

Abstract
Long-term depression (LTD) at parallel fibre synapses on a cerebellar Purkinje cell has been regarded as a cellular basis for motor learning. Although Ca2+/calmodulin-dependent protein kinase II (CaMKII) has been implicated in the LTD induction as an important Ca2+-sensing molecule, the underlying signalling mechanism remains unclear. Here, we attempted to explore the potential signalling pathway underlying the CaMKII involvement in LTD using a systems biology approach, combined with validation by electrophysiological and FRET imaging experiments on a rat cultured Purkinje cell. Model simulation predicted the following cascade as a candidate mechanism for the CaMKII contribution to LTD: CaMKII negatively regulates phosphodiesterase 1 (PDE1), subsequently facilitates the cGMP/protein kinase G (PKG) signalling pathway and down-regulates protein phosphatase 2A (PP-2A), thus supporting the LTD-inducing positive feedback loop consisting of mutual activation of protein kinase C (PKC) and mitogen-activated protein kinase (MAPK). This model suggestion was corroborated by whole-cell patch clamp recording experiments. In addition, FRET measurement of intracellular cGMP concentration revealed that CaMKII activation causes sustained increase of cGMP, supporting the signalling mechanism of LTD induction by CaMKII. Furthermore, we found that activation of the cGMP/PKG pathway by nitric oxide (NO) can support LTD induction without activation of CaMKII. Thus, this study clarified interaction between NO and Ca2+/CaMKII, two important factors required for LTD.
Key points
Long-term depression (LTD) at parallel fibre synapses on a cerebellar Purkinje cell has been regarded as a cellular basis for motor learning.
Although Ca2+/calmodulin-dependent protein kinase II (CaMKII) has been implicated in the LTD induction and motor learning, the underlying molecular mechanism remains unclear.
By combined application of simulation and experiments, we have attempted to explore the potential signalling pathway underlying the CaMKII involvement in LTD. Our data show that CaMKII supports the LTD-inducing signalling pathway consisting of other protein kinases such as protein kinase C and mitogen-activated protein kinase.
The gating of the LTD-inducing pathway by CaMKII is mediated by negative regulation of phosphodiesterase 1, and the resultant facilitation of the cGMP/protein kinase G (PKG) pathway. As a result, protein phosphatase 2A activity was suppressed, supporting the LTD induction.
In addition, nitric oxide-mediated cGMP/PKG activation compensated for the lack of CaMKII activation in LTD induction.
This study provides a comprehensive understanding of elaborate intracellular signalling mechanisms for LTD regulation.
Introduction
Synaptic plasticity has been regarded as a basis for learning and memory, and its regulatory mechanisms have been extensively studied (Ito, 2001; Kandel, 2001). In many forms of synaptic plasticity, Ca2+/calmodulin-dependent protein kinase II (CaMKII) plays critical roles (Kano et al. 1996; Hansel et al. 2001; Kawaguchi & Hirano, 2002; Lisman et al. 2002; Gaiarsa et al. 2002; Colbran & Brown, 2004; Elgersma et al. 2004). At hippocampal excitatory synapses, it has been suggested that CaMKII-mediated positive regulation of AMPA-type glutamate receptors (AMPARs) underlies long-term potentiation (LTP) (Lee et al. 2000; Correia et al. 2008). In contrast, CaMKII is involved in long-term depression (LTD) in a cerebellar Purkinje cell, although the mechanism is unknown (Hansel et al. 2006). Here, we studied a signalling role of CaMKII in cerebellar LTD using combined application of whole-cell patch clamp recording and systems biology simulation.
Cerebellar LTD is induced by coupled activation of a climbing fibre (CF) and parallel fibres (PFs), which has been regarded as a cellular basis for motor learning (Ito, 2001; Hansel et al. 2001; Dean et al. 2010). Extensive studies have shown that the LTD induction is regulated by complex signalling cascades (Linden & Connor, 1991; Lev-Ram et al. 1997; Kawasaki et al. 1999; Wang & Linden, 2000; Ito, 2001; Chung et al. 2003; Feil et al. 2003; Launey et al. 2004). A positive feedback signalling loop based on mutual activation of protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) cascades plays a central role in the LTD induction (Kuroda et al. 2001; Tanaka et al. 2007; Tanaka & Augustine, 2008). The LTD induction by the PKC–MAPK positive feedback loop is controlled by protein phosphatase 2A (PP-2A), which is regulated by the nitric oxide (NO) and cGMP/protein kinase G (PKG) pathway. The involvement of CaMKII in LTD was also reported using αCaMKII knockout mice (Hansel et al. 2006). The ablation of αCaMKII causes LTD impairment in juvenile mice, and conversion of LTD into LTP in adult mice, accompanied by impaired adaptation of reflex ocular movement. Knockout mice lacking βCaMKII also exhibit ataxia and LTD impairment (van Woerden et al. 2009). Thus, CaMKII was suggested to play a pivotal role in LTD induction as a Ca2+-sensing signalling molecule. However, the detailed mechanism of CaMKII contribution to LTD has remained unknown.
To explore the role of CaMKII in LTD, we extended a kinetic simulation model for signalling cascades of LTD by adding a molecular network regulating CaMKII activity, and tried thereby to obtain insight into the target of CaMKII in the cascades (see Fig. 1). By simulation we examined the influence of CaMKII activity on LTD and found that the CaMKII-mediated negative regulation of Ca2+/CaM-dependent phosphodiesterase 1 (PDE1) is a candidate mechanism to support LTD induction through promotion of the cGMP/PKG signalling pathway. In addition, the model predicted an interaction between CaMKII- and NO-mediated regulations of the cGMP/PKG pathway in LTD induction. Cellular experiments validated the model suggestion that CaMKII gates LTD by enhancing the cGMP/PKG pathway through PDE1 regulation.
Figure 1. Signalling cascades regulating LTD with extension of CaMKII regulating pathways in a Purkinje cell.
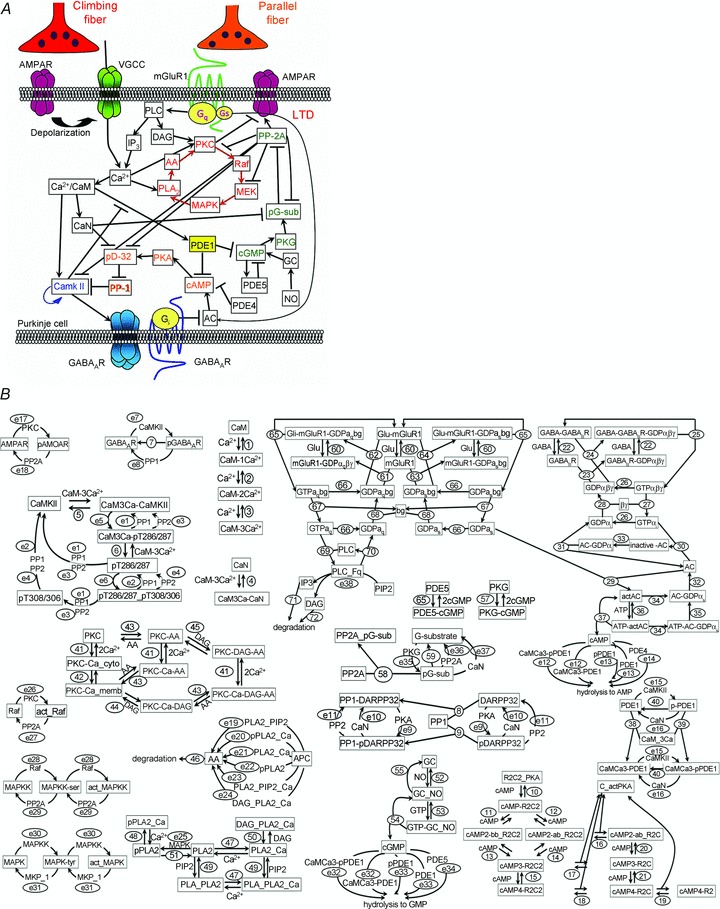
A, the Ca2+-triggered positive feedback reaction consisting of MAPK, MEK, Raf, PKC, arachidonic acid (AA), and PLA2 (red colour) plays a pivotal role in the LTD induction, which is controlled by the activity of PP-2A (green). AC, adenylyl cyclase; CaN, calcineurin; DAG, diacylglycerol; GC, guanylyl cyclase; pD-32, phospho-DARPP-32; pG-sub, phospho-G-substrate; PLC, phospholipase C; VGCC, voltage-gated Ca2+ channel. B, detailed diagrams for molecular reactions of the LTD model with extension of the CaMKII regulating pathways. Kinetics of each numbered reaction are listed in Tables 1–3.
Methods
Culture
Experimental procedures were performed in accordance with the principles of UK regulations, the policies on the use of animals and humans in neuroscience research in the USA, and the guidelines regarding care and use of animals for experimental procedures of Kyoto University, and were approved by the local committee for handling experimental animals in the Graduate School of Science, Kyoto University.
A primary culture of cerebellar neurons was prepared from newborn Wistar rats of both sexes (Kawaguchi & Hirano, 2007). Briefly, rat pups were decapitated on day of birth (P0), and cerebella were dissected, followed by treatment with 0.1% trypsin. Cells were then dissociated by trituration and cultured in a defined medium. Whole-cell patch clamp recordings, Förster resonance energy transfer (FRET) imaging and immunocytochemistry were performed 2–3 weeks after preparation of the culture.
Electrophysiology
Methods used for electrophysiological experiments were similar to those in previous studies (Kawaguchi et al. 2011). Briefly, whole-cell patch clamp recording from a cultured Purkinje cell was performed with an amplifier (EPC9, HEKA) in a solution containing (in mm) 145 NaCl, 5 KOH, 2 CaCl2, 1 MgCl2, 10 Hepes, and 10 glucose (pH 7.3) at room temperature (20–24°C). The solution contained bicuculline (20 μm, Tocris Cookson) and tetrodotoxin (TTX, 1 μm, Wako, Japan) to inhibit GABAergic IPSCs and action potentials, respectively. A patch pipette used to record from a Purkinje cell was filled with an internal solution (pH 7.3, adjusted by CsOH) containing (in mm) 121 CsCl, 33 KCl, 1.4 EGTA, 10 Hepes, 2 Mg-ATP and 0.2 Na-GTP. The membrane potential of a Purkinje cell was held at −70 mV. Only recordings with an input resistance of more than 100 MΩ and a series resistance of less than 25 MΩ were accepted. To minimize the voltage clamp error, the glutamate response amplitude at the beginning of experiments was set at around 300 pA. Series resistance and input resistance were monitored every 2 min and experiments were terminated when a change of more than 20% was detected. The method for iontophoretic application of glutamate was similar to that in previous studies (Tsuruno & Hirano, 2007). A glass pipette containing 10 mm glutamate and 10 mm Hepes (pH 7.4) was aimed at a proximal dendrite, and 20 ms positive voltage pulses were applied every 20 s. Nodularin (10 nm), KN62 (5 μm), 8-methoxymethyl-IBMX (8-MM-IBMX, 20 μm), KT5823 (1 μm), KT5720 (1 μm) (all from Calbiochem), diethylamine nitric oxide (DEANO, 3 or 10 μm, Invitrogen, Carlsbad, CA, USA) or 8-Br-cGMP (20 μm, Tocris) was applied to the bath 5–10 min before recording. Autocamtide-2-related inhibitory peptide II (AIPII, 1 μm, Calbiochem), fostriecin (50 nm, Calbiochem), protein phosphatase inhibitor-2 (100 nm, New England Biolabs, MA, USA), 8-CPT-cGMP (50 μm, Biolog, Germany), or Sp-cAMP (10 μm, Calbiochem) was applied intracellularly through a patch pipette.
Immunocytochemistry
Cultured neurons were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized with 0.5% Tween 20, then blocked with 2% skimmed milk, and finally labelled with primary and secondary antibodies. The following antibodies were used: a mouse monoclonal antibody against calbindin D28 (1:500, Swant, Marly, Switzerland), a rabbit polyclonal antibody (pAb) against active CaMKII autophosphorylated at Thr286 (1:500, Promega), and Alexa 488- or 568-conjugated pAb against rabbit or mouse IgG (1:400, Molecular Probes). Fluorescence images were obtained with a confocal laser microscope (FV1000 imaging system, Olympus, Japan), and analysed using ImageJ software (NIH, USA). The high K+-containing solution for conditioning treatment was prepared by replacing 50 mm Na+ with K+ in the normal external solution. Cultured cerebellar neurons were treated with the 50 mm K+-containing solution for 10 s in the presence of TTX (1 μm) and SCH50911 (10 μm, a selective antagonist for GABAB receptors). The latter was used to avoid the potential indirect suppression of CaMKII activation by GABAB receptors (Kawaguchi & Hirano, 2000, 2002). Then, neurons were washed with the normal external solution until fixation at 30 min after the onset of the conditioning treatment. The averaged fluorescence signals for active CaMKII in the area positive for calbindin, a molecular marker of Purkinje cells, were compared.
Imaging
[Ca2+]i was measured with a Ca2+ imaging system (Aquacosmos, Hamamatsu Photonics, Japan) mounted on an upright microscope (BX50WI, Olympus) using fura-4F (50 μm, Invitrogen). Fura-4F was loaded into a Purkinje cell through a patch pipette, and excited alternately at 340 nm and 380 nm for 120 ms. Each fluorescence image was recorded at 2 Hz, and the fluorescence ratio (the fluorescence excited at 340 nm divided by that at 380 nm) was calculated.
FRET images were obtained from a Purkinje cell transfected with a cGES-DE5 probe using an upright IX71 microscope equipped with the FV1000 fluorescence imaging system (Olympus). The cGES-DE5 probe was excited at 440 nm, and the emission between 460 and 500 nm (for cyan fluorescent protein: CFP) and that between 515 and 615 nm (for Venus, a variant of yellow fluorescent protein: YFP) were recorded. The fluorescence intensities of ECFP and Venus in a thick shaft of proximal dendrite were measured. Then, the fluorescence ratio (Venus/CFP) was calculated.
DNA construction and transfection
The cGMP monitoring probe based on FRET was constructed with essentially identical design to cGES-DE5 (Nikolaev et al. 2006). The cDNA of rat phosphodiesterase 5(PDE5) was cloned by PCR of the cDNA template obtained by reverse transcription of mRNAs prepared from rat cerebellar culture. cDNA encoding the cGMP binding regions of PDE5 (amino acid residues E144 to A298), that encoding Venus, and that encoding ECFP, were inserted in tandem into the pCAGplay expression vector at the BglII/PstI site, the EcoRI/BglII site, and the PstI/XhoI site, respectively (Kawaguchi & Hirano, 2007). The expression plasmid (50 ng μl−1 total) encoding cGES-DE5 or the constitutively active form of the calcineurin α subunit (CA-CaN) (Fujii & Hirano, 2002) was injected into the nucleus of a Purkinje cell through a sharp glass pipette. The imaging experiments were performed 2–3 days after the injection.
Simulation
A computational model of signalling cascades regulating LTD (Kuroda et al. 2001; Tanaka et al. 2007) and one of rebound potentiation (RP) that we had developed previously (Kitagawa et al. 2009; Kawaguchi et al. 2011) were integrated into a single framework by linking overlapping molecules and their reactions with some modifications as explained briefly below. The signalling cascades and the detailed reaction diagrams of the integrated model are shown in Fig. 1.
Here, two systems biology models for LTD and RP were integrated by linking the molecular reactions affected by Ca2+, PP-2A, calcineurin, PDE1 and metabotropic glutamate receptor 1 (mGluR1), as explained below. First, Ca2+-mediated activations of Ca2+/CaM, phospholipase A2 (PLA2), and PKC were incorporated. It should be noted that inositol trisphosphate (IP3)-mediated release of Ca2+ from the internal store was omitted here because the Ca2+ concentration was used as an input to the model in this study. Second, dephosphorylation of dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), G-substrate, CaMKII, MAPK/ERK kinase (MEK), Raf and AMPARs by PP-2A were incorporated as similar reactions. Third, dephosphorylation of DARPP-32 and G-substrate by calcineurin and PP-2A were incorporated. Fourth, PDE1-mediated hydrolysis of cAMP and cGMP were incorporated. Fifth, in addition to mGluR1-mediated Gq-type of G protein activation, coupling mGluR1 to Gs-type of G protein was incorporated with 1% efficiency compared to the Gq coupling (Tateyama & Kubo, 2006). Previous studies demonstrated that mGluR1 activation facilitates the RP induction not through the Gq pathway but through the Gs/cAMP/PKA pathway (Sugiyama et al. 2008). All of the parameters (40 for molecular concentration, 121 for molecular interactions, and 70 for enzymatic reactions) used for simulation are listed in Tables 1–3.
Table 1.
Molecular concentration
| Molecule | Concentration (μm) | Previous version | Notes and references |
|---|---|---|---|
| Ca2+ | 0.1 | Same | See Kitagawa et al. (2009). |
| Glu | 0.001 | — | Assumed to produce a basal cAMP concentration around 0.14 μm (Sugiyama et al. 2008). |
| GABA | 0.01 | Same | See Kitagawa et al. (2009). |
| NO | 0.05 | — | Assumed to produce the basal cGMP concentration around 50 nm (Francis et al. 2010). |
| CaM | 60 | Same | See Kitagawa et al. (2009). |
| CaN | 10 | Same | See Kawaguchi et al. (2011). |
| CaMKII | 20 | Same | See Kitagawa et al. (2009). |
| PKC | 2 | 1 | The PKC amount was assumed to be slightly larger. |
| PP-1 | 0.4 | 0.54 | Assumed to balance the level of CaMKII phosphorylation. |
| PP-2A | 0.4 | 0.2 | PP-2A amount was increased to inhibit CaMKII, AMPARs, DARPP-32, G-substrate, MEK and Raf. |
| AMPAR | 1 | 0.5 | The ratio of phosphorylated and dephosphorylated AMPARs reflects the LTD establishment. Thus, the absolute amount of AMPARs is not important here. |
| GABAAR | 1 | Same | See Kitagawa et al. (2009). |
| DARPP-32 | 0.7 | 1.1 | According to the decrease of PP-1 in the model, amount of DARPP-32 was also reduced. |
| R2C2-PKA | 0.25 | Same | See Kawaguchi et al. (2011). |
| GABABR | 0.5 | Same | See Kitagawa et al. (2009). |
| GDPαiβγ | 1.5 | Same | See Kawaguchi et al. (2011). |
| AC | 0.02 | Same | See Kawaguchi et al. (2011). |
| cAMP | 0.1 | Same | See Kitagawa et al. (2009). |
| ATP | 2000 (constant) | Same | See Kitagawa et al. (2009). |
| PDE1 | 0.6 | Same | See Kawaguchi et al. (2011). |
| PDE4 | 0.6 | 0.4 | To enable rapid regulation of cAMP concentration, the amount of PDE4 was increased. |
| PDE5 | 0.6 | Newly defined | The amount of PDE5 was assumed to be similar to that of PDE1 or PDE4 in a Purkinje cell. |
| G-substrate | 0.7 | 10.8 | Because the affinity between phospho-G substrate and PP-2A was set much higher based on Endo et al. (2003), the amounts of PKG and G-substrate were reduced. |
| PKG | 0.1 | 2.445 | |
| cGMP | 0 | Same | Kuroda et al. (2001). |
| GTP | 100 | Same | Kitagawa et al. (2009). |
| GC | 0.02 | 3 | The amount of soluble guanylyl cyclase was assumed to be identical to that of adenylyl cyclase, producing a basal cGMP concentration around 50 nm (Francis et al. 2010). |
| Raf | 0.2 | 0.5 | The amounts of proteins in MAPK cascades were slightly reduced to balance the strength of the positive feedback loop and that of PP-2A activity. |
| MAPKK | 0.1 | 0.5 | The amounts of proteins in MAPK cascades were slightly reduced to balance the strength of the positive feedback loop and that of PP-2A activity. |
| MAPK | 0.2 | 1 | The amounts of proteins in MAPK cascades were slightly reduced to balance the strength of the positive feedback loop and that of PP-2A activity. |
| MKP-1 (MAPK phosphatase 1) | 0.005 | 0.0032 | The amounts of proteins in MAPK cascades were slightly reduced to balance the strength of the positive feedback loop and that of PP-2A activity. |
| PLA2 | 0.4 | Same | Tanaka et al. (2007). |
| AA | 0 | Same | Kuroda et al. (2001). |
| APC (aracholonyl-phosphatidylcholine) | 3 | 30 | Tanaka et al. (2007). |
| PIP2 (phosphati-dylinositol 4,5-bisphosphate) | 3 | 10 | Tanaka et al. (2007). |
| DAG | 0 | Same | Kuroda et al. (2001). |
| mGluR1 | 0.5 | 0.19524 | Adjusted to be identical to that of GABABRs. |
| Gq | 1.5 | 0.89513 | Adjusted to be identical to that of Gi protein. |
| Gs | 1.5 | — | Adjusted to be identical to that of Gi protein. |
| PLC | 0.2 | 0.8 | Assumption |
Table 3.
Kinetics of enzymatic reactions
| ID | Km (μm) | kcat (s−1) | Previous version | Notes and references | ||
|---|---|---|---|---|---|---|
| e1, e2 | 3 | 2 | Same | See Kitagawa et al. (2009). | ||
| e3, e4 | 5 | 0.2 | 10 | 0.2 | PP-2A mediated dephosphorylation of CaMKII was assumed to be slightly stronger compared with Kawaguchi et al. (2011). | |
| e5 | — | 11 | 11 | See Kawaguchi et al. 2011. | ||
| e6 | — | 0.5 | 0.5 | See Kawaguchi et al. 2011. | ||
| e7 | 11 | 1 | Same | See Kitagawa et al. (2009). | ||
| e8 | 3 | 2 | Same | See Kitagawa et al. (2009). | ||
| e9 | 2.4 | 2.7 | Same | See Kitagawa et al. (2009). | ||
| e10 | 1.6 | 0.5 | Same | See Kawaguchi et al. (2011). | ||
| e11 | 5 | 2 | 2 | 0.4 | Dephosphorylation of DARPP-32, AMPARs, G-substrate, Raf and MAPKK were assumed to have similar kinetics. | |
| e12 | 12 | 5 | Same | See Kawaguchi et al. (2011). | ||
| e13 | 12 | 0.1 | Same | See Kitagawa et al. (2009). | ||
| e14 | 2 | 0.05 | Same | See Kitagawa et al. (2009). | ||
| e15 | 11 | 2 | Same | See Kawaguchi et al. (2011). | ||
| e16 | 2 | 0.5 | Same | See Kawaguchi et al. (2011). | ||
| e17 | 3.5 | 0.05 | Same | Tanaka et al. (2007). | ||
| e18 | 5 | 2 | 15.7 | 6 | Dephosphorylation of DARPP-32, AMPARs, G-substrate, Raf and MAPKK were assumed to have similar kinetics. | |
| e19 | 20 | 11.04 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e20 | 20 | 120 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e21 | 20 | 54 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e22 | 20 | 120 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e23 | 20 | 36 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e24 | 20 | 60 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e25 | 25.6 | 20 | Same | Tanaka et al. (2007) | ||
| e26 | 11.5 | 0.0335 | Same | Kuroda et al. (2001); Tanaka et al. (2007). | ||
| e27 | 5 | 2 | 15.7 | 6 | Dephosphorylation of DARPP-32, AMPARs, G-substrate, Raf and MAPKK were assumed to have similar kinetics. | |
| e28 | 0.398 | 0.105 | Same | Tanaka et al. (2007). | ||
| e29 | 5 | 2 | 15.7 | 6 | Dephosphorylation of DARPP-32, AMPARs, G-substrate, Raf and MAPKK were assumed to have similar kinetics. | |
| e30 | 0.0463 | 0.15 | Same | Tanaka et al. (2007). | ||
| e31 | 0.16667 | 1 | Same | Tanaka et al. (2007). | ||
| e32 | 2 | 15 | Newly defined | Francis et al. (2010). | ||
| e33 | 2 | 0.1 | Assumption | Francis et al. (2010). | ||
| e34 | 2 | 3 | 2 | 3.87 | Poppe et al. (2008); Francis et al. (2010). | |
| e35 | 0.2 | 2 | 0.2 | 0.72 | Hall et al. (1999). | |
| e36 | 5 | 2 | Newly defined | Hall et al. (1999). | ||
| e37 | 1.6 | 0.5 | Newly defined | CaN-mediated dephosphorylation of phospho-G substrate was assumed to take place with identical kinetics to that of phospho-DARPP-32, which are similar to the reports by King et al. (1984). | ||
| e38 | 5 | 48 | Same | Kuroda et al. (2001). | ||
Table 2.
Kinetics of molecular interactions
| ID | kf (μm−1 s−1) | kb (s−1) | Previous version | Notes and references | ||
|---|---|---|---|---|---|---|
| 1 | 4 | 100 | 4 | 80 | Modified to slightly decrease the amount of Ca2+/calmodulin complex in response to Ca2+ increase. Kawaguchi et al. (2011). | |
| 2 | 40 | 640 | Same | Modified to slightly decrease the amount of Ca2+/calmodulin complex in response to Ca2+ increase. Kawaguchi et al. (2011). | ||
| 3 | 40 | 700 | Same | Modified to slightly decrease the amount of Ca2+/calmodulin complex in response to Ca2+ increase. Kawaguchi et al. (2011). | ||
| 4 | 400 | 3 | Same | See Kawaguchi et al. (2011). | ||
| 5 | 400 | 8 | Same | See Kawaguchi et al. (2011). | ||
| 6 | 400 | 0.24 | Same | See Kawaguchi et al. (2011). | ||
| 7 | 0.04 | — | Same | See Kawaguchi et al. (2011). | ||
| 8 | 0.5 | 0.5 | Same | See Kitagawa et al. (2009). | ||
| 9 | 500 | 0.5 | Same | See Kitagawa et al. (2009). | ||
| 10 | 2 | 0.75 | Same | See Kitagawa et al. (2009). | ||
| 11 | 1 | 1.5 | Same | See Kitagawa et al. (2009). | ||
| 12 | 10 | 7.5 | Same | See Kitagawa et al. (2009). | ||
| 13 | 20 | 7.5 | Same | See Kitagawa et al. (2009). | ||
| 14, 20 | 1 | 0.75 | Same | See Kitagawa et al. (2009). | ||
| 15 | 10 | 15 | Same | See Kitagawa et al. (2009). | ||
| 16, 17 | 0.005 (s−1) | 5 (μm−1 s−1) | Same | See Kitagawa et al. (2009). | ||
| 18 | 6 (s−1) | 5 (μm−1 s−1) | Same | See Kitagawa et al. (2009). | ||
| 19 | 3 (s−1) | 10 (μm−1 s−1) | Same | See Kitagawa et al. (2009). | ||
| 21 | 10 | 7.5 | Same | See Kitagawa et al. (2009). | ||
| 22 | 1 | 2 | Same | See Kitagawa et al. (2009). | ||
| 23 | 0.2 | 0.1 | Same | See Kitagawa et al. (2009). | ||
| 24 | 10 | 0.1 | Same | See Kitagawa et al. (2009). | ||
| 25 | 0.2 | — | 0.25 | — | Two-step G protein activation process was simplified to a single step reaction with similar kinetics. | |
| 26 | 0.066667 | — | Same | See Kitagawa et al. (2009). | ||
| 27 | 0.01 | 1 | Same | See Kitagawa et al. (2009). | ||
| 28 | 0.7 | 0.0013 | Same | See Kitagawa et al. (2009). | ||
| 29, 30 | 10 | 0.02 | Same | See Kitagawa et al. (2009). | ||
| 31, 32 | 0.1 | 0 | Same | See Kitagawa et al. (2009). | ||
| 33, 34 | 0.33333 | 0 | Same | See Kitagawa et al. (2009). | ||
| 35 | 10 | 0 | Same | See Kitagawa et al. (2009). | ||
| 36 | 0.2 | 48 | Same | See Kitagawa et al. (2009). | ||
| 37 | 12 | 0 | Same | See Kitagawa et al. (2009). | ||
| 38 | 600 | 0.06 | Same | See Kawaguchi et al. (2011). | ||
| 39 | 600 | 0.36 | Same | See Kawaguchi et al. (2011). | ||
| 40 | 0.2 | 0 | Same | See Kitagawa et al. (2009). | ||
| 41 | 1 | 10 | 0.25 | 1 | Slater et al. (1999). | |
| 42 | 1 | 0.05 | 1 | 0.1 | Assumption (Tsuruno and Hirano, 2007). | |
| 43 | 0.2 | 10 | Same | Kuroda et al. (2001). | ||
| 44 | 2 | 0.02 | 0.3 | 0.015 | Ananthanarayanan et al. (2003). | |
| 45 | 4 | 0.02 | 0.4 | 0.2 | O’Flaherty et al. (2001). | |
| 46 | 0.5 | — | 0.4 | — | Slightly higher degradation kinetics was assumed compared with Kuroda et al. (2001). | |
| 47 | 0.1 | 0.1 | 0.01 | 0.1 | Ajay & Bhalla (2004). | |
| 48 | 6 | 0.1 | 6 | 0.1 | Ajay & Bhalla (2004). | |
| 49 | 0.0012 | 0.48 | Same | Kuroda et al. (2001). | ||
| 50 | 0.05 | 0.1 | Same | Kuroda et al. (2001). | ||
| 51 | 0.17 | — | Same | Kuroda et al. (2001). | ||
| 52 | 1 | 0.01 | 0.01 | 0.0025 | EC50 of NO-mediated cGMP production is around 2–10 nm (Friebe & Koesling, 2003). The affinity of GTP with NO-stimulated GC (Km) is around 50 μm (Garthwaite, 2005). | |
| 53 | 0.735 | 29.4 | 3.5 | 29.4 | EC50 of NO-mediated cGMP production is around 2–10 nm (Friebe & Koesling, 2003). The affinity of GTP with NO-stimulated GC (Km) is around 50 μm (Garthwaite, 2005). | |
| 54 | 7.35 | — | Same | Kuroda et al. (2001). | ||
| 55 | 0.673 | Same | Kuroda et al. (2001). | |||
| 56 | 1 | 0.25 | Newly defined | cGMP binding to the two allosteric sites of PDE5 has an apparent Kd∼1 μm (Niino et al. 2010). | ||
| 57 | 2 | 0.1 | 2 | 0.1 | Kuroda et al. (2001); Poppe et al. (2008); Francis et al. (2010). | |
| 58 | 10 | 0.06 | 1 | 0.27 | Endo et al. (2003). | |
| 59 | 0.2 | — | 0.0001 | Assumption. | ||
| 60 | 1 | 10 | — | Masu et al. (1991); Tateyama & Kubo (2006). | ||
| 61 | 0.2 | 0.1 | — | Assumed to be identical to Gi binding to GABABRs. | ||
| 62 | 10 | 0.1 | — | Assumed to be identical to Gi binding to GABABRs. | ||
| 63 | 0.002 | 0.1 | — | mGluR1 couples to not only Gq but also Gs (Tateyama & Kubo, 2006). It was previously shown that mGluR1 facilitates the RP induction through Gs/cAMP/PKA pathway, not through Gq pathway, in a Purkinje cell (Sugiyama et al. 2008). Thus, Gs coupling to mGluR1 was incorporated in this model, assuming 100 times weaker coupling efficiency compared with the Gq coupling. | ||
| 64 | 0.1 | 0.1 | — | mGluR1 couples to not only Gq but also Gs (Tateyama & Kubo, 2006). It was previously shown that mGluR1 facilitates the RP induction through Gs/cAMP/PKA pathway, not through Gq pathway, in a Purkinje cell (Sugiyama et al. 2008). Thus, Gs coupling to mGluR1 was incorporated in this model, assuming 100 times weaker coupling efficiency compared with the Gq coupling. | ||
| 65 | 0.2 | — | Assumed to be identical to Gi reactions. | |||
| 66 | 0.066667 | — | — | Assumed to be identical to Gi reactions. | ||
| 67 | 0.01 | 1 | — | Assumed to be identical to Gi reactions. | ||
| 68 | 0.7 | 0.0013 | — | Assumed to be identical to Gi reactions. | ||
| 69 | 2.52 | 0.1 | Same | Kuroda et al. (2001). | ||
| 70 | 0.066667 | — | — | Assumed to be identical to Gs and Gi reactions. | ||
| 71 | 10 | — | Same | Kuroda et al. (2001). | ||
| 72 | 10 | — | Same | Kuroda et al. (2001). | ||
In the model, biochemical reactions in the signalling cascades were represented either as binding-dissociation reactions or as enzymatic reactions. For example, a binding reaction in which A and B bind to form complex AB is expressed as the following equation:
| (1) |
where kf and kb are the rate constants for the forward and backward processes. These rate constants are determined by the dissociation constant Kd and time constant τ. Kd is defined as kb/kf, and τ reflects the velocity of the reaction toward equilibrium. The reaction is represented as a differential equation:
| (2) |
Enzymatic reactions were expressed with the Michaelis–Menten formulation:
| (3) |
where S, E and P are substrate, enzyme and product, respectively. The Michaelis constant Km is defined as Km = (k1 + kcat)/k1. The maximum enzyme velocity Vmax is expressed as Vmax = kcat × [E]total, where [E]total is the total concentration.
We performed the simulation using CellDesigner 4.2 (Funahashi et al. 2003). Ordinary differential equations (ODEs) were numerically solved by the SOSlib (SBML ODE Solver Library). We started simulation experiments after the model reached equilibrium in the basal condition.
Statistics
Data are presented as mean ± SEM. Statistical significance was assessed by unpaired Student’s t test or by one-way ANOVA followed by the post hoc Dunnett's T3 test, unless otherwise stated.
Results
LTD impairment by CaMKII inhibition depends on protein phosphatase activity
The balance between protein kinases and phosphatases has been suggested to be critical for controlling the LTD establishment in a Purkinje cell (Coesmans et al. 2004; Launey et al. 2004; Belmeguenai & Hansel, 2005). Therefore, we first considered that the failure of LTD induction resulting from inhibition of CaMKII might be affected by protein phosphatases. To test this possibility, we performed whole-cell patch clamp recording experiments on a rat cultured Purkinje cell (see Methods; Kawaguchi & Hirano, 2007). We recorded AMPAR-mediated current responses to glutamate which was iontophoretically applied to a secondary dendrite (∼50 μm from the soma). LTD was induced by six consecutive stimulations (applied every 20 s) consisting of glutamate application and depolarization pulses (0 mV for 1 s) (Fig. 2A–C), causing potent Ca2+ increase in a broad area of the dendritic trees (see Fig. 2E and F; Tsuruno & Hirano, 2007). As shown in Fig. 2A and B, the conditioning persistently depressed the amplitude of AMPAR responses (54 ± 6% at 30 min, P < 0.001, paired Student's t test). Consistently with previous electrophysiological studies (Hansel et al. 2006), LTD was impaired by CaMKII inhibition with AIPII (1 μm), a peptide inhibitor (95 ± 3%, P < 0.001 compared with the control, unpaired Student's t test). The impairment of LTD by CaMKII inhibition with AIPII was rescued by additional inhibition of PP-2A and PP-1 using nodularin (10 nm) (57 ± 5%, P < 0.001 compared with AIPII alone) (Fig. 2A and B). In addition, another CaMKII inhibitor KN62 (5 μm) also suppressed the LTD establishment (102 ± 4%, P < 0.001), which was also abolished by additional inhibition of protein phosphatases with nodularin (57 ± 7%, P < 0.001) (Fig. 2C). Neither AIPII nor nodularin affected the depolarization-induced Ca2+ increase (Fig. 2G) or the basal glutamate responses (AIPII, 104 ± 23%, n = 8 cells; nodularin, 100 ± 6%, n = 5 cells, data not shown). To address which of PP-2A or PP-1 mediates the LTD impairment by CaMKII inhibition, we selectively suppressed either PP-2A or PP-1 with a specific inhibitor fostriecin (50 nm) or Inhibitor-2 (100 nm), respectively (Belmeguenai & Hansel, 2005). Inhibition of PP-2A by fostriecin rescued the LTD induction from suppression by CaMKII inhibition (67 ± 4%, P < 0.005), whereas that of PP-1 by Inhibitor-2 did not (100 ± 7%, P > 0.5) (Fig. 2D). Thus, the requirement of CaMKII activity for LTD induction was controlled by the activity of PP-2A.
Figure 2. LTD impairment by CaMKII inhibition was abolished by inhibition of PP-2A.
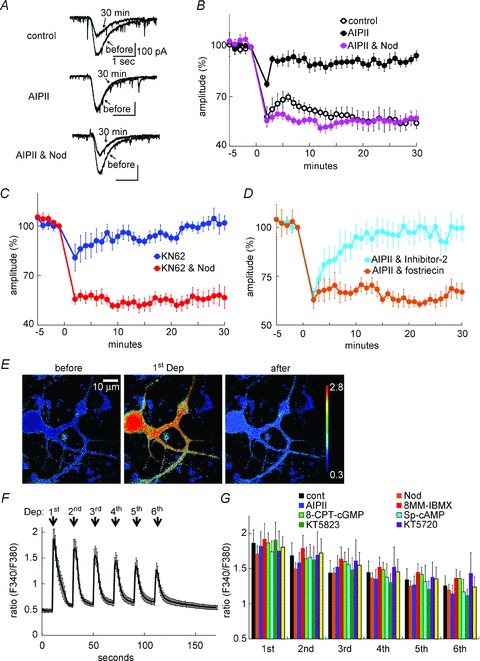
A and B, representative current traces (A) and time courses of amplitudes (B) of current response to glutamate before and after the coupled conditioning stimulation in the presence or absence of AIPII and nodularin. n = 5 for each. C and D, time courses of amplitudes of glutamate responses before and after the conditioning stimulation in the presence of KN62 and nodularin (C) or of AIPII and either fostriecin or Inhibitor-2 (D). n = 5 for each. E and F, representative images (E) and time course (F) of fluorescence ratio (F340/F380) of fura-4F before and after the conditioning depolarization. The conditioning depolarization consisted of 6 sets of depolarization pulses (0 mV for 1 s) applied every 20 s. n = 8 cells. G, peak fluorescence ratios of fura-4F in the secondary dendrite during each depolarization pulse in the absence or presence of an indicated pharmacological agent. n = 8 for each.
Systems biology models for LTD extended with CaMKII regulation mechanism
Next, we attempted to clarify the mechanism by which protein phosphatases are involved in the CaMKII contribution to LTD. To do this, we used a computational model of LTD. The signalling cascades regulating LTD at excitatory synapses were previously modelled and systematically analysed, but those analyses lacked CaMKII (Kuroda et al. 2001; Kotaleski et al. 2002; Doi et al. 2005; Ogasawara et al. 2007; Tanaka et al. 2007). On the other hand, signalling pathways related to CaMKII in a Purkinje cell were previously modelled as key regulators of RP, a form of LTP at inhibitory synapses (Kano et al. 1992, 1996; Kawaguchi & Hirano, 2000, 2002, 2007; Kitagawa et al. 2009; Kawaguchi et al. 2011). The RP model demonstrated how its induction is dynamically regulated by patterns of [Ca2+]i increase through CaMKII regulation (Kitagawa et al. 2009; Kawaguchi et al. 2011). To investigate how CaMKII affects LTD, we extended the LTD model by simply adding signalling cascades regulating RP. The model considered a single compartment without spatial segregation of excitatory synapses from inhibitory ones, because CaMKII is abundantly localized at excitatory postsynaptic spines as well (see Fig. 3B). The combined signalling cascades of the integrated model are shown in Fig. 1A.
Figure 3. Simulation of CaMKII-dependent LTD induction through gating of MAPK–PKC positive feedback loop.
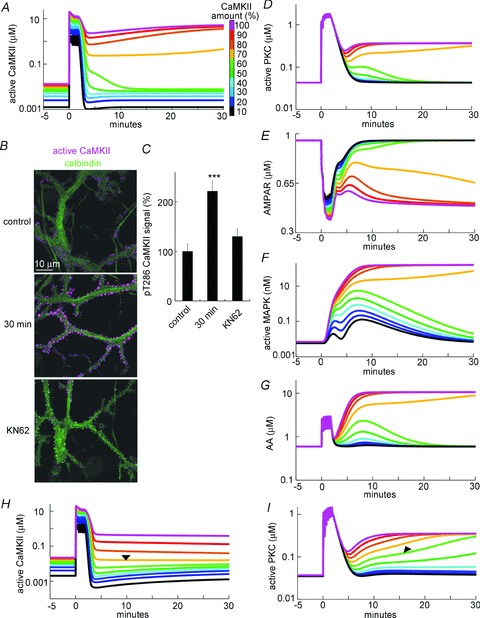
A and D–G, simulated time courses of the amount of active CaMKII (A), active PKC (D), non-phosphorylated AMPARs (E), active MAPK (F), and AA (G) before and after the conditioning stimulation consisting of 6 sets of [Ca2+]i (2 μm) and glutamate (5 μm) increases for 4 s. B, representative immunofluorescence images of active CaMKII autophosphorylated at Thr286 (magenta) and calbindin (green), a molecular marker of Purkinje cells, before (control) or 30 min after the conditioning treatment with 50 mm K+-containing external solution in the presence or absence of KN62. C, relative immunofluorescence signals for active CaMKII. ***P < 0.001 by Dunnett's test. n = 15 cells for each. H and I, simulated time courses of the amount of active CaMKII (H) and PKC (I) in the basal [Ca2+]i (0.075 μm) before and after the conditioning stimulation consisting of 6 sets of [Ca2+]i (2 μm) and glutamate (1 μm) for 4 s. Arrowheads indicate the persistent PKC activation (I) without sustained CaMKII activation (H).
The core component of the LTD induction mechanism is the Ca2+-triggered positive feedback loop consisting of PKC, Raf, MEK, MAPK, PLA2 and arachidonic acid (AA) (Kuroda et al. 2001; Tanaka et al. 2007; Tanaka & Augustine, 2008). Once triggered, the positive feedback loop prolongs PKC activation (Tanaka & Augustine, 2008), resulting in the long-term increase of phosphorylation of the AMPA-type glutamate receptor GluA2. As a consequence, the balance between endocytosis and exocytosis of AMPARs changes, resulting in the establishment of LTD (Wang & Linden, 2000; Chung et al. 2003). The PKC–MAPK positive feedback pathway and GluA2 phosphorylation is negatively regulated by the activity of PP-2A. Although the role of CaMKII in LTD remains unknown, its critical role in RP at inhibitory synapses has been demonstrated (Kano et al. 1992, 1996). CaMKII undergoes a positive feedback reaction by autophosphorylation at its own Thr286 residue (Lisman et al. 2002; Colbran & Brown, 2004). In addition, CaMKII supports its own activity by negative regulation of PDE1, which hydrolyses cAMP and cGMP (Hashimoto et al. 1989; Kitagawa et al. 2009). The suppression of PDE1 by CaMKII is expected to facilitate cGMP signalling, which is involved in the regulation of LTD (Hall et al. 1999; Ito, 2001). Thus, we hypothesized that the indirect modulation of cGMP by CaMKII might explain, at least partially, the CaMKII contribution to LTD. To test this idea, the modified new LTD model was constructed by linking the molecular reactions by several signalling components such as Ca2+, PP-2A, calcineurin, PDE1 and mGluR1, which have been reported or are likely to be involved in both LTD and RP (Aiba et al. 1994; Kawaguchi & Hirano, 2002; Launey et al. 2004; Belmeguenai & Hansel, 2005; Sugiyama et al. 2008) (for details, see Methods). Precise molecular reaction diagrams and parameters are shown in Fig. 1B and Tables 1–3.
First we confirmed that LTD is established through PKC–MAPK cascade activation by Ca2+ and glutamate inputs in this new model. The signalling network was stimulated by the conditioning stimulation consisting of six sets (with 20 s intervals) of Ca2+ increases (from basal 0.1 to 2 μm) and mGluR1 activation by glutamate increase (from 0.001 to 5 μm) for 4 s. As shown in Fig. 3A (magenta trace), the conditioning persistently activated CaMKII in the simulation model (from 0.013 to 5.11 μm at 30 min after the stimuli). Sustained activation of CaMKII through autophosphorylation at the Thr286 residue was experimentally supported by immunocytochemistry on a cultured Purkinje cell: conditioning treatment with a 50 mm K+-containing external solution for 10 s increased the active CaMKII autophosphorylated at Thr286 for longer than 30 min, particularly at spines where excitatory synapses are formed (221 ± 21% at 30 min, P < 0.0001) (Fig. 3B and C). In line with previous studies (Kuroda et al. 2001; Tanaka et al. 2007), the model simulation also exhibited sustained activation of a PKC–MAPK positive feedback loop upon the transient Ca2+ and glutamate stimulation (PKC, 0.04 to 0.37 μm; MAPK, 0.007 to 160 nm; AA, 0.59 to 10.6 μm), causing a sustained decrease of non-phosphorylated AMPARs (0.95 to 0.49 μm), which represents the functional AMPAR level in the model. Thus, the combined inputs of Ca2+ and glutamate to the model could establish LTD through triggering the PKC–MAPK positive feedback loop.
Using simulation, we analysed whether the LTD-inducing positive feedback loop was affected by CaMKII. When the amount of CaMKII was reduced to less than 60%, the persistent CaMKII activation was impaired (Fig. 3A). Importantly, in parallel with the failure of CaMKII activation, the PKC–MAPK positive feedback loop was also suppressed as the amount of CaMKII was reduced (Fig. 3D–G). Thus, our model suggested that whether CaMKII is effectively activated or not has a strong impact on the LTD-inducing PKC–MAPK positive feedback reaction. This simulation result is consistent with our patch clamp experimental results (see Fig. 2) and previous studies suggesting that CaMKII is required for the LTD induction (Hansel et al. 2006).
It should be noted that the persistent CaMKII activity upon a transient Ca2+ increase was not necessarily indispensable for sustained activation of the PKC–MAPK positive feedback loop, although they tended to behave in parallel in most cases. As shown in Fig. 3H, reducing the basal Ca2+ level (to 0.075 from 0.1 μm) weakened the high stable state of CaMKII activity, and the CaMKII activity returned to the basal level immediately after the stimulation when the amount of CaMKII was slightly decreased, consistent with the previous studies (Kitagawa et al. 2009; Kawaguchi et al. 2011). In spite of the failure to maintain the CaMKII activity, the active PKC exhibited a persistent increase, which still depended on the amount of CaMKII in the model (Fig. 3I). Thus, our model indicated that the PKC–MAPK positive feedback loop is by itself able to sustain its activation without persistent CaMKII activation. CaMKII activity during and soon after the Ca2+ and glutamate inputs seems to control the onset of the PKC–MAPK positive feedback loop.
We next examined whether the weakening of the PKC–MAPK positive feedback loop by the reduction of CaMKII might depend on phosphatase activity, and if so, what type of phosphatase is involved. The signalling cascades modelled here contain PP-1, PP-2A and PP-2B (also known as calcineurin: CaN). It was tested whether alteration of the amount of each phosphatase affects the CaMKII-dependent regulation of the PKC–MAPK loop. As shown in Fig. 4A–C, reduction of PP-2A to 50% markedly rescued the activation of the PKC–MAPK pathway and hence the LTD induction estimated by the fraction of non-phosphorylated AMPARs, even if 90% of CaMKII was removed. In contrast, the reduction of PP-1 or calcineurin could not rescue LTD. These simulation results accorded with the experimental results (see Fig. 2), supporting the idea that the impairment of LTD by CaMKII inhibition involves protein phosphatase, especially PP-2A.
Figure 4. PP-2A suppression recovered LTD under CaMKII suppression.
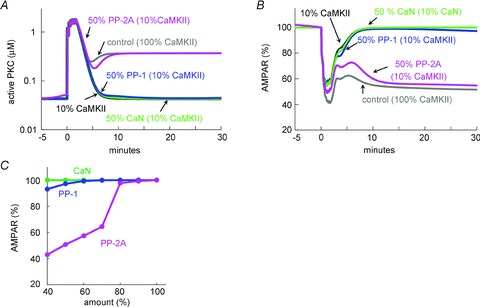
A and B, simulated time courses of amount of active PKC (A) and non-phosphorylated AMPARs (B) before and after the conditioning [Ca2+]i and glutamate increase in a model in which the amount of CaMKII is reduced to 10% without or with additional 50% reduction of PP-2A, PP-1, or calcineurin (CaN). C, relation between the amount of PP-2A, PP-1 or CaN and non-phosphorylated AMPAR fraction at 30 min after the conditioning stimulation.
PDE1 mediates the LTD impairment by CaMKII inhibition
We next attempted to examine whether the PDE1 regulation by CaMKII is responsible for the CaMKII dependency of LTD. Using the model, we evaluated the sensitivity of signalling components to the CaMKII activity level between 0.1 and 1 μm, the concentration range around which CaMKII seemed to influence the PKC–MAPK positive feedback loop (see Fig. 3). We arbitrarily altered the CaMKII activity from 0.001 to 10 μm in the model, and calculated the resultant changes of other molecular activities at equilibrium. As sensitive components, the theoretical analysis clearly highlighted the PKC–MAPK positive feedback loop and two protein phosphatase pathways bifurcating from PDE1: PP-1 regulation by the cAMP/protein kinase A (PKA)/pDARPP-32 pathway, and PP-2A regulation by the cGMP/PKG/pG-substrate pathway (Fig. 5A). CaMKII directly phosphorylates PDE1 and thereby reduces its affinity for Ca2+/CaM to about one-sixth, consequently increasing the cAMP and cGMP concentrations (Hashimoto et al. 1989). Increase in cAMP causes enhanced phosphorylation of DARPP-32 by PKA, resulting in the inhibition of PP-1. On the other hand, the facilitated cGMP/PKG/pG-substrate pathway is expected to suppress PP-2A (Hall et al. 1999), providing a supportive effect on the PKC–MAPK positive feedback loop. Indeed, simulation showed that the activities of PP-2A and PDE1 were concurrently modulated after the Ca2+ and glutamate stimulation in a manner dependent on the CaMKII amount (Fig. 5B and C). Thus, our model analysis suggested that CaMKII activity influences the PP-2A activity through negative regulation of PDE1.
Figure 5. PDE1 is critical for the CaMKII-mediated gating of LTD.
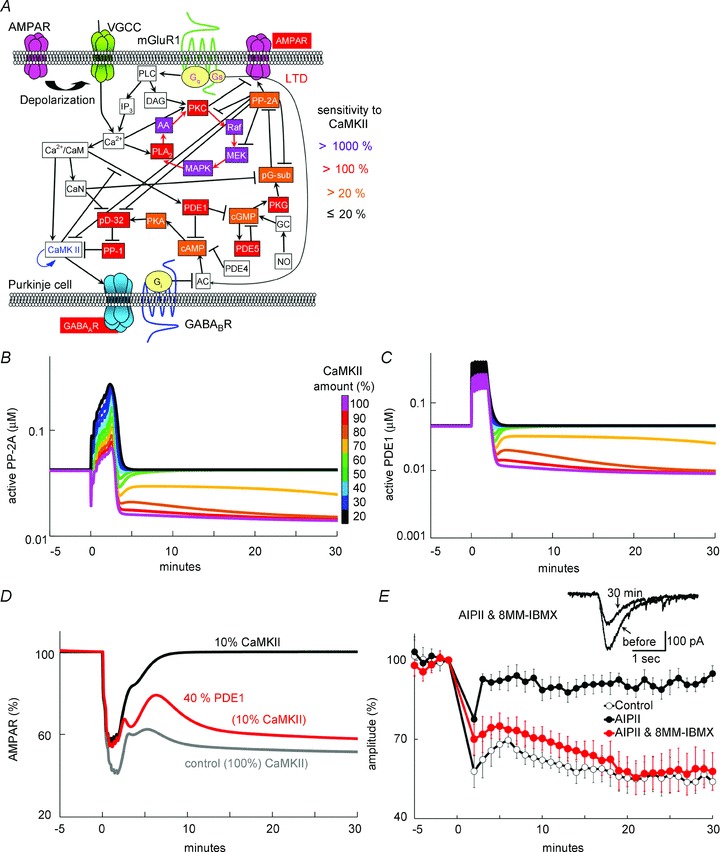
A, signalling components sensitive to the CaMKII activity highlighted by simulation analysis. The CaMKII activity was arbitrarily altered from 0.001 to 10 μm, and the activity level of other signalling molecules were calculated. Highlighted components exhibited large activity changes when the CaMKII activity was altered between 0.1 and 1 μm. The colour indicates the magnitude of activity changes: magenta, >1000%; red, >100%; orange >20%. B and C, simulated time courses of active PP-2A (B) and active PDE1 (C) in response to the LTD induction by the [Ca2+]i increase and glutamate stimulation, in the presence of various amounts of CaMKII. D, simulated time courses of non-phosphorylated AMPARs before and after the LTD induction with or without reduction of the amount of PDE1 in addition to CaMKII reduction. E, time courses of glutamate response amplitudes (n = 5 for each) before and after the LTD stimulation with AIPII and 8-MM-IBMX. Data without (control) and those with AIPII (AIPII, same as those in Fig. 2B) are presented for comparison. Representative current traces in the presence of both AIPII and 8-MM-IBMX are shown in insets.
To test whether the negative regulation of PDE1 contributes to the CaMKII-mediated LTD regulation, the amount of PDE1 was reduced to 40% in addition to the reduction of CaMKII to 10% in the model. Simulation showed that the impairment of LTD by CaMKII reduction was rescued by the additional PDE1 reduction (Fig. 5D). Supporting this simulation, in the presence of a specific PDE1 inhibitor, 8-MM-IBMX (20 μm), the LTD induction was not suppressed by CaMKII inhibition with AIPII (58 ± 7%, P < 0.005 compared with AIPII alone) (Fig. 5E). 8-MM-IBMX did not affect the depolarization-induced Ca2+ increase (Fig. 2G) or the basal glutamate responses (101 ± 2%, n = 5 cells). Thus, we concluded that PDE1 mediates the regulation of LTD by CaMKII.
Involvement of the cGMP/PKG pathway in the CaMKII-mediated LTD regulation
We next examined whether the cGMP/PKG pathway or the cAMP/PKA pathway is the downstream mediator of LTD gating by CaMKII. The concentration of cGMP or cAMP was arbitrarily increased in the model, and the resultant effect on the LTD impairment by a CaMKII decrease was examined. As shown in Fig. 6A and B, the increase of cGMP rescued LTD even if the CaMKII amount was set at 10%, whereas that of cAMP did not. This discrepancy between the effect of cGMP and cAMP on the LTD rescue supports the idea that the cGMP pathway predominantly controls the LTD induction through PP-2A regulation.
Figure 6. cGMP/PKG pathway mediates the CaMKII-mediated LTD regulation.
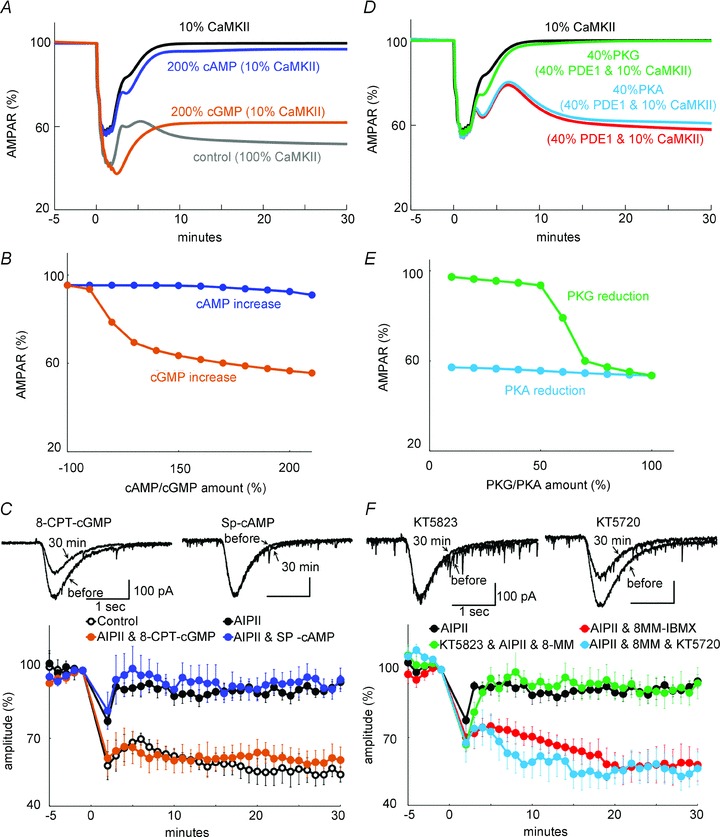
A, simulated time courses of the amount of non-phosphorylated AMPARs before and after an increase in [Ca2+]i and glutamate under reduced CaMKII. The amount of cAMP or cGMP was doubled. B, relation between the simulated AMPAR amount (at 30 min) and the altered amount of cGMP/cAMP. C, time courses of glutamate response amplitudes before and after the LTD conditioning stimuli in the absence or presence of AIPII, and 8-pCPT-cGMP or Sp-cAMP. n = 5 for each. Data without (control) and with AIPII (AIPII, same as those in Fig. 2B) are presented for comparison. D, simulated time courses of amount of non-phosphorylated AMPARs under reduced CaMKII. The amount of PKG or PKA was reduced to 40% in addition to PDE1 reduction to 40%. E, relation between the simulated AMPAR amount (at 30 min) and the altered amount of PKG/PKA. F, time courses of glutamate response amplitudes before and after the LTD conditioning stimuli in the absence or presence of AIPII, 8-MM-IBMX and KT5823 or KT5720. n = 5 for each. Data with AIPII alone or that with AIPII and 8-MM-IBMX (same as those in Figs 2B and 5E) are presented for comparison.
The rescue of LTD by increasing cGMP was experimentally verified by whole-cell patch clamp recording. Application of 8-CPT-cGMP (50 μm), a cGMP analogue, through a recording pipette abolished the impairment of LTD by AIPII (60 ± 6%, P < 0.005 compared with AIPII alone) (Fig. 6C). Extracellular application of 8-Br-cGMP, a membrane-permeable cGMP analogue, also abolished the suppression of LTD by CaMKII inhibition (56 ± 7%, P < 0.005 compared with AIPII alone) (not shown). In contrast, Sp-cAMP (10 μm) did not rescue the impairment of LTD by AIPII (96 ± 5%, P > 0.8 compared with AIPII alone) (Fig. 6C). Neither 8-CPT-cGMP nor Sp-cAMP affected the depolarization-induced Ca2+ increase (Fig. 2G) or the basal glutamate response (8-CPT-cGMP, 104 ± 13%, n = 8; Sp-cAMP, 98 ± 21%, n = 8). Thus, cGMP plays a predominant role downstream of PDE1 in the CaMKII-mediated LTD regulation.
Next, we tested the involvement of PKG as a downstream mediator of cGMP. As the amount of PKG, but not of PKA, was reduced in the model, the PDE1 reduction failed to rescue LTD from the impairment by CaMKII reduction (Fig. 6D and E). In accordance with the simulation result, the rescue of LTD by PDE1 inhibitor 8-MM-IBMX was prevented by additional inhibition of PKG with KT5823 (94 ± 7%, P < 0.01) (Fig. 6F). In contrast, a selective PKA inhibitor, KT5720, did not affect the LTD rescue by PDE1 inhibition (55 ± 7%, P > 0.8) (Fig. 6F). Neither KT5823 nor KT5720 affected the depolarization-induced Ca2+ increase (Fig. 2G) or the basal glutamate response (KT5823, 104 ± 8%, n = 5; KT5720, 102 ± 3%, n = 5). Taking all these results together, CaMKII supports the LTD induction through facilitation of the cGMP/PKG pathway by negatively regulating PDE1.
CaMKII-mediated facilitation of cGMP signalling
Our simulation and electrophysiological analysis demonstrated the functional interactions of CaMKII and the cGMP/PKG pathway in LTD (see Figs 2 and 6), which seems to be mediated by PDE1 (see Fig. 5). To directly show the causal relationship between activation of CaMKII and cGMP signalling, we attempted to detect the increase of cGMP by CaMKII activation using the FRET technique. Previous studies developed a FRET probe for cGMP imaging named cGES-DE5, consisting of the cGMP association region of PDE5 (GAF domain) and two fluorophores, YFP and CFP (Nikolaev et al. 2006). We constructed essentially the same probe with the exception that Venus was substituted for YFP (Fig. 7A). The structural change of the GAF domain resulting from cGMP binding causes more efficient FRET between Venus and CFP. We first confirmed that cGES-DE5 is useful for detecting a change of cGMP concentration in a living cultured Purkinje cell. To increase the cGMP concentration, soluble guanylyl cyclase was activated by a NO donor, DEANO (10 μm). DEANO application to the cGES-DE5-transfected Purkinje cell caused a gradual decrease of the fluorescence of CFP accompanied by a compensatory increase in that of Venus, resulting in a marked increase of the fluorescence ratio (Venus/CFP) (Fig. 7B–D). Thus, the cGMP increase was detected by the FRET change of cGES-DE5 in a Purkinje cell. As shown in Fig. 7E, DEANO application (10 μm), which is expected to saturate the cGMP binding to the probe, increased the fluorescence ratio (Venus/CFP) by 22.6 ± 1.3% at 20 min. Furthermore, the slight increase of cGMP caused by a selective PDE1 inhibitor, 8-MM-IBMX, was also detected as a 3.8 ± 1.0% increase of the fluorescence ratio (Fig. 7F).
Figure 7. FRET imaging of cGMP in a Purkinje cell.
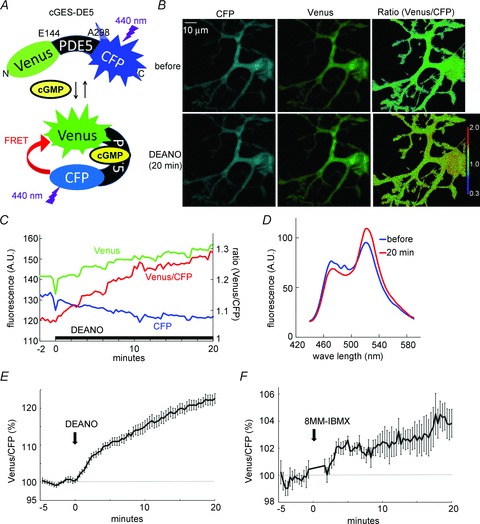
A, the FRET probe for cGMP imaging, cGES-DE5, consists of Venus and CFP linked by the cGMP-binding region of PDE5 (E144–A298). Binding of cGMP to the probe enhances the FRET efficiency between CFP and Venus. B and C, representative images (B) and intensity change (C) of fluorescence signals for CFP, Venus and fluorescence ratio (Venus/CFP) in a cultured Purkinje cell before and after the DEANO application. D, fluorescence emission spectrum of the cGES-DE5 probe before and 20 min after the DEANO application. E and F, time courses of FRET increase of cGED-DE5 in response to application of DEANO (E) or 8-MM-IBMX (F). n = 8 for each.
We next attempted to detect the cGMP increase, if any, caused by CaMKII-mediated negative regulation of PDE1. Whole-cell patch clamp recording was performed from a cGES-DE5-expressing Purkinje cell, and conditioning depolarization (0 mV, 500 ms, 5 times at 0.5 Hz) was applied to cause a sufficient Ca2+ increase for CaMKII activation. The conditioning depolarization persistently increased the fluorescence ratio (Venus/CFP) (105.3 ± 0.8% at 25 min, P < 0.005 by paired t test) (Fig. 8A and B). The sustained FRET increase was suppressed by inhibition of CaMKII with AIPII (100.5 ± 1.1%, P < 0.005 compared with the control condition) (Fig. 8B). Thus, the cGMP concentration persistently increased after a brief depolarization in a manner dependent on CaMKII. These FRET experimental results were reproduced by simulation monitoring the amount of cGMP binding to cGES-DE5, which was incorporated into the model with a dissociation constant of 1 μm for cGMP binding (Nikolaev et al. 2006) (Fig. 8C). Thus, CaMKII activation by a transient Ca2+ elevation persistently increases cGMP in a Purkinje cell.
Figure 8. Imaging of CaMKII-dependent sustained increase of cGMP in a Purkinje cell.
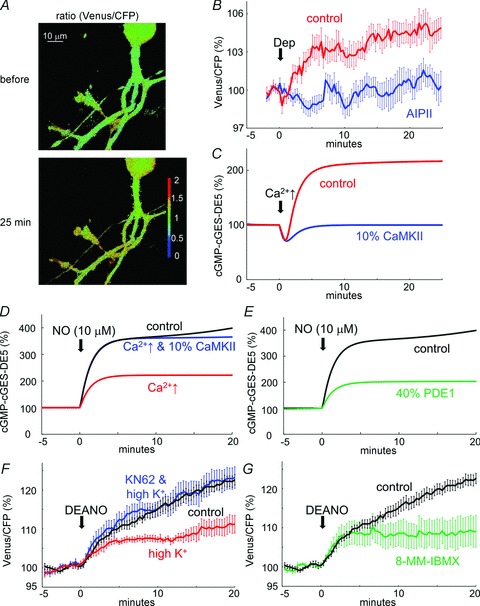
A and B, representative images (A) and time courses (B) of fluorescence ratio (Venus/CFP) before and after the conditioning depolarization of a cultured Purkinje cell with or without AIPII. n = 10 for each. C, simulated time courses of cGMP-bound cGES-DE5 probe protein amount before and after the conditioning Ca2+ increase (2 μm for 10 s) with or without reduction of CaMKII amount. D and E, simulated time courses of change of cGMP-bound cGES-DE5 probe in response to NO stimulation without or with CaMKII reduction to 10%. Prior transient Ca2+ stimulation was applied (D) or the amount of PDE1 was reduced to 40% in advance (E) in the model. F and G, time courses of FRET efficiency changes upon DEANO application to a Purkinje cell with or without prior high K+ treatment in the presence or absence of KN62 (F) or pretreatment with 8-MM-IBMX (G). n = 8 for each.
To further confirm the cGMP increase by CaMKII activation, we examined whether the depolarization- induced FRET increase affects the cGMP increase caused by NO input. A cultured Purkinje cell was pre-treated with 50 mm K+-containing external solution for 10 s, and CaMKII was persistently activated (see Fig. 3B and C). When DEANO was applied at 20 min after the CaMKII activation pre-treatment, a smaller FRET increase was caused (111.2 ± 2.3%, P < 0.005) (Fig. 8F). On the other hand, in a Purkinje cell treated with high K+ together with a CaMKII inhibitor, KN62 (5 μm), DEANO caused a similar FRET increase to that in control cells without prior K+ treatment (123.2 ± 2.8%, P > 0.85) (Fig. 8F). Thus, CaMKII activation by K+ treatment seemingly reduced the subsequent DEANO-mediated FRET increase, probably through increasing the cGMP-bound cGES-DE5 probe, then reducing the remaining working range of the FRET change of the cGES-DE5 probe. These experimental results were supported by the simulation shown in Fig. 8D. Pre-activation of the signalling cascades by a brief Ca2+ increase (to 2 μm from 0.1 μm for 10 s) weakened the subsequent association of cGES-DE5 with cGMP upon NO stimulation (10 μm) depending on the CaMKII amount (Fig. 8D). Furthermore, the reduction of PDE1 to 40% in the model also weakened the NO-stimulated cGMP association with cGES-DE5 to an extent similar to that caused by a prior Ca2+ increase (Fig. 8E). Accordingly, pretreatment with a PDE1 inhibitor, 8-MM-IBMX, weakened the subsequent FRET increase by DEANO (109.1 ± 4.1%, P < 0.05) (Fig. 8G). To summarize, not only NO input but also CaMKII activation increases cGMP through negative regulation of PDE1.
Dynamic interaction of CaMKII and NO in the LTD induction
Our data indicate that CaMKII gates the LTD induction by PDE1 inhibition through the cGMP/PKG pathway (Fig. 9A). Previous studies have also suggested LTD gating by NO through the cGMP/PKG pathway (Lev-Ram et al. 1997; Ogasawara et al. 2007). Thus, we hypothesized that LTD induction might be regulated by synergistic actions of the Ca2+-activated CaMKII activity and NO inputs.
Figure 9. LTD regulation by synergistic actions of CaMKII and NO.
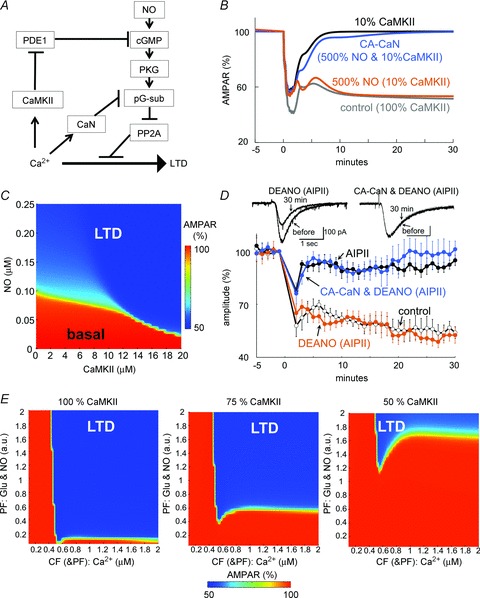
A, signalling scheme for interactions of Ca2+-activated CaMKII and calcineurin, and the NO-mediated activation of cGMP pathway contributing to LTD. B, simulated time courses of non-phosphorylated AMPARs before and after the LTD conditioning with or without CaMKII reduction to 10%, NO increase to 500%, and/or replacement of 10% calcineurin by the constitutively active form (CA-CaN). C, relation of CaMKII amount and basal NO required for LTD establishment. The amount of non-phosphorylated AMPARs 30 min after the conditioning stimulation was plotted in a colour scale as a function of basal NO concentration and CaMKII amount. D, time courses of glutamate response amplitudes before and after the conditioning stimulation in the presence of AIPII and DEANO in a cultured Purkinje cell with or without transfection of CA-CaN. n = 5 for each. Data without (control) and with AIPII (AIPII, same as those in Fig. 2B) are presented for comparison. E, simulated CaMKII-dependent dynamic relation between the intensity of Ca2+ increase primarily reflecting CF inputs and the intensity of glutamate and NO inputs reflecting PF inputs. The amount of non-phosphorylated AMPARs 30 min after the conditioning stimulation with different amounts of CaMKII was plotted in a colour scale against the strengths of PF and CF (& PF) inputs.
In accord with this idea, our simulation showed that the LTD induction was robustly rescued by increasing the basal NO level, even if the CaMKII amount was reduced (Fig. 9B and C). This simulation result was validated by whole-cell recording. In the presence of DEANO (3 μm), CaMKII inhibition by AIPII failed to impair LTD (53 ± 5%, P < 0.001) (Fig. 9D), suggesting that NO can compensate for the low activity of CaMKII. When applied at 3 μm, DEANO did not affect the depolarization-induced Ca2+ increase (Fig. 2G) or the basal glutamate response (103 ± 4%, n = 5 cells). Thus, the balance between Ca2+ increase and NO input determines whether LTD is induced or not through dynamic interplay of PDE1 regulation by CaMKII and the activation of soluble guanylyl cyclase by NO.
The signalling pathways shown in Fig. 9A suggest that the NO- and CaMKII-regulated cGMP/PKG pathway might be counteracted by calcineurin-mediated dephosphorylation of pG-substrate. In accordance with this idea, transfection of a constitutively active form of calcineurin (CA-CaN), which lacks an auto-inhibitory pseudo-substrate region (Fujii & Hirano, 2002), suppressed the LTD rescue by NO from impairment by CaMKII inhibition (101 ± 5%, P < 0.001) (Fig. 9D). Model simulation also showed that replacement of 10% of calcineurin by a constitutively active form abolished the NO-mediated LTD rescue (Fig. 9B). Taking all these results together, the LTD induction is gated by coordinated interactions of Ca2+-activated CaMKII and calcineurin, together with NO inputs.
Finally, we analysed by simulation how the amount of CaMKII affects the relationship between CF and PF inputs required for LTD establishment, in order to obtain insights into the possible functional consequence of interplay between NO and CaMKII. As shown in Fig. 9E, stronger inputs consisting of NO and glutamate mainly reflecting PF activity could compensate for the CaMKII reduction. In contrast, the magnitude of the Ca2+ increase primarily caused by CF input effective for LTD was not altered by the amount of CaMKII. Thus, our model suggested that the relative dependence of LTD on inputs from PFs and a CF might dynamically change according to the amount/activity of CaMKII.
Discussion
Although CaMKII is essential for LTD, its underlying mechanism has remained unclear. Using combined application of model prediction and experimental validation, this study revealed a signalling mechanism by which CaMKII contributes to LTD at PF synapses on a cerebellar Purkinje cell. The LTD induction was gated by CaMKII through facilitating the cGMP/PKG pathway by down-regulation of PDE1 (see Figs 5 and 6). PKG phosphorylates G-substrate, which suppresses the PP-2A activity counteracting the LTD induction (Hall et al. 1999). Taking advantage of FRET imaging of intracellular cGMP in a cultured Purkinje cell, we demonstrated that the CaMKII activation by a transient Ca2+ elevation persistently increased cGMP, most likely through negative regulation of PDE1 (see Fig. 8). Furthermore, NO rescued the LTD induction from impairment by CaMKII inhibition (see Fig. 9). Thus, LTD might be dynamically controlled by patterns of inputs from a CF and PFs, influencing CaMKII activity through Ca2+ and NO, respectively.
Molecular mechanism of LTD
Extensive studies during the past two decades have clarified the pivotal roles of signalling molecules involved in LTD, such as those constituting the PKC–MAPK positive feedback loop, regulating PP-2A activity via the NO/cGMP pathway, and those involved in endocytosis of AMPARs (Linden & Connor, 1991; Lev-Ram et al. 1997; Kawasaki et al. 1999; Wang & Linden, 2000; Ito, 2001; Hansel et al. 2001; Chung et al. 2003; Feil et al. 2003; Launey et al. 2004; Tanaka & Augustine, 2008). More recently the involvement of CaMKII in LTD was reported using αCaMKII knockout mice (Hansel et al. 2006). In contrast to the pivotal role of αCaMKII in hippocampal LTP, the ablation of αCaMKII in the cerebellum causes LTD impairment in juvenile mice, and conversion of LTD into LTP in adult mice. Furthermore, αCaMKII knockout mice show impaired adaptation of reflex ocular movement. Knockout mice lacking βCaMKII also exhibit ataxia and LTD impairment (van Woerden et al. 2009). Despite its importance, the detailed signalling mechanism of the CaMKII contribution to LTD has remained unknown. This study demonstrates that the CaMKII activity plays a permissive role in LTD induction by supporting the PKC–MAPK positive feedback loop through suppression of PP-2A (see Figs 2–4). Our model and experimental results indicated that the negative regulation of PDE1 by CaMKII enhances the cGMP/PKG signalling pathway which down-regulates PP-2A (see Figs 5, 6 and 8), contributing to LTD. Our data do not necessarily rule out the possibility that the LTD gating by CaMKII is also due to phosphorylation of other substrates, such as AMPARs and their associating proteins in postsynaptic density, although phosphorylation of AMPARs and their scaffolding proteins rather augments excitatory synaptic transmission in most cases (Lee et al. 2000; Correia et al. 2008).
The mutual activation of PKC and MAPK cascades would be a mechanism ensuring the establishment of LTD through effective endocytosis of phosphorylated AMPARs (Wang & Linden, 2000; Chung et al. 2003). The LTD induction relies on the PKC and MAPK activities for a few tens of minutes after the conditioning stimulation (Tanaka & Augustine, 2008). Thus, the efficiency of the PKC–MAPK positive feedback loop for a while after the induction is most likely to be critical. Prolonged PKC activation in Purkinje cells was previously demonstrated in slice preparations, as translocation to the plasma membrane after transient Ca2+ increase and glutamate input (Tanaka & Augustine, 2008). Consistently, cultured Purkinje cells also exhibited sustained translocation of fluorescently tagged PKC to membranous regions including dendritic spines after K+ and glutamate treatment, although the signal was rather weak (authors’ unpublished observations). The modest sustained translocation might be due to the limited amount of arachidonic acid produced by the PKC–MAPK cascades compared with the large amount of overexpressed PKC protein, which might have been responsible for the failure to detect the sustained translocation of PKC during the LTD induction in previous studies (Tsuruno & Hirano, 2007).
At PF synapses, LTP is postsynaptically induced by PF stimulation alone through facilitated transport of AMPARs toward the cell surface (Lev-Ram et al. 2002; Coesmans et al. 2004). This facilitated AMPAR transport seems to be mediated by S-nitrosylation of the N-ethylmaleimide-sensitive factor (NSF) by NO, which is released depending on PF activity (Lev-Ram et al. 2002; Kakegawa & Yuzaki, 2005). Activation of protein phosphatases by a small increase in the postsynaptic intracellular Ca2+ concentration is also required for LTP (Coesmans et al. 2004; Belmeguenai & Hansel, 2005; Schonewille et al. 2010), probably through suppressing the PKC–MAPK positive feedback loop which induces LTD. If Ca2+ increases sufficiently for CaMKII activation, as shown here by simulation (see Fig. 3), the activity of the PKC–MAPK positive feedback loop works and overcomes the LTP-inducing process. Interestingly, the ablation of βCaMKII was reported to convert LTP into LTD upon the LTP-inducing PF stimulation, suggesting that the role of CaMKII is to control the threshold of signalling activity for LTD (van Woerden et al. 2009). This idea is consistent with our conclusion that CaMKII is not necessarily required, but supports the LTD induction through promoting cGMP signalling (see Figs 2 and 6). Our simulation suggested that a smaller Ca2+ increase was more favourable for LTD when the CaMKII amount was reduced (see Fig. 9E). An interesting issue to be addressed in a future study would be whether ablation of βCaMKII, which has a higher affinity for Ca2+/CaM, restricts the appropriate Ca2+ increase for LTD to lower levels such as that caused by activation of PFs only, resulting in the conversion of LTP into LTD. Alternatively, the specific role of βCaMKII, such as association with F-actin, might be responsible for the inversed regulation of LTP/LTD. In any case, the gating role of CaMKII in the LTD establishment through the PDE1 pathway provides a clue to the comprehensive understanding of LTP/LTD regulation by CaMKII at PF synapses.
Kinetic simulation model of signalling cascades of LTD
Modelling of signalling cascades of cellular phenomena such as synaptic plasticity is useful for the intuitive understanding of the dynamic behaviour of a whole system (Bhalla & Iyengar, 1999; Kuroda et al. 2001; Doi et al. 2005; Ogasawara et al. 2007; Tanaka et al. 2007; Kitagawa et al. 2009; Kotaleski & Blackwell, 2010; Kawaguchi et al. 2011). Here, we simply combined the model for LTD and that for RP by linking signalling components such as Ca2+ and cyclic nucleotides in a single compartment without spatial separation. It should be noted that the present model omitted several signalling components, such as IP3-mediated Ca2+ release from the internal store, because Ca2+ was controlled as an input here. In spite of the simplicity, the model suggested that PDE1 is a key molecule linking several core signalling pathways for LTD regulation, which was supported experimentally (see Fig. 5). Thus, LTD at excitatory synapses and RP at inhibitory synapses might concurrently take place at neighbouring sites due to the diffusive property of key signalling molecules such as cGMP. Incorporating the spatial regulation of each molecular component into the signalling framework might enable the spatial interactions of LTD and RP to be studied precisely. Previous studies showed that LTD and RP are induced in a similar manner characterized by the leaky temporal integration of Ca2+ signals (Tanaka et al. 2007; Kawaguchi et al. 2011). The similarity might be ascribed to the CaMKII-mediated PDE1 inhibition simultaneously controlling PP-1 and PP-2A, which regulate RP and LTD, respectively.
Functional significance of CaMKII-mediated regulation of LTD and RP
Motor learning ability has been correlated with LTD in many kinds of transgenic mice with alteration of signalling molecules, including CaMKII (Aiba et al. 1994; De Zeeuw et al. 1998; Feil et al. 2003; Hansel et al. 2006; Takeuchi et al. 2008; van Woerden et al. 2009). However, normal motor learning in LTD-impaired conditions has also been reported (Welsh et al. 2005; Schonewille et al. 2011). Thus, even if LTD is suppressed, motor learning ability might be ensured through a robust mechanism brought about by multiple forms of synaptic plasticity in the cerebellar neuronal circuits (Hansel et al. 2001; Dean et al. 2010). Among them, RP is likely to be a compensatory mechanism because of its similar Ca2+ dependency to LTD and their common suppressive effect on Purkinje cell activity. Thus, the CaMKII-mediated signalling for coordinated regulation of LTD and RP may be essential for learning ability. CaMKII has also been implicated in severe mental retardation in Angelman syndrome, which is characterized by impaired learning, seizures and ataxia (Weeber et al. 2003). Genetic manipulation of αCaMKII at inhibitory autophosphorylation sites Thr305/306 with replacement by Ala was shown to rescue the ataxia and other deficits in model mice of the syndrome (van Woerden et al. 2007). Misregulation of CaMKII might cause unfavourable control of LTD/LTP at PF synapses and RP at inhibitory synapses on a Purkinje cell, resulting in motor discoordination. Thus, our integrated model and the present results such as the LTD rescue by pharmacological manipulation of NO input might be useful in the development of the treatment of motor deficits in Angelman syndrome patients.
Acknowledgments
We thank Drs Elizabeth Nakajima, Takeshi Sakaba, Mitsuharu Midorikawa and Yoshiaki Tagawa for critical reading of the manuscript and helpful comments. This work was supported by grants from MEXT, Japan, to S.K. and T.H., and from the Takeda Science Foundation to S.K., and by the Global COE program A06 of MEXT, Japan, to Kyoto University. The authors have no competing financial interests.
Glossary
- AA
arachidonic acid
- AC
adenylyl cyclase
- AMPAR
AMPA-type glutamate receptor
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CA-CaN
constitutively active calcineurin
- CF
climbing fibre
- DAG
diacylglycerol
- DARPP-32
dopamine and cAMP-regulated phosphoprotein of 32 kDa
- FRET
Förster resonance energy transfer
- GC
guanylyl cyclase
- LTD
long-term depression
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- mGluR1
metabotropic glutamate receptor 1
- NO
nitric oxide
- NSF
N-ethylmaleimide-sensitive factor
- PDE1
phosphodiesterase 1
- PF
parallel fibre
- PKA
protein kinase A
- PKC
protein kinase C
- PKG
protein kinase G
- PLA2
phospholipase A2
- PLC
phospholipase C
- PP-1
protein phosphatase 1
- PP-2A
protein phosphatase 2A
- RP
rebound potentiation
Author contributions
S.K. designed and performed the experiments and simulations; S.K. and T.H. interpreted the data and wrote the manuscript. Both authors approved the final version. This work was predominantly performed in the Graduate School of Science, Kyoto University.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Ajay SM, Bhalla US. A role for ERKII in synaptic pattern selectivity on the time-scale of minutes. Eur J Neurosci. 2004;20:2671–2680. doi: 10.1111/j.1460-9568.2004.03725.x. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan B, Stahelin RV, Digman MA, Cho W. Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J Biol Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J Neurosci. 2005;25:10768–10772. doi: 10.1523/JNEUROSCI.2876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signalling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C. Bidirectional parallel fibre plasticity in the cerebellum under climbing fibre control. Neuron. 2004;44:691–700. doi: 10.1016/j.neuron.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Correia SS, Bassani S, Brown TC, Lisé MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11:457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Dean P, Porrill J, Ekerot CF, Jörntell H. The cerebellar microcircuit as an adaptive filter: experimental and computational evidence. Nat Rev Neurosci. 2010;11:30–43. doi: 10.1038/nrn2756. [DOI] [PubMed] [Google Scholar]
- Doi T, Kuroda S, Michikawa T, Kawato M. Inositol,1,4,5- trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J Neurosci. 2005;25:950–961. doi: 10.1523/JNEUROSCI.2727-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Sweatt JD, Giese KP. Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci. 2004;24:8410–8415. doi: 10.1523/JNEUROSCI.3622-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Nairn AC, Greengard P, Ito M. Thr123 of rat G-substrate contributes to its action as a protein phosphatase inhibitor. Neurosci Res. 2003;45:79–89. doi: 10.1016/s0168-0102(02)00199-2. [DOI] [PubMed] [Google Scholar]
- Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M, Konnerth A, De Zeeuw CI, Hofmann F. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. Cell Biol. 2003;163:295–302. doi: 10.1083/jcb.200306148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res. 2003;93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- Fujii H, Hirano T. Calcineurin regulates induction of late phase of cerebellar long-term depression in rat cultured Purkinje neurons. Eur J Neurosci. 2002;16:1777–1788. doi: 10.1046/j.1460-9568.2002.02235.x. [DOI] [PubMed] [Google Scholar]
- Funahashi A, Tanimura N, Morohashi M, Kitano H. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. Biosilico. 2003;1:159–162. [Google Scholar]
- Gaiarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Dynamics of cellular NO-cGMP signalling. Front Biosci. 2005;10:1868–1880. doi: 10.2741/1666. [DOI] [PubMed] [Google Scholar]
- Hall KU, Collins SP, Gamm DM, Massa E, DePaoli-Roach AA, Uhler MD. Phosphorylation-dependent inhibition of protein phosphatase-1 by G-substrate. A Purkinje cell substrate of the cyclic GMP-dependent protein kinase. J Biol Chem. 1999;274:3485–3495. doi: 10.1074/jbc.274.6.3485. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D’Angelo E. Beyond parallel fibre LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GH, Andreev D, De Zeeuw CI, Elgersma Y. αCaMKII is essential for cerebellar LTD and motor learning. Neuron. 2006;51:835–843. doi: 10.1016/j.neuron.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Sharma RK, Soderling TR. Regulation of Ca2+/calmodulin- dependent cyclic nucleotide phosphodiesterase by the autophosphorylated form of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1989;264:10884–10887. [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kano M, Kano M, Fukunaga K, Konnerth A. Ca2+-induced rebound potentiation of γ-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci U S A. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kakegawa W, Yuzaki M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc Natl Acad Sci U S A. 2005;102:17846–17851. doi: 10.1073/pnas.0508910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABAB receptors in a Purkinje neuron. Neuron. 2000;27:339–347. doi: 10.1016/s0896-6273(00)00041-6. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Nagasaki N, Hirano T. Dynamic impact of temporal context of Ca2+ signals on inhibitory synaptic plasticity. Scientific Rep. 2011;1:143. doi: 10.1038/srep00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Fujii H, Gotoh Y, Morooka T, Shimohama S, Nishida E, Hirano T. Requirement for mitogen-activated protein kinase in cerebellar long term depression. J Biol Chem. 1999;274:13498–13502. doi: 10.1074/jbc.274.19.13498. [DOI] [PubMed] [Google Scholar]
- King MM, Huang CY, Chock PB, Nairn AC, Hemmings HC, Jr, Chan KF, Greengard P. Mammalian brain phosphoproteins as substrates for calcineurin. J Biol Chem. 1984;259:8080–8083. [PubMed] [Google Scholar]
- Kitagawa Y, Hirano T, Kawaguchi S. Prediction and validation of a mechanism to control the threshold for inhibitory synaptic plasticity. Mol Syst Biol. 2009;5:280. doi: 10.1038/msb.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaleski JH, Lester D, Blackwell KT. Subcellular interactions between parallel fibre and climbing fibre signals in Purkinje cells predict sensitivity of classical conditioning to interstimulus interval. Integr Physiol Behav Sci. 2002;37:265–292. doi: 10.1007/BF02734249. [DOI] [PubMed] [Google Scholar]
- Kotaleski JH, Blackwell KT. Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci. 2010;11:239–251. doi: 10.1038/nrn2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Schweighofer N, Kawato M. Exploration of signal transduction pathways in cerebellar long-term depression by kinetic simulation. J Neurosci. 2001;21:5693–5702. doi: 10.1523/JNEUROSCI.21-15-05693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launey T, Endo S, Sakai R, Harano J, Ito M. Protein phosphatase 2A inhibition induces cerebellar long-term depression and declustering of synaptic AMPA receptor. Proc Natl Acad Sci U S A. 2004;101:676–681. doi: 10.1073/pnas.0302914101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY. Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron. 1997;18:1025–1038. doi: 10.1016/s0896-6273(00)80340-2. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci U S A. 2002;99:8389–8393. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Niino Y, Hotta K, Oka K. Blue fluorescent cGMP sensor for multiparameter fluorescence imaging. PLoS One. 2010;5:e9164. doi: 10.1371/journal.pone.0009164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Gambaryan S, Lohse MJ. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat Methods. 2006;3:23–25. doi: 10.1038/nmeth816. [DOI] [PubMed] [Google Scholar]
- O’Flaherty JT, Chadwell BA, Kearns MW, Sergeant S, Daniel LW. Protein kinases C translocation responses to low concentrations of arachidonic acid. J Biol Chem. 2001;276:24743–24750. doi: 10.1074/jbc.M101093200. [DOI] [PubMed] [Google Scholar]
- Ogasawara H, Doi T, Doya K, Kawato M. Nitric oxide regulates input specificity of long-term depression and context dependence of cerebellar learning. PLoS Comput Biol. 2007;3:e179. doi: 10.1371/journal.pcbi.0020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signalling pathways. Nat Methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron. 2010;67:618–628. doi: 10.1016/j.neuron.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, Linden DJ, Huganir RL, De Zeeuw CI. Reevaluating the role of LTD in cerebellar motor learning. Neuron. 2011;70:43–50. doi: 10.1016/j.neuron.2011.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater SJ, Milano SK, Stagliano BA, Gergich KJ, Ho C, Mazurek A, Taddeo FJ, Kelly MB, Yeager MD, Stubbs CD. Synergistic activation of protein kinase Cα, -βI, and -γ isoforms induced by diacylglycerol and phorbol ester: roles of membrane association and activating conformational changes. Biochemistry. 1999;38:3804–3815. doi: 10.1021/bi982778r. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Kawaguchi S, Hirano T. mGluR1-mediated facilitation of long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. Eur J Neurosci. 2008;27:884–896. doi: 10.1111/j.1460-9568.2008.06063.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Khiroug L, Santamaria F, Doi T, Ogasawara H, Ellis-Davies GC, Kawato M, Augustine GJ. Ca2+ requirements for cerebellar long-term synaptic depression: role for a postsynaptic leaky integrator. Neuron. 2007;54:787–800. doi: 10.1016/j.neuron.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Augustine GJ. A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron. 2008;59:608–620. doi: 10.1016/j.neuron.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Ohtsuki G, Yoshida T, Fukaya M, Wainai T, Yamashita M, Yamazaki Y, Mori H, Sakimura K, Kawamoto S, Watanabe M, Hirano T, Mishina M. Enhancement of both long-term depression induction and optokinetic response adaptation in mice lacking delphilin. PLoS One. 2008;3:e2297. doi: 10.1371/journal.pone.0002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateyama M, Kubo Y. Dual signalling is differentially activated by different active states of the metabotropic glutamate receptor 1α. Proc Natl Acad Sci U S A. 2006;103:1124–1128. doi: 10.1073/pnas.0505925103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruno S, Hirano T. Persistent activation of protein kinase Cα is not necessary for expression of cerebellar long-term depression. Mol Cell Neurosci. 2007;35:38–48. doi: 10.1016/j.mcn.2007.01.016. [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Hoebeek FE, Gao Z, Nagaraja RY, Hoogenraad CC, Kushner SA, Hansel C, De Zeeuw CI, Elgersma Y. βCaMKII controls the direction of plasticity at parallel fibre-Purkinje cell synapses. Nat Neurosci. 2009;12:823–825. doi: 10.1038/nn.2329. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Yamaguchi H, Zeng XH, Kojo M, Nakada Y, Takagi A, Sugimori M, Llinás RR. Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proc Natl Acad Sci U S A. 2005;102:17166–17171. doi: 10.1073/pnas.0508191102. [DOI] [PMC free article] [PubMed] [Google Scholar]