

Abstract
Staphylococcus aureus exhibits a strong capacity to attach to abiotic or biotic surfaces and form biofilms, which lead to chronic infections. We have recently shown that Esp, a serine protease secreted by commensal Staphylococcus epidermidis, disassembles preformed biofilms of S. aureus and inhibits its colonization. Esp was expected to degrade protein determinants of the adhesive and cohesive strength of S. aureus biofilms. The aim of this study was to elucidate the substrate specificity and target proteins of Esp and thereby determine the mechanism by which Esp disassembles S. aureus biofilms. We used a mutant Esp protein (EspS235A) with defective proteolytic activity; this protein did not disassemble the biofilm formed by a clinically isolated methicillin-resistant S. aureus (MRSA) strain, thereby indicating that the proteolytic activity of Esp is essential for biofilm disassembly. Esp degraded specific proteins in the biofilm matrix and cell wall fractions, in contrast to proteinase K, which is frequently used for testing biofilm robustness and showed no preference for proteolysis. Proteomic and immunological analyses showed that Esp degrades at least 75 proteins, including 11 biofilm formation- and colonization-associated proteins, such as the extracellular adherence protein, the extracellular matrix protein-binding protein, fibronectin-binding protein A, and protein A. In addition, Esp selectively degraded several human receptor proteins of S. aureus (e.g., fibronectin, fibrinogen, and vitronectin) that are involved in its colonization or infection. These results suggest that Esp inhibits S. aureus colonization and biofilm formation by degrading specific proteins that are crucial for biofilm construction and host-pathogen interaction.
INTRODUCTION
Staphylococcus aureus is a major cause of community- and hospital-associated infectious diseases in humans, ranging from minor skin infections, such as furuncles and boils, to severe infections, such as pneumonia, osteomyelitis, endocarditis, and sepsis (1). Colonization of the nose by S. aureus represents a major risk factor for these infections (2). In recent years, the rapid emergence of virulent strains resistant to many antibiotics, such as methicillin-resistant S. aureus (MRSA), has become a major problem worldwide (3). S. aureus exhibits a strong capacity to attach to the surface of implanted medical devices and forms multilayered communities of bacteria called biofilms (4).
Biofilm formation proceeds in two distinct phases: primary attachment and proliferation (5–7). In the human body, the attachment directly to the surface of implanted medical devices or human extracellular matrix (ECM) and plasma proteins, such as fibronectin (Fn), fibrinogen (Fg), and vitronectin (Vn), is the first step of S. aureus biofilm formation (8). Then, proliferation proceeds through the production of biofilm matrices that contribute to bacterial accumulation in multiple layers. In staphylococcal biofilms, cells are embedded in extracellular matrices composed of proteins, polysaccharides (polysaccharide intercellular adhesin [PIA] or poly-N-acetylglucosamine [PNAG]), extracellular DNA (eDNA), and, presumably, host factors. Some surface proteins, such as major autolysin (Atl) (9, 10), biofilm-associated protein (Bap) (11), clumping factors (ClfA and ClfB) (12), ECM protein-binding protein (Emp) (13), Fn-binding proteins (FnBPA and FnBPB) (14, 15), S. aureus surface proteins (SasC and SasG) (16, 17), and staphylococcal protein A (Spa) (18), have been identified in S. aureus as being important in cell-to-cell and cell-to-surface interactions. Recently, β-toxin, a neutral sphingomyelinase, was found to have a structural role in S. aureus biofilm matrix, in cooperation with eDNA from lysed cells (19). Staphylococcal biofilms display extraordinary resistance to antimicrobial killing, limiting the efficacy of antibiotic therapy, and surgical intervention is often required to remove infected tissues or medical devices, such as catheters and orthopedic implants. S. aureus is therefore a frequent cause of biofilm-associated infections, which are a tremendous burden on our health care system. The prevention of biofilm-associated infections of S. aureus is a major goal of hospital infection control.
Recently, we have reported that Esp, a serine protease produced by a Gram-positive commensal bacterium, Staphylococcus epidermidis, inhibits S. aureus nasal colonization and biofilm formation and disassembles preformed S. aureus biofilms (20). This biofilm-degrading enzyme is effective against biofilms formed by MRSA and vancomycin-intermediate S. aureus (VISA). These findings attracted us to develop an innovative strategy for controlling the colonization and infection of the pathogen by using Esp or its derivatives. However, how Esp inhibits nasal colonization and biofilm formation of S. aureus and disassembles preformed biofilms has largely been unknown. Since Esp is an extracellular serine protease (20–23), we hypothesized that Esp degrades various proteins, including determinants for the adhesive (adhesion to abiotic or biotic surfaces) and cohesive (bacterium to bacterium) strength of S. aureus biofilms. The goal of the present study was to elucidate target proteins of Esp, including S. aureus surface proteins and host ECM proteins which are crucial for S. aureus colonization and biofilm formation.
MATERIALS AND METHODS
Proteins.
Fn and hemoglobin (Hg) from human, papain from Carica papaya, proteinase K from Tritirachium album, thermolysin from Bacillus thermoproteolyticus, Vn from human plasma, and protein A from S. aureus were purchased from Sigma. Fg from human plasma was from Wako. Recombinant S. epidermidis Esp was purified as previously described (23). Dispersin B from Aggregatibacter actinomycetemcomitans was gifted from Karen LoVetri (Kane Biotech Inc.).
Bacterial strains.
Bacterial strains used in this study are listed in Table S2 in the supplemental material. E. coli strains were grown at 37°C in LB medium containing appropriate antibiotics (100 μg/ml ampicillin). S. aureus strains were grown at 37°C in brain heart infusion (BHI) medium (Difco) or BHI medium supplemented with 1% glucose (BHIG).
Plasmid construction.
The efb gene was amplified from S. aureus MR23 genomic DNA using a primer set, Efb-BamHI-F and Efb-His-HindIII-R (see Table S3 in the supplemental material), and cloned into pNCMO2 (TaKaRa) using BamHI and HindIII restriction sites. The resulting plasmid was termed pEfb-His (see Table S2 in the supplemental material). The eap gene was amplified from S. aureus MR23 genomic DNA using a primer set, Eap-PstI-brevi-F and Eap-His-XbaI-R (see Table S3 in the supplemental material), and cloned into pNCMO2 using PstI and XbaI restriction sites. The resulting plasmid was named pEap-His (see Table S2 in the supplemental material). The S. aureus MR23 eap gene containing the 5′ untranslated region (UTR) was ligated with the gfpuv gene by splicing by overlap extension PCR (SOE-PCR) (24). In the first PCR, the eap and gfpuv genes were amplified using primer sets Eap-5′UTR-PstI-F/Eap-GFP-R and Eap-GFP-F/GFP-XbaI-R, respectively. The second PCR was performed to amplify the gfp-fused eap gene using these amplified fragments as the templates and a primer set: Eap-5′UTR-PstI-F/GFP-XbaI-R. The resulting fragment was digested by PstI and XbaI and was then cloned into pNCMO2 using PstI and XbaI restriction sites. The gfpuv gene was amplified from pGFPuv (Clontech). The DNA fragment containing sarA promoter P1 of S. aureus SH1000 was also ligated with the gfpuv gene by SOE-PCR using primers PsarA-PstI-F, PsarA-GFP-R, PsarA-GFP-F, and GFP-HindIII-R, and the resulting fragment was cloned into pNCMO2 using PstI and HindIII restriction sites. The constructed plasmids were named pEap-GFP and pP1GFP, respectively (see Table S2 in the supplemental material).
Purification of recombinant proteins.
Recombinant S. aureus proteins, including the extracellular adherence protein (Eap) fused with a C-terminal His6 tag and extracellular fibrinogen-binding protein (Efb) fused with a C-terminal His6 tag, were purified using a Brevibacillus expression-secretion system as previously described (23), with a minor modification for Eap. Eap was associated on the surface of Brevibacillus choshinensis cells and was therefore extracted with 1 M NaCl. The extract was subjected to Ni-affinity column chromatography. Purified recombinant proteins were dialyzed against phosphate-buffered saline (PBS) buffer containing 10% glycerol and stored at −80°C. Protein concentrations were determined using a protein assay kit (Bio-Rad).
Biofilm formation.
Single colonies of S. aureus strains were grown in 5 ml BHI medium at 37°C by shaking overnight. The overnight cultures were 1,000-fold diluted in 5 ml BHIG medium and then placed in 35-mm-diameter polystyrene dishes (Nunc). The cultures were incubated at 37°C for 24 h under static conditions. After the incubation, the dishes were gently shaken and the culture media were discarded. The dishes were then washed twice with PBS and dried under atmosphere.
Cell aggregation and biofilm formation of B. choshinensis cells were tested as follows: B. choshinensis cells harboring pNCMO2 (an empty vector), pEap-His, or pEfb-His (see Table S2 in the supplemental material) were grown in MT medium (1% glucose, 1% peptone, 0.5% meat extract, 0.2% yeast extract, 0.001% FeSO4 · 7H2O, 0.001% MnSO4 · 4H2O, 0.0001%, ZnSO4, 20 mM MgCl2) supplemented with 10 μg/ml neomycin (MTNm) at 30°C under shaking conditions at 150 rpm for 4 days. The cultures were incubated in standing glass tubes at room temperature for 3 h, and cell aggregations were observed. For the biofilm formation assay, these strains were grown in MTNm at 30°C under shaking conditions at 150 rpm for 2 days. The cultures were then diluted 100-fold in fresh MTNm, and the diluted cultures (200 μl) were incubated at 30°C under static conditions in 96-well polystyrene plates (Corning) for 2 days. After the incubation, the plates were gently shaken to remove the deposited bacteria and the culture media were discarded. After twice washing with PBS, biofilms were stained with 0.25% safranin and eluted with 70% ethanol (200 μl). The absorbance at 492 nm of the ethanol extract was measured using a microplate reader (Tecan). Assays were performed in triplicate.
Biofilm destruction assay.
Biofilms of various S. aureus strains formed in 35-mm-diameter polystyrene dishes were incubated with the indicated proteases, 5 μg/ml Esp, 5 μg/ml thermolysin, 5 μg/ml proteinase K, and 5 μg/ml papain, in 100 mM Tris-HCl (pH 8.0) at 37°C for the indicated periods. After the incubation, the dishes were gently shaken and the supernatants were discarded. The dishes were then washed twice with 10 mM Tris-HCl (pH 8.0) and dried under atmosphere. The pictures of the dishes were taken by digital camera. Profiles of the fractionated proteins were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) with Coomassie brilliant blue (CBB) staining.
Isolation of biofilm matrix proteins, cell wall-anchoring proteins, and cytoplasmic proteins.
S. aureus proteins were fractionated into biofilm matrix, cell wall, and cytoplasmic fractions. Biofilms of various S. aureus strains formed in the test dish were detached by mixing with washing buffer composed of 10 mM Tris-HCl (pH 8.0) and protease inhibitor cocktail (Nacalai) and transferred into a test tube. If required, the biofilm was mechanically peeled away from the surface of the test dish. The yielded biofilm was shaken by vortexing and centrifuged at 5,000 × g for 10 min (washing fraction). The supernatant was transferred into a new test tube. The pellet was dissolved in a matrix extraction buffer composed of 10 mM Tris-HCl (pH 8.0), 1 M NaCl, and protease inhibitor cocktail, and the mixture was incubated at 25°C for 30 min with gentle rotation (biofilm matrix fraction). After the incubation, the mixture was centrifuged at 5,000 × g for 10 min and the supernatant was transferred into a new test tube. The fractionated proteins were analyzed by SDS-PAGE. If required, dispersin B (0.2 mg/ml) was added to the biofilm matrix fraction to check for components of the insoluble materials in the fraction. The pellet was dissolved in a cell wall-anchored protein extraction buffer composed of 10 mM Tris-HCl (pH 8.0), 25% sucrose, 0.2 mg/ml lysostaphin, and protease inhibitor cocktail and incubated at 37°C for 30 min under static conditions. After the incubation, the mixture was centrifuged at 15,000 × g for 10 min and the supernatant was transferred into a new test tube (cell wall fraction). For the analysis of cytoplasmic proteins, the pellet was dissolved in 10 mM Tris-HCl (pH 8.0) and protease inhibitor cocktail and sonicated. After centrifugation at 15,000 × g for 10 min, cytoplasmic soluble proteins were recovered (cytoplasmic fraction).
Cell surface proteins, which were degraded by various proteases, were also fractionated from S. aureus biofilms and analyzed by SDS-PAGE as described above.
Scanning electron microscopy.
Structures of S. aureus SH1000 biofilms with or without 1 M NaCl treatment and the NaCl-extracted biofilm matrix were observed by scanning electron microscopy according to the method previously reported (25).
Cell aggregation and biofilm promotion by addition of the NaCl extract from S. aureus biofilms.
S. aureus MS4 was grown in BHI medium at 37°C overnight. The overnight culture was diluted 50-fold in BHI medium supplemented with or without 1 M NaCl extract (100 μl) from SH1000 biofilms as described above. NaCl (1 M, 100 μl) was also used instead of the NaCl extract. These diluted cultures were incubated at 37°C for 1 h in 35-mm-diameter polystyrene dishes. After the incubation, the dishes were gently shaken and the culture media were discarded. The plates were then washed twice with PBS buffer and dried under atmosphere. The remaining biofilms were stained with 0.1% crystal violet.
Identification of proteins.
Cell surface proteins (biofilm matrix and cell wall fractions) isolated from S. aureus MR23 biofilms were separated by one-dimensional (1D) SDS-PAGE. The major protein bands with apparent molecular masses of 60 kDa and 15 kDa were identified by N-terminal amino acid sequence analyses as described previously (20). Top-down proteomic analysis was also performed to identify proteins that are degraded by Esp (details are described in the supplemental material).
Promotion of S. aureus biofilm formation by exogenously supplemented recombinant proteins.
Single colonies of S. aureus MS4 were grown in 5 ml BHI medium at 37°C overnight. The overnight cultures were 1,000-fold diluted in 5 ml BHIG medium supplemented with the indicated concentrations (0.05 to 1 μM) of purified Eap and Efb, and the diluted cultures were then placed into 96-well polystyrene plates. The plates were incubated at 37°C for 24 h under static conditions. After the incubation, the plates were gently shaken and the culture media were discarded. The plates were then washed twice with PBS buffer and dried under atmosphere. The remaining biofilms were stained with 0.1% crystal violet, and the absorbance at 595 nm was monitored using a microplate reader (Tecan). Assays were performed in triplicate.
Western blotting.
Eap in the biofilm matrix fraction of S. aureus MR23 was detected by Western blotting. Anti-Eap polyclonal antibody (primary antibody; Abcam) and goat anti-rabbit IgG-horseradish peroxidase (HRP; secondary antibody; Bio-Rad) were diluted 1,000- and 200,000-fold, respectively, in Tris-buffered saline (TBS)–Tween (TBS-T; 20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.1% [vol/vol] Tween 20). Samples were separated by SDS-PAGE and transferred to an Immobilon-P membrane (Millipore Corp.). To block unspecific interactions with IgG-binding proteins, the membrane was incubated for 1 h with 5% (vol/vol) goat serum (Sigma) dissolved in TBS buffer containing 0.3% (wt/vol) bovine serum albumin (BSA; Sigma) as previously described (26). Spa in S. aureus MR23 biofilm matrix and cell wall fractions was detected using rabbit IgG (Sigma) and goat anti-rabbit IgG-HRP (Bio-Rad). The primary antibody and secondary antibody were diluted 20,000-fold in TBS-T. Samples were separated and transferred as mentioned above. The membranes were incubated for 1 h with 3% BSA (Sigma) in TBS-T. The membrane was treated with the primary antibody at 25°C for 1 h and then washed twice with TBS-T. Subsequently, the membrane was incubated in TBS-T containing the secondary antibody. After washing three times with TBS-T, the immunoreactive signals were detected with an ECL-plus detection kit (GE Healthcare).
Degradation of purified proteins by Esp in vitro.
Purified staphylococcal proteins, including Eap and Spa, and human proteins, including Fg, Fn, Vn, and Hg (1 μM each), were incubated with or without 1 μM Esp in PBS buffer at 37°C. At the indicated time points, aliquots (10 μl) of the solutions were collected and mixed with 10 μl of 2× SDS sample buffer (150 mM Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 10% 2-mercaptoethanol). Proteins were analyzed by SDS-PAGE (12% gel) and visualized with CBB staining. If required, the band intensity on the gel was estimated using an ImageQuant system (GE Healthcare).
Fluorescence microscopy.
S. aureus RN4220 (27) was used for modification of recombinant plasmids generated in E. coli. S. aureus MR23 was transformed with pEap-GFP or pP1GFP, both of which were extracted and purified from RN4220. These transformants were inoculated into BHI medium containing 50 μg/ml neomycin and cultured at 37°C overnight. The overnight cultures were 1,000-fold diluted in BHIG medium supplemented with 50 μg/ml neomycin and further incubated at 37°C under static conditions. At the indicated times, cell morphology and localization of Eap-GFP were observed by fluorescence microscopy with a green fluorescent protein (GFP) filter.
RESULTS
A high concentration of sodium chloride triggers the detachment of biofilm matrix from S. aureus cells.
Fractionation of cell surface proteins in S. aureus biofilms is necessary to determine which proteins are degraded by Esp and thereby to figure out how Esp destructs S. aureus biofilms. The cell surface proteins are composed of cell surface-associated proteins, including biofilm matrix proteins and cell wall-anchored proteins harboring an LPXTG motif. To date, cell wall-anchored proteins of S. aureus biofilms have been extensively examined by proteomic approaches (28–30), but biofilm matrix proteins are largely unknown. It has been reported that a high concentration of salt (1 M NaCl) induces the release of TasA, a biofilm matrix protein, from Bacillus subtilis cells (31). Here, we also used 1 M NaCl to detach biofilm matrix components from S. aureus cells. First, we prepared biofilms formed by S. aureus SH1000 (32), a laboratory strain frequently used for biofilm assays, and compared the surface structure of intact biofilms and that of 1 M NaCl-treated biofilms by scanning electron microscopy. As shown in Fig. 1A, SH1000 cells were embedded in adherent materials in the biofilm. After gently mixing with 1 M NaCl, surface-adherent materials were removed and the cell surface became more obvious (Fig. 1B). In addition, networks of fibrillar structure were confirmed in a 1 M NaCl-extracted sample by scanning electron microscopy (Fig. 1C). Notably, several proteins and insoluble materials that were stacked in the well of an SDS-polyacrylamide gel were observed in the biofilm matrix fraction (Fig. 1D, lane 3). The insoluble materials were degraded by dispersin B (Fig. 1E), a polysaccharide-degrading enzyme (33), but not proteinase K and DNase I, indicating that the biofilm matrix of SH1000 contains polysaccharides. This is consistent with the susceptibility of the SH1000 biofilm to dispersin B. Next, we examined whether 1 M NaCl extracts from S. aureus SH1000 are responsible for biofilm formation and cell aggregation. The extracts were added into the culture of S. aureus MS4, a clinically isolated methicillin-sensitive S. aureus (MSSA) strain. MS4 alone formed less biofilm, and buffer containing 1 M NaCl did not affect its biofilm formation (Fig. 1F). The addition of 1 M NaCl extract from SH1000 biofilms triggered the cell aggregation and partial biofilm formation of MS4 (Fig. 1F), suggesting that the treatment with a high concentration of NaCl enabled us to detach and yield S. aureus biofilm matrices. In addition, as described below, biofilm matrix proteins were identified by this technique combined with mass spectrometry. This is the first time that biofilm matrix proteins have been identified using a systems biology approach.
Fig 1.
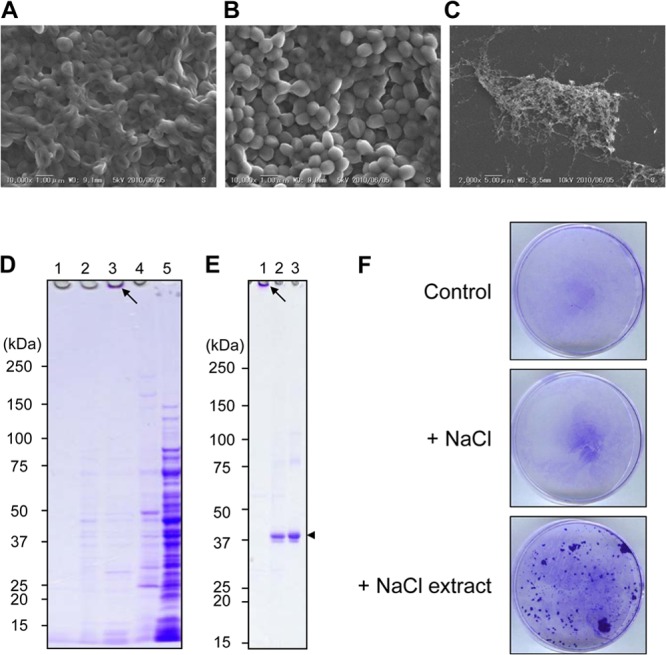
Isolation of biofilm matrix proteins and cell wall-anchored proteins from S. aureus biofilms. The surface structures of biofilms produced by S. aureus SH1000 without (A) and with (B) 1 M NaCl treatment were observed by scanning electron microscopy. (C) The structure of the NaCl-extracted biofilm matrices from the SH1000 biofilm was also analyzed. (D) The culture supernatant fraction (lane 1), 10 mM Tris-HCl (pH 8.0) washing fraction (lane 2), 1 M NaCl-extracted fraction (lane 3), cell wall fraction obtained by lysostaphin treatment (lane 4), and cytoplasmic fraction yielded after sonication (lane 5) were dissolved by SDS-PAGE and stained with CBB. An arrow indicates insoluble materials stacked on the well. (E) A 1 M NaCl extract from the SH1000 biofilm was treated without (lane 1) or with (lane 2) 0.2 mg/ml dispersin B and then subjected to SDS-PAGE. Dispersin B was also subjected to SDS-PAGE as a control (lane 3). The arrow indicates insoluble materials stacked on the well, and the arrowhead represents dispersin B. In panels D and E, molecular sizes are given at the left. (F) The effect of 1 M NaCl extracts from biofilms on cell aggregation and attachment to an abiotic surface was examined. S. aureus MS4, which forms less biofilm, was incubated at 37°C for 1 h in the absence or presence of a 1 M NaCl extract from SH1000 biofilms. Cells attached to polystyrene dishes were stained with crystal violet. NaCl solution (1 M) instead of 1 M NaCl extract was added as a negative control.
Profiles of biofilm matrix proteins and cell wall-anchored proteins of S. aureus clinical isolates.
We examined the profiles of biofilm matrix proteins and cell wall-anchored proteins from a laboratory MSSA strain (SR6, a derivative of JCM 2874), clinically isolated MRSA strains (MR10 and MR23), and MSSA strains (MS4 and MS10) by 1D SDS-PAGE analyses. Interestingly, the profiles of proteins in the biofilm matrix (Fig. 2A) and cell wall fractions (Fig. 2B) were distinct among the strains tested. The biofilm matrices of SR6 and MR10, both of which form a dispersin B-sensitive biofilm (F. Sato et al. unpublished data), contained insoluble materials (Fig. 2A), as seen in the SH1000 biofilm (Fig. 1D, lane 3). Remarkably, the biofilm matrix of MR23, which forms proteinase K-susceptible and dispersin B- and DNase I-resistant biofilms (Sato et al. unpublished), contained two abundant proteins with molecular masses of approximately 60 kDa and 15 kDa (Fig. 2A). The CBB-stained protein bands were then dissected and subjected to N-terminal amino acid sequence analysis. Homology searches based on the retrieved amino acid sequences (60-kDa band, A-A-K-P-L-D-K-S-S-S-X-L; 15-kDa band, S-E-G-Y-G-P-X/P-E-K-/E-K-P/K-V/P-V; X represents any amino acid) revealed that the predominant 60-kDa and 15-kDa proteins recovered were the extracellular adhesion protein (Eap), also known as the major histocompatibility complex (MHC) class II analog protein (Map), and the extracellular fibrinogen-binding protein (Efb), respectively. In contrast to the biofilm matrices from SH1000, SR6, and MR10, the biofilm matrix of MR23 lacked insoluble materials stacked on the top of an SDS-polyacrylamide gel. In addition, MS4 and MS10, which form proteinase K- and DNase I-sensitive and dispersin B-resistant biofilms, also did not contain them. Therefore, the existence of the insoluble materials stacked on the top of the gel could be one of the hallmarks of biofilm matrices composed of polysaccharides. To identify Esp target proteins, strain MR23 was used as described below, since this strain forms the most robust biofilm among the proteinaceous biofilm producers examined.
Fig 2.
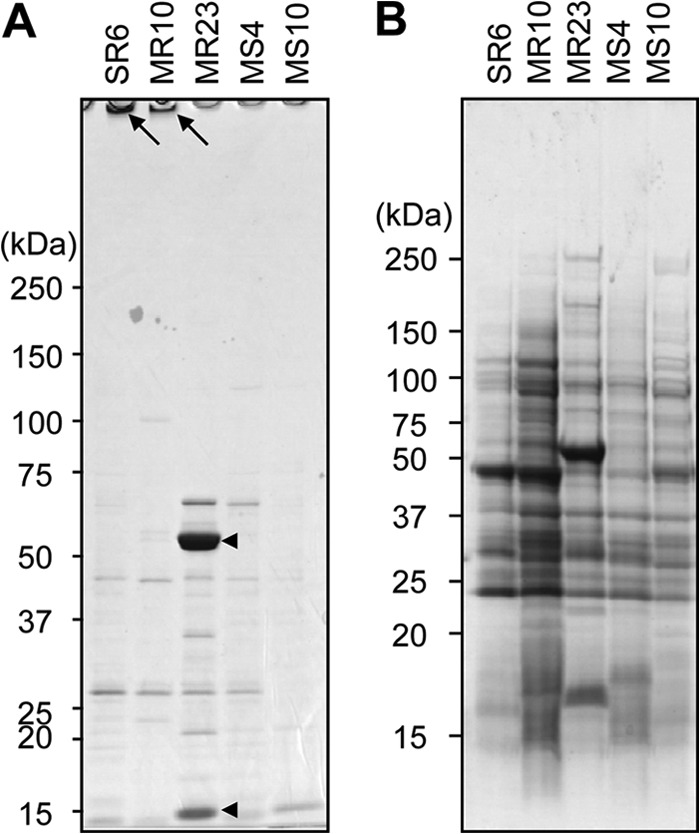
Profiles of biofilm matrix and cell wall-anchored proteins isolated from S. aureus biofilms. (A) Biofilm matrices were extracted from the indicated S. aureus strains by 1 M NaCl treatment and analyzed by SDS-PAGE. Arrows, insoluble materials stacked on the well; arrowheads, Eap (60 kDa) and Efb (15 kDa), which were determined by N-terminal amino acid sequencing. (B) Cell wall-anchored proteins were isolated from the indicated S. aureus strains after lysostaphin treatment and centrifugation. The supernatants were analyzed by SDS-PAGE. Molecular sizes are given at the left.
Degradation of specific proteins is required for Esp-dependent S. aureus biofilm disassembly.
Previously, we showed using amidinophenylmethanesulfonyl fluoride (APMSF), a serine protease inhibitor, that proteolytic activity is involved in biofilm disassembly by Esp purified from S. epidermidis culture supernatant (20). To more carefully examine whether proteolytic activity is involved in biofilm disassembly by Esp, we used a mutant Esp protein (EspS235A), which lacks proteolytic activity (34) (see Fig. S1A in the supplemental material). As shown in Fig. 3A and Fig. S1B in the supplemental material, wild-type Esp (EspWT) disassembled biofilms formed by MR23, while EspS235A did not, thereby indicating that the proteolytic activity of Esp is essential for biofilm disassembly. Next, we treated preformed MR23 biofilms using various proteases, including thermolysin (metalloprotease), proteinase K (serine protease), and papain (cysteine protease), in addition to Esp (serine protease). MR23 biofilms were disassembled by the addition of all proteases tested (Fig. 3A). Cell surface proteins (biofilm matrix and cell wall fractions) degraded by these proteases were examined according to the procedure described above (Fig. 1 and 2). Interestingly, proteinase K and papain degraded proteins in the biofilm matrix fraction almost completely, whereas EspWT and thermolysin degraded certain specific proteins (Fig. 3B). These differences were not due to the level of proteolytic activity (see Fig. S2 in the supplemental material). Of note, EspWT degraded certain proteins in the biofilm matrix fraction (200 kDa, 85 kDa, and 70 kDa) (Fig. 3C). Degradation of cell wall-anchored proteins by these proteases showed similar patterns; EspWT and thermolysin degraded several specific proteins, while proteinase K and papain hydrolyzed many more proteins (Fig. 3D; see Fig. S3 in the supplemental material). EspWT significantly degraded a protein with a high molecular mass (200 kDa) in the cell wall fraction (Fig. 3D).
Fig 3.

Biofilm destruction triggered by various proteases. (A) Biofilms produced by MR23 were destructed by wild type Esp (EspWT), protease-defective mutant Esp (EspS235A), and the indicated proteases (5 μg/ml) at 37°C for 24 h. Biofilms without protease treatment (control) are also shown. (B) Proteins in the biofilm matrix fractions with or without treatment with the indicated proteases were isolated and analyzed by SDS-PAGE, as shown in Fig. 1. (C) The time-dependent degradation of proteins in the biofilm matrix fraction by EspWT was analyzed. As a control, PBS was added instead of EspWT, and the biofilm was incubated at 37°C for 8 h. Molecular sizes are given at the left. (D) The time-dependent degradation of cell wall-anchored proteins by EspWT was also analyzed as described for panel C. Arrowheads in panels C and D, proteins that were significantly degraded by EspWT.
Esp degrades biofilm-associated proteins in the biofilm matrix fraction of S. aureus.
Proteins, including ones specifically degraded by EspWT in the MR23 biofilm, were identified by a proteomic analysis (see Fig. S4 in the supplemental material). Proteins in the biofilm matrix fractions before and after treatment with EspWT were subjected to one-dimensional SDS-PAGE. All visible protein bands in the sample without the treatment with EspWT were cut out, and the proteins were extracted and identified by nano-liquid chromatography/tandem mass spectrometry (nano-LC-MS/MS) (see Fig. S4A in the supplemental material). Reference gels for the sample treated with Esp were also analyzed (see Fig. S4B in the supplemental material). Identified proteins with similar migration in SDS-polyacrylamide gels were compared between these two samples (e.g., see band 1A versus 1B and band 2A versus 2B in Fig. S4 in the supplemental material), and proteins that were identified only in the sample without the treatment with EspWT were determined to be target proteins of Esp. As summarized in Table S1 in the supplemental material, EspWT degraded at least 74 proteins in the biofilm matrix, including 10 secreted proteins harboring a secretion signal, 8 membrane proteins, 3 cell wall-anchoring proteins containing an LPXTG motif, 45 cytoplasmic proteins, and 8 proteins whose localization has not been determined. Among the proteins that were identified as targets of Esp in the biofilm matrix fraction, Atl (9, 10), β-toxin (19), Emp (13), FnBPA (14, 15), and Spa (18) have been found to be responsible for S. aureus biofilm formation (Table 1). Spa and FnBPA, cell wall-anchoring proteins harboring an LPXTG motif, are thought to be released from the cell wall by cell lysis, proteolytic processing, or unknown mechanisms and attach to the cell surface by noncovalent bonds during biofilm development. These biofilm-associated proteins play important roles in biofilm development as the glue in cell-to-cell and cell-to-surface interactions. It is therefore reasonable to consider that a primary mechanism of S. aureus biofilm disassembly triggered by Esp is proteolytic degradation.
Table 1.
Biofilm- or infection-associated proteins that are degraded by Esp in S. aureus MR23 biofilm
| Protein name | Description | LPXTG motifa | Length (no. of amino acids) | Molecular mass (Da) | Extracellular function | Reference(s) or source |
|---|---|---|---|---|---|---|
| SdrD | Serine-aspartate repeat-containing protein D | + | 1,381 | 149,448 | Adhesion, abscess formation | 35, 36 |
| Atl | Bifunctional autolysin | − | 1,248 | 136,751 | Biofilm formation, adhesion | 9, 10, 37 |
| FnBPA | Fibronectin-binding protein A | + | 1,018 | 111,707 | Biofilm formation, adhesion | 10, 14, 15, 38 |
| Eap (Map) | Extracellular adherence protein (MHC class II analog protein) | − | 689 | 77,047 | Biofilm formation, adhesion, anti-inflammatory, antiangiogenic, abscess formation | This study, 36, 39–42 |
| Spa | Immunoglobulin G-binding protein A | + | 520 | 56,833 | Biofilm formation, immune evasion, abscess formation | 18, 35, 43 |
| Sbi | Immunoglobulin-binding protein | − | 436 | 50,012 | Complement evasion | 44 |
| IsdA | Iron (Fe2+) ABC superfamily ATP binding cassette transporter | + | 350 | 38,793 | Adhesion, abscess formation | 35, 36, 45 |
| Emp | Extracellular matrix protein-binding protein | − | 340 | 38,495 | Biofilm formation, adhesion, abscess formation | 13, 36, 46 |
| Hlb | β-Hemolysin/β-toxin | − | 340 | 33,036 | Biofilm formation | 19 |
| SceD | Probable transglycosylase | − | 231 | 24,077 | Nasal colonization | 47 |
| Efb | Fibrinogen-binding protein | − | 165 | 18,764 | Adhesion, immune evasion | 48 |
+, presence; −, absence.
Eap forms a biofilm matrix architecture and contributes to biofilm development.
Eap was the most abundant protein in the biofilm matrix fraction of MR23, which was identified by N-terminal amino acid sequencing (Fig. 2A). Eap was also determined to be a target of Esp, since it was identified in the sample of the biofilm matrix fraction without EspWT treatment (see bands 1A, 2A, 6A, 7A, 12A, and 13A in Fig. S4 in the supplemental material) but not in that with the treatment (see bands 1B, 2B, 6B, 7B, 12B, and 13B in Fig. S4 in the supplemental material; summarized in Table S1 in the supplemental material). However, the band intensity of Eap stained with CBB was changed slightly from before to after the EspWT treatment (compare band 4A to band 4B in Fig. S4 in the supplemental material). To assess more precisely whether EspWT degrades Eap, we examined its degradation in vitro using purified recombinant Eap (Fig. 4A and B) and in the biofilm using an anti-Eap polyclonal antibody (Fig. 4C and D). Eap was gradually degraded within 24 h in vitro and in the biofilm, indicating that Eap is a target of Esp.
Fig 4.

Esp-dependent degradation of Eap in vitro and in S. aureus biofilm. (A) Purified recombinant Eap (1 μM) was incubated in the presence of EspWT at 37°C for 24 h. As a control, PBS was added instead of EspWT. (B) Band intensities on the gel (A) were estimated using the ImageQuant system. Data points represent the means and standard deviations of results from three independent experiments. The standard deviation is less than the size of the symbol if no error bars are seen. (C) The time-dependent degradation of Eap by EspWT in the biofilm was analyzed by SDS-PAGE with CBB staining and Western blotting using an anti-Eap polyclonal antibody. As a control, PBS was added instead of Esp. Molecular sizes are given at the left. (D) Band intensities on the X-ray film (C) were estimated using ImageQuant. The means and standard deviations of triplicate determinations are represented.
To address the role of Eap in biofilm formation, we used a Brevibacillus choshinensis expression-secretion system which enables us to produce a particular protein in the extracellular environment (23). As shown in Fig. 5, extracellular production of recombinant Eap fused to a C-terminal His6 tag triggered cell aggregation and biofilm formation of B. choshinensis cells, whereas Efb (48), the other abundant protein in the biofilm matrix fraction of MR23 (Fig. 2A), did not. A large amount of recombinant Eap was purified from the cell-associated fraction (Fig. 5C), indicating that Eap was predominantly associated with the surface of B. choshinensis cells. In addition, the purified recombinant Eap was supplemented into medium at the beginning of biofilm formation of S. aureus MS4, which forms less biofilm and also shows less extracellular proteolytic activity. As shown in Fig. 5D, Eap stimulated the biofilm formation of S. aureus MS4 in a dose-dependent manner, while recombinant Efb only slightly affected the biofilm formation. These results clearly indicate that Eap potentially contributes to biofilm formation not only by S. aureus but also by B. choshinensis.
Fig 5.
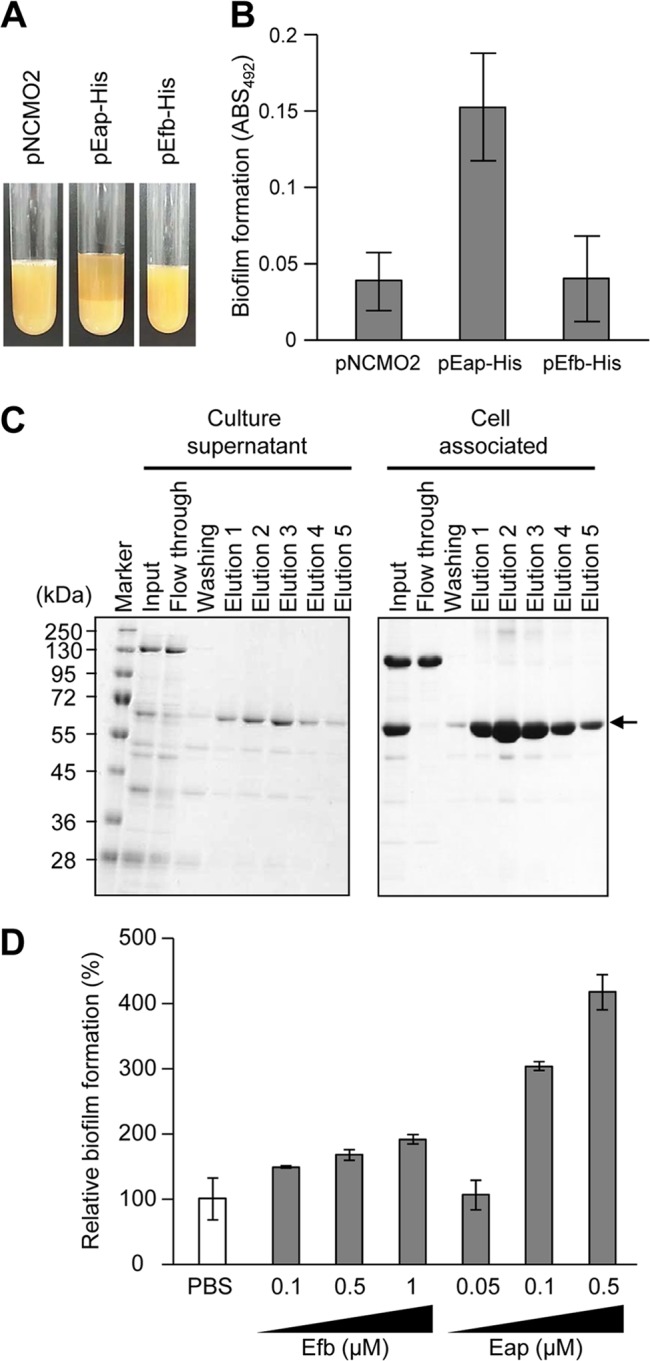
Stimulation of biofilm formation by heterologous expression or supplementation of recombinant Eap. (A) Aggregation of Brevibacillus choshinensis cells harboring pEap-His or pEfb-His was observed. B. choshinensis cells harboring pNCMO2 (an empty vector) were used as a control. (B) The biofilm formation of B. choshinensis cells harboring pEap-His was compared with that of B. choshinensis cells harboring pNCMO2 or pEfb-His. Biofilms were stained with safranin and estimated by measuring the absorbance at 492 nm (ABS492). The means and standard deviations of triplicate determinations are represented. (C) C-terminally His-tagged Eap was purified from culture supernatant and cell-associated fractions using a nickel-affinity resin. Arrow, recombinant Eap. Molecular sizes are given at the left. (D) The indicated concentrations of purified Eap and Efb were supplemented into BHIG medium at the initial stage of biofilm formation by S. aureus MS4, which forms less biofilm under the tested conditions. After incubation at 37°C for 24 h, biofilms were stained with crystal violet and estimated by measuring the absorbance at 595 nm. As a control, PBS was used instead of Eap (defined as 100%). The means and standard deviations of triplicate determinations are represented.
In order to visualize the cellular localization of Eap in S. aureus biofilms, C-terminal green fluorescent protein-fused Eap (Eap-GFP) was expressed in MR23 biofilms. As a control, GFPuv was also expressed using the same plasmid. GFPuv spread inside the cell (Fig. 6A), whereas Eap-GFP localized on the cell surface after 1 day (Fig. 6B). A similar cellular localization pattern of Eap was observed by immunofluorescence microscopy (see Fig. S5 in the supplemental material). Interestingly, an increased amount of Eap-GFP localized on the cell surface and formed biofilm matrix structures after 2 days. To our knowledge, this is the first time that biofilm matrix architectures composed of Eap have been visualized during S. aureus biofilm formation.
Fig 6.
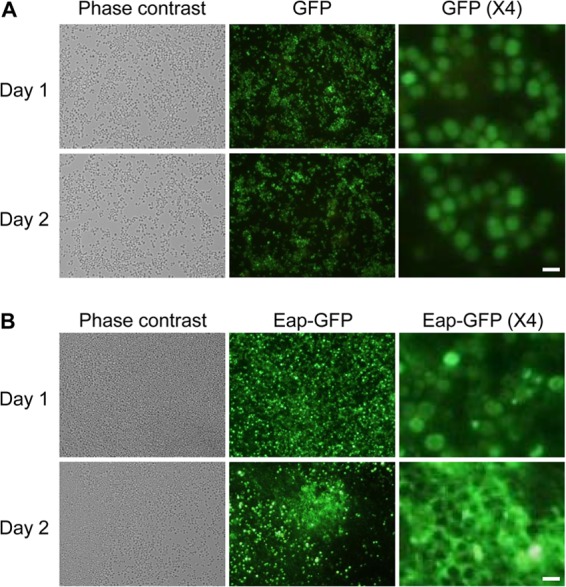
Eap is a component of the S. aureus biofilm matrix. The cell morphology and GFP fluorescence of S. aureus MR23 cells harboring pP1GFP (A) and pEap-GFP1 (B) were observed during biofilm formation (day 1 and day 2) by phase-contrast and fluorescence microscopy. Fourfold-magnified images taken under a GFP filter are also shown on the right. Bars, 1 μm.
Esp degrades specific proteins in the cell wall fraction of S. aureus.
Proteins degraded by EspWT in the cell wall fraction of MR23 biofilms were also identified by a similar procedure for the biofilm matrix fraction. As shown in Fig. S6 in the supplemental material, FnBPA and the Ser-Asp repeat-containing protein D (SdrD) (35) were identified as target proteins of Esp. Spa was detected as a major cell wall-anchored protein, but no significant degradation was observed after the EspWT treatment (see Fig. S6 in the supplemental material). In contrast, non-cell wall-anchored Spa (in the biofilm matrix fraction) was degraded by EspWT, as mentioned above (see Table S1 in the supplemental material). This discrepancy suggested that cell wall-anchored Spa is not degraded by Esp in the biofilm. To address this possibility more precisely, Western blotting was performed using cell wall and biofilm matrix fractions from MR23 biofilms treated with EspWT. The Spa in the biofilm matrix fraction was degraded immediately after the addition of EspWT, while that in the cell wall fraction was not (Fig. 7A). Purified Spa was also degraded by EspWT in vitro (Fig. 7B and C), suggesting that cell wall-anchored Spa is protected in the biofilm from the proteolytic action of Esp, presumably by a mechanism in which its processing sites are embedded in cell wall or biofilm matrix materials.
Fig 7.
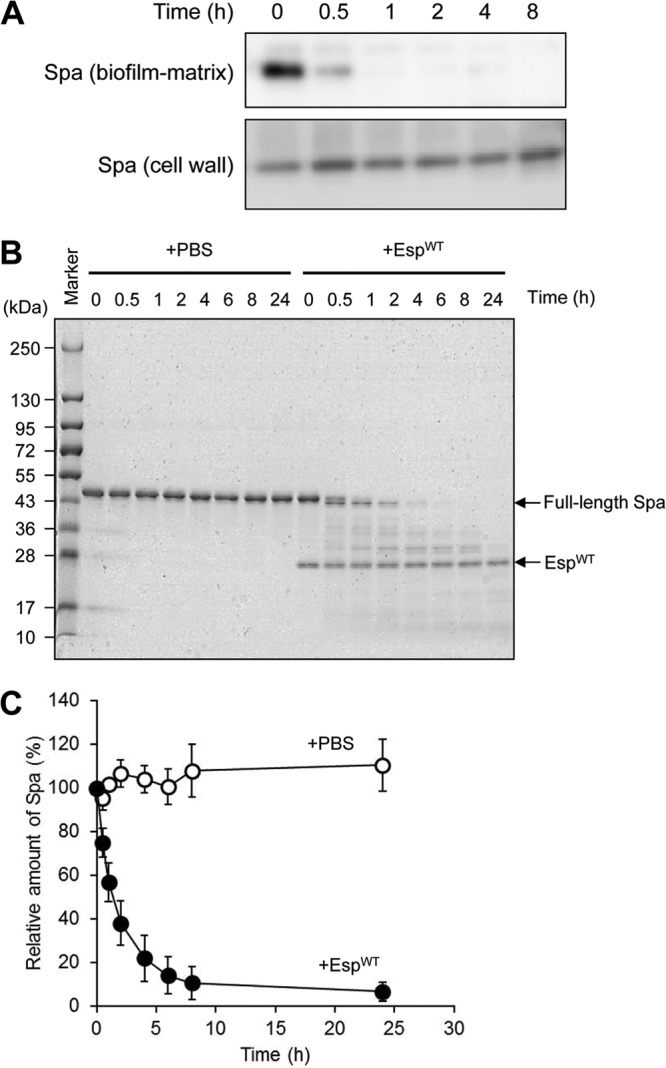
Degradation of Spa by Esp. (A) Degradation of Spa in the biofilm matrix and cell wall-anchored fractions from EspWT-treated biofilms of S. aureus MR23 were examined by Western blotting. (B) Purified Spa was degraded by EspWT in vitro. Molecular sizes are given at the left. (C) Band intensities on the gel (B) were estimated using ImageQuant. Data points represent the means and standard deviations of results from three independent experiments. The standard deviation is less than the size of the symbol if no error bars are seen.
Esp degrades human ECM and plasma proteins important for host-pathogen interaction.
S. aureus is a pathogen of medical device-associated infection. Host ECM and plasma proteins cover the devices soon after insertion, and thus, the specific interaction between these proteins and S. aureus surface proteins is important for colonization and infection. We have reported that Esp not only disassembles S. aureus biofilms but also inhibits its nasal colonization (20). To elucidate how S. aureus colonization is inhibited and how Esp affects the host-pathogen interaction, we tested whether several human proteins are degraded by Esp. Recently, it has been reported that the presence of Hg in nasal secretions contributes to S. aureus nasal colonization via prevention of the agr quorum-sensing system (49). We first asked whether Hg is degraded by EspWT in vitro. As shown in Fig. 8A, no degradation of Hg was observed. We next tested whether major human ECM and plasma proteins, such as Fn, Fg, and Vn, are degraded by EspWT in vitro, since they are key receptors for S. aureus colonization on human epithelial and endothelial cells (50–52). Fn was degraded into various fragments (Fig. 8B), while Vn was partially processed to a 40-kDa fragment (Fig. 8C). In the case of Fg, which is composed of 2 sets of 3 chains designated the α, β, and γ subunits (53), only α chains were digested by EspWT (Fig. 8D).
Fig 8.
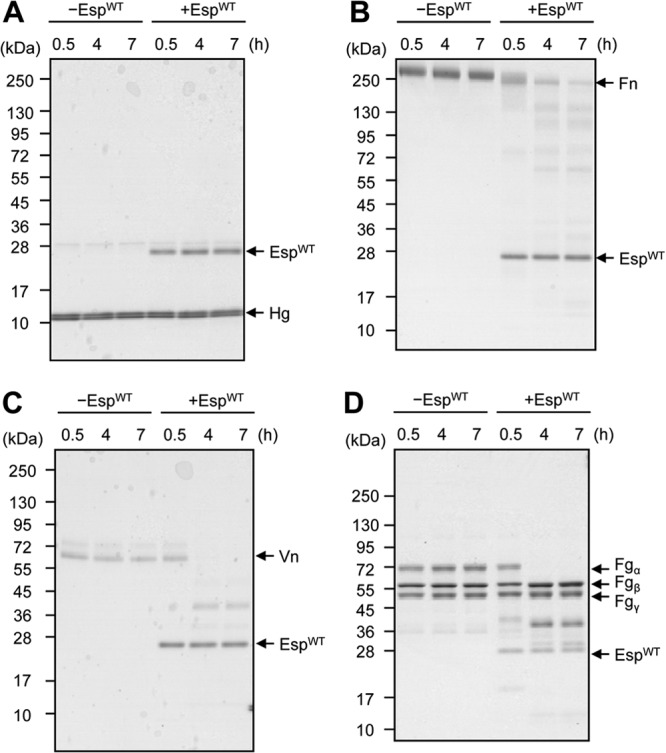
Degradation of staphylococcal receptor proteins of humans by EspWT. Human ECM and plasma proteins, including Hg (A), Fn (B), Vn (C), and Fg (D), were incubated with or without EspWT in PBS buffer at 37°C. At the indicated time points, proteins were analyzed by SDS-PAGE. Molecular sizes are given at the left.
Several biofilm-associated proteins (Atl, Emp, FnBPA, Eap, and Spa) and adhesins (Efb, IsdA, and SdrD), all of which interact with host ECM and plasma proteins (18, 35, 37–40, 45, 46, 48), were degraded by Esp. In addition, SceD, a protein essential for colonization of S. aureus in a cotton rat model (47), was also degraded by Esp. These results together suggest that this protease could inhibit nasal colonization of S. aureus presumably via degradation of S. aureus surface proteins and host receptors that are crucial for host-pathogen interactions.
DISCUSSION
Once a biofilm is fully developed, it can be disassembled to single cells or larger clusters through both mechanical and active processes (54). Biofilm disassembly is crucial for the dissemination of bacteria to other colonization sites and triggered by several factors, such as enzymes (proteases, glycosidases, and DNases) and surfactants (phenol-soluble modulins) that degrade or solubilize adhesive molecules in the biofilm matrix (55–57). An in-depth understanding of biofilm disassembly mechanisms could not only provide fundamental insights into bacterial lifestyles associated with infectious diseases but also lead to treatment options. To date, several approaches to disassemble and eliminate biofilms formed by various bacterial species have been reported. These approaches use biofilm matrix-degrading enzymes, such as bovine DNase I (58), dispersin B (59), and lysostaphin (60), and small molecules, such as d-amino acids (61) and norspermidine (62).
In the present study, we focused on extracellular serine protease Esp produced by commensal S. epidermidis and molecular mechanisms of Esp-dependent disassembly of pathogenic S. aureus biofilms (20). We identified 75 cell surface-associated proteins (in the biofilm matrix and cell wall fractions) as targets of Esp in the biofilm of MRSA (see Table S1 in the supplemental material). They include several known biofilm-associated proteins (Table 1), such as Atl (9, 10), β-toxin (19), Emp (13), FnBPA (14, 15), and Spa (18). They play important roles in biofilm development as the glue in cell-to-cell or cell-to-surface interactions. It has previously been shown that Eap contributes to the biofilm development of S. aureus Newman under certain conditions (e.g., iron-depleted conditions and serum-containing conditions) (13, 40). In these studies, an S. aureus eap-deletion mutant was used to evaluate the role of Eap in biofilm development. Considering the fact that biofilm formation is a multifactorial event, a single-gene deletion could be compensated for by other factors, and thus, no phenotype may be observed. In the case of extracellular proteins, supplementation of the purified protein into the culture of biofilm-negative strains is an efficacious approach. In this study, we found that supplementation of recombinant Eap increased the biofilm formation of S. aureus MS4, a biofilm-negative strain (Fig. 5D). In addition, heterologous expression of specific genes in biofilm-negative bacterial species can be an alternative strategy. Using a Brevibacillus expression-secretion system, we also proved that Eap stimulates cell aggregation and biofilm formation and it strongly bound to the cell surface (Fig. 5A to C). Furthermore, Eap formed a structural framework in the biofilm matrix (Fig. 6B; see Fig. S5 in the supplemental material). Our proteomic and immunological analyses revealed that Esp degrades Eap (Fig. 4; see Table S1 in the supplemental material), in addition to the other biofilm-associated proteins mentioned above. It is therefore reasonable to consider that a primary mechanism of S. aureus biofilm disassembly triggered by Esp is proteolytic degradation of these biofilm-associated proteins.
Several biofilm-associated proteins, such as Atl (37), Emp (46), FnBPA (38), Eap (39), and Spa (18), as well as adhesins, such as Efb (48), IsdA (45), and SdrD (35), interact with host ECM and plasma proteins. They therefore play an important role in the infection of host cells by S. aureus. Our results indicate that Esp degrades these surface proteins of S. aureus (Table 1) and human ECM and plasma proteins, such as Fn, Fg, and Vn (Fig. 8). In addition, SceD, a protein essential for nasal colonization of S. aureus in cotton rats (47), is also degraded by Esp (Table 1; see Table S1 in the supplemental material). These results together suggest that Esp inhibits nasal colonization of S. aureus presumably via degradation of S. aureus surface proteins and host receptors that are crucial for the pathogen-host interaction.
Surface proteins of S. aureus are known to be important for causing infectious diseases in humans, ranging from minor skin infections to severe infections (1). SdrD, Spa, IsdA, and Emp are necessary for abscess formation (36). Of note, Eap has drawn the attention of researchers because of its importance in various aspects of infection (e.g., anti-inflammatory and antiangiogenic properties and abscess formation) (41, 42). Immunoglobulin-binding proteins Spa and Sbi contribute to immune evasion by forming complexes with VH3-type IgM on the surface of B cells, pathogen-specific antibodies, or complement factor 3d (43, 44). Efb is also associated with immune evasion through the binding to the α chain of complement factor 3 (63). Our data clearly indicate that Esp degrades these surface proteins and toxins, such as β-toxin and coagulase (Table 1; see Table S1 in the supplemental material), suggesting that Esp might be applicable for the prevention or treatment of infectious diseases caused by this pathogen. However, we should pay attention to the use of Esp for therapeutic purposes, since the safety of this protease has not yet been established. Development of a vaccine targeting proteins that are degraded by Esp and also contribute to S. aureus biofilm formation and colonization of human skin and the human nasal cavity could be an alternative approach.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Research Activity Start-up to S.S. from the Japan Society for the Promotion of Science (JSPS), by a Grant-in-Aid for Young Scientists (B) to S.S. from JSPS, by a grant to Y.M. from the Uehara Memorial Foundation, by a grant to Y.M. from The Science Research Promotion Fund, by a grant to Y.M. from MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016, and by a grant to S.S. from the Joint Usage/Research Center for Developmental Medicine, IMEG, Kumamoto University.
We acknowledge Yasuaki Hiromasa (Department of Biochemistry, Kansas State University) for mass spectrometry analyses, Teru Ogura (Department of Molecular Cell Biology, Kumamoto University, Japan) for sending E. coli strains and plasmids, Karen LoVetri (Kane Biotech Inc.) for gifting dispersin B, and Fumiya Sato (Division of Infectious Disease and Control, The Jikei University School of Medicine, Japan) for providing clinically isolated S. aureus strains. We also thank Hitomi Shinji for discussion, Satomi Yamada, Takuya Kitamura, Rintarou Shigemori, and Kensuke Sekiguchi for experimental support, and Yumi Tokoro for secretary work.
Footnotes
Published ahead of print 11 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01672-12.
REFERENCES
- 1. Archer GL, Climo MW. 2001. Staphylococcus aureus bacteremia—consider the source. N. Engl. J. Med. 344:55–56 [DOI] [PubMed] [Google Scholar]
- 2. von Eiff C, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N. Engl. J. Med. 344:11–16 [DOI] [PubMed] [Google Scholar]
- 3. Zetola N, Francis JS, Nuermberger EL, Bishai WR. 2005. Community-acquired meticillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 5:275–286 [DOI] [PubMed] [Google Scholar]
- 4. Otto M. 2008. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 322:207–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 6. Götz F. 2002. Staphylococcus and biofilms. Mol. Microbiol. 43:1367–1378 [DOI] [PubMed] [Google Scholar]
- 7. O'Gara JP. 2007. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 270:179–188 [DOI] [PubMed] [Google Scholar]
- 8. Patti JM, Allen BL, McGavin MJ, Höök M. 1994. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48:585–617 [DOI] [PubMed] [Google Scholar]
- 9. Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 259:260–268 [DOI] [PubMed] [Google Scholar]
- 10. Houston P, Rowe SE, Pozzi C, Waters EM, O'Gara JP. 2011. Essential role for the major autolysin in the fibronectin-binding protein-mediated Staphylococcus aureus biofilm phenotype. Infect. Immun. 79:1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cucarella C, Solano C, Valle J, Amorena B, Lasa I, Penadés JR. 2001. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 183:2888–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDevitt D, Francois P, Vaudaux P, Foster TJ. 1994. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 11:237–248 [DOI] [PubMed] [Google Scholar]
- 13. Johnson M, Cockayne A, Morrissey JA. 2008. Iron-regulated biofilm formation in Staphylococcus aureus Newman requires ica and the secreted protein Emp. Infect. Immun. 76:1756–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Neill E, Pozzi C, Houston P, Humphreys H, Robinson DA, Loughman A, Foster TJ, O'Gara JP. 2008. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 190:3835–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vergara-Irigaray M, Valle J, Merino N, Latasa C, García B, Ruiz de Los Mozos I, Solano C, Toledo-Arana A, Penadés JR, Lasa I. 2009. Relevant role of fibronectin-binding proteins in Staphylococcus aureus biofilm-associated foreign-body infections. Infect. Immun. 77:3978–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corrigan RM, Rigby D, Handley P, Foster TJ. 2007. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 153:2435–2446 [DOI] [PubMed] [Google Scholar]
- 17. Schroeder K, Jularic M, Horsburgh SM, Hirschhausen N, Neumann C, Bertling A, Schulte A, Foster S, Kehrel BE, Peters G, Heilmann C. 2009. Molecular characterization of a novel Staphylococcus aureus surface protein (SasC) involved in cell aggregation and biofilm accumulation. PLoS One 4:e7567 doi:10.1371/journal.pone.0007567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, Lopez JA, Foster TJ, Penadés JR, Lasa I. 2009. Protein A-mediated multicellular behavior in Staphylococcus aureus. J. Bacteriol. 191:832–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huseby MJ, Kruse AC, Digre J, Kohler PL, Vocke JA, Mann EE, Bayles KW, Bohach GA, Schlievert PM, Ohlendorf DH, Earhart CA. 2010. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc. Natl. Acad. Sci. U. S. A. 107:14407–14412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y. 2010. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465:346–349 [DOI] [PubMed] [Google Scholar]
- 21. Dubin G, Chmiel D, Mak P, Rakwalska M, Rzychon M, Dubin A. 2001. Molecular cloning and biochemical characterization of proteases from Staphylococcus epidermidis. Biol. Chem. 382:1575–1582 [DOI] [PubMed] [Google Scholar]
- 22. Moon JL, Banbula A, Oleksy A, Mayo JA, Travis J. 2001. Isolation and characterization of a highly specific serine endopeptidase from an oral strain of Staphylococcus epidermidis. Biol. Chem. 382:1095–1099 [DOI] [PubMed] [Google Scholar]
- 23. Sugimoto S, Iwase T, Sato F, Tajima A, Shinji H, Mizunoe Y. 2011. Cloning, expression and purification of extracellular serine protease Esp, a biofilm degrading enzyme, from Staphylococcus epidermidis. J. Appl. Microbiol. 111:1406–1415 [DOI] [PubMed] [Google Scholar]
- 24. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 25. Sugimoto S, Yamanaka K, Nishikori S, Miyagi A, Ando T, Ogura T. 2010. AAA+ chaperone ClpX regulates dynamics of prokaryotic cytoskeletal protein FtsZ. J. Biol. Chem. 285:6648–6657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pasztor L, Ziebandt AK, Nega M, Schlag M, Haase S, Franz-Wachtel M, Madlung J, Nordheim A, Heinrichs DE, Götz F. 2010. Staphylococcal major autolysin (Atl) is involved in excretion of cytoplasmic proteins. J. Biol. Chem. 285:36794–36803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Novick RP. 1967. Properties of a cryptic high frequency transducing phage in Staphylococcus aureus. Virology 33:155–166 [DOI] [PubMed] [Google Scholar]
- 28. Gatlin CL, Pieper R, Huang ST, Mongodin E, Gebregeorgis E, Parmar PP, Clark DJ, Alami H, Papazisi L, Fleischmann RD, Gill SR, Peterson SN. 2006. Proteomic profiling of cell envelope-associated proteins from Staphylococcus aureus. Proteomics 6:1530–1549 [DOI] [PubMed] [Google Scholar]
- 29. Muthukrishnan G, Quinn GA, Lamers RP, Diaz C, Cole AL, Chen S, Cole AM. 2011. Exoproteome of Staphylococcus aureus reveals putative determinants of nasal carriage. J. Proteome Res. 10:2064–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nandakumar R, Nandakumar MP, Marten MR, Ross JM. 2008. Proteome analysis of membrane and cell wall associated proteins from Staphylococcus aureus. J. Proteome Res. 4:250–257 [DOI] [PubMed] [Google Scholar]
- 31. Romero D, Aguilar C, Losick R, Kolter R. 2010. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. U. S. A. 107:2230–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaplan JB, Velliyagounder K, Ragunath C, Rohde H, Mack D, Knobloch JK, Ramasubbu N. 2004. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J. Bacteriol. 186:8213–8220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohara-Nemoto Y, Ono T, Shimoyama Y, Kimura S, Nemoto TK. 2008. Homologous and heterologous expression and maturation processing of extracellular glutamyl endopeptidase of Staphylococcus epidermidis. Biol. Chem. 389:1209–1217 [DOI] [PubMed] [Google Scholar]
- 35. Corrigan RM, Miajlovic H, Foster TJ. 2009. Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 9:22 doi:10.1186/1471-2180-9-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23:3393–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell. Microbiol. 12:1746–1764 [DOI] [PubMed] [Google Scholar]
- 38. Fröman G, Switalski LM, Speziale P, Höök M. 1987. Isolation and characterization of a fibronectin receptor from Staphylococcus aureus. J. Biol. Chem. 262:6564–6571 [PubMed] [Google Scholar]
- 39. Palma M, Haggar A, Flock JI. 1999. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J. Bacteriol. 181:2840–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thompson KM, Abraham N, Jefferson KK. 2010. Staphylococcus aureus extracellular adherence protein contributes to biofilm formation in the presence of serum. FEMS Microbiol. Lett. 305:143–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Athanasopoulos AN, Economopoulou M, Orlova VV, Sobke A, Schneider D, Weber H, Augustin HG, Eming SA, Schubert U, Linn T, Nawroth PP, Hussain M, Hammes HP, Herrmann M, Preissner KT, Chavakis T. 2006. The extracellular adherence protein (Eap) of Staphylococcus aureus inhibits wound healing by interfering with host defense and repair mechanisms. Blood 107:2720–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT. 2002. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 8:687–693 [DOI] [PubMed] [Google Scholar]
- 43. Kim HK, Thammavongsa V, Schneewind O, Missiakas D. 2012. Recurrent infections and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 15:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burman JD, Leung E, Atkins KL, O'Seaghdha MN, Lango L, Bernadó P, Bagby S, Svergun DI, Foster TJ, Isenman DE, van den Elsen JM. 2008. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J. Biol. Chem. 283:17579–17593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clarke SR, Wiltshire MD, Foster SJ. 2004. IsdA of Staphylococcus aureus is a broad spectrum, iron-regulated adhesin. Mol. Microbiol. 51:1509–1519 [DOI] [PubMed] [Google Scholar]
- 46. Hussain M, Becker K, von Eiff C, Schrenzel J, Peters G, Herrmann M. 2001. Identification and characterization of a novel 38.5-kilodalton cell surface protein of Staphylococcus aureus with extended-spectrum binding activity for extracellular matrix and plasma proteins. J. Bacteriol. 183:6778–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stapleton MR, Horsburgh MJ, Hayhurst EJ, Wright L, Jonsson IM, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J. Bacteriol. 189:7316–7325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palma M, Shannon O, Quezada HC, Berg A, Flock JI. 2001. Extracellular fibrinogen-binding protein, Efb, from Staphylococcus aureus blocks platelet aggregation due to its binding to the alpha-chain. J. Biol. Chem. 276:31691–31697 [DOI] [PubMed] [Google Scholar]
- 49. Pynnonen M, Stephenson RE, Schwartz K, Hernandez M, Boles BR. 2011. Hemoglobin promotes Staphylococcus aureus nasal colonization. PLoS Pathog. 7:e1002104 doi:10.1371/journal.ppat.1002104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheung AL, Krishnan M, Jaffe EA, Fischetti VA. 1991. Fibrinogen acts as a bridging molecule in the adherence of Staphylococcus aureus to cultured human endothelial cells. J. Clin. Invest. 87:2236–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chhatwal GS, Preissner KT, Müller-Berghaus G, Blobel H. 1987. Specific binding of the human S protein (vitronectin) to streptococci, Staphylococcus aureus, and Escherichia coli. Infect. Immun. 55:1878–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Proctor RA, Mosher DF, Olbrantz PJ. 1982. Fibronectin binding to Staphylococcus aureus. J. Biol. Chem. 257:14788–14794 [PubMed] [Google Scholar]
- 53. McKee PA, Mattock P, Hill RL. 1970. Subunit structure of human fibrinogen, soluble fibrin, and cross-linked insoluble fibrin. Proc. Natl. Acad. Sci. U. S. A. 66:738–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boles BR, Horswill AR. 2011. Staphylococcal biofilm disassembly. Trends Microbiol. 19:449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lauderdale KJ, Boles BR, Cheung AL, Horswill AR. 2009. Interconnections between sigma B, agr, and proteolytic activity in Staphylococcus aureus biofilm maturation. Infect. Immun. 77:1623–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, Cheung GY, Otto M. 2012. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. U. S. A. 109:1281–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rice KC, Mann EE, Endres JL, Weiss EC, Cassat JE, Smeltzer MS, Bayles KW. 2007. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 104:8113–8118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mann EE, Rice KC, Boles BR, Endres JL, Ranjit D, Chandramohan L, Tsang LH, Smeltzer MS, Horswill AR, Bayles KW. 2009. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4:e5822 doi:10.1371/journal.pone.0005822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Donelli G, Francolini I, Romoli D, Guaglianone E, Piozzi A, Ragunath C, Kaplan JB. 2007. Synergistic activity of dispersin B and cefamandole nafate in inhibition of staphylococcal biofilm growth on polyurethanes. Antimicrob. Agents Chemother. 51:2733–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu JA, Kusuma C, Mond JJ, Kokai-Kun JF. 2003. Lysostaphin disrupts Staphylococcus aureus and Staphylococcus epidermidis biofilms on artificial surfaces. Antimicrob. Agents Chemother. 47:3407–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. 2010. d-Amino acids trigger biofilm disassembly. Science 328:627–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kolodkin-Gal I, Cao S, Chai L, Böttcher T, Kolter R, Clardy J, Losick R. 2012. A self-produced trigger for biofilm disassembly that targets exopolysaccharide. Cell 149:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Lee LY, Höök M, Haviland D, Wetsel RA, Yonter EO, Syribeys P, Vernachio J, Brown EL. 2004. Inhibition of complement activation by a secreted Staphylococcus aureus protein. J. Infect. Dis. 190:571–579 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.