplasmic localization of PA28β and PA200 in our experiments; however, longer exposures might have revealed some fraction of these proteins within the nucleus. Knockdown of PA28α (Fig. 2B), PA28β (Fig. 2B), or PA200 (Fig. 2C) failed to inhibit Daxx degradation by pp71, whereas treatment with lactacystin, a specific inhibitor of the 20S catalytic core, stabilized Daxx. These data indicate that the immunoproteasome (PA28αβ) and PA200 are not required for pp71-mediated Daxx proteasomal degradation. Note that PA200 knockdown experiments were performed in nonpermissive 293 cells, as we were unable to achieve detectable knockdown of this proteasome activator in fibroblasts.
Fig 1.
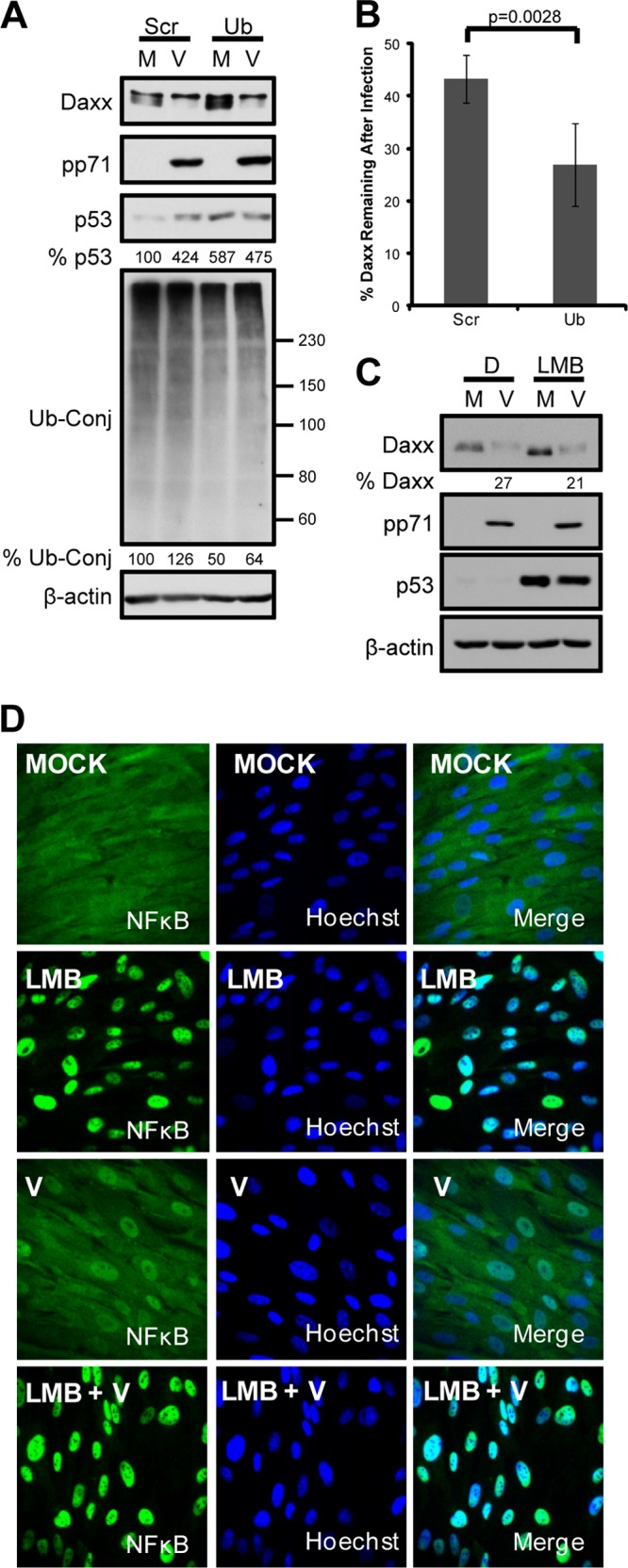
pp71-mediated Daxx degradation is both nuclear and ubiquitin independent. (A) Human fibroblasts (HFs) were transfected with scrambled (Scr) or ubiquitin-specific (Ub) siRNAs for 96 h and then mock infected (M) or infected with HCMV at an MOI of 1 (V) for 8 h. Seventy-two hours prior to infection, the cells were incubated in medium containing 0.1% serum. Cell lysates were harvested and analyzed with immunoblotting. p53 and ubiquitin conjugate bands between 60 and 150 kDa were quantified with ImageQuant, normalized to the results for β-actin, and are presented as a percentage of the mock-infected, scramble control. (B) HFs transfected as described above for 72 h were infected as described above for 6 h and then analyzed by immunoblot using the LI-COR Odyssey Fc. Daxx bands normalized to the results for β-actin and converted to percentages of the results for mock-infected samples were averaged from four independent biological replicates and are presented with standard deviations. Statistical analysis indicates significant difference between the values. (C) HFs were pretreated with leptomycin B (LMB) or dimethyl sulfoxide (DMSO) (D) for 2 h and then mock infected (M) or infected (V) with UV-inactivated HCMV at an MOI of 3 for 6 h. Lysates analyzed with immunoblotting were quantified with ImageQuant 5.2 software. p53 stabilization illustrates successful inhibition of CRM1, as nuclear export is needed for degradation. (D) Cells from the experiment whose results are shown in panel C were processed for the indirect immunofluorescence staining of NF-κB (green) in order to confirm effective inhibition of CRM1-mediated nuclear export. Nuclei were counterstained with Hoechst (blue).
Fig 2.

Cytoplasmic proteasome activators and PA28γ are dispensable for pp71-mediated Daxx degradation. (A) HF lysates were separated into nuclear (N) and cytoplasmic (C) fractions and analyzed with immunoblotting. Tubulin and lamin A/C were used to determine the effectiveness of fractionation. WT and PA200-null mouse B cells were used to define the PA200-specific band, denoted with an asterisk. (B) HFs were transfected with siRNAs targeting the indicated PA28 isoforms or a scrambled control (Scr). Forty-eight hours later, they were mock infected (M) or infected (V) with UV-inactivated virus at an MOI of 3. Cells were treated with lactacystin (L) or DMSO (D) at the start of viral infection. Lysates harvested 6 h postinfection were analyzed by immunoblotting and quantified with ImageQuant. (C) Stable PA200 knockdown (sh#1, sh#2) or parental control (shC) cells were transfected with constructs expressing hemagglutinin (HA)-tagged WT pp71 (71), an HA-tagged mutant of pp71 (D) unable to bind to Daxx (25), or an empty vector control (E) for 24 h. Lysates were analyzed with immunoblotting, and bands were quantified with ImageQuant. (D) Subconfluent wild-type (WT), PA28γ-null (KO [knockout]), or PA28αβγ-null mouse embryo fibroblasts (MEFs) were mock infected (M) or infected with wild-type (V) or a pp71-null virus (Δ71) at an MOI of 5. Lysates collected 9 h postinfection were analyzed with immunoblotting and quantified with ImageQuant. pp65 levels were examined to illustrate equal entry between samples infected with wild-type and pp71-null viruses. (E) Lysates from wild-type (WT) and PA28γ-null MEFs (KO) were analyzed with immunoblotting. Tubulin was used as a loading control. (F) Lysates from wild-type (WT) and PA28αβγ-null MEFs (KO) were analyzed with immunoblotting. (G) Subconfluent wild-type (WT) and PA28αβγ-null (KO) MEFs were cultured in 0.1% serum-containing medium for 72 h and then mock transduced (M) or transduced with 30,000 particles per cell of recombinant adenoviruses that express either nothing (R) or the HCMV pp71 (71) or UL69 (69) protein. Lysates harvested 24 h postransduction were analyzed with immunoblotting. Expression of hemagglutinin (HA)-tagged pp71 and UL69 was monitored with antibodies against HA.
PA28γ is dispensable for pp71-mediated Daxx degradation.
While PA28γ mediates the ubiquitin-independent degradation of both cellular and viral targets (15–17, 31), knockdown of PA28γ failed to inhibit pp71-mediated degradation of Daxx, which was prevented by lactacystin (Fig. 2B). Likewise, infection with wild-type but not pp71-null HCMV induced Daxx degradation in embryonic fibroblasts derived from PA28γ (Fig. 2D and E) and PA28αβγ (Fig. 2D and F) knockout mice (PA200 knockout MEFs are not available and, thus, were not examined). In addition, PA28αβγ knockout MEFs supported pp71-mediated Daxx degradation after transduction with a pp71-expressing recombinant adenovirus (Fig. 2G) but not one expressing the HCMV UL69 tegument protein. Thus, the proteasome activator previously implicated in ubiquitin-independent degradation, PA28γ, is dispensable for the pp71- and proteasome-mediated degradation of Daxx.
The 19S RP is required for pp71 to degrade Daxx and Rb.
The remaining proteasome activator, the 19S RP, is required for ubiquitin-dependent proteolysis and can be biochemically divided into base and lid subcomplexes (32). The base consists of six ATPases that associate with the 20S core (Rpt1 to -6), two non-ATPase subunits (Rpn1 and Rpn2), and two ubiquitin receptors (Rpn10 and Rpn13), while the lid is comprised of eight subunits (Rpn3, -5, -6, -7, -8, -9, -11, and -12) (33–35). Depletion of certain 19S RP subunits, like Rpn1, can inhibit 19S RP function and stabilize substrates that are degraded by the 26S proteasome (15, 36). Daxx degradation during HCMV infection was inhibited in Rpn1-depleted cells where p53, a 26S proteasome substrate, was stabilized (Fig. 3A). Daxx was also stabilized by lactacystin treatment or depletion of the β5 chymotrypsinlike subunit of the 20S core. Importantly, under conditions where 19S RP-dependent substrates like p53 are stabilized in Rpn1-depleted cells, p21, a substrate that is constitutively degraded through a 19S RP-independent, 20S-dependent process (15), is still degraded (Fig. 3B), indicating that 19S RP subunit depletion does not affect all proteasomal degradation. At least three individual subunits of the 19S RP with independent functions (Rpn1, a structural scaffold [37], Rpt2, an ATPase [38], and Rpn11, a deubiquitinase [39]) are each required for pp71-mediated Daxx degradation (Fig. 3C, D, E, and F). Inhibition of 19S RP function by Rpn1 depletion or inhibition of 20S function by lactacystin does not impair the ability of pp71 to enter the nucleus (Fig. 4A and B) or associate with Daxx (Fig. 5). The punctate appearance of pp71 in cells either depleted of Rpn1 or treated with lactacystin likely results from its previously described localization to promyelocytic leukemia nuclear bodies in the absence of Daxx degradation, which, as shown above, is inhibited under these conditions. Thus, we conclude that the 26S proteasome is responsible for pp71-mediated Daxx degradation.
Fig 3.

The 19S RP is required for ubiquitin-independent, pp71-mediated Daxx degradation. (A) HFs were transfected with two independent siRNAs targeting the 19S Rpn1 subunit (R1.1 or R1.2), one targeting the 20S β5 subunit (β5), or scrambled control (Scr) for 72 h. Controls were treated with lactacystin (L) or DMSO (D) at the start of mock (M) or HCMV (V) infections at an MOI of 1. Lysates collected 8 h later were analyzed with immunoblotting and quantified with ImageQuant. Levels of p53 were examined to demonstrate successful inhibition of proteasome function. (B) HFs were transfected with a scrambled control (Scr) or Rpn1-specific siRNA (R1.2) for 72 h. Cells were incubated in the presence or absence of cycloheximide (50 μg/ml) (CHX) for 4 h. Lysates were collected and analyzed with immunoblotting using the LI-COR Odyssey Fc imaging system. p53 and p21 bands were quantified with LI-COR Image Studio software, normalized to the levels of β-actin, and reported as percentages of the results for mock-treated samples. (C) HFs were transfected with siRNAs for 72 h and infected with UV-inactivated virus at an MOI of 1. Lysates collected 6 h postinfection were analyzed with immunoblotting. Daxx bands were quantified with ImageQuant 5.2 software, normalized to the levels of β-actin, and are reported as percentages of the results for mock-infected samples. (D) Data from four biological replicates for which representative results are shown in panel C were averaged and are presented with standard deviations. Asterisks indicate a significant difference (P < 0.05) between the compared values. (E) Cell lysates from the experiment for which representative results are shown in panel C were also analyzed with the LI-COR Odyssey Fc imaging system. Daxx bands were quantified with LI-COR Image Studio software, normalized to the results for β-actin, and reported as percentages of the results for mock-infected samples. (F) Data from three biological replicates for which representative results are shown in panel E were averaged and are presented with standard deviations. Asterisks indicate a significant difference (P < 0.05) between the compared values.
Fig 4.
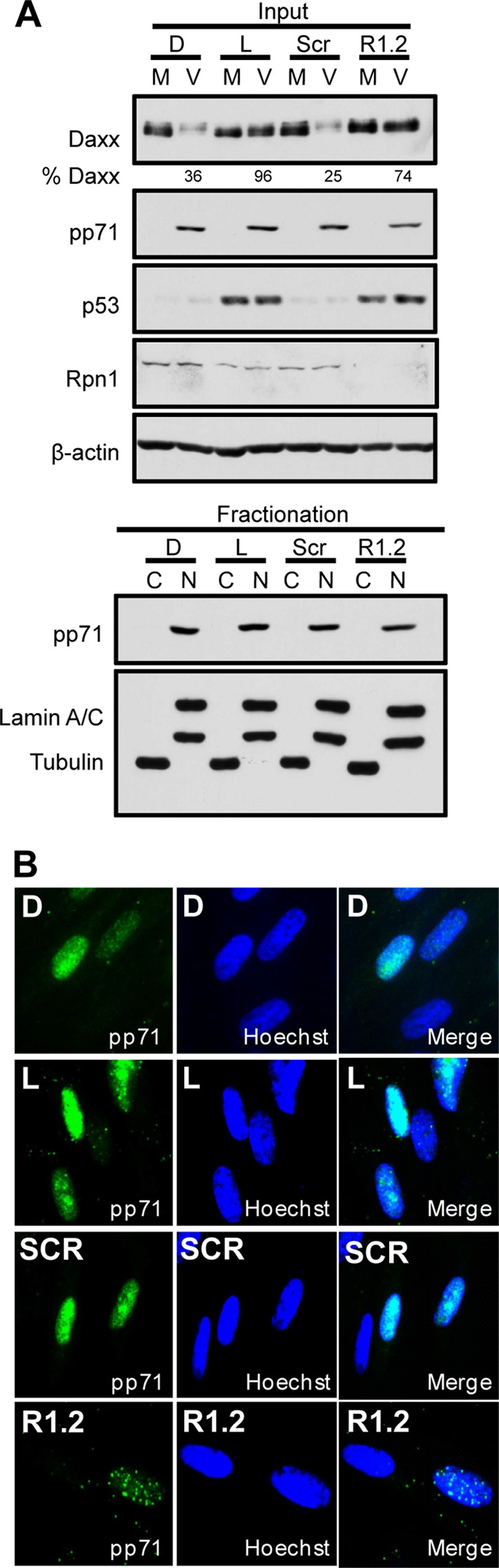
Knockdown of Rpn1 does not impair pp71 nuclear localization. (A) HFs were transfected with siRNAs targeting the 19S Rpn1 subunit (R1.2) or a scrambled control (Scr) or treated with lactacystin (L) or DMSO (D) at the start of infection. HFs were mock infected (M) or infected (V) with an MOI of 1 at 72 h posttransfection. Lysates collected 3 h postinfection (Input) were subjected to nuclear (N) and cytoplasmic (C) fractionation and analyzed with immunoblotting. Lamin A/C and tubulin indicate the efficiency of subcellular fractionation. (B) HFs transfected with a scrambled control (Scr) or an Rpn1-specific siRNA (R1.2) or treated with lactacystin (L) or DMSO (D) were infected with an MOI of 1 for 8 h. Coverslips obtained from each treatment group were stained for pp71 (green) and nuclei (Hoechst, blue).
Fig 5.
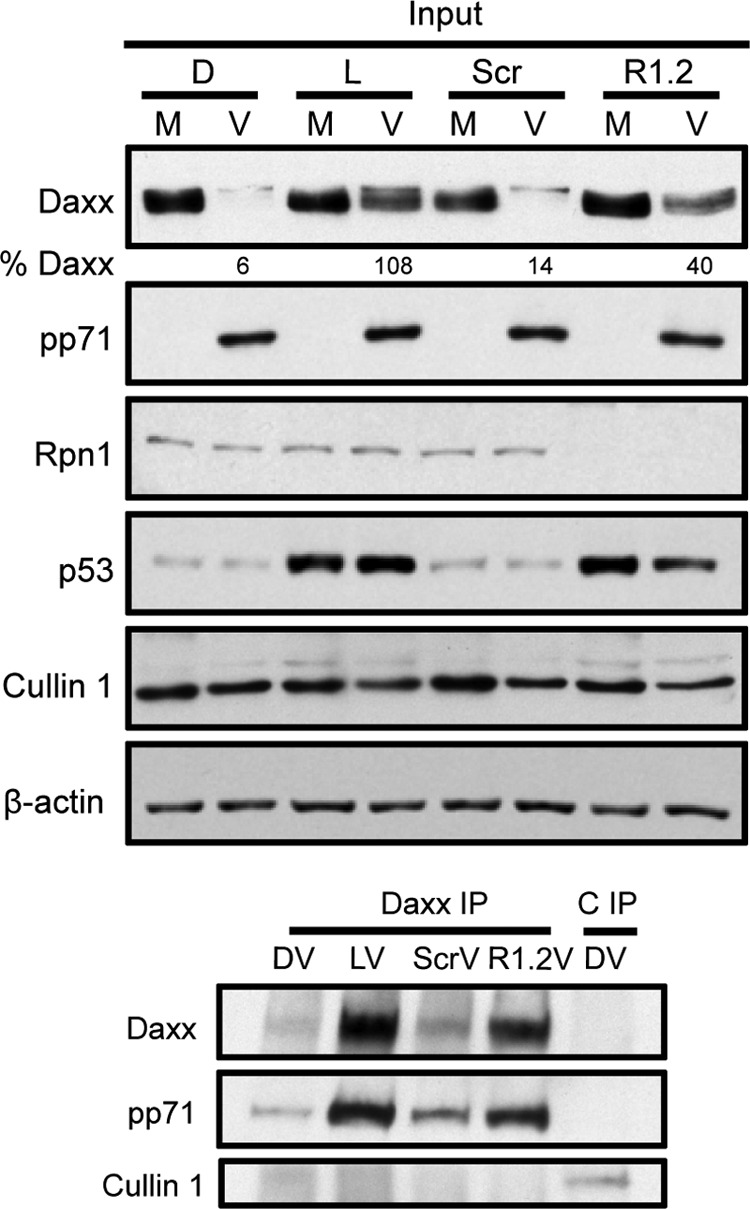
Knockdown of Rpn1 does not impair pp71 interaction with Daxx. Rpn1 (R1.2) or scrambled control (Scr) siRNAs were transfected into HFs that were then mock infected (M) or infected (V) at an MOI of 3 with UV-inactivated virus for 2 h. In parallel, samples treated with DMSO (D) or lactacystin (L) were also infected. Lysates were subjected to immunoprecipitation (IP) with Daxx antibodies. An additional Cullin1 IP (C IP) represents a specificity control. Input and IP isolations were analyzed with immunoblotting.
At the start of HCMV infection, pp71 degrades not only Daxx but also the hypophosphorylated form of the tumor suppressor Rb (12) without its prior ubiquitination (14). Residual hypophosphorylated Rb not degraded by pp71 is later phosphorylated by UL97 (12). In cells transfected with a scrambled siRNA, pharmacological inhibition of UL97 with Gö6976 failed to stabilize Rb during HCMV infection, whereas proteasome inhibition did (Fig. 6A). This indicates that within the time frame of these experiments, the loss of hypophosphorylated Rb occurs through pp71-mediated degradation, not UL97-mediated phosphorylation. Similar to Daxx, Rb was degraded in control cells but remained near mock levels in HCMV-infected cells previously depleted of Rpn1 (Fig. 6A), Rpt2, or Rpn11 (Fig. 6B). In total, these results suggest that, although Daxx and Rb degradation by pp71 are ubiquitin-independent processes, degradation of these substrates during HCMV infection requires the 19S RP complex.
Fig 6.
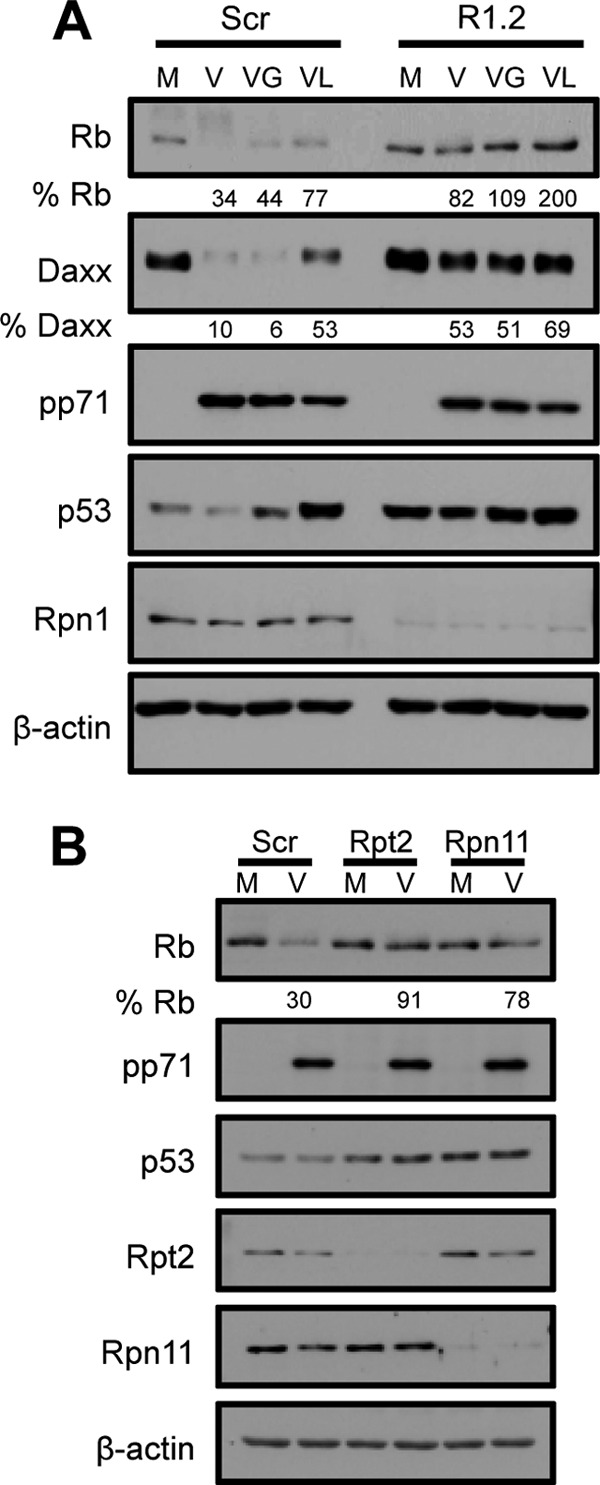
The 19S RP is required for the degradation of Rb by pp71 during HCMV infection. (A) HFs were transfected with a scrambled control (Scr) or Rpn1-specific siRNA (R1.2) for 72 h and then mock infected (M) or infected (V) at an MOI of 1. Forty-eight hours prior to infection, the cells were incubated in medium containing 0.1% serum. At the start of infection, cells were treated with Gö6976 (VG) (an inhibitor of UL97-mediated phosphorylation), lactacystin (VL), or DMSO (V). Lysates harvested 9 h postinfection were analyzed with immunoblotting and quantified with ImageQuant. (B) HFs were transfected with a scrambled control siRNA or one specific for the indicated 19S subunit. Twenty-four hours after transfection, HFs were incubated in 0.1% serum-containing medium for 48 h and then mock infected (M) or infected (V) with HCMV at an MOI of 1. Lysates collected 3 h postinfection were analyzed with immunoblotting and quantified with ImageQuant.
DISCUSSION
Viruses commandeer proteasome pathways to combat cellular immune defenses and promote viral replication (4, 7, 40). Most viral and cellular substrates are degraded through the canonical ubiquitin-dependent pathway where target substrates are covalently modified with ubiquitin molecules and delivered to the proteasome for degradation. There is, however, a growing realization that substrates can be degraded by the proteasome without prior ubiquitination in uninfected and virus-infected cells (8, 41). Interestingly, the majority of substrates degraded in this manner, both in uninfected and virally infected cells, reflect proteins that have confirmed or suspected roles in tumorigenesis (8). HCMV may be oncomodulatory (42), and it utilizes ubiquitin-independent, proteasome-dependent degradation to promote viral replication through degradation of the tumor suppressors Daxx and Rb by the viral pp71 protein. Whether there is significance to this unusual mode of protein degradation, as opposed to the more common ubiquitin-dependent option, for viral replication or oncomodulation is not appreciated.
Like Rb, Daxx proteins sometimes migrate as a series of distinct bands on polyacrylamide gels, and pp71 appears to preferentially target the fastest-migrating forms for degradation. For Rb, these clearly represent hypophosphorylated proteins (9). For Daxx, it is unclear whether or not faster-migrating bands represent unmodified proteins or, perhaps, one of the differentially spliced forms recently described (43). While Daxx sumoylation is stimulated by pp71, this posttranslational modification is not required for its degradation (44).
Here, we show that base and lid components of the 19S RP are required for pp71-mediated degradation of both Daxx and Rb, indicating that pp71 uses a proteasome species that is predominantly associated with ubiquitin-dependent degradation to degrade its substrates. The requirement for a 19S RP ATPase (Rpt2) might be expected, as substrate unfolding appears necessary for translocation into the narrow 20S channel harboring the protease active sites. As Rpn1 chaperones the assembly of an Rpt ATPase subcomplex and is required for the accumulation of Rpt2 (Fig. 3C) (37), its requirement is also not surprising. However, the requirement for Rpn11, a lid deubiquitinase, was unexpected. Perhaps the overall structure and/or function of the 19S RP is required for pp71-mediated degradation of Daxx and Rb.
How the 19S RP facilitates the ubiquitin-independent degradation of pp71 substrates is unknown. The 19S RP is also implicated in the ubiquitin-independent in vitro degradation of ornithine decarboxylase (ODC), as this is carried out by purified 26S but not 20S proteasomes (45) and is inhibited by the addition of polyubiquitin chains (46) that presumably compete with the substrate for 19S availability or function. Interestingly, we observed an enhancement of Daxx degradation during HCMV infection when ubiquitin was depleted (Fig. 1B). Perhaps depletion of the free-ubiquitin pool and a decrease in ubiquitinated substrates increased 26S proteasome availability for pp71-mediated degradation of Daxx. ODC degradation also requires a binding partner (antizyme) to unmask a cryptic degron within the carboxy terminus of the protein (46). In vivo degradation of a reporter fused to this degron required 19S RP function (36), but mechanistic features of this process have not been described. While pp71 could be playing an antizymelike role in the degradation of Daxx and Rb, degrons within these substrates have not been identified. Thus, the role for the 19S RP in the ubiquitin-independent degradation of antizyme and pp71 substrates remains to be defined.
Supplementary Material
ACKNOWLEDGMENTS
We thank Phil Balandyk (UW—Madison) for expert technical assistance, Ron Kopito (Stanford) for assistance with the PA200 experiments, and Martin Rechsteiner (University of Utah), Lance Barton (Austin College), and Barry Sleckman (Washington University) for generously providing materials.
This work was supported by National Institutes of Health grant AI074984 (to R.F.K.) and training grant T32 CA009135 (L.L.W.). R.F.K. is a Vilas Fellow and Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Published ahead of print 13 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03301-12.
REFERENCES
- 1. Stadtmueller BM, Hill CP. 2011. Proteasome activators. Mol. Cell 41:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jung T, Catalgol B, Grune T. 2009. The proteasomal system. Mol. Aspects Med. 30:191–296 [DOI] [PubMed] [Google Scholar]
- 3. Krogan NJ, Lam MHY, Fillingham J, Keogh M-C, Gebbia M, Li J, Datta N, Cagney G, Buratowski S, Emili A, Greenblatt JF. 2004. Proteasome involvement in the repair of DNA double-strand breaks. Mol. Cell 16:1027–1034 [DOI] [PubMed] [Google Scholar]
- 4. Loureiro J, Ploegh HL. 2006. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv. Immunol. 92:225–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muratani M, Tansey WP. 2003. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4:192–201 [DOI] [PubMed] [Google Scholar]
- 6. Nakayama KI, Nakayama K. 2006. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6:369–381 [DOI] [PubMed] [Google Scholar]
- 7. Blanchette P, Branton PE. 2009. Manipulation of the ubiquitin-proteasome pathway by small DNA tumor viruses. Virology 384:317–323 [DOI] [PubMed] [Google Scholar]
- 8. Hwang J, Winkler L, Kalejta RF. 2011. Ubiquitin-independent proteasomal degradation during oncogenic viral infections. Biochim. Biophys. Acta 1816:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee SH, Kalejta RF, Kerry J, Semmes OJ, O'Connor CM, Khan Z, Garcia BA, Shenk T, Murphy E. 2012. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 109:9575–9580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799 [DOI] [PubMed] [Google Scholar]
- 13. Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338 [DOI] [PubMed] [Google Scholar]
- 14. Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. U. S. A. 100:3263–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. 2007. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGγ proteasome. Mol. Cell 26:843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O'Malley BW. 2007. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGγ-proteasome pathway. Mol. Cell 26:831–842 [DOI] [PubMed] [Google Scholar]
- 17. Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ, O'Malley BW. 2006. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 124:381–392 [DOI] [PubMed] [Google Scholar]
- 18. Barton LF, Runnels HA, Schell TD, Cho Y, Gibbons R, Tevethia SS, Deepe GS, Monaco JJ. 2004. Immune defects in 28-kDa proteasome activator γ-deficient mice. J. Immunol. 172:3948–3954 [DOI] [PubMed] [Google Scholar]
- 19. Cantrell SR, Bresnahan WA. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khor B, Bredemeyer AL, Huang CY, Turnbull IR, Evans R, Maggi LB, Jr, White JM, Walker LM, Carnes K, Hess RA, Sleckman BP. 2006. Proteasome activator PA200 is required for normal spermatogenesis. Mol. Cell Biol. 26:2999–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 97:14506–14511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109–9120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hwang J, Saffert RT, Kalejta RF. 2011. Elongin B-mediated epigenetic alteration of viral chromatin correlates with efficient human cytomegalovirus gene expression and replication. mBio 2(2):e00023–11 doi:10.1128/mBio.00023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hofmann H, Sindre H, Stamminger T. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fabre B, Lambour T, Delobel J, Amalric F, Monsarrat B, Burlet-Schiltz O, Bousquet-Dubouch M-P. 13 December 2012. Subcellular distribution and dynamics of active proteasome complexes unraveled by a workflow combining in vivo complex cross-linking and quantitative proteomics. Mol. Cell. Proteomics [Epub ahead of print.] doi:10.1074/mcp.M112.023317 mcp.M112.023317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rechsteiner M, Hill CP. 2005. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15:27–33 [DOI] [PubMed] [Google Scholar]
- 28. Soza A, Knuehl C, Groettrup M, Henklein P, Tanaka K, Kloetzel P-M. 1997. Expression and subcellular localization of mouse 20S proteasome activator complex PA28. FEBS Lett. 413:27–34 [DOI] [PubMed] [Google Scholar]
- 29. Ustrell V, Hoffman L, Pratt G, Rechsteiner M. 2002. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 21:3516–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu Y, Wang L, Zhou P, Wang G, Zeng Y, Wang Y, Liu J, Zhang B, Liu S, Luo H, Li X. 2011. Regulation of REG[gamma] cellular distribution and function by SUMO modification. Cell Res. 21:807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suzuki R, Moriishi K, Fukuda K, Shirakura M, Ishii K, Shoji I, Wakita T, Miyamura T, Matsuura Y, Suzuki T. 2009. Proteasomal turnover of hepatitis C virus core protein is regulated by two distinct mechanisms: a ubiquitin-dependent mechanism and a ubiquitin-independent but PA28gamma-dependent mechanism. J. Virol. 83:2389–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. da Fonseca PC, He J, Morris EP. 2012. Molecular model of the human 26S proteasome. Mol. Cell 46:54–66 [DOI] [PubMed] [Google Scholar]
- 33. Elsasser S, Chandler-Militello D, Müller B, Hanna J, Finley D. 2004. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279:26817–26822 [DOI] [PubMed] [Google Scholar]
- 34. Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78:477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. 2008. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, Richard McCombie W, Elledge SJ, Hannon GJ. 2004. A resource for large-scale RNA-interference-based screens in mammals. Nature 428:427–431 [DOI] [PubMed] [Google Scholar]
- 37. Kaneko T, Hamazaki J, Iemura S, Sasaki K, Furuyama K, Natsume T, Tanaka K, Murata S. 2009. Assembly pathway of the mammalian proteasome base subcomplex is mediated by multiple specific chaperones. Cell 137:914–925 [DOI] [PubMed] [Google Scholar]
- 38. Rubin DM, Glickman MH, Larsen CN, Dhruvakumar S, Finley D. 1998. Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 17:4909–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, Koonin EV, Deshaies RJ. 2002. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298:611–615 [DOI] [PubMed] [Google Scholar]
- 40. Petroski M. 2008. The ubiquitin system, disease, and drug discovery. BMC Biochem. 9:S7 doi:10.1186/1471-2091-9-S1-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jariel-Encontre I, Bossis G, Piechaczyk M. 2008. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 1786:153–177 [DOI] [PubMed] [Google Scholar]
- 42. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. 2012. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86:854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wethkamp N, Hanenberg H, Funke S, Suschek CV, Wetzel W, Heikaus S, Grinstein E, Ramp U, Engers R, Gabbert HE, Mahotka C. 2011. Daxx-β and Daxx-γ, two novel splice variants of the transcriptional co-repressor Daxx. J. Biol. Chem. 286:19576–19588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hwang J, Kalejta RF. 2009. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 83:6591–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murakami Y, Matsufuji S, Kameji T, Hayashi S, Igarashi K, Tamura T, Tanaka K, Ichihara A. 1992. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360:597–599 [DOI] [PubMed] [Google Scholar]
- 46. Zhang M, Pickart CM, Coffino P. 2003. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J. 22:1488–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.