

Abstract
The genes required for 3-hydroxybenzoate and gentisate catabolism in Corynebacterium glutamicum are closely clustered in three operons. GenR, an IclR-type regulator, can activate the transcription of genKH and genDFM operons in response to 3-hydroxybenzoate and gentisate, and it can repress its own expression. Footprinting analyses demonstrated that GenR bound to four sites with different affinities. Two GenR-binding sites (DFMn01 and DFMn02) were found to be located between positions −41 and −84 upstream of the −35 and −10 regions of the genDFM promoter, which was involved in positive regulation of genDFM transcription. The GenR binding site R-KHn01 (located between positions −47 and −16) overlapped the −35 region of the genKH promoter sequence and is involved in positive regulation of its transcription. The binding site R-KHn02, at which GenR binds to its own promoter, was found within a footprint extending from position −44 to −67. It appeared to be involved in negative regulation of the activity of the genR promoter. A consensus motif with a 5-bp imperfect palindromic sequence [ATTCC-N7(5)-GGAAT] was identified among all four GenR binding sites and found to be necessary to GenR regulation through site-directed mutagenesis. The results reveal a new regulatory function of the IclR family in the catabolism of aromatic compounds.
INTRODUCTION
Isocitrate lyase regulator-type (IclR-type) transcriptional regulators are widespread in both Gram-positive and Gram-negative bacteria. They are involved in carbon metabolism, multidrug resistance, virulence, quorum-sensing signals, and sporulation (1). They have also been found in Archaea, indicating a wide taxonomic distribution (1). Members of the IclR family include repressors, activators, and dual-function proteins that act as activators of certain genes and as repressors of others (including autoregulation). The physiological roles of some of these proteins have been elucidated for a limited number of these proteins using genetic and biochemical approaches. The best-characterized example in this family is the glyoxylate shunt repressor IclR in Escherichia coli, which regulates the aceBAK operon encoding enzymes in its acetate utilization and autologous repression of iclR gene transcription (2–4). Another example is TtgV in Pseudomonas putida which is a repressor for the expression of the ttgDEF and ttgGHI operons encoding multidrug efflux pumps (1, 5). TtgV crystallizes as a tetramer and undergoes large conformational changes at the monomeric, dimeric, and tetrameric levels for binding to its operator, revealing a general model for cooperative DNA binding of tetrameric gene regulators (6). In the microbial degradation of aromatic compounds, a number of IclR family members were found to play roles in the regulation of their catabolic pathways (1, 7). Among the IclR family members identified in this category, PobR activates the expression of the pobA gene in the p-hydroxybenzoate pathway and represses itself in Acinetobacter sp. strain ADP1 (8, 9). PcaR and PcaU are involved in the positive regulation of protocatechuate degradation, but both negatively regulate themselves in Pseudomonas putida (10, 11) and Acinetobacter sp. strain ADP1 (12, 13). MhpR positively regulates the expression of enzymes involved in the degradation of 3-(3-hydroxyphenyl) propionic acid in E. coli (14, 15). Up until now, no consensus sequence has been found in the architecture of the DNA binding sites among the characterized regulators in the IclR family. The common mechanism of the binding of these IclR-type regulators to their promoters has not been revealed either.
The gentisate (2, 5-dihydroxybenzoate [GEN]) pathway is one of the most important ring cleavage pathways in the catabolism of a large number of aromatic compounds, including 3-hydroxybenzoate (3-HBA) (16, 17), naphthalene, salicylate, 3,6-dichloro-2-methoxybenzoate, and xylenol. The naphthalene catabolic genes of Ralstonia sp. strain U2 and Polaromonas naphthalenivorans CJ2 are all organized in a single operon, and a LysR-type transcriptional regulator NagR, divergently transcribed from its structural genes, activates the expression of nag catabolic genes (including those for gentisate catabolism) in the presence of the inducer salicylate (18, 19). The degradation of 3-HBA in Klebsiella pneumoniae M5a1 is accomplished by enzymes encoded by the mhbTDHIM operon, which is positively regulated by a LysR-type regulator, MhbR (20, 21).
Corynebacterium glutamicum ATCC 13032 utilizes 3-HBA or GEN as its sole carbon and energy source, and its 3-HBA degradation via the GEN pathway has been characterized at both the biochemical and genetic levels, as shown in Fig. 1A (16, 22, 23). The ring cleavage oxidation of GEN is catalyzed by genD-encoded gentisate 1,2-dioxygenase (GDO). The ring fission product maleylpyruvate is isomerized to fumarylpyruvate by genM-encoded mycothiol-dependent maleylpyruvate isomerase. Then fumarylpyruvate is hydrolyzed to maleate and pyruvate by GenF, a fumarylpyruvate hydrolase. GenK actively transports gentisate to facilitate the utilization of GEN by this strain (22). GenH is a 3-hydroxybenzoate 6-hydroxylase (3-HBA 6-hydroxylase) catalyzing the NADH-dependent hydroxylation of 3-HBA to GEN in vitro (23).
Fig 1.

(A) Catabolic pathway of 3-hydroxybenzoate and gentisate in Corynebacterium glutamicum ATCC 13032, together with the catabolic reaction catalyzed by the gen gene products in vivo. The boxed diagram shows the GenK-mediated gentisate transportation. (B) Organization of the gen cluster and the physical locations of RT-PCR products for transcriptional analysis, promoters for activity detection, and probes for EMSAs. The region of the DNA fragment is indicated. (52).
GenR (formerly Ncgl2921) has been proposed as an IclR-type regulator involved in 3-HBA and GEN assimilation by disruption and complementation in C. glutamicum RES167 (16). In this study, we demonstrated that GenR is an IclR-type regulator with two types of activation and one type of autorepression, representing a novel regulatory mechanism in the IclR family.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture media.
The bacterial strains and plasmids used in the present study are listed in Table 1, and primers are listed in Table S1 in the supplemental material. C. glutamicum strains were grown in LB medium supplemented with 2 g liter−1 of glucose or in mineral salt medium (MM) (26). The mixture was kept at pH 8.4 and 30°C and supplemented with 0.05 g liter−1 of yeast extract and appropriate carbon sources. Aromatic compounds were added at a final concentration of 2 mM as carbon and energy sources. For the generation of mutants and maintenance of C. glutamicum, brain heart infusion broth medium (BHIBM) was used (27). When required for selection, antibiotics were added at the following concentrations: kanamycin, 50 μg ml−1 for E. coli and 25 μg ml−1 for C. glutamicum; chloramphenicol, 30 μg ml−1 for E. coli and 10 μg ml−1 for C. glutamicum; ampicillin, 100 μg ml−1 for E. coli; nalidixic acid, 50 μg ml−1 for C. glutamicum.
Table 1.
Bacteria and plasmids involved in this study
| Bacteria or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Bacteria | ||
| C. glutamicum strains | ||
| RES167 | Restriction-deficient mutant of ATCC 13032 Δ(cglIM-cglIR-cglIIR) | 24 |
| RES167 ΔgenR | Fragment of DNA encoding amino acids 52–243 of GenR (formerly Ncg12921) was deleted | 16 |
| E. colistrains | ||
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Gibco BRL |
| Rosetta(DE3)/pLysS | Camr; F− ompT hsdSB(rB− mB−) gal dcm (DE3), pLysSRARE vector (Camr) | Novagen |
| Trans1-T1 phage resistant | F− ϕ80(lacZ)ΔM15 ΔlacX74 hsd R(rK− mK+) ΔrecA1398 endA1 tonA | TransGen Biotech |
| Plasmids | ||
| pMD18-T | Ampr, lacZα; cloning vector for sequencing | TaKaRa |
| pET-28a (+) | Expression vector; Kanr, C/N-terminal His tag/thrombin/T7 tag, T7 lac promoter, T7 transcription start, f1 origin, lacI | Novagen |
| pXMJ19 | Camr, Ptac lacq, pBL1 oriVCg., pK18, oriVEc., trrnB of E. coli, E. coli-C. glutamicum shuttle vector | 25 |
| pZWHJ001 | Camr, pXMJ19 carrying genR(formerly ncg12921) to generate complementation for genR | This study |
| pZWHJ002 | Camr, pBL1, oriVCg, pK18, oriVEc., trrnB of E. coli, lacZ; promoterless derivative of pXMJ19 with lacZ digested with the enzyme combination NarI/HindIII; deletion of Ptac lacq; shuttle vector | This study |
| pZWHJ003 | Kanr, 777-bp PCR fragment of genR cloned into pET-28a(+) HindIII/XhoI site, His tag banding N-terminal GenR | This study |
| pZWHJ005 | Kanr; cloning Ptac promoter from pXMJ19 into the XbaI site of pET-28a-ncgl2921n to enhance the expression of regulator GenR; His tag banding N-terminal GenR | This study |
| pZWHJ006 | Camr; derivative of pCH19 with 463-bp promoter of genDFMoperon for detecting promoter activation | This study |
| pZWHJ007 | Camr; derivative of pCH19 with 457-bp promoter of genR operon for detecting promoter activation | This study |
| pZWHJ008 | Camr; derivative of pCH19 with 427-bp promoter of genKH operon for detecting promoter activation | This study |
| pZWHJ009 | Camr; pZWHJ006 with site DFMn01 mutated | This study |
| pZWHJ010 | Camr; pZWHJ006 with site DFMn02 mutated | This study |
| pZWHJ011 | Camr; pZWHJ006 with sites DFMn01 and DFMn02 mutated | This study |
| pZWHJ012 | Camr; pZWHJ007 with site R-KHn01 mutated | This study |
| pZWHJ013 | Camr; pZWHJ008 with site R-KHn01 mutated | This study |
| pZWHJ014 | Camr; pZWHJ007 with site R-KHn02 mutated | This study |
| pZWHJ015 | Camr; pZWHJ008 with site R-KHn02 mutated | This study |
oriVEc, E. colioriV; oriVCg., C. glutamicum oriV.
Construction of plasmids and strains.
Plasmid purification, DNA manipulation, and agarose gel electrophoresis were carried out as described previously (28). C. glutamicum cells were transformed by electroporation according to the method described by Eggeling and Reyes (27). genR was amplified using primers ncgl2921e3 and ncgl2921e4 and digested with BamHI and HindIII (Fermentas, Lithuania) before being cloned into pXMJ19 (25) to produce plasmid pZWHJ001. Plasmid pZWHJ002 was constructed by replacing lacI and Ptac with an lacZ fragment of Rosetta(DE3)/pLysS at the EheI and PstI (Fermentas) sites of plasmid pXMJ19. The probable promoter regions DFMa, Ra, and KHa were amplified using the primers listed in Table S1 in the supplemental material from the C. glutamicum genome and digested with BamHI and HindIII before being cloned into pZWHJ002 to produce plasmids pZWHJ006, pZWHJ007 and pZWHJ008, respectively.
The genR gene was amplified using primers ncgl2921e1 and ncgl2921e2, digested with BamHI and HindIII, and inserted into pET28a(+) to produce plasmid pZWHJ003. The Ptac promoter was amplified using primers ptacr3 and ptacr4 from vector pXMJ19 and digested with XbaI. It was then cloned into plasmid pZWHJ003 to generate plasmid pZWHJ005.
The GenR binding sites DFMn01 and DFMn02, subregions of the DFMn binding site region, and R-KHn01 and R-KHn02, subregions of the R-KHn binding site region, were mutated using the In-Fusion Advantage PCR cloning method with pZWHJ006, pZWHJ007, and pZWHJ008 as the templates. To mutate site DFMn01, 10-bp substitutions (ATTCCN7GGAAT to GGCAAN7AACGG) were generated using the mutagenic primers pDFMnmut01 and pDFMnmut02. The PCR fragment was amplified using TransStart Fast Pfu DNA Polymerase (TransGen, Beijing, China) and joined using an In-Fusion Advantage PCR cloning kit (Clontech, Beijing, China) to replace the wild-type fragments of pZWHJ006, generating pZWHJ009. In the same manner, plasmid pZWHJ010 was constructed by replacing ATTCCN5GGAAA with GGCAAN5AACGG at site DFMn02 on pZWHJ006. Constructs pZWHJ012 and pZWHJ013 were constructed by replacing TTTCCN7GGAAT with GGCAAN7AACGG at the R-KHn01 sites on pZWHJ007 and pZWHJ008, respectively. Constructs pZWHJ014 and pZWHJ015 were made by replacing ATCACN7AGAAT with GGCAAN7AACGG at the R-KHn02 site on pZWHJ007 and pZWHJ008, respectively. Construct pZWHJ011, containing mutated sites DFMn01 and DFMn02 (designated DFMn01m and DFMn02m, respectively) was obtained by replacing the ATTCCN5GGAAA fragment with the GGCAAN5AACGG fragment in pZWHJ009.
Enzyme assays.
β-Galactosidase activity was determined in Miller units, as previously described (29, 30). The 3-HBA 6-hydroxylase activity and GDO activity were determined as described previously (31, 32). Protein concentration was determined according to the Bradford method (33). The 14C-labeled gentisate uptake assays were performed as described previously (22).
GenR overexpression, purification, and immunoblotting.
N-terminal His-tagged GenR (His6-GenR) was expressed by E. coli Rosetta(DE3)pLysS carrying pZWHJ005, containing promoters T7 and Ptac upstream of genR. Overexpression of the cloned genR gene was achieved by transforming the pZWHJ005 plasmid into E. coli Rosetta(DE3)/pLysS (Novagen, Madison, WI). The transformed cells were grown at 37°C to an optical density at 600 nm (OD600) of 0.8 in 1 liter of LB medium. Isopropyl-β-d-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.1 mM, and the cultures were incubated at 16°C for another 16 h. His6-GenR from the supernatant was purified using Ni-nitrilotriacetic acid (NTA)-agarose chromatography (Novagen). Protein was monitored using SDS-PAGE. The purified protein from pZWHJ005 was detected as a single 27-kDa band by Coomassie blue staining and SDS-PAGE. This was verified by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) mass spectrometry with a coverage score of 56% (data not shown).
RNA preparation and transcription analysis.
Total RNA from C. glutamicum was isolated using the hot phenol method (34). For transcription analysis, total RNA was digested with 1 U g−1 of recombinant DNase I (TaKaRa) for 1 h at 37°C, and 1 μg of RNA was reversely transcribed with PrimeScript Reverse Transcriptase (TaKaRa). The resulting cDNA was amplified using reverse transcription-PCR (RT-PCR) and quantitative real-time PCR (RT-qPCR), using the primers listed in Table S1 in the supplemental material. RT-qPCR was performed in a CFX Connect Real-Time PCR Detection System (Bio-Rad) in 25-μl reaction volumes using iQ SYBR green Supermix (Bio-Rad) with the primers listed in Table S1 in the supplemental material at a final concentration of 200 nM each. All samples were run in triplicate in three independent experiments each. Relative expression levels were estimated using the 2−ΔΔCT (where CT is threshold cycle) method, and the 16S rRNA gene served as a reference for normalization (35).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously (36). Gels were dried and exposed in a cassette (Yuehua, Shantou, China) using a phosphor storage screen (PerkinElmer, Boston, MA). Images of radioactive filters were obtained and quantified with a Cyclone Plus Storage Phosphor System and OptiQuant image analysis software (PerkinElmer).
Primer extension and S1 nuclease protection analysis.
Primer extension reactions were performed as described previously (37), using 20 to 50 μg of RNA as the template, 32P-labeled oligonucleotides, and PrimeScript Reverse Transcriptase (TaKaRa). The samples were run on 6% polyacrylamide-urea sequencing gels next to the corresponding sequencing ladder with an AccuPower DNA Sequencing Kit (Bioneer, Seoul, South Korea). After electrophoresis, the gels were dried and exposed to Kodak X-ray film.
S1 nuclease protection analysis was performed as described previously (38). The genKH probe was generated by PCR using the radiolabeled pegenKH04 and unlabeled pgenKH02 oligonucleotides.
DNase I footprinting.
Footprinting assays were performed as described previously (39). The primers were also used in primer extension assays.
Nucleotide sequence accession number.
The DNA sequence of pZWHJ002 was submitted to the GenBank under accession number KC465961.
RESULTS
3-HBA and GEN stimulate their catabolic pathways.
Previous biochemical and genetic characterizations indicated that the expression of genDFM genes (formerly ncgl2918-ncgl2919-ncgl2920) for 3-HBA and GEN catabolism was induced by 3-HBA and GEN in C. glutamicum (16). In the current study, the expression of the genKH genes (formerly ncgl2922-ncgl2923) was also investigated under 3-HBA and GEN induction. C. glutamicum cells were found to exhibit 3-HBA 6-hydroxylase (encoded by genH) at a specific activity of 0.23 U/mg of protein in 3-HBA and 0.21 U/mg of protein in GEN. They expressed gentisate transporter (GenK) at an activity of 2.48 ± 0.12 nmol/min/mg of cell dry weight in 3-HBA or 2.37 ± 0.15 nmol/min/mg in GEN. In this way, both 3-HBA and GEN served as inducers of the expression of each gene encoding enzymes in the 3-HBA and GEN catabolism. In order to better define the transcription of their catabolic genes, the organization of the gen cluster was identified by RT-PCR from the strain RES167. RT-PCR performed with RNA purified from RES167 cells grown in 3-HBA generated products that extended across the boundaries between each gen gene. The presence of amplified DNA fragments obtained with each primer pair suggested that the genR gene was transcribed monocistronically (see Fig. S1 in the supplemental material). The genDFM genes and genKH genes constituted one operon each. The transcriptional arrangement of these regions was then more precisely analyzed by RT-qPCR. As shown in Fig. 2A, the transcription of genDFM and genKH was increased significantly from that in RES167 cells grown in 3-HBA/GEN. The level of genDFM mRNA expression was seven times higher in GEN-grown cells and 14-fold higher in 3-HBA-grown cells than in glucose-grown cells; genKH mRNA expression was 18-fold higher in cells grown in GEN and 36-fold higher in cells grown in 3-HBA than in cells grown in glucose. This is consistent with the levels of 3-HBA- and GEN-induced enzyme activity, as shown above. To further confirm that 3-HBA and GEN induce specific pathways in strain RES167, transcriptional and translational lacZ genes fused to the probable promoter region of each operon were constructed as described in Materials and Methods. The results showed that the increased levels of β-galactosidase activity were attributed to genDFM and genKH promoters in the 3-HBA- and GEN-induced cells. The enhanced β-galactosidase activity observed under induction (Fig. 2C) was consistent with the increased mRNA levels in the cells induced by 3-HBA and GEN, demonstrating that 3-HBA and GEN induce the expression of the genDFM and genKH operons in this pathway. The effects of the genKH promoter were slightly more pronounced than those of the genDFM promoter (Fig. 2A and C), as indicated by the results of both RT-qPCR and β-galactosidase analyses.
Fig 2.

Transcriptional analyses of genDFM, genR, and genKH of strains RES167 and RES167 ΔgenR in response to Glu (glucose), GEN, and 3-HBA. (A and B) qRT-PCR analyses examining the transcription of genDFM, genR, and genKH (indicated as DFM, R, and KH, respectively, on the x axis) in strains RES167 and RES167 ΔgenR. RNA samples were isolated from strains RES167 and RES167 ΔgenR induced on MM with 2 mM Glu, GEN, or 3-HBA overnight as described in Materials and Methods. Cultures grown on MM with GEN or 3-HBA were collected for RNA isolation. The levels of gene expression in each sample were calculated as the fold expression ratio after normalization to 16S rRNA gene transcript levels. The values are averages of two independent RT-qPCR experiments. Error bars indicate standard deviations. (C and D) β-Galactosidase activity driven by genDFM, genR, and genKH promoters. Strains RES167 and RES167 ΔgenR, both containing pZWHJ006, pZWHJ007, and pZWHJ008, were grown in triplicate in BHIBM and transferred into MM at a 1.0% inoculum containing 2 mM Glu, GEN, or 3-HBA for induction of β-galactosidase activity overnight. The mutant RES167 ΔgenR was first grown in BHIBM until mid-log phase and then harvested and transferred to MM for induction of β-galactosidase activity. β-Galactosidase activity was assessed using a Miller assay. The data are derived from at least three independent measurements, and error bars indicate standard deviations.
Role of the genR gene in the 3-HBA and GEN pathway.
It has been proposed that GenR may activate the expression of genDFM genes in C. glutamicum, based on genR disruption and complementation (16). In the current study, the biochemical activity of proteins encoded by genKH was also assayed in the genR-disrupted mutant and its complemented strain. No 3-HBA 6-hydroxylase activity was detected in the mutant RES167 ΔgenR, but specific activities of 0.23 and 0.31 U/mg of protein were detected in strain RES167 and the complemented strain RES167 ΔgenR/pZWHJ001 under 3-HBA induction, respectively. In an uptake assays using 14C-labeled gentisate, strain RES167 exhibited transport activity of 2.48 ± 0.12 nmol/min/mg of cell dry weight under 3-HBA induction. However, the mutant RES167 ΔgenR lost nearly all ability to transport its targets. The complemented strain retained this ability, showing an activity of 2.34 ± 0.49 nmol/min/mg of cell dry weight.
Transcription of each operon was also analyzed in the mutant RES167 ΔgenR. Strain RES167 ΔgenR was transformed with plasmids pZWHJ006 and pZWHJ008 and induced by 3-HBA and GEN for expression of β-galactosidase activity. As shown in Fig. 2D, no activity was detected in the mutant strains containing pZWHJ006 or pZWHJ008. Strains RES167 and RES167 ΔgenR, which contained plasmid pZWHJ007 with the genR promoter region Ra (Fig. 1), were used to detect the effects of GenR on its own expression. The level of β-galactosidase activity was found to be similar in the wild-type strain. This indicated that genR transcription was not enhanced by induction with growth substrates 3-HBA and GEN, as shown in Fig. 2C. β-Galactosidase activity was approximately four times higher in strain RES167 ΔgenR than in the wild-type strain, indicating that the genR promoter was negatively regulated by its own gene product, GenR (Fig. 2C and D). RT-qPCR was also performed to further assess the effects of GenR on the expression of the 3-HBA and GEN catabolic operons. As shown in Fig. 2A, transcription of genR was not increased in the wild-type strain under the 3-HBA- and GEN-induced conditions. However, genR transcription increased by 4-fold in the mutant RES167 ΔgenR (Fig. 2A and B), indicating that GenR represses the expression of genR.
All of these results indicate that GenR is involved in the activation of the 3-HBA and GEN catabolic pathways under 3-HBA- and GEN-induced conditions, that it represses its own transcription, and that it is unaffected by 3-HBA and GEN.
Identification of the transcription start sites of genDFM, genR, and genKH operons.
In order to identify the promoter regions responsible for the regulation of the expression of the gen genes, their transcription initiation sites (TSSs) were identified using primer extension and high-resolution S1 mapping of mRNA from the wild-type strain grown under 3-HBA-induced conditions. As shown in Fig. 3, the TSS of genDFM was located by primer extension analysis. It was found to begin on a residue A, 108 bp upstream of the genD putative start codon (ATG) (Fig. 3A, lane P). The TSS of genR was localized to a guanine residue 27 bp upstream of the genR putative start codon (ATG) (Fig. 3B, lane P). Mapping the 5′ end of the genKH transcriptional unit resulted in the unambiguous identification of a TSS using primer extension analysis (Fig. 3C, lane P). High-resolution S1 nuclease protection analysis (38) was then used to identify the genKH TSS, which produced a single band. This allowed a definite identification of a T residue as the TSS, 20 bp upstream of the genKH putative start codon (ATG) (Fig. 3C, lane S1). Subsequent analysis of the area upstream of genDFM and the intergenic region between the TSSs of genR and genKH revealed the −10 and −35 regions of the respective promoters, which are underlined and marked in Fig. 3D and E.
Fig 3.

Determination of the transcription start sites of genDFM, genR, and genKH operons. 32P-labeled pegenDFM01, pegenR01, and pegenKH04 primers for the genDFM operon, genR operon, and genKH operon were used to identify the transcription start sites of the three operons using primer extension analysis and high-resolution S1 mapping of wild-type RNA. Amounts of 20 to 50 μg of total RNA were obtained from strain RES167 grown in 3-HBA. Lanes P in A, B, and C show the product of primer extension. Lane S1 in C shows the product of high-resolution S1 mapping. The nucleotide sequence around the TSS (+1) is indicated by brackets, and the 5′ ends are enlarged and in boldface. (D and E) The location of the sequences upstream of the genDFM operon and the genR-genKH promoter regions are shown in detail. The first codon of each gene is given in italics. The TSSs are shown by +1 and arrows. The putative −10 and −35 promoter sequences are underlined, the regulator GenR primary binding sites are highlighted in gray, and their palindromic sequences are in boldface.
The position of the TSSs relative to the genR and genKH operons indicated that the transcriptional units are separated by 20 bp. This arrangement requires superimposition of the −10-bp and −35-bp RNA polymerase (RNAP) binding sites for the transcription of the genR and genKH operons (Fig. 3B and C).
GenR binds the upstream regions of the genDFM, genR, and genKH operons.
In order to investigate the manner in which GenR regulates the transcription of the three operons, we performed electrophoretic mobility shift assays (EMSAs) using the 233-bp DFMan probe of the upstream region of genDFM and the 226-bp R-KHan probe spanning the genR and genKH operons as targets. The locations of these probes are shown in Fig. 1B. GenR showed distinct patterns of binding to these regulatory regions at protein concentrations ranging from 32 to 260 nM, and a 130 nM saturating concentration of His6-GenR for the formation of a single, stable protein-DNA complex was used to further examine the specificity of binding to probes (Fig. 4). GenR formed a principal protein-DNA complex with the DFMan probe only in the presence of inducers 3-HBA and GEN (Fig. 4A). This was consistent with in vivo observations of 3-HBA- and GEN-induced transcription of the genDFM operon. Interestingly, GenR always formed a single, principal protein-DNA complex with the R-KHan fragment (Fig. 4B), regardless of the presence or absence of the inducers 3-HBA and GEN. EMSAs with unlabeled specific and nonspecific competitor DNA were used as controls for DFMan (see Fig. S2A in the supplemental material) and R-KHan (see Fig. S2B).
Fig 4.
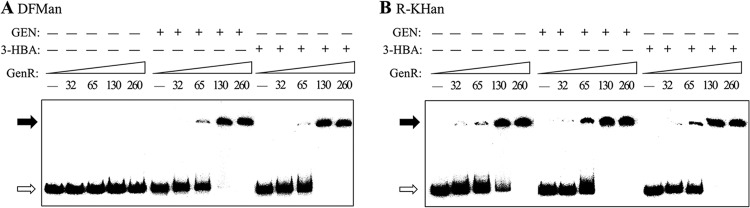
Electrophoretic mobility shift assays for determination of GenR binding sites upstream of the genDFM, genR, and genKH operons. (A and B) Effects of 3-HBA and GEN on the binding affinity of GenR for the upstream regions of genDFM, genR, and genKH operons. The probes for genDFM (DFMan in A), genR, and genKH operons (R-KHan in B) were incubated with increasing amounts of His6-GenR while the amounts of GEN and 3-HBA remained constant. The concentrations (nM) of purified His6-GenR are indicated by numbers in parentheses, either with (+) or without (−) 300 nM GEN and 3-HBA. Glucose served as a control. Each lane contained 0.5 ng (0.3 to 0.4 nM) of 32P-labeled DFMan and R-KHan probes. The free probes are indicated by open arrows, and the retarded DNA fragments are indicated by solid arrows.
To determine the regulatory role of GenR in the expression of gen clusters, the DNA binding sites of each operon were identified. DNase I footprinting analyses revealed that His6-GenR protected a large region (designated DFMn) from −41 to −84 bp relative to the genDFM TSS (Fig. 5A). This can be divided to two subregions, DFMn01 (from −41 to −61) and DFMn02 (from −62 to −84). The footprint on the R-KH promoter region covered two regions, one (designated R-KHn01) from +28 to −4 bp relative to the genR TSS and −47 to −16 bp relative to the genKH TSS and the other region (designated R-KHn02) from −44 to −67 bp relative to the genR TSS and +25 to +48 bp relative to the genKH TSS. The sequencing results produced using 32P-labeled primers pegenR01 and pegenKH04 are shown in Fig. 5B and C. Nevertheless, R-KHn01 was found to be protected at the lowest concentration of GenR (32 nM) but R-KHn02 was not. These results imply that His6-GenR has preferentially bound to R-KHn01 over R-KHn02. This is consistent with the results of competitive EMSAs (Fig. 6C and D).
Fig 5.

Identification of the GenR binding sites upstream of genDFM, genR, and genKH operons by DNase I footprinting analysis. The reaction mixtures contained approximately 200 ng of end-labeled PCR products. These were amplified with prnagRD02 primer and 32P-labeled pegenDFM01 primer (A), pgenR01 and 32P-labeled pegenR01 (B), or pgenKH02 and 32P-labeled pegenKH04 (C). Before DNase I treatment, labeled DNA was preincubated with His6-GenR for 30 min in the presence of 300 nM 3-HBA. Standards were generated by sequencing with 32P-labeled primers pegenDFM01, pegenR01, and pegenKH04, respectively. The concentrations (μM) of purified His6-GenR and the GenR-protected sequences are indicated. The nucleotide sequence around the TSS (+1) is shown by solid arrows, and the GenR binding sites are represented by brackets. Arrows indicate the direction of transcription.
Fig 6.
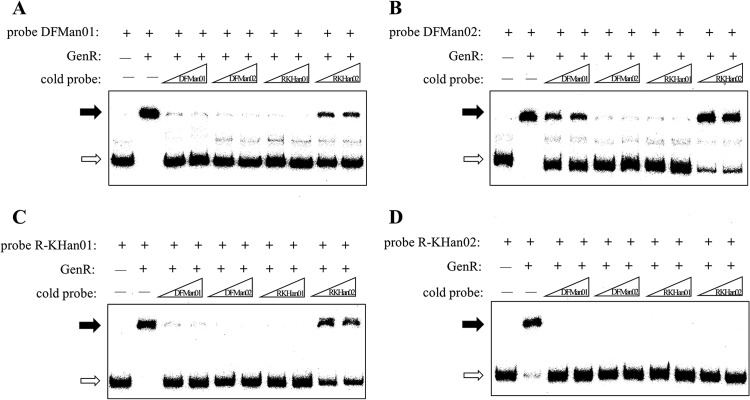
Comparison of the relative affinity of GenR for the different binding sites. The free probes are indicated by open arrows, and the retarded DNA fragments are indicated by solid arrows. (A) EMSA of His6-GenR with 32P-labeled probe DFMan01 and unlabeled probes (DFMan01, DFMan02, R-KHan01, and R-KHan02). (B) EMSA of His6-GenR with 32P-labeled probe DFMan02 and unlabeled probes. (C) EMSA of His6-GenR with 32P-labeled probe R-KHan01 and unlabeled probes. (D) EMSA of His6-GenR with 32P-labeled probe R-KHan02 and unlabeled probes. Labeled probe and the unlabeled competitors were incubated with 130 nM His6-GenR and 300 nM 3-HBA for 30 min at 25°C. Each lane contained 0.5 ng (0.3 to 0.4 nM) of 32P-labeled probe.
Bioinformatics analysis of the DNA regions that interact with GenR was performed using the motif-based sequence analysis tool MEME (40). Results showed a consensus sequence in the form of an imperfect palindrome [ATTCC-N7(5)-GGAAT] among GenR binding sites. This consensus sequence was different from those found to interact with other IclR-type regulators, such as PcaU (12), MhpR (15), PcaR (11), and PobR (8).
Affinity of GenR for different target sites.
Of four target regions detected for His6-GenR, competitive EMSAs (41) were performed to examine the affinity of GenR for binding to different target sites. Binding reaction mixtures containing a 100-fold or 150-fold molar excess of competitor DNA of probes DFMan01 spanning site DFMn01, DFMan02 (for site DFMn02), and R-KHan01 (for site R-KHn01) were incubated with His6-GenR for 30 min at 25°C. The labeled probe R-KHan02 was dissociated from the complex of His6-GenR and R-KHan02, using competitive binding to its unlabeled counterpart. This occurred not only when the cold probe R-KHan02 was at a concentration 100-fold or 150-fold higher than that of the labeled probe R-KHan02 but also when the other three cold probes were at the same concentrations (Fig. 6D). DFMan01 containing the GenR binding site DFMn01, DFMan02 (for site DFMn02), and R-KHan01 (for site R-KHn01) were used as probes. A His6-GenR-DNA complex corresponding to DFMan01, DFMan02, and R-KHan01 was partially dissociated using R-KHan02 as a cold probe, but the degree to which the His6-GenR-DNA complex corresponded to DFMan01, DFMan02, and R-KHan01 was increased by using DFMan01, DFMan02, and R-KHan01 as cold probes (Fig. 6A, B, and C). These results indicated that the affinity of His6-GenR to site R-KHan02 was the weakest among the four sites. A slight difference in affinity was observed between the His6-GenR-DNA complex containing probe DFMan01 and the complex containing R-KHan01 (Fig. 6A and C). In addition, binding reaction mixtures containing a 25-fold or 50-fold molar excess of competitor DNA were created to evaluate the affinity of His6-GenR to DFMan01 and R-KHan01 probes. Results showed that the affinity of His6-GenR to site DFMan01 was stronger than to site R-KHan01 (see Fig. S3B and C in the supplemental material). The different intensities of GenR-DNA complexes indicated that GenR probably has the most affinity for DFMn02, followed by DFMn01, R-KHn01, and R-KHn02, in that order.
GenR was shown to bind to sites DFMn01 and DFMn02 and to R-KHn01 and R-KHn02 with different levels of affinity. This raised the question of whether GenR binds to either pair via the formation of a higher-order complex. His6-GenR was found to bind to both DFMn01 and DFMn02 and to both R-KHn01 and R-KHn02. These binding arrangements were examined using EMSAs. However, the formation of the His6-GenR-DNA higher-order complexes was not observed in either case when the concentration of His6-GenR was gradually increased (Fig. 4A and B).
Role of the conserved sequence in GenR binding and transcriptional regulation of genDFM, genR, and genKH operons.
An imperfect palindromic ATTCC-N7(5)-GGAAT sequence was shown to be in the binding site of GenR, but it did not match the general consensus sequence found in other primary binding sites of the IclR-type regulator (1). To determine the importance of the palindromic ATTCC-N7(5)-GGAAT sequences in the GenR-binding sites, site-directed mutation was introduced into each GenR-binding site by changing the sites to GGCAA-N7(5)-AACGG. Probes containing either wild-type sites or mutated sites were used with purified His6-GenR in EMSAs. The data shown in Fig. 7A to D indicated that the consensus sequence ATTCC-N7(5)-GGAAT was an essential determinant of GenR binding activity.
Fig 7.

Mutational analyses of the four GenR binding sites. (A to D) EMSAs using the wild-type and mutated DNA fragments. Probes DFMan01, DFMan02, R-KHan01, and R-KHan02 contained the intact GenR binding sites DFMn01, DFMn02, R-KHn01, and R-KHn02, respectively. Probes DFMan01m, DFMan02m, R-KHan01m, and R-KHan02m contained the mutated sites described in Materials and Methods. The amounts (nM) of GenR used in lanes 1 to 5 are indicated. The free probes are indicated by open arrows, and the retarded DNA fragments are indicated by solid arrows. (E) β-Galactosidase activity driven by genDFM, genR, and genKH promoters with GenR binding sites in strain RES167. DFMa, Ra, and KHa are the wild-type GenR binding sites. DFMan01m, DFMan02m, DFMan12m, Ran02m, and KHan02m are the mutated binding sites. The β-galactosidase activity analyses were performed as described in the text. The data are derived from at least three independent measurements, and error bars indicate standard deviations.
In order to analyze the effects of mutation at DFMn01m, DFMn02m, R-KHn01m, and R-KHn02m on the transcription of each individual operon, the mutated consensus sequence [GGCAA-N7(5)-AACGG] was introduced into pZWHJ006 (containing the genDFM promoter), pZWHJ007 (containing the genR promoter), and pZWHJ008 (containing the genKH promoter) to replace its natural counterparts. This produced pZWHJ009, pZWHJ010, pZWHJ011, pZWHJ014, and pZWHJ015. These were transformed into strain RES167. Because previous DNase I footprinting and S1 mapping results indicated that the −10 and −35 regions of the transcription of genKH and the GenR binding site R-KHn01 overlapped, only pZWHJ014 and pZWHJ015 (both containing the mutated site R-KHn02m) were analyzed for β-galactosidase activity. As shown in Fig. 7E, approximately 60% β-galactosidase activity was lost in the presence of promoters containing DFMn01m or DFMn02m, and the activity was completely lost in the double mutant DFMn12m. A 4-fold β-galactosidase activity increase was observed with the genR promoter containing mutated site R-KHn02m (Fig. 7E). It was set at a similar level to the genR knockout strain (Fig. 2D). However, the level of activity was similar to that observed in the presence of the genKH promoter containing the mutated site R-KHn02m or its wild-type counterpart (Fig. 2C). This suggests that the transcription of genDFM was positively regulated by the GenR binding sites DFMn01 and DFMn02 and that the transcription of genR was negatively regulated by the GenR binding site R-KHn02. The transcription of genKH was not affected by the mutated GenR binding site R-KHn02 (Fig. 7E), indicating that it is positively regulated by R-KHn01 rather than R-KHn02.
DISCUSSION
The gen genes required for degradation of 3-HBA and GEN in C. glutamicum can be divided into three divergent and closely clustered operons (Fig. 1B). GenR positively regulates the expression of genDFM and genKH in the presence of 3-HBA and GEN in a GenR-dependent manner (Fig. 2). But it represses the expression of its own encoding gene (genR) without being affected by 3-HBA and GEN (Fig. 2). Like that of PobR (8) and PcaR (11), the autoregulation of GenR was shown to be independent of the coinducer molecules glucose, 3-HBA, and GEN. The genR operon encoding an IclR-type regulatory protein is located between the genDFM and genKH operons of the structural genes. This is notably different from the usual arrangement of operons encoding the GEN catabolic pathways in strains U2 (19) and M5a1 (21) or the operons that IclR family members use to regulate the degradation of aromatic compounds (1). NagR and MhbR have been reported to regulate the expression of enzymes involved in naphthalene and 3-HBA catabolic pathways, respectively, via gentisate. Both belong to the large family of LysR-type transcriptional regulators (19, 21). In addition to regulating their own expression, members of the IclR family generally regulate the expression of a single operon of structural genes, but they tend to do so in a different manner. Among the representative members of the IclR family (Fig. 8), GenR was found to be distinct from other members, showing that a peculiar arrangement and the formation of this arrangement can lead to the transcription of the three operons evaluated here.
Fig 8.

Localization of the regulator binding sites of PobR, PcaU, MhpR, PcaR, and GenR. TSSs and transcriptional directions of the target genes are indicated with the thin arrows. The −10 and −35 regions are marked, and the regulator primary binding sites are highlighted in black squares. The large arrows indicate the direction of transcription of each operon (filled, structural genes; open, regulatory genes).
IclR-type regulators typically take one of three formations when they mediate regulatory circuits. (i) The regulator uses the same binding sites to activate the transcription of the target genes and repress its own gene transcription, as shown in Fig. 8 for PobR (8) and PcaU (12). (ii) The binding sites activate transcription of the target genes only when the regulatory gene is constitutively expressed, as shown in Fig. 8 for MhpR (15). (iii) The regulator uses two binding sites for activation of the transcription of the target genes of a given operon and another binding site for repression of its own gene transcription, as shown in Fig. 8 for PcaR (11). However, none of these three phenomena applies to the GenR system, in which four binding sites regulate the transcription of three operons. The association of the GenR-DNA complex with probes containing site DFMn01, DFMn02, or R-KHn01 was affected by 3-HBA and GEN, and the probes containing site R-KHn02 was not (Fig. 4A and B; see also Fig. S3A in the supplemental material). Both GenR binding sites DFMn01 and DFMn02 are necessary for positive regulation of the transcription of genDFM, whereas the single binding site R-KHn01 is required for upregulation of the transcription of genKH. The GenR binding site R-KHn02 appeared to be involved in negative regulation of the activity of the genR promoter.
Members of the IclR family exert their diverse regulatory mechanisms by using different forms of binding. PobR binds to a single operator site in the pobA-pobR intergenic region containing three 8-bp inverted repeat sequences (8). The regulator PcaU binds a 45-bp sequence of three perfect 10-bp repeats, including two palindromic repeats and one direct repeat (12). PcaR binds to its own promoter at site R1 and at sites R2 and R1 of the pcaIJ promoter. A similar 15-bp target, R1, is centered in the −10 region of two promoters (11). The MhpR protects a region centered at position −58 with respect to the TSS of the Pa promoter, with a 17-bp imperfect palindromic motif (15). In the current study, a consensus motif [ATTCC-N7(5)-GGAAT] with a 5-bp imperfect palindromic sequence was identified within each of four GenR binding sites. This site was notably different from sites of the other IclR-type regulators, such as PobR (8), PcaU (12), PcaR (11), and MhpR (15). This palindromic motif in GenR binding sites is probably involved in sequence-specific recognition. Site-directed mutagenesis indicated that the palindromic sequence ATTCC-N7(5)-GGAAT was essential to GenR binding activity (Fig. 7A to D) and to expression of genDFM, genR, and genKH (Fig. 7E).
The three general mechanisms used for simple activation in bacterial regulatory studies have been summarized in two reviews (42, 43). In class I activation, the activator binds to sites located upstream of the promoter −35 element and recruits RNAP through direct interaction with the RNAP. In class II activation, the activator binds to sites that overlap the −35 element and contacts domain 4 of the RNAP σ subunit, which results in recruitment of RNAP to the promoter. This can affect other steps in the initiation process. Class III activation takes place in cases in which the binding of an activator may alter the conformation of the promoter and so enable the interaction of RNAP with the promoter −10 and/or −35 element. The NtrC-family PhhR is a global regulator which acts as both a repressor and an activator in Pseudomonas putida (44). Among the 13 PhhR regulons, the activator is potentially able to work via a class I- or a class II-like mechanism (45). The Escherichia coli catabolite activator protein (CAP) is also a global regulator that activates transcription at more than 100 promoters, which are generalizable to class I and class II CAP-dependent promoters (46, 47). Until the present study, no two of these mechanisms had ever been found to occur in a single IclR-family regulator. Here, both class I and class II activation of GenR was observed. GenR was found to bind three sites upstream of genDFM and genKH and to activate transcription of these genes. On the genKH promoter sequences, the GenR binding site R-KHn01 (located from position −47 to −16) was found to occlude the RNAP binding site from the −35 region (Fig. 5B and C), exhibiting a class II type activation. Although this overlap between the regulator binding site and the binding site of RNAP is unusual among bacterial transcriptional activators (42, 43), such interaction has been established in the studies of several regulatory proteins, including CAP (46, 47), PhhR (45), PcaR (11), MerR (48), and SoxR (49). The two GenR binding sites (DFMn01 and DFMn02) located from −41 to −84 are upstream of the −35 and −10 regions of the genDFM promoter (Fig. 5A), which is subject to class I activation. This presents a novel type of regulation for a single catabolic pathway.
Given the relatively higher affinity of DFMn02 to GenR than DFMn01, it can be proposed that GenR binds first to the more upstream binding site DFMn02, nucleating the occupation of the entire site at higher GenR levels, leading to the obstruction of RNAP binding. A similar case was reported for the involvement of JadR1 in jadomycin B biosynthesis in Streptomyces venezuelae (50). When a regulator binds to multiple sites for one regulated process, binding sometimes takes place at different rates at different sites. This is thought to be dependent on the affinity between the regulator and the binding sites (41, 51). Based on the relative affinities of GenR for different binding sites (Fig. 6), it is possible that GenR binds to R-KHn02 in the absence of 3-HBA and GEN and then to the R-KHn01 in the presence of both. When 3-HBA and GEN are present, GenR may first bind to DFMn02 and then to DFMn01. At these two sites, GenR can recruit RNAP to the promoter region of genDFM and then initiate transcription. It can be concluded that the binding of GenR to R-KHn01, DFMn02, and DFMn01 is critical to the transcriptional regulation of genDFM and genKH in the presence of 3-HBA and GEN induction.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant 31271333) and the National Key Basic Research Program of China (973 Program, grant 2012CB725202).
We are grateful to Shuang-Jiang Liu for strain RES167 ΔgenR. We thank Huarong Tan and Steve Busby for useful discussions.
Footnotes
Published ahead of print 25 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02216-12.
REFERENCES
- 1. Molina-Henares AJ, Krell T, Guazzaroni ME, Segura A, Ramos JL. 2006. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30:157–186 [DOI] [PubMed] [Google Scholar]
- 2. Yamamoto K, Ishihama A. 2003. Two different modes of transcription repression of the Escherichia coli acetate operon by IclR. Mol. Microbiol. 47:183–194 [DOI] [PubMed] [Google Scholar]
- 3. Gui LZ, Sunnarborg A, Pan B, LaPorte DC. 1996. Autoregulation of IclR, the gene encoding the repressor of the glyoxylate bypass operon. J. Bacteriol. 178:321–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sunnarborg A, Klumpp D, Chung T, Laporte DC. 1990. Regulation of the glyoxylate bypass operon: cloning and characterization of iclR. J. Bacteriol. 172:2642–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guazzaroni ME, Teran W, Zhang XD, Gallegos MT, Ramos JL. 2004. TtgV bound to a complex operator site represses transcription of the promoter for the multidrug and solvent extrusion TtgGHI pump. J. Bacteriol. 186:2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu D, Fillet S, Meng CX, Alguel Y, Kloppsteck P, Bergeron J, Krell T, Gallegos MT, Ramos J, Zhang XD. 2010. Crystal structure of TtgV in complex with its DNA operator reveals a general model for cooperative DNA binding of tetrameric gene regulators. Genes Dev. 24:2556–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tropel D, van der Meer JR. 2004. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 68:474–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiMarco AA, Ornston LN. 1994. Regulation of P-hydroxybenzoate hydroxylase synthesis by PobR bound to an operator in Acinetobacter calcoaceticus. J. Bacteriol. 176:4277–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiMarco AA, Averhoff B, Ornston LN. 1993. Identification of the transcriptional activator pobR and characterization of its role in the expression of pobA, the structural gene for p-hydroxybenzoate hydroxylase in Acinetobacter calcoaceticus. J. Bacteriol. 175:4499–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romero-Steiner S, Parales RE, Harwood CS, Houghton JE. 1994. Characterization of the pcaR regulatory gene from Pseudomonas putida, which is required for the complete degradation of p-hydroxybenzoate. J. Bacteriol. 176:5771–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo Z, Houghton JE. 1999. PcaR-mediated activation and repression of pca genes from Pseudomonas putida are propagated by its binding to both the −35 and the −10 promoter elements. Mol. Microbiol. 32:253–263 [DOI] [PubMed] [Google Scholar]
- 12. Popp R, Kohl T, Patz P, Trautwein G, Gerischer U. 2002. Differential DNA binding of transcriptional regulator PcaU from Acinetobacter sp. strain ADP1. J. Bacteriol. 184:1988–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trautwein G, Gerischer U. 2001. Effects exerted by transcriptional regulator PcaU from Acinetobacter sp. strain ADP1. J. Bacteriol. 183:873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manso I, Torres B, Andreu JM, Menendez M, Rivas G, Alfonso C, Diaz E, Garcia JL, Galan B. 2009. 3-Hydroxyphenylpropionate and phenylpropionate are synergistic activators of the MhpR transcriptional regulator from Escherichia coli. J. Biol. Chem. 284:21218–21228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torres B, Porras G, Garcia JL, Diaz E. 2003. Regulation of the mhp cluster responsible for 3-(3-hydroxyphenyl)propionic acid degradation in Escherichia coli. J. Biol. Chem. 278:27575–27585 [DOI] [PubMed] [Google Scholar]
- 16. Shen XH, Jiang CY, Huang Y, Liu ZP, Liu SJ. 2005. Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71:3442–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones DCN, Cooper RA. 1990. Catabolism of 3-Hydroxybenzoate by the gentisate pathway in Klebsiella pneumoniae M5a1. Arch. Microbiol. 154:489–495 [DOI] [PubMed] [Google Scholar]
- 18. Jeon CO, Park M, Ro HS, Park W, Madsen EL. 2006. The naphthalene catabolic (nag) genes of Polaromonas naphthalenivorans CJ2: evolutionary implications for two gene clusters and novel regulatory control. Appl. Environ. Microbiol. 72:1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jones RM, Britt-Compton B, Williams PA. 2003. The naphthalene catabolic (nag) genes of Ralstonia sp strain U2 are an operon that is regulated by NagR, a LysR-type transcriptional regulator. J. Bacteriol. 185:5847–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Y, Gao XL, Wang SH, Liu H, Williams PA, Zhou NY. 2012. MhbT is a specific transporter for 3-hydroxybenzoate uptake by Gram-negative bacteria. Appl. Environ. Microbiol. 78:6113–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin L-X, Liu H, Zhou N-Y. 2010. MhbR, a LysR-type regulator involved in 3-hydroxybenzoate catabolism via gentisate in Klebsiella pneumoniae M5a1. Microbiol. Res. 165:66–74 [DOI] [PubMed] [Google Scholar]
- 22. Xu Y, Wang SH, Chao HJ, Liu SJ, Zhou NY. 2012. Biochemical and molecular characterization of the gentisate transporter GenK in Corynebacterium glutamicum. PLoS One 7:e38701 doi:10.1371/journal.pone.0038701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Y-F, Zhang J-J, Wang S-H, Zhou N-Y. 2010. Purification and characterization of the ncgl2923-encoded 3-hydroxybenzoate 6-hydroxylase from Corynebacterium glutamicum. J. Basic Microbiol. 50:599–604 [DOI] [PubMed] [Google Scholar]
- 24. Tauch A, Kirchner O, Loffler B, Gotker S, Puhler A, Kalinowski J. 2002. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr. Microbiol. 45:362–367 [DOI] [PubMed] [Google Scholar]
- 25. Jakoby M, Ngouoto-Nkili CE, Burkovski A. 1999. Construction and application of new Corynebacterium glutamicum vectors. Biotechnol. Tech. 13:437–441 [Google Scholar]
- 26. Konopka A. 1993. Isolation and characterization of a subsurface bacterium that degrades aniline and methylanilines. FEMS Microbiol. Lett. 111:93–99 [Google Scholar]
- 27. Eggeling L, Reyes O. 2005. 23 Experiments, p 535–566. In Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL [Google Scholar]
- 28. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Tanaka Y, Okai N, Teramoto H, Inui M, Yukawa H. 2008. Regulation of the expression of phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) genes in Corynebacterium glutamicum R. Microbiology 154:264–274 [DOI] [PubMed] [Google Scholar]
- 31. Wang LH, Hamzah RY, Yu Y, Tu SC. 1987. Pseudomonas cepacia 3-hydroxybenzoate 6-hydroxylase: induction, purification, and characterization. Biochemistry 26:1099–1104 [DOI] [PubMed] [Google Scholar]
- 32. Lack L. 1959. The enzymic oxidation of gentisic acid. Biochim. Biophys. Acta 34:117–123 [DOI] [PubMed] [Google Scholar]
- 33. Bradford MM. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 34. Lin-Chao S, Bremer H. 1986. Effect of the bacterial-growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol. Gen. Genet. 203:143–149 [DOI] [PubMed] [Google Scholar]
- 35. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 36. Hellman LM, Fried MG. 2007. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2:1849–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boorstein WR, Craig EA. 1989. Primer extension analysis of RNA. Methods Enzymol. 180:347–369 [DOI] [PubMed] [Google Scholar]
- 38. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 39. Galas DJ, Schmitz A. 1978. DNAse footprinting: a simple method for detection of protein-DNA binding specificity. Nucleic Acids Res. 5:3157–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 41. Pan YY, Liu G, Yang HY, Tian YQ, Tan HR. 2009. The pleiotropic regulator AdpA-L directly controls the pathway-specific activator of nikkomycin biosynthesis in Streptomyces ansochromogenes. Mol. Microbiol. 72:710–723 [DOI] [PubMed] [Google Scholar]
- 42. Browning DF, Busby SJW. 2004. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2:57–65 [DOI] [PubMed] [Google Scholar]
- 43. Lee DJ, Minchin SD, Busby SJW. 2012. Activating transcription in bacteria. Annu. Rev. Microbiol. 66:125–152 [DOI] [PubMed] [Google Scholar]
- 44. Herrera MC, Duque E, Rodriguez-Herva JJ, Fernandez-Escamilla AM, Ramos JL. 2010. Identification and characterization of the PhhR regulon in Pseudomonas putida. Environ. Microbiol. 12:1427–1438 [DOI] [PubMed] [Google Scholar]
- 45. Herrera MC, Krell T, Zhang XD, Ramos JL. 2009. PhhR binds to target sequences at different distances with respect to RNA polymerase in order to activate transcription. J. Mol. Biol. 394:576–586 [DOI] [PubMed] [Google Scholar]
- 46. Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199–213 [DOI] [PubMed] [Google Scholar]
- 47. Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. 1996. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell 87:1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caslake LF, Ashraf SI, Summers AO. 1997. Mutations in the alpha and sigma-70 subunits of RNA polymerase affect expression of the mer operon. J. Bacteriol. 179:1787–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hidalgo E, Demple B. 1997. Spacing of promoter elements regulates the basal expression of the soxS gene and converts SoxR from a transcriptional activator into a repressor. EMBO J. 16:1056–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L, Tian X, Wang J, Yang H, Fan K, Xu G, Yang K, Tan H. 2009. Autoregulation of antibiotic biosynthesis by binding of the end product to an atypical response regulator. Proc. Natl. Acad. Sci. U. S. A. 106:8617–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weiss V, Claveriemartin F, Magasanik B. 1992. Phosphorylation of nitrogen regulator-I of Escherichia coli induces strong cooperative binding to DNA essential for activation of transcription. Proc. Natl. Acad. Sci. U. S. A. 89:5088–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns B, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Kramer R, Linke B, McHardy A, Meyer F, Mockel B, Pfefferle W, Puhler A, Rey D, Ruckert C, Rupp O, Sahm H, Wendisch V, Wiegrabe I, Tauch A. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.