

Abstract
EGFR and Hippo signaling both control growth, and when dysregulated contribute to tumorigenesis. We find that EGFR activates the Hippo pathway transcription factor Yorkie, and demonstrate that Yorkie is required for the influence of EGFR on cell proliferation in Drosophila. EGFR regulates Yorkie through an influence of its Ras-MAPK branch on the Ajuba LIM protein Jub. Jub is epistatic to EGFR and Ras for Yorkie regulation, Jub is subject to MAPK-dependent phosphorylation, and EGFR-Ras-MAPK signaling enhances Jub binding to the Yorkie kinase Warts, and the adaptor protein Salvador. An EGFR-Hippo pathway link is conserved in mammals, as activation of EGFR or RAS activates the Yorkie homologue YAP, and EGFR-RAS-MAPK signaling promotes phosphorylation of the Ajuba family protein WTIP, and also enhances WTIP binding to the Warts and Salvador homologues LATS and WW45. Our observations implicate the Hippo pathway in EGFR-mediated tumorigenesis and identify a molecular link between these pathways.
INTRODUCTION
Tumor formation often involves the inappropriate activation of regulatory pathways that play crucial roles in controlling growth and cell fate decisions during normal development. One such pathway is Epidermal Growth Factor Receptor (EGFR) signaling, which has been implicated in several types of human cancers, including brain, breast, and lung (Normanno et al., 2006; Sibilia et al., 2007). Distinct downstream signal transduction mechanisms for EGFR signaling have been identified and subject to intensive study. One of the best known involves the G protein Ras, and its activation of the Raf kinase, which phosphorylates and activates MEK (MAPK-ERK Kinase), which then phosphorylates and activates ERK (Extra cellular signal Related Kinase) (Schlessinger, 2004). ERK is a member of the MAPK (Mitogen Activated Protein Kinase) family, and can modulate the activity of transcription factors by phosphorylating them. However, other classes of substrates have been identified, and the respective contributions of different downstream targets to growth promotion by EGFR-Ras-MAPK signaling remain unclear. Here, we identify a mechanism involving Hippo pathway regulation that contributes to growth control by EGFR-Ras-MAPK signaling.
Hippo signaling has emerged over the last decade as a major determinant of organ growth. First identified in Drosophila, its role in growth control and tumorigenesis is conserved in mammals (reviewed in Bao et al., 2011; Pan, 2010; Reddy and Irvine, 2008; Staley and Irvine, 2012). The core of the Hippo pathway comprises two kinases – Hippo (Hpo, Mst in vertebrates) and Warts (Wts, Lats in vertebrates), and their co-factors Salvador (Sav, WW45 in vertebrates) and Mob As Tumor Suppressor (Mats, Mob in vertebrates). Hpo, Sav and Mats activate Wts, and Wts then phosphorylates the transcriptional co-activator Yorkie (Yki, Yap and Taz in vertebrates). Phosphorylation downregulates Yki by preventing it from accumulating in the nucleus. Nuclear Yki interacts with DNA binding proteins, and promotes transcription of genes involved in promoting cell proliferation and inhibiting apoptosis. One remarkable feature of Hippo signaling is its responsiveness to a variety of distinct upstream regulatory inputs (reviewed in Codelia and Irvine, 2012; Irvine, 2012; Staley and Irvine, 2012). Previously characterized upstream inputs include the Fat cadherin pathway, proteins that respond to cell-cell contact or cell density (e.g. Crumbs, Echinoid, Merlin, E-cadherin) and proteins that influence apical-basal polarity. Hippo signaling can also be influenced by a variety of manipulations that affect the F-actin cytoskeleton.
The contributions of different upstream inputs into Hippo signaling can vary depending upon the tissue-type and developmental context. Hippo signaling plays an important role in controlling normal glial cell numbers, as inappropriate activation of Yki leads to excess glial cells, whereas downregulation of Yki leads to loss of glial cells (Reddy and Irvine, 2011). This role in controlling glial growth is conserved, because mutation of the upstream regulator Merlin leads to glial cell tumors in humans (McClatchey et al., 1998; Merel et al., 1995; Orr et al., 2011). EGFR is also implicated in glial cell tumors, as gliomas often have elevated EGFR signaling, and activation of EGFR promotes glial cell proliferation in Drosophila (Hatanpaa et al., 2010; Nagane et al., 2001; Read et al., 2009; Witte et al., 2009). Here, we demonstrate that EGFR signaling constitutes an additional input into Hippo pathway regulation, and provide evidence that this linkage is important for glial cell growth. We further characterize the connection between these pathways, demonstrating that Ras-MAPK signaling mediates the influence of EGFR on Hippo signaling, and that the Ajuba LIM domain protein Jub is a key target of MAPK signaling within the Hippo pathway. Moreover, we show that an EGFR-Ras-MAPK-Ajuba link is conserved in mammalian cells. Together, these studies define a conserved link between EGFR/Ras and Hippo signaling in growth control through Ajuba family proteins, with important implications for diverse cancers.
RESULTS
EGFR activation promotes glial growth through a Yki-dependent mechanism
Hippo signaling plays a key role in regulating glial cell numbers, as increased activation of Yki is sufficient to promote proliferation of glial cells in the developing Drosophila eye disc and brain, whereas decreasing Yki activity reduces glial cell numbers (Reddy and Irvine, 2011). Activation of EGFR signaling has also been reported to be sufficient to promote increased Drosophila glial cell proliferation (Read et al., 2009; Witte et al., 2009). This prompted us to conducted genetic tests to investigate the epistatic relationship between Hippo and EGFR signaling in controlling glial cell proliferation.
EGFR signaling can be constitutively activated by expressing an isoform in which the extracellular ligand-binding domain has been replaced by the dimerization domain from phage lambda (EGFRλ) (Queenan et al., 1997). We expressed EGFRλ specifically in glial cells by using the UAS-Gal4 system to drive its expression under the control of a Gal4 line expressed in glial cells (repo-Gal4) (Fig. 1A,E, H). As reported previously, in flies expressing repo-Gal4 and UAS-EGFRλ transgenes, glial cell numbers and glial cell proliferation are increased (Fig. 1B,F,I) (Read et al., 2009; Witte et al., 2009). Reduction of Yki levels, using RNAi mediated by expression of UAS-hairpin transgenes, suppressed these effects of activated-EGFR (Fig. 1D,G,J), indicating that EGFR-promoted glial growth is Yki-dependent.
Figure 1. Relationship between EGFR and Yki in controlling glial growth.

(A-D) Third instar eye discs, stained for Repo (red) and Elav (blue), from larvae expressing repo-Gal4 UAS-mCD8:GFP (green) and A) control, B) UAS-EGFRλ, white arrows highlight extra glial cells in the optic stalk C) UAS-Yki-RNAi, D) UAS-EGFRλ Yki-RNAi UAS. (E-G) Brain lobes stained for glial nuclei with anti-Repo (red) from larvae expressing repo-Gal4 UAS-dcr2 UAS-mCD8:GFP (green) and E) control, F) UAS-EGFRλ, white bar highlights extra glial cells, G) UAS-EGFRλ UAS-Yki-RNAi. (H-J) Third instar eye discs, labeled with EdU (magenta) from larvae expressing repo-Gal4 UAS-dcr2 UAS-mCD8:GFP (green) and H) control, I) UAS-EGFRλ, white arrow highlights extra glial cell proliferation, J) UAS-EGFRλ Yki-RNAi. (K-N) Third instar eye discs, stained for Repo (red) and Elav (blue), from larvae expressing repo-Gal4 UAS-mCD8:GFP (green) and K) UAS-EGFR-RNAi, L) UAS-YkiS168A UAS-EGFR-RNAi M) UAS-YkiS168A N) UAS-EGFRλ UAS-YkiS168A. In this and subsequent figures, panels marked by prime shows the single channel of the stain to the left.
Complementary to the tumorigenic effects of EGFR activation, we found that reduction of EGFR levels by RNAi greatly reduced glial cell numbers, thus establishing a normal requirement for EGFR signaling in Drosophila glia (Fig. 1K). This reduction in glial cell numbers could be partially suppressed by expression of an activated form of Yki in glial cells (YkiS168A, Fig. 1L), resulting in a phenotype intermediate between the loss of glial cells associated with EGFR RNAi (Fig. 1K), and the gain of glial cells associated with activated-Yki (Fig. 1M) (Reddy and Irvine, 2011). The micro RNA gene bantam plays an important role in controlling glial cell numbers (Reddy and Irvine, 2011), and can be regulated by both Hippo and EGFR signaling (Herranz et al., 2012), which can synergize to induce a strong glial overgrowth phenotype (Fig. 1N), comparable to that induced by direct over-expression of bantam (Reddy and Irvine, 2011). Nonetheless, the observations that yki RNAi suppresses the increased glial cell numbers of EGFRλ and that activated-Yki suppresses the decreased glial cell numbers of EGFR RNAi, together raised the possibility that Yki could be controlled downstream of EGFR to regulate glial cell numbers.
EGFR promotes Yki activity
We next examined interactions between Hippo and EGFR signaling in wing imaginal discs in order to investigate whether the genetic relationship between them observed in glial cells also occurs in other tissues, and to determine whether this well-characterized model system could be used to better define the relationship between these two pathways. Transgenes were expressed under the control of engrailed-Gal4 (en-Gal4), which drives expression in posterior cells. As expression of activated-EGFR under en-Gal4 caused embryonic lethality, we used a conditional system, in which Gal4 activity is blocked by a temperature-sensitive form of Gal80 (Lee and Luo, 1999). In addition to EGFRλ, we also expressed EGFR activated by a point mutation in the cytoplasmic domain, EGFRA887T (Lesokhin et al., 1999). Both forms of activated-EGFR increased cell proliferation in wing discs, as revealed by EdU labeling (Fig. 2A and data not shown). This increased cell proliferation was blocked by RNAi-mediated downregulation of Yki (Fig. 2B), indicating that, as in glial cells, activated-EGFR requires Yki to promote cell proliferation.
Figure 2. EGFR signaling regulates Yki activity.

(A-G) Third instar wing discs, labeled with EdU (magenta) (A&,B), or β-gal from an ex-lacZ transgene (red) (C-G) and from larvae expressing en-Gal4 UAS-GFP (green) and A) UAS EGFRA887T, B) UAS-EGFRA887T UAS-Yki-RNAi, C) control, D) UAS-EGFRA887T , E) UAS-EGFRA887T UAS-Yki-RNAi, F) UAS-YkiS168A, G) UAS-YkiS168A UAS-EGFR-RNAi. H) Wing disc stained for Yki (red) and DNA (Hoechst, blue) from en-Gal4 UAS-GFP (green) UAS-EGFRA887T. Higher magnifications of single channels of Yki and DNA stains from boxed regions 1 and 2 are shown in black and white at right. I) Vertical section through a portion of the wing disc shown in H. J) Western blots (anti-phospho Ser 168 Yki, anti-V5) on material precipitated using anti-V5 beads from S2 cells transfected to express Yki:V5, and where indicated (MAPK*) pUAS -RasV12, pUAS-Phl, pUAS-Dsor and pUAST-rl constructs, and a Myc:Wts expression plasmid. The histogram at bottom shows quantitation of Yki phosphorylation in transfected cells from multiple experiments, error bars indicate s.d. each of the means is distinct based on one way ANOVA. See also Fig. S1.
To investigate whether this genetic requirement could reflect an influence of EGFR on Yki activity, we examined expression of expanded (ex) using an ex-lacZ reporter, which is a well-established downstream target of Yki (Hamaratoglu et al., 2006). Importantly, ex is not regulated by Capicua (Herranz et al., 2012), which is responsible for mediating a Yki-independent effect of EGFR signaling on bantam (Herranz et al., 2012). ex-lacZ was increased by expression of activated EGFR (Figs 2D, S1A, I). Moreover, yki RNAi reversed this effect on ex-lacZ (Fig. 2E,S1C). In complementary experiments, EGFR RNAi had no effect on ex-lacZ expression induced by activated-Yki (Fig. 2G). Confirmation that EGFR activation induces activation of Yki was provided by direct examination of Yki localization. Yki is normally predominantly cytoplasmic in wing discs, but activation of Yki is associated with partial translocation of Yki into the nucleus (Dong et al., 2007; Oh and Irvine, 2008). Expression of activated-EGFR increased Yki nuclear localization (Fig. 2H,I); this effect was comparable to that induced by mutation of upstream tumor suppressors of the Hippo pathway (Dong et al., 2007; Oh and Irvine, 2008).
EGFR regulates Yki through a Ras-Raf-MAPK pathway
EGFR signaling involves multiple downstream branches, but one of the most important downstream effectors is the G protein Ras. An oncogenic, activated form of Ras (RasV12) can induce cell proliferation in both wing discs and glial cells (Karim and Rubin, 1998; Read et al., 2009). We found that this stimulation of proliferation is Yki-dependent (Fig 3A-F). Moreover, RasV12 also increased both ex-lacZ expression, and nuclear Yki (Fig. 3G,H,J). To further strengthen the connection between EGFR-Ras signaling and Yki activation, we confirmed that an additional target of Yki, fj-lacZ (Cho et al., 2006), is upregulated by RasV12 (Fig. 3K), and we also confirmed that expression of RasV12 decreases Yki phosphorylation in wing discs (Fig. 3I), without disturbing the epithelial structure of disc cells (Fig. S2). These observations imply that EGFR signaling acts through Ras to modulate Yki activity. This conclusion was confirmed by the observation that downregulation of Ras by RNAi could suppress the influence of activated-EGFR on ex-lacZ (Fig. 3M).
Figure 3. EGFR regulates Yki through a Ras-Raf-MAPK pathway.

Third instar brain lobes (A,D), eye discs (B,E) or wing discs (C,F) stained for Repo (red) or EdU (magenta) from larvae expressing (A,B,D,E) repo-Gal4 UAS-mCD8:GFP (green) or (C,F) en-Gal4 UAS-GFP (green) and A-C) UAS RasV12, or D-F) UAS RasV12 UAS-Yki-RNAi. G,H) Wing disc stained for Yki (red) and DNA (Hoechst, blue) from en-Gal4 UAS-GFP (green) UAS-RasV12. Higher magnifications of single channels of Yki and DNA stains from boxed regions 1 and 2 are shown at right, H shows vertical sections through the disc. I) Western blot on lysates of wing discs expressing RasV12 (UAS-RasV12 tub-Gal4 tub-Gal80ts, induced for 3 days at 29 °C), probed with anti-Yki and anti-phospho168 Yki antibodies. (J, L-Q) Third instar wing discs, stained for β-gal expressed by ex-lacZ (red) from larvae expressing en-Gal4 UAS-GFP (green) and J) UAS-RasV12, L) UAS-RasV12 UAS-Yki-RNAi, M) UAS-EGRλ UAS-Ras-RNAi, N) UAS-RasV12S35, O) UAS-RasV12C40, P) UAS-RasV12G37, Q) UAS-RasV12 UAS-rl-RNAi. K) Wing discs, stained for β-gal expressed by fj-lacZ (cyan) from larvae expressing en-Gal4 UAS-mRFP (membrane bound RFP, red) and UAS-RasV12. See also Fig. S2.
Mutations in a Ras effector loop that preferentially impair the influence of Ras on distinct downstream pathways have been created and characterized (Bergmann et al., 1998; Karim and Rubin, 1998; Rodriguez-Viciana et al., 1997). RasV12G37 and RasV12C40 isoforms are deficient in their ability to interact with and activate Raf kinase, whereas RasV12S35 retains the ability to activate Raf, but, at least in mammalian cells, has been shown to be deficient in Ral GDS and PI3K activation. RasV12S35 increased ex-lacZ expression and wing growth, whereas RasV12G37 and RasV12C40 had no effect (Fig. 3N-P). Together, these results implicate Raf as the key downstream signaling branch involved in the influence of EGFR on Yki activity. Raf acts through the MAPK cascade that activates ERK. Drosophila ERK is encoded by the rolled (rl) gene, and the influence of RasV12 on Yki activity was suppressed by rl RNAi (Fig. 3Q). Thus, EGFR regulates Yki through a Ras-Raf-MAPK pathway. Consistent with this, we also found that the increased phosphorylation of Yki induced by over-expression of Wts in cultured Drosophila S2 cells could be suppressed by co-transfection of plasmids inducing MAPK activation (Fig. 2J).
EGFR regulation of Yki activity requires Jub
Genetic epistasis experiments were performed to investigate how EGFR signaling impinges on Hippo signaling. The observations that EGFR and Ras promote nuclear translocation of Yki, and dephosphorylation of Ser168 (Figs 2J, 3I) implies that they act at or upstream of Wts, as phosphorylation by Wts is the main factor influencing Yki localization, and Wts is the kinase responsible for Ser168 phosphorylation (Dong et al., 2007; Oh and Irvine, 2008). Components of the Hippo pathway that inhibit Wts activity and act at or upstream of Wts include Ras association family member (Rassf) (Polesello et al., 2006), Zyxin (Zyx) (Rauskolb et al., 2011), and Ajuba LIM protein (Jub) (Das Thakur et al., 2010), each of which were examined for their ability to suppress the activation of Yki by EGFR or Ras.
Rassf associates with Ras and Hpo, and antagonizes Hpo activation (Polesello et al., 2006), but downregulation of Rassf failed to reverse Yki activation by EGFR or Ras (Fig. 4A), nor did over-expression of Rassf impair Yki activation (Fig. S1). Zyx is a Zyxin family LIM domain protein that binds Wts and acts in Fat-Hippo signaling in conjunction with Dachs to down regulate Wts (Rauskolb et al., 2011), but depletion of Zyx had no effect on the ability of EGFR or Ras to activate Yki (Fig. 4B). Jub is an Ajuba family LIM domain protein that binds Wts and Sav (Das Thakur et al., 2010), and which has previously been shown to be required for multiple upstream branches of Hippo signaling, as Jub mutation suppresses both fat and ex mutations (Rauskolb et al., 2011). We found that jub was epistatic to EGFR and Ras, as downregulation of Jub by RNAi led to decreased ex-lacZ expression even in the presence of activated-EGFR or RasV12 (Fig. 4C,D). Thus, activation of Yki by EGFR-Ras signaling is Jub-dependent.
Figure 4. Jub-dependent regulation of Yki by EGFR-Ras-MAPK.

(A-D) Third instar wing discs, stained for β-gal expressed by ex-lacZ (red) from larvae expressing en-Gal4 UAS-GFP (green) and A) UAS-EGFRA887T UAS-Rassf-RNAi, B) UAS-EGFRA887T UAS-Zyx-RNAi, C) UAS-EGFRA887T UAS-Jub-RNAi, D) UAS-RasV12 UAS-Jub-RNAi. (E-J) Western blot analysis of MAPK dependent phosphorylation and stability of Jub proteins in S2 cells. MAPK activation (indicated by MAPK*) was achieved by co-transfection of pUAS -RasV12, pUAS-Phl, pUAS-Dsor and pUAST-rl constructs. E) Cells were transfected to express full-length Flag:Ajuba proteins and MAPK* and blots were probed with anti-Flag. Samples were run on 5% SDS-PAGE gels with 30μl of 5mM phos-tag. F) Cells were transfected to express full-length Flag:Ajuba and MAPK* and blots were probed with anti-Flag. Where indicated samples were treated with phosphatase (CIP) and samples were run on 5% SDS-PAGE gels with 40μl of 5mM phos-tag. G) Cells were transfected to express N-terminal fragments of Flag:Ajuba proteins and MAPK* and blots were probed with anti-Flag. Samples were run on on 6% SDS-PAGE gels with 40μl of 5mM phos-tag. H) Ajuba stabilization by activated RAS signaling. Cells were transfected to express Flag:Ajuba and MAPK*. Blots were probed with anti-Flag, anti-V5 (GFP- as tansfection efficiency control) and as a loading control, anti-Actin. I) Ajuba-LIM domain phosphorylation by MAPK signaling. Cells were transfected to express C-terminal fragment of Flag:Ajuba and MAPK* and blots were probed with anti-Flag. J) Cells were transfected to express N-terminal fragments of Flag:Ajuba proteins and MAPK* and blots were probed with anti-Flag. Where indicated an MEK inhibitor (PD98059) was added and samples were run on on 6% SDS-PAGE gels with 40μl of 5mM phos-tag. K, L) Western blots on Phos-tag gels run on Jub-N or Jub-C protein samples subject to in vitro phosphorylation using ERK kinase, and CIP phosphatase treatment, where indicated.
EGFR-Ras-MAPK signaling promotes Jub phosphorylation
The observation that jub is epistatic to EGFR and Ras raised the possibility that Jub might be regulated by EGFR-Ras-MAPK signaling. We investigated this by expressing Jub in S2 cells. When Ras-MAPK signaling was activated by expression of RasV12 and MAPK pathway kinases (Raf/Phl, MEK/Dsor and ERK/Rl), Jub phosphorylation was elevated on multiple sites. This phosphorylation was particularly evident using Phos-tag gels, which contain a phosphate-binding moiety that retards the mobility of phosphorylated proteins (Kinoshita et al., 2006)(Fig. 4E). Further confirmation of Jub phosphorylation was provided by the reversal of this mobility shift by phosphatase treatment (Fig. 4F). Preliminary mapping experiments indicate that there are multiple ERK-dependent sites on Jub, as both a C-terminal fragment, which contains the three LIM domains, and an N-terminal fragment, were subject to a Ras-MAPK induced mobility shift (Fig. 4G,I). The increased phosphorylation induced by Ras-MAPK activation was reversed by treatment with PD98059, an MEK inhibitor, consistent with ERK-dependent phosphorylation of Jub (Fig. 4J). The ability of ERK to phosphorylate Jub was further confirmed by in vitro kinase reactions, which could shift a fraction of N- or C-terminal Jub polypeptides to higher mobility, phosphorylated forms (Fig. 4K,L). In addition to promoting phosphorylation of Jub, Ras-MAPK activation consistently led to increased levels of Jub protein (Fig. 4H), suggesting that Jub was stabilized by MAPK activity.
EGFR-Ras-MAPK signaling promotes Jub binding to Sav and Wts
Jub promotes Yki activation, and binds to Sav and Wts (Das Thakur et al., 2010). These observations suggest that Jub binding to Sav and Wts inhibits Wts kinase activity to increase levels of non-phosphorylated (ie active) Yki. Thus, the observed increase in levels of Jub upon MAPK activation could contribute to increased Wts inactivation. However, we wondered whether post-translational modification of Jub might also affect the interaction of Jub with these binding partners. This was examined through co-immunoprecipitation experiments. Assaying the relative amount of Sav or Wts precipitated by Jub revealed that Ras-MAPK activation led to a three-fold increase in Jub-Sav binding, and a two-fold increase in Jub-Wts binding (Fig. 5). Thus, Ras-MAPK activation also enhances the physical interaction of Jub to Sav and Wts.
Figure 5. MAPK increases Jub binding to Sav and Wts.
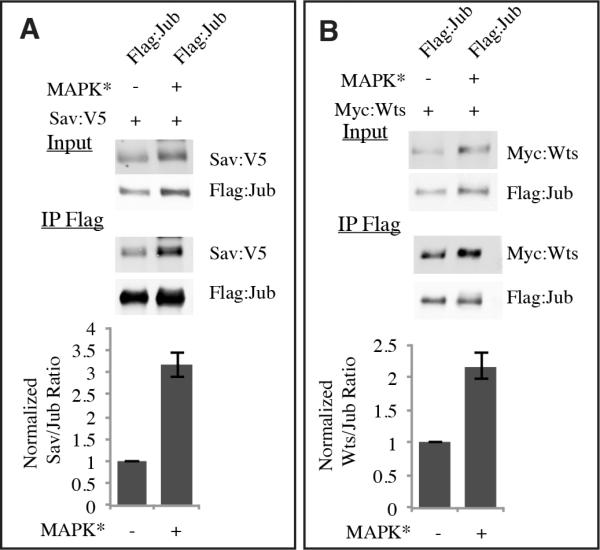
Western blots, probed with anti-Flag, and anti-V5 (A), or anti-Myc (B) on samples from S2 cells co-transfected to express Flag: Jub and Sav:V5 (A), or Myc:Wts (B). MAPK* indicates MAPK activation as in Fig. 4. Upper panels (Input) show blots on cell lysate, lower panels (IP Flag) show the blots on material precipitated by anti-Flag beads. Histograms, representing average results from 3 (Sav) or 4 (Wts) independent experiments, below quantify precipitation of Sav (A) or Wts (B) by Jub, normalized to the average ratio in cells without MAPK activation. Error bars indicate s.e.m., and the means are significantly different by paired t test.
EGFR-RAS-MAPK signaling regulates YAP and WTIP in mammalian cells
To determine whether the cross talk between EGFR and Hippo signaling identified in Drosophila is conserved, we examined the influence of activated-EGFR and RAS on YAP in two human cell lines, U87MG (a glial cell line) and HEK293T (a kidney cell line, results not shown). In both cell lines, expression of activated-EGFR (Greulich et al., 2005) or activated-RAS (HRASV12) increased YAP levels, and decreased YAP phosphorylation at Ser127 (Fig. 6A-C). Both of these effects are consistent with decreased LATS activity, because in mammals, in addition to controlling the subcellular localization of YAP (via Ser127 phosphorylation), LATS also controls the stability of YAP (via Ser384 phosphorylation) (Zhao et al., 2010). We further confirmed that activated-RAS could promote both nuclear localization of YAP, and the expression of YAP target genes (Fig. 6D, G).
Figure 6. Regulation of YAP by EGFR-RAS-MAPK signaling.

A-C,E) Western blots, probed with indicated antibodies on samples from U87MG cells co-transected with constructs to express the indicated proteins. A) YAP stabilization by activated RAS signaling: Cells were transfected to express Flag:YAP2, and where indicated HRasV12 , 2+ indicates DNA concentration was doubled. Blots were probed with anti-Flag, anti-diphospho-ERK (dpERK, indicates activated ERK) and as a loading control, anti-Actin. Where indicated an MEK inhibitor (PD98059) was added. B) YAP phosphorylation by activated EGFR or RAS. Cells were transfected to express Flag:YAP2, and where indicated HRasV12 or activated-EGFR. YAP was immunoprecipitated using anti-Flag beads, and probed with anti-Flag for total YAP and anti-YAP-phosphoSer127 (pYAP). Histogram, representing average results from 3 independent experiments, shows phospho-YAP/total YAP ratio from blots, both HRASV12 (P=0.017) and EGFR* (P=.0.28) have significantly different ratios from control cells. C) YAP stabilization and phosphorylation by RAF-MAPK signaling. Cells were transfected to express Flag:YAP2, and where indicated HRasV12, HRasV12C40, HRasV12G37, HRasV12S35, activated-RAF (RAF*) or activated-MEK (MEK*). Blots were probed with anti-Flag, and as a loading control, anti-GAPDH. YAP was immunoprecipitated using anti-Flag beads, and probed with anti-Flag for total YAP and anti-YAP-phosphoSer127 (pYAP). Histogram, representing average results from 4 independent experiments, shows Phospho-YAP/total YAP ratio from blots. All of the RAS/MAPK activation plasmids significantly change the pYAP/YAP ratio from that in control samples by pair-wise t test. D) Fold change in mRNA levels of the YAP target genes CTGF and BIRC3, assayed by quantitative reverse transcriptase PCR between U87MG cells expressing or not expressing HRASV12, normalized to GAPDH levels. E) Influence of kinase inhibitors on YAP levels and phosphorylation. Cells were transfected to express Flag:YAP2, and where indicated HRasV12. The top two panels show cell lysate, probed with anti-Flag or anti-GAPDH (loading control) and anti-YAP-phosphoS127. The bottom two panels show YAP immunoprecipitated using anti-Flag beads, and probed with anti-Flag for total YAP and anti-YAP-phosphoS127 (pYAP). Histogram, representing average results from 3 independent experiments, shows Phospho-YAP/total YAP ratio from blots. The pYAP/YAP ratio is significantly lower than the no HRASV12 control in all cases (by pairwise t test), but only the MEK inhibitor significantly (P=0.013) increased the ratio compared to RAS activation without inhibitors. F,G) U87MG cells transfected with YAP2:EGFP (green), and in (G) with HRASV12. Nuclei were stained using DAPI (blue). Panels marked by prime show YAP2:EGFP alone, and arrows point to selected nuclei to show the increased nuclear localization in the presence of activated RAS. Error bars in B,C,D, and E indicate standard deviation.
To examine signal transduction downstream of RAS, we again used both effector loop mutations (McFall et al., 2001; Rodriguez-Viciana et al., 1997) and kinase inhibitors. In contrast to Drosophila, where Ras effector loop mutations, in vivo RNAi, and kinase inhibitors in cultured cells all indicated that Ras acts through the MAPK cascade, characterization of human cells suggests that multiple signal transduction mechanisms might contribute to the influence of RAS on YAP. Thus, none of three RAS effector loop mutations could block RASV12 from stabilizing YAP (Fig. 6C). An MEK inhibitor also reduced, but did not abolish, the influence of RAS activation on YAP (Fig. 6A,E). Thus, EGFR signaling also modulates Hippo signaling in mammals, but the links between them might be more complex than in Drosophila. Nonetheless, the influence of effector loop mutations and kinase inhibitors imply that an EGFR-RAS-MAPK connection to Hippo signaling is conserved. This conclusion was further supported by the observation that expression of activated forms of RAF or MEK (Boehm et al., 2007) increased YAP levels and reduced YAP phosphorylation (Fig. 6C).
Mammals contain three Ajuba family proteins: Ajuba, LIM domain containing 1 (LIMD1), and Wilms tumor protein 1-interacting protein (WTIP). In co-transfection experiments, phosphorylation of WTIP was obviously increased by activated RAS or MEK (Fig. 7A), whereas Ajuba or LIMD1 were not visibly affected (not shown). Activated RAS or MEK also appeared to stabilize WTIP, as the levels of transfected protein were significantly increased, whereas Ajuba and LIMD1 again were not significantly affected (Fig. 7B). Thus, amongst the three mammalian Ajuba family protein, it appears that the influence of EGFR-RAS-MAPK signaling is preferentially targeted to WTIP, and we could confirm that WTIP is directly phosphorylated by ERK using in vitro kinase reactions (Fig. 7C). Moreover, activation of RAS increased binding of WTIP to both the Sav homologue WW45, and the Wts homologue LATS1 (Fig. 7E-H). Thus, we infer that there exists a conserved biochemical mechanism mediating cross-talk between EGFR and Hippo signaling which is effected through MAPK-dependent phosphorylation of an Ajuba family protein, and which changes its stability and its binding to Sav and Wts homologues.
Figure 7. Regulation of WTIP by EGFR-RAS-MAPK signaling.

A) Increased WTIP phosphorylation (*phospho-WTIP) by Ras-MAPK signaling. Western blot on lysates of cells co-transfected to express WTIP:V5, HRasV12 or MEK DD, as indicated. Lysates were run on a Phos-tag gel, and probed with anti-V5. NS denotes non-specific band. B) Stabilization of Ajuba family proteins by RAS-MAPK signaling. Cells were transfected to express GFP:V5, and, as indicated, Ajuba:V5, LIMD1:V5, WTIP:V5, HRasV12 or MEK DD (activated MEK). Cell lysates were probed with anti-V5. Histogram, representing average results from 3 independent experiments, quantifies Ajuba family protein levels normalized to GFP, only for effects of activated RAS or MEK on WTIP were differences statistically significant by t test, error bars indicate s.d. C) Western blot (anti-V5) on affinity-purified V5-tagged WTIP, subject to in vitro kinase reaction using ERK, phosphatase treatment using CIP, where indicated, and then separated on Phos-tag gels. D) Western blot (anti-V5) on samples from U87MG cells transfected to express WTIP:V5, WTIPS422A:V5 and HRASV12 as indicated. Samples were run on a 7% phos-tag gel. E) Ajuba family protein binding to WW45. Cells co-transfected with WW45:GFP and, as indicated, Ajuba:V5, LIMD1:V5, WTIP:V5, GFP:V5, or HRasV12 were probed with anti-GFP (upper panel). Lower panels show blots on material precipitated by anti-V5 beads. F) Ajuba family protein binding to Lats1. Cells co-transfected with Myc:LATS1 and, as indicated, Ajuba:V5, LIMD1:V5, WTIP:V5, GFP:V5, or HRasV12 were probed with anti-Myc (upper panel). Lower panels show blots on material precipitated by anti-V5 beads. G) Histogram, representing average results from 3-5 independent experiments, quantifies LATS1 binding to WTIP normalized to the average ratio in control experiments. error bars show s.e.m. H) Histogram, representing average results from 3-5 independent experiments, quantifies WW45 binding to WTIP normalized to the average ratio in control experiments. error bars show s.e.m. I,J) Western blots showing representative examples of the influence of WTIPS422A mutation on the ability of activated RAS to influence binding of WTIP to WW45 (I) and (LATS1 (J), experiments were otherwise identical to those shown in E and F.
All Ajuba family proteins contain multiple potential MAPK phosphorylation sites, but few are conserved between Drosophila Jub and mammalian WTIP. However, Jub, WTIP, and LIMD1, but not Ajuba, each have a predicted MAPK near the C terminus, within the last LIM domain. To evaluate the potential functional significance of this site, we mutated this Ser residue to Ala with WTIP (WTIPS422A). Indeed, this mutation impaired HRASV12-induced phosphorylation of WTIP in cultured cells (Fig. 7D). Moreover, it eliminated the ability of HRASV12 to increase the binding of WTIP to LATS1 and WW45 (Fig. 7G-J), thereby directly linking Ras-MAPK dependent phosphorylation of WTIP to the modulation of its binding to LATS1 and WW45.
DISCUSSION
As inappropriate activation of EGFR-RAS signaling is implicated in some of the most common and deadly cancers, including breast cancer, lung cancer, pancreatic cancer and glioblastoma, elucidating the mechanisms by which EGFR-RAS signaling can promote tumor growth has great clinical significance (Normanno et al., 2006; Sibilia et al., 2007). Hippo signaling normally functions as an important inhibitor of growth, and impairment of Hippo signaling has also been linked to diverse cancers (Bao et al., 2011; Pan, 2010). Motivated by the observation that EGFR and Hippo signaling are both linked to glial cell tumors (Hatanpaa et al., 2010; Lau et al., 2008; Nagane et al., 2001; Orr et al., 2011), and that their ability to promote glial cell proliferation is conserved in Drosophila (Read et al., 2009; Reddy and Irvine, 2011; Witte et al., 2009), we investigated the relationship between these pathways in controlling glial growth. These studies led us to the unexpected discovery of a direct functional link between them. The EGFR-RAS-MAPK branch of EGFR signaling induces post-translational modification and altered activity of a Hippo pathway component, Jub/WTIP, which is essential for the influence of EGFR on glial cell proliferation. Moreover, the influence of EGFR signaling on Yki/YAP activity could be readily observed in both glial and epithelial tissues in Drosophila, and in both glial and epithelial human cell lines.
The first description of upstream regulators of Hippo signaling occurred less than seven years ago, with reports that fat, ex, and Merlin could regulate Hippo signaling (Bennett and Harvey, 2006; Cho et al., 2006; Hamaratoglu et al., 2006; Silva et al., 2006; Tyler and Baker, 2007; Willecke et al., 2006). One of the remarkable features of Hippo signaling revealed by subsequent studies is the diverse array of additional upstream regulatory inputs into Hippo signaling, including several components of cell-cell junctions, regulators of cell polarity and cellular stress, F-actin accumulation, mechanical tension, and cross-talk with other signaling pathways (reviewed in Codelia and Irvine, 2012; Irvine, 2012; Staley and Irvine, 2012). Hippo signaling thus functions as an integrator of diverse growth-regulatory signals, to which we can now add receptor tyrosine kinase signaling mediated through Ras. One implication of the diverse inputs into Hippo signaling is that the measureable effect of any one input is dependent upon the status of the others. For example, activation of Ras is sufficient to activate Yki in wing discs, but knockdown of Ras-MAPK signaling had little effect on Yki activity in wing discs (Supplementary Figure S1), presumably due to the number and activity of additional factors that influence Hippo signaling there.
In addition to the regulation of Yki through MAPK and Ajuba family proteins, EGFR and Hippo signaling are also integrated at other levels. Yorkie/Yap regulate the expression of ligands for EGFR signaling (Zhang et al., 2009), which creates the potential for positive feedback regulation between these pathways. It is also noteworthy that Ajuba has been observed to influence MAPK activity (Goyal et al., 1999; Jamora et al., 2005), and hence could be considered a regulatory component of both Hippo and EGFR signaling. In Drosophila, a key target of Yki signaling for growth control is the microRNA gene bantam. EGFR can also independently regulate bantam through repression of the transcriptional repressor protein Capicua, which is itself a target of bantam (Herranz et al., 2012). Another potential point of cross-talk is Merlin, which is a regulator of Hippo signaling, but which has also been identified as an inhibitor of EGFR signaling, leading to some controversy as to whether the growth control functions of Merlin are mediated principally by Hippo signaling or EGFR signaling (Yi and Kissil, 2010). Our observation that both pathways could converge on regulation of Yki/YAP suggests at least a partial resolution to this controversy.
While in Drosophila we found that the influence of EGFR was mediated through the Ras-MAPK pathway, in cultured mammalian cells the connection between these pathways was more complex, as both effector loop mutations and kinase inhibitor studies suggest that other downstream effector branches, including PI3Kinase-dependent signaling, could also participate in modulating YAP. This observation is intriguing in light of recent studies that reported a connection between insulin signaling and Yki/YAP activity mediated through an as yet unknown mechanism that involves PI3Kinase signaling (Strassburger et al., 2012).
Investigations into the mechanism by which EGFR and Hippo cross-talk led us to Jub, which is genetically required for the influence of EGFR on Yki. Jub/WTIP are both subject to MAPK-dependent phosphorylation. Sequence analysis identifies several consensus MAPK sites in each protein, and it is possible that indirect effects of MAPK on other kinases could also contribute to Jub/WTIP phosphorylation. Nonetheless, mutagenesis implicates a single site near the C-terminus as playing a key role in both MAPK-dependent phosphorylation of WTIP, and MAPK-dependent stimulation of binding to LATS and WW45. Thus, we suggest that phosphorylation of this site triggers a conformational change that influences the ability of WTIP to bind LATS and WW45.
Although the precise biochemical mechanism by which Ajuba family proteins act within the Hippo pathway has not yet been determined, it presumably involves their binding to Sav and Wts. As the jub mutant phenotype is opposite to wts, and wts is epistatic to jub (Das Thakur et al., 2010; Rauskolb et al., 2011), a likely scenario is that Jub functions as an inhibitor of Wts. This could occur by Jub preventing Wts activation by Sav, Hpo, and Mats, preventing Wts phosphorylation of Yki, or altering the stability or sub-cellular localization of Wts. In any case, we infer that increased binding of Jub/WTIP to Wts/LATS and Sav/WW45 results in stronger inhibition of Wts activity by Jub/WTIP. Our binding studies relied on over-expressed proteins, and it will be important in the future to investigate quantitatively the degree to which this modulation of binding accounts for the observed influence of EGFR signaling on Yki/YAP activity in vivo. Nonetheless, our genetic and biochemical studies together clearly implicate Jub/WTIP as a crucial point of cross talk between EGFR and Hippo signaling.
EXPERIMENTAL PROCEDURES
Drosophila genetics
Expression of UAS lines was achieved using repo-Gal4 (for glia) or en-Gal4 (for wing discs). Additional experiments (not shown) utilized hs-Flp[122]; act>y+>gal-4 UAS-GFP (AyGal4) for Gal4 expression in clones. RNAi was induced including UAS-Dicer2 (Dietzl et al., 2007), and using the following UAS-hairpin transgenes: yki RNAi (vdrc104523), EGFR RNAi (JF01368), rl RNAi (HMS00173), Ras RNAi (HMS01294), jub RNAi (HMS00714) Zyx RNAi (NIG32018)(Rauskolb et al., 2011). Over-expression experiments used UAS-EGFRλ (Queenan et al., 1997), UAS-EGFRA887T (Lesokhin et al., 1999), UAS-Ras V12, UAS-Ras V12S35, UAS-Ras V12C40, UAS-Ras V12G37 (Karim and Rubin, 1998), UAS-yki:FlagS168A (Cho et al., 2006). Expanded was monitored using ex-LacZ (Hamaratoglu et al., 2006).
Histology and imaging
Tissue was fixed and stained as described previously (Reddy and Irvine, 2011), using mouse anti-Repo (1:400), rat anti-Elav (1:400) (DSHB), rabbit anti-Yki (1:400), goat anti–bgal (1:1,000, Biogenesis). EdU labeling and detection was performed using Click-iT EdU Alexa Flour Imaging kits (Invitrogen) with Alexa-594 azide. Dissected larvae were incubated with EdU (20 μM) at room temperature for 10 minutes, washed with PBS, and fixed with 4% paraformaldehyde in PBS. Numbers of glial cells in eye discs were counted manually within the Elav-staining region of eye discs. All the eye imaginal discs used for each genotype were staged based on number of rows of Elav positive cells (~10 rows) in eye discs.
Tissue culture Reagents and plasmids
S2 cells were cultured with Schneider's Drosophila medium (Invitrogen) and 10% FBS (Sigma). U87MG glioma cell line was maintained in DMEM with 10% serum. U87MG cells were maintained in 2% serum after transfection. Plasmids used for S2 cells experiments: Full length Ajuba, (1-729aa), Ajuba-N-terminal (1-466aa), Ajuba-C-terminal (LIM domain, 467-729aa) constructs were generated by PCR from cDNA plasmids with N-terminal Flag tag and inserted into pUAST vectors. pAW-Gal4, Myc-tagged Wts, V5-tagged GFP, Flag-Tagged GFP and V5-tagged Sav plasmids have been described previously (Bennett and Harvey, 2006; Reddy and Irvine, 2011; Silva et al., 2006). Coding regions of pUAST-Rolled, pUAST-Phl, pUAST-Dsor with HA tag were amplified from cDNAs and cloned into pUAST vector. cDNA encoding RasV12 was amplified by PCR from UAS-transgenic flies and inserted into pUAST vector. Mammalian (human) constructs used in this study include: pcDNA-GFP:V5, pcDNA3-Jub:V5, pcDNA3-LIMD1:V5, pcDNA3-hWTIP:V5 and pcDNA3-Myc:Lats1 (Zhao et al., 2010). Following constructs were obtained from Addgene: CMV2-YAP2 (19045), pBabe puro EGFR L858R (11012), pBabe puro H-RasV12(9051), pBabe puro H-RasV12S35(18746), pBabe puro H-RasV12G37(18745), pBabe puro H-RasV12C40 (18747), pBabe puro MEK-DD (15268), pBabe puro BRAF-V600E (15269), and pEGFP -hWW45 (19052). MEK-DD is constitutive active form of MEK in which Raf dependent regulatory phosphorylation sites (S218 and S222) were substituted by aspartic acid residues (Goyal et al., 1999). Raf-V600E is an active form of BRAF with somatic mutation in kinase domain (V600E), observed in human cancers (Davies et al., 2002). Pharmacological inhibitors were added into culture plates during and after transfection. PDK1 inhibitor (OSU-03012, Selleck chemicals) at 3μM/ml concentration, MEK inhibitor (PD98059-Millipore) 50mM concentration, PI3K inhibitor (Wortmannin, Cell Signaling) at 1 μM concentration and p38 inhibitor (SB 202190, Sigma) at 1 μM concentration. WTIP Ser 422 was mutated to Ala using a site-specific mutagenesis kit (Stratagene) according to the manufacturers instructions.
Co-immunoprecipitation and Western blotting
For co-immunoprecipitations, transient transfections were performed with equal amounts of DNA (0.5 μg per construct, plus empty vector to keep total amounts of DNA used constant) using Cellfectin (Invitrogen) for S2 cells or Lipofectamine 2000 (Invitrogen) for mammalian cells in 6 well plates according to the manufacturer's protocol. Co-immunoprecipitation assays were performed as described previously (Silva et al., 2006). In brief, cells were harvested 48 h later in RIPA lysis buffer (50 mM Tris–HCl, pH 7.5; 150 mM NaCl; 1% NP40; 0.5% Sodium deoxycholate; 0.1% SDS; 1 mM EDTA) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Calbiochem). Cell lysates (500 μg for each sample) were incubated with 10 μl anti-FLAG beads (Sigma) at 4 °C for 2 h followed by five washes in RIPA buffer. Beads were then boiled in Laemmli sample buffer at 100 °C for five minutes and loaded onto SDS-PAGE gels. Western blotting was performed using rabbit anti-V5 (1:4000, Bethyl Labs), mouse anti-Flag (1:10000 Sigma), rabbit anti-Yki (1:3000), rabbit anti-phospho Yki (gift of D. Pan, 1:300 on disc lysates, 1:2000 on S2 cell IPs), rabbit anti-mouse anti-V5 (1:10000, Invitrogen), rabbit anti-Myc (1:1000 Santacruz) mouse anti-Actin (1:5000, Calbiochem), mouse anti-GAPDH (1:10000) and rabbit anti-phospho Yap (S127) (1:3000, Cell Signaling). Western blotted proteins were visualized and quantified using using fluorescent conjugated secondary antibodies from Odyssey and Licor Imaging system. To detect phosphorylated proteins in SDS-PAGE, we used Phos-Tag AAL-107 (FMS Laboratory) according to the manufacturer's instructions. GraphPad Prism software was used for statistical analysis of band intensities.
In-viro kinase Assays
Cells from one 6 well plate were lysed in RIPA buffer with protease and phosphatase inhibitor cocktails. Tagged proteins were immunoprecipiated using either anti-V5 or anti-Flag agarose beads. Beads were then washed three times in RIPA buffer, followed by two washes in 20mM Tris (pH 7.5), 150mM Nacl with EDTA. One tenth of the immunoprecipitated beads were suspended in 20 μL kinase buffer (25mM Tris (pH 7.5), 5mM glycerol phosphate, 2mM DTT, 10mM MgCl2 with phosphotase inhibitor cocktail) with 100mM ATP and 0.5μg of active ERK1 (Millipore) and incubated at 30°C for 30 minutes.
RT PCR
Total RNA was extracted from cells in 6 well plates using TriZol Plus RNA Kit (Invitrogen). cDNA was synthesized using Superscript II reverse transcriptase (Invitogen), and quantitative PCR was performed using QuantiTect SYBR Green PCR Kit (Qiagen) on Cephaid Smart Cycler II. GAPDH (Glyceraldehyde-3-phosphate Dehydrogenase) was used as an internal control. The Primer sequences for RT-PCR were:
| GAPDH-FW | GGAGCGAGATCCCTCCAAAAT |
| GAPDH-RV | GGCTGTTGTCATACTTCTCATGG |
| CTGF-FW | ACCGACTGGAAGACACGTTTG |
| CTGF-RV | CCAGGTCAGCTTCGCAAGG |
| BIRC3-FW | TTTCCGTGGCTCTTATTCAAACT |
| BIRC3-RV | GCACAGTGGTAGGAACTTCTCAT |
Supplementary Material
ACKNOWLEDGEMENTS
We thank G. Sun and B. Gumbiner for communication of unpublished observations, D. Pan, S. Blair, J. Thomas, the Developmental Studies Hybridoma Bank, and the Bloomington stock center for antibodies and Drosophila stocks. This research was supported by the Howard Hughes Medical Institute and NIH grant R01GM078620.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bao Y, Hata Y, Ikeda M, Withanage K. Mammalian Hippo pathway: from development to cancer and beyond. Journal of biochemistry. 2011;149:361–379. doi: 10.1093/jb/mvr021. [DOI] [PubMed] [Google Scholar]
- Bennett FC, Harvey KF. Fat Cadherin Modulates Organ Size in Drosophila via the Salvador/Warts/Hippo Signaling Pathway. Curr Biol. 2006;16:2101–2110. doi: 10.1016/j.cub.2006.09.045. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, McCall K, Steller H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell. 1998;95:331–341. doi: 10.1016/s0092-8674(00)81765-1. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. Delineation of a Fat tumor suppressor pathway. Nat Genet. 2006;38:1142–1150. doi: 10.1038/ng1887. [DOI] [PubMed] [Google Scholar]
- Codelia VA, Irvine KD. Hippo signaling goes long range. Cell. 2012;150:669–670. doi: 10.1016/j.cell.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Das Thakur M, Feng Y, Jagannathan R, Seppa MJ, Skeath JB, Longmore GD. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Current biology. CB. 2010;20:657–662. doi: 10.1016/j.cub.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal RK, Lin P, Kanungo J, Payne AS, Muslin AJ, Longmore GD. Ajuba, a novel LIM protein, interacts with Grb2, augments mitogen-activated protein kinase activity in fibroblasts, and promotes meiotic maturation of Xenopus oocytes in a Grb2- and Ras-dependent manner. Molecular and cellular biology. 1999;19:4379–4389. doi: 10.1128/mcb.19.6.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS medicine. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz H, Hong X, Cohen SM. Mutual repression by bantam miRNA and Capicua links the EGFR/MAPK and Hippo pathways in growth control. Current biology. CB. 2012;22:651–657. doi: 10.1016/j.cub.2012.02.050. [DOI] [PubMed] [Google Scholar]
- Irvine KD. Integration of intercellular signaling through the Hippo pathway. Seminars in cell & developmental biology. 2012;23:812–817. doi: 10.1016/j.semcdb.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, Lee P, Kocieniewski P, Azhar M, Hosokawa R, Chai Y, Fuchs E. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS Biology. 2005;3:e11. doi: 10.1371/journal.pbio.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development. 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Lau YK, Murray LB, Houshmandi SS, Xu Y, Gutmann DH, Yu Q. Merlin is a potent inhibitor of glioma growth. Cancer Res. 2008;68:5733–5742. doi: 10.1158/0008-5472.CAN-08-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lesokhin AM, Yu SY, Katz J, Baker NE. Several levels of EGF receptor signaling during photoreceptor specification in wild-type, Ellipse, and null mutant Drosophila. Developmental biology. 1999;205:129–144. doi: 10.1006/dbio.1998.9121. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, Jacks T. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998;12:1121–1133. doi: 10.1101/gad.12.8.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall A, Ulku A, Lambert QT, Kusa A, Rogers-Graham K, Der CJ. Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Molecular and cellular biology. 2001;21:5488–5499. doi: 10.1128/MCB.21.16.5488-5499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merel P, Hoang-Xuan K, Sanson M, Moreau-Aubry A, Bijlsma EK, Lazaro C, Moisan JP, Resche F, Nishisho I, Estivill X, et al. Predominant occurrence of somatic mutations of the NF2 gene in meningiomas and schwannomas. Genes Chromosomes Cancer. 1995;13:211–216. doi: 10.1002/gcc.2870130311. [DOI] [PubMed] [Google Scholar]
- Nagane M, Lin H, Cavenee WK, Huang HJ. Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer letters. 2001;162(Suppl):S17–S21. doi: 10.1016/s0304-3835(00)00648-0. [DOI] [PubMed] [Google Scholar]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Oh H, Irvine KD. In vivo regulation of Yorkie phosphorylation and localization. Development. 2008;135:1081–1088. doi: 10.1242/dev.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr BA, Bai H, Odia Y, Jain D, Anders RA, Eberhart CG. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. Journal of neuropathology and experimental neurology. 2011;70:568–577. doi: 10.1097/NEN.0b013e31821ff8d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello C, Huelsmann S, Brown NH, Tapon N. The Drosophila RASSF homolog antagonizes the hippo pathway. Current biology. CB. 2006;16:2459–2465. doi: 10.1016/j.cub.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan AM, Ghabrial A, Schupbach T. Ectopic activation of torpedo/Egfr, a Drosophila receptor tyrosine kinase, dorsalizes both the eggshell and the embryo. Development. 1997;124:3871–3880. doi: 10.1242/dev.124.19.3871. [DOI] [PubMed] [Google Scholar]
- Rauskolb C, Pan G, Reddy BV, Oh H, Irvine KD. Zyxin links fat signaling to the hippo pathway. PLoS biology. 2011;9:e1000624. doi: 10.1371/journal.pbio.1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 2009;5:e1000374. doi: 10.1371/journal.pgen.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy BV, Irvine KD. The Fat and Warts signaling pathways: new insights into their regulation, mechanism and conservation. Development. 2008;135:2827–2838. doi: 10.1242/dev.020974. [DOI] [PubMed] [Google Scholar]
- Reddy BV, Irvine KD. Regulation of Drosophila glial cell proliferation by Merlin-Hippo signaling. Development. 2011;138:5201–5212. doi: 10.1242/dev.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation; research in biological diversity. 2007;75:770–787. doi: 10.1111/j.1432-0436.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H. The Tumor-Suppressor Gene fat Controls Tissue Growth Upstream of Expanded in the Hippo Signaling Pathway. Curr Biol. 2006;16:2081–2089. doi: 10.1016/j.cub.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Staley BK, Irvine KD. Hippo signaling in Drosophila: recent advances and insights. Developmental dynamics : an official publication of the American Association of Anatomists. 2012;241:3–15. doi: 10.1002/dvdy.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Developmental biology. 2012;367:187–196. doi: 10.1016/j.ydbio.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Tyler DM, Baker NE. Expanded and fat regulate growth and differentiation in the Drosophila eye through multiple signaling pathways. Dev Biol. 2007;305:187–201. doi: 10.1016/j.ydbio.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke M, Hamaratoglu F, Kango-Singh M, Udan R, Chen CL, Tao C, Zhang X, Halder G. The Fat Cadherin Acts through the Hippo Tumor-Suppressor Pathway to Regulate Tissue Size. Curr Biol. 2006;16:2090–2100. doi: 10.1016/j.cub.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Witte HT, Jeibmann A, Klambt C, Paulus W. Modeling glioma growth and invasion in Drosophila melanogaster. Neoplasia. 2009;11:882–888. doi: 10.1593/neo.09576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Kissil JL. Merlin in organ size control and tumorigenesis: Hippo versus EGFR? Genes & Development. 2010;24:1673–1679. doi: 10.1101/gad.1964810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, Brugge JS, Dyson NJ, Haber DA. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nature cell biology. 2009;11:1444–1450. doi: 10.1038/ncb1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.