

Abstract
Oxidative stress has been implicated in the development of vascular disease and in the promotion of endothelial dysfunction via the reduction in bioavailable nitric oxide (NO•). Glutathione (GSH) is a tripeptide thiol antioxidant that is utilized by glutathione peroxidase (GPx) to scavenge reactive oxygen species (ROS) such as hydrogen peroxide and phospholipid hydroperoxides. Relatively frequent single nucleotide polymorphisms (SNPs) within the 5’ promoters of the GSH synthesis genes GCLC and GCLM are associated with impaired vasomotor function as measured by decreased acetylcholine-stimulated coronary artery dilation and with increased risk of myocardial infarction. Although the influence of genetic knockdown of GPx on vascular function has been investigated in mice, no work to date has been published on the role of genetic knock down of GSH synthesis genes on vascular reactivity. We therefore investigated the effects of targeted disruption of Gclm in mice and the subsequent depletion of GSH on vascular reactivity, NO• production, aortic nitrotyrosine protein modification, and whole genome transcriptional responses as measured by DNA microarray. Gclm−/+ and Gclm−/− mice had 72% and 12%, respectively, of WT aortic GSH content. Gclm−/+ mice had a significant impairment in acetylcholine (ACh)-induced relaxation in aortic rings as well as increased aortic nitrotyrosine protein modification. Surprisingly, Gclm−/− aortas showed enhanced relaxation compared to Gclm−/+ aortas, as well as increased NO• production. Although aortic rings from Gclm−/− mice had enhanced ACh-relaxation, they have a significantly increased sensitivity to phenylephrine (PE)-induced contraction. Alternatively, the PE response of Gclm−/+ aortas was nearly identical to that of their WT littermates. In order to examine the role of NO• or other potential endothelium derived factors in differentially regulating vasomotor activity, we incubated aortic rings with the NO• synthase inhibitor L-NAME or physically removed the endothelium prior to PE treatment. L-NAME treatment and endothelium removal enhanced PE-induced contraction in WT and Gclm−/+ mice, but this effect was severely diminished in Gclm−/− mice, indicating a potentially unique role for GSH in mediating vessel contraction. Whole genome assessment of aortic mRNA in Gclm−/− and WT mice revealed altered expression of genes within the canonical Ca2+ signaling pathway, which may have a role in mediating these observed functional effects. These findings provide additional evidence that the de novo synthesis of GSH can influence vascular reactivity and provide insights regarding possible mechanisms by which SNPs within GCLM and GCLC influence the risk of developing vascular diseases in humans.
Keywords: Glutathione, glutamate cysteine ligase, GCLM knockout mouse, nitric oxide, vascular reactivity
Introduction
Endothelium derived relaxing factors (EDRFs), especially nitric oxide (NO•) and its associated low molecular weight nitrosothiols (e.g. nitrosocysteine and nitrosoglutathione), have critical roles in the regulation of vascular tone, control of blood pressure, inhibition of platelet aggregation, and the onset of thrombosis (Loscalzo, 2001). Vascular oxidative stress and its effect on endothelial thiol redox status influence endothelial production of NO•. When tetrahydrobiopterin (BH4), a cofactor for endothelial NO• synthase (eNOS), becomes oxidized, the homodimeric structure of eNOS is compromised (Chalupsky and Cai, 2005, Chen et al., 2010, He et al., 2010, Jones et al., 2010, Xia et al., 1998, Xie et al., 2010) resulting in two eNOS monomers, a process termed ‘uncoupling’. eNOS requires its dimeric structure to properly transfer an electron donated from NADPH across its reductase domain to catalyze the reaction between L-arginine and oxygen (O2), to produce L-citrulline and NO• at its oxygenase domain (Brocq et al., 2008). The uncoupling of eNOS eliminates NO• synthesis. However, when eNOS is uncoupled, electrons donated from NADPH are still capable of transferring across its reductase domain, which can subsequently combine with available O2 to produce superoxide radical (O2•−) (Xia et al., 1998).
Because vascular oxidative stress can result in cardiovascular dysfunction, the role of antioxidant enzymes in modulating vascular reactivity continues to be investigated. Loscalzo and Stamler, and colleagues have produced a series of seminal works revealing the importance of NADPH generation by the pentose phosphate shunt, and glutathione peroxidases (GPx) 1 and 3 in maintaining the reduced cellular environment required for normal eNOS function and NO• synthesis (Espinola-Klein et al., 2007, Jin et al., 2011, Leopold et al., 2007, Maron et al., 2009, Stamler et al., 1988, Weiss et al., 2001). These studies have highlighted the importance of the antioxidant thiol glutathione (GSH) in vascular function by preventing BH4 oxidation and eNOS uncoupling. GSH can react with NO• to form S-nitrosoglutathione (GSNO), which also has vasoactive properties (Lima et al., 2010). In these discussions of the importance of NADPH, GSH, and GPx activities in preserving normal vascular function, emphasis has been placed on the maintenance of an appropriate balance between the reduced (GSH) and oxidized (GSSG) forms of glutathione.
GPx catalyzes the reduction of H2O2 to H2O, a reaction that results in the formation of GSSG (Griffith, 1999). Reduction of GSSG to GSH by glutathione reductase (GRx) is NADPH-dependent, and is critical for maintaining a reduced state in the cell. Because of their important roles in preventing vascular oxidative stress, these redox cycling events have received much attention. While impairments in this redox cycling have been investigated, little attention has been paid to the potentially important role of de novo GSH synthesis in vascular disease. GSH is a tripeptide composed of glutamate, cysteine, and glycine and is synthesized in a two-step process. In the first step, glutamate is ligated with cysteine to form γ-glutamylcysteine (γ-GC). Formation of γ-GC is the rate-limiting step in GSH synthesis and is catalyzed by the heterodimeric enzyme glutamate cysteine ligase (GCL). γ-GC is rapidly ligated with glycine by GSH synthase (GS) to form GSH. GCL is composed of catalytic (GCLC) and modifier (GCLM) subunits. GCL activity is determined by the expression of GCLC and GCLM mRNA and protein, as well as by stabilization of the GCL holoenzyme by oxidation (Botta et al., 2008, Krejsa et al., 2010, McConnachie et al., 2007).
Although few studies have investigated the role of de novo GSH synthesis in mediating vascular function in animal models, Nakamura and colleagues reported that in a Japanese population, relatively common single nucleotide polymorphisms (SNPs) in the 5’ promoter regions of both GCLC and GCLM genes are associated with increased risk of myocardial infarction (MI) (Koide et al., 2003, Nakamura et al., 2002). In addition, the investigators observed that patients with the -588CT SNP in GCLM (frequency of 19.2% of control population) have a compromised vasodilatory response to acetylcholine (ACh) infusion, as measured by an increase in coronary blood flow (Nakamura et al., 2003). Nakamura and colleagues also showed that endothelial cells transfected with a promoter-reporter construct containing this SNP have greater than a 50% reduction in GCLM promoter activity and an overall increased sensitivity to oxidative stress in vitro (Nakamura et al., 2002). Because this SNP occurs with a relatively high frequency, investigation of the potential association of cardiovascular disease and compromised GSH synthesis is crucial. Following this work by Nakamura et al., others have investigated the effects of L-buthionine-[S,R]-sulfoximine (BSO), a potent inhibitor of GCLC and GCL, on vascular reactivity in rodents (Denniss et al., 2011, Ford et al., 2006). Ford et al. (2006) demonstrated that a 10-day treatment with BSO in drinking water produced impairment in ACh-mediated aortic ring relaxation and enhanced sensitivity to phenylephrine (PE)-induced contraction in rats. Alternatively, Denniss et al. (2011) demonstrated that 10-day treatment with BSO did not influence ACh-mediated vessel relaxation of the common carotid artery in rats, indicating that GSH depletion does not have a universal effect on vascular function. Since a genetic component influencing GCLM expression has been found to impair vessel function and NO• synthesis in humans, it is desirable to further investigate this association in a mouse model of compromised de novo GSH synthesis.
We developed a mouse model of compromised GSH synthesis by genetic disruption of Gclm (Botta et al., 2008, McConnachie et al., 2007). GSH is essential for embryonic development as it has been previously demonstrated that deletion of Gclc results in embryonic lethality in mice (Shi et al., 2000). In Gclm null mice (Gclm−/−), GCLC has severely compromised enzymatic activity, resulting in roughly 5-10% of normal levels of GSH across most tissues (McConnachie et al., 2007). Mice heterozygous for Gclm (Gclm−/+) have roughly 80-95% of normal GSH stores across tissues. As GSH stores are so dramatically depleted in Gclm−/− mice, it has been hypothesized that these mice will exhibit extreme sensitivity to toxicants that induce injury via oxidative stress (Dalton et al., 2004, Botta et al., 2008, McConnachie et al., 2007). Indeed, if mitigation of toxicant injury specifically requires GSH, then injury is dramatically exacerbated in Gclm−/− mice (i.e. acetaminophen induced liver injury) (McConnachie et al., 2007). However, several reports have indicated that Gclm−/− mice exhibit responses that are either no different than WT controls, or in some cases are more protected (Haque et al., 2010, Johansson et al., 2010, Weldy et al., 2011). The prevailing hypothesis for this counterintuitive finding is that with such severe GSH depletion, alternative antioxidant enzymes are up-regulated in Gclm−/− mice and thus compensate for the low GSH levels. Although this finding of an adaptive response is interesting, it has little clinical correlation since humans with 5’ promoter polymorphisms in GCL genes have only moderately compromised GSH levels. Nonetheless, cells from people with the C-588T SNP do show a compromised ability to up-regulate GCLM when subjected to oxidative stress. Recently, we reported for the first time that Gclm−/+ mice are more sensitive to lung inflammation following diesel exhaust particulate exposure when compared to WT and Gclm−/− mice (Weldy et al., 2011). This finding was significant because it showed that slight impairment in GSH synthesis could lead to increased sensitivity to pro-oxidants, and suggested that the Gclm−/+ mouse may be a valuable tool for investigating the role of de novo GSH synthesis in vascular disease. Gclm−/+ mice may serve as a model that is more relevant to the human condition in which GCL 5’ promoter polymorphisms have been shown to predispose to compromised vasomotor function and cardiovascular disease.
In this report, we investigate for the first time the influence of genetic disruption of the GSH synthesis gene Gclm on aortic GSH and GSSG content, vascular reactivity, NO• synthesis, aortic nitrotyrosine protein modification, cGMP production, aortic ROS, and whole genome expression changes in the aorta as measured by DNA microarray in mice. Because GSH has been shown to influence vascular reactivity, we hypothesized that the loss of Gclm and the resulting decrease in GSH would compromise ACh-mediated vessel relaxation and NO• synthesis, and increase sensitivity to PE-stimulated vessel contraction.
Material and Methods
Experiments were performed using Gclm WT, Gclm−/+, and Gclm−/− mice backcrossed for at least 10 generations onto the C57BL/6 background. Mice were bred and housed in a modified specific pathogen free (SPF) vivarium at the University of Washington. All animal experiments were approved by the University of Washington Institutional Animal Care and Use Committee. Littermates were genotyped as previously described (McConnachie et al., 2007).
Analysis of GCLC and GCLM protein level by Western immunoblot
Mice were sacrificed using CO2 narcosis, followed by cervical dislocation. Twenty seven aortas (9 WT, 9 Gclm−/+, and 9 Gclm−/−) were quickly excised and homogenized in TES/SB (20 mM Tris, pH 7.4, 1 mM EDTA, 250 mM sucrose, 20 mM serine, and 1 mM boric acid). Aortas from 3 mice of the same genotype were homogenized together and GCLM and GCLC protein levels were detected with the use of rabbit polyclonal antisera raised against ovalbumin conjugates of peptides specific to each subunit, using previously described procedures (Thompson et al., 1999). The optical density of GCLC and GCLM specific bands on x-ray films were assessed using Image J software and normalized to β-actin protein levels in 3 samples for each genotype, representing 9 animals for each genotype.
GSH and GSSG analysis by HPLC
From the same aorta homogenate used for GCLC/GCLM Western blot analysis, immediately after homogenization 100 μL were removed and diluted 1:1 with 10% 5-sulfosalicylic acid to stabilize GSH and precipitate proteins. The homogenates were incubated on ice for 10 min, and then centrifuged at 15,600 X G in a microcentrifuge for 2 min to obtain deproteinated supernatants. Concentrations of GSH and GSSG normalized to protein level present in the original homogenate were determined by high-pressure liquid chromatography (HPLC) using a modification of previously described methods (Eaton and Hamel, 1994, Thompson et al., 1999). Briefly, for GSH measurements, supernatant was mixed with monobromobimane (MBB) to derivatize GSH and measured by HPLC with fluorescence detection. For GSSG measurements, 2-vinylpiridine was added to the supernatant to remove all GSH. Residual 2-vinylpiridine was then removed with chloroform extraction, and the remaining GSSG was reduced to GSH with tris(2-carboxyethyl)phosphine (TCEP; 10μM), derivatized with MBB and measured by HPLC as above. With each n representing a homogenate of 3 aortas, we obtained an n of 3, 3, and 5 for WT, Gclm−/+, and Gclm−/− mice respectively.
Vascular Reactivity
Aortas from male WT, Gclm−/+, and Gclm−/− mice were cut into 3-mm rings and transferred to an organ bath containing 6 ml of physiological saline solution (119 mM NaCl, 4.7 mM KCl, 2.4 mM MgSO4, 1.2 mM KH2PO4, 3.3 mM CaCl2, 25 mM NaHCO3, 30 μM EDTA, 6 mM dextrose), equilibrated with 95% O2 and 5% CO2. Buffer was maintained at 37°C, pH 7.4. Aortic rings were hung with wire to a force transducer (Model 610M, Danish Myo Technology, Aarhus, Denmark), and the transducer was interfaced to a Powerlab 8/26 recorder for measurement of isometric force. Rings were placed under an initial tension of 20 mN and equilibrated for 1 hr. Ring contraction was measured using PE hydrochloride (Sigma-Aldrich) and potassium-physiological salt solution (KPSS), and endothelium-dependent and -independent relaxations were measured using ACh and sodium nitroprusside, respectively. An n of 10, 12, and 9 were obtained for WT, Gclm−/+, and Gclm−/− mice respectively. Vessel relaxation was expressed as the percentage of contraction induced by phenylephrine. Endothelial removal was achieved by gently rolling an aortic ring around a 27 g needle. N3-nitro-L-arginine methyl ester (L-NAME) treatment was used to inhibit NOS activity and was achieved by incubating aortic rings in 100 μM L-NAME for 30 min prior to pre-contraction. For L-NAME and endothelium removal studies, and n of 5, 7, and 5 were obtained for WT, Gclm−/+, and Gclm−/− mice respectively.
Detection of aortic NO• production by Fe(DETC)2 spin trap and ESR
Aortic NO• production was detected in female WT, Gclm−/+, and Gclm−/− mice by methods previously described (Khoo et al., 2004). Briefly, aortas were quickly removed and cleaned of all perivascular adipose tissue and incubated in a Krebs/HEPES buffer (99 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4; 1 mM KH2PO4, 1.9 mM CaCl2, 25 mM NaHCO3, 11.1 mM glucose, 20 mM HEPES) adjusted to pH 7.4. Aortas were then incubated at 37 °C in the non-colloidal iron diethyldithiocarbamate (Fe/DETC) spin trap (Preparation of colloid Fe(DETC)2: Sodium DETC (3.6 mg) and FeSO4 7H20 (2.25 mg) were dissolved under argon gas in 10 ml of ice cold Krebs-Hepes buffer) with 5 μM ACh for 90 min. Immediately after incubation, 3 aortas of the same genotype were combined and placed into a 1 ml syringe (with the end cut off) and frozen in liquid nitrogen. The frozen pellet was then pressed out of the syringe and stored at -80°C until NO• detection by electron spin resonance (ESR) spectroscopy. ESR studies were performed on a table-top x-band spectrometer Miniscope (Magnettech, Berlin, Germany). Measurements were taken on samples placed in a Dewar tube and kept in liquid nitrogen. Instrument settings were: biofield 3275, sweep 115G, microwave frequency 9.78 Ghz, microwave power 20 mW, and a kinetic time of 10 min. An n of 5 was obtained for each genotype, each n representing a pellet of 3 combined aortas, thus 15 aortas from each genotype were assessed.
Measurement of aortic cGMP
A total of 24 mice male mice were used for the assessment of aortic cGMP. Eight aortas were collected from each WT, Gclm−/+, and Gclm−/− genotype, following collection of the aorta, aortas were immediately snap frozen and preserved at -80 °C for subsequent analysis. Aortas were weighed, and then immediately homogenized directly into 5% trichloroacetic acid. cGMP was measured by cGMP Enzyme Immunoassay (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s protocol.
Aortic nitrotyrosine assessment by immunofluorescence
Three aortas, including the perivascular adipose tissues, from WT, Gclm−/+, and Gclm−/− mice were collected, embedded in optimal cutting temperature (OCT) compound (Tissue-Tek, Sakura Finetek, Torrance, CA), and frozen over dry ice in 70% ethanol in standard disposable vinyl cryomolds. Aortas were cross-sectioned in 10 μm sections using a cryomicrotome and mounted on VWR Superfrost Plus Micro Slides (VWR International, Radnor, PA). Sections underwent a 1hr block using 10% goat serum/1% BSA, a 2hr incubation in rabbit anti-nitrotyrosine antibody at a 5 μg/ml working concentration (catalog # 06-284, Upstate Cell Signaling-Millipore, Billerica, MA), and a 1 hr incubation with goat anti rabbit Alexa 555 secondary at 1:400. Sections were fixed in 4% PFA for 5 min and cover-slipped using aqueous mounting medium with anti-fading agents (Biomeda Corp., Foster City, CA). Images were taken with a Nikon Diaphot microscope using epifluorescence illumination. Green excitation was used and images were taken at emission wavelengths from 600-700 nm using Nuance Multispectral camera (Cambridge Research Inc., Cambridge, MA) with a 10 nm step with an exposure time of 1 sec for each step. Images were compiled as a cube, and mean fluorescent intensity with background subtraction was determined using NIH Image J. Four sections per aorta were used to determine mean fluorescent intensity for each aorta.
Measurement of aortic ROS by L-012 chemiluminescence
Nine aortas were used for the assessment of ROS generation by the sensitive luminol-derived marker L-012. Aortas were collected from 3 WT, Gclm−/+, and Gclm−/− female mice, placed into Krebs/HEPES buffer (composition described above), perivascular adipose tissue cleaned away, and 3 aortic rings of 4 mm were cut per aorta. The aortic rings were placed into a white opaque 96 well flat bottom microtiter plate, and 2 rings were incubated in 100 μl of Krebs/HEPES buffer alone while 1 ring was incubated in Krebs/HEPES buffer with L-NAME (500 μM) for 10 min. One hundred μL of L-012, diluted to 200 μM with Krebs/HEPES buffer, was then added to each well to achieve a final concentration of 100 μM L-012 and 250 μM L-NAME. Chemiluminescence was measured on a Plate Lumino luminescence spectrometer (Stratec, Birkenfeld, Germany) at 5 and 20 min and relative light units (RLU) were expressed as RLU/min. For the wells not incubated with L-NAME, RLU/min values were averaged. RLU/min values from L-NAME treated aortic rings were compared to this averaged value.
Affymetrix GeneChip Whole Transcript Sense Target Labeling
Whole aortas were collected from four WT, Gclm−/+, and Gclm−/− mice and incubated in RNAlater for at least 12 hr at 4°C, then stored at -20°C. RNA was isolated from aortic tissues using the RNeasy Kit following the manufacturer’s protocol (Qiagen, Inc., Valencia, CA). Integrity of RNA samples was assessed with an Agilent 2100 Bioanalyzer. Only samples passing this stringent quality control were processed. Processing of the RNA samples was carried out according to the Affymetrix GeneChip Whole Transcript Sense Target labeling protocol (for details see http://www.affymetrix.com/index.affx). Briefly, double-stranded cDNA is synthesized with random hexamers tagged with a T7 promoter sequence. The double-stranded cDNA was subsequently used as a template and amplified by T7 RNA polymerase producing many copies of antisense cRNA. In the second cycle of cDNA synthesis, random hexamers are used to prune reverse transcription of the cRNA from the first cycle to produce single-stranded DNA in the sense orientation. The DNA was labeled by terminal deoxynucleotidyl transferase (TdT) with the Affymetrix® proprietary DNA Labeling Reagent covalently linked to biotin. The biotin labeled DNA fragments were hybridized to the array, washed and stained with fluorescent anti-streptavidin biotinylated antibody. Following an additional wash step, the arrays are scanned with an Affymetrix GeneChip® 3000 scanner. Image generation and feature extraction is performed using Affymetrix GeneChip Command Console (AGCC) software.
Microarray data analysis of Affymterix Mouse Gene 1.0 ST arrays
Raw microarray data were processed and analyzed with tools from Bioconductor (Gentleman et al., 2004). Data were normalized using the Robust Multichip Average (RMA) method (Irizarry et al., 2003) from the Bioconductor Affy package. Genes with significant evidence for differential expression were identified using the limma software package (Smyth, 2004). The limma methodology calculates a p-value for each gene using a modified t test in conjunction with an empirical Bayes method to moderate the standard errors of the estimated log fold changes. This method draws strength across genes for more robust and accurate detection of differentially expressed genes. Such an adjustment has repeatedly been shown to avoid an excess of false positives when identifying differentially expressed genes (Allison et al., 2006). Using the p-values from limma, we used the Bioconductor package q-value (Dabney and Storey, 2006, Tusher et al., 2001) to estimate the false discovery rate associated with the list of differentially expressed genes. This methodology allowed us to address the problem of multiple hypotheses testing without resorting to an excessively conservative approach that controls the family-wise error, such as a Bonferroni correction. Differentially expressed genes were further investigated using the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood, CA).
Statistical Analyses
Data were analyzed using Prism (Graphpad Software, La Jolla, CA). Data were subjected to ANOVA followed by a Dunnett’s post-hoc test. All error bars in figures represent standard error of the mean (SEM). *, **, *** = Significant difference from the matched control at p-values of < 0.05, 0.01 and 0.001, respectively. Vascular reactivity data were analyzed by repeated-measurement 2-way ANOVA. Concentration-response curves were fitted with a nonlinear regression program (GraphPad Prism) to obtain values of maximal effect, which were compared by 1-way ANOVA.
Results
Aortic GCLC and GCLM protein content in WT, Gclm−/+, and Gclm−/− mice
We have previously reported that in comparison to WT mice, Gclm−/+ mice have roughly mice have no detectable 50% of total GCLM protein level in the liver and kidney whereas Gclm−/−GCLM (McConnachie et al., 2007). Alternatively, total protein level of GCLC is increased to 1.8 fold in liver and kidney of Gclm−/− mice, while Gclm−/+ mice exhibit an intermediate phenotype. In agreement with our previous reports, GCLM protein is absent in the aorta of Gclm−/− mice. In Gclm−/+ mice GCLM protein is reduced to approximately half of that in WT mice (Figure 1). Although the levels of GCLM protein within the aorta follows a trend that is expected across genotypes, GCLC protein level is not increased in either Gclm−/+ or Gclm−/− mice (Figure 1).
FIGURE 1.
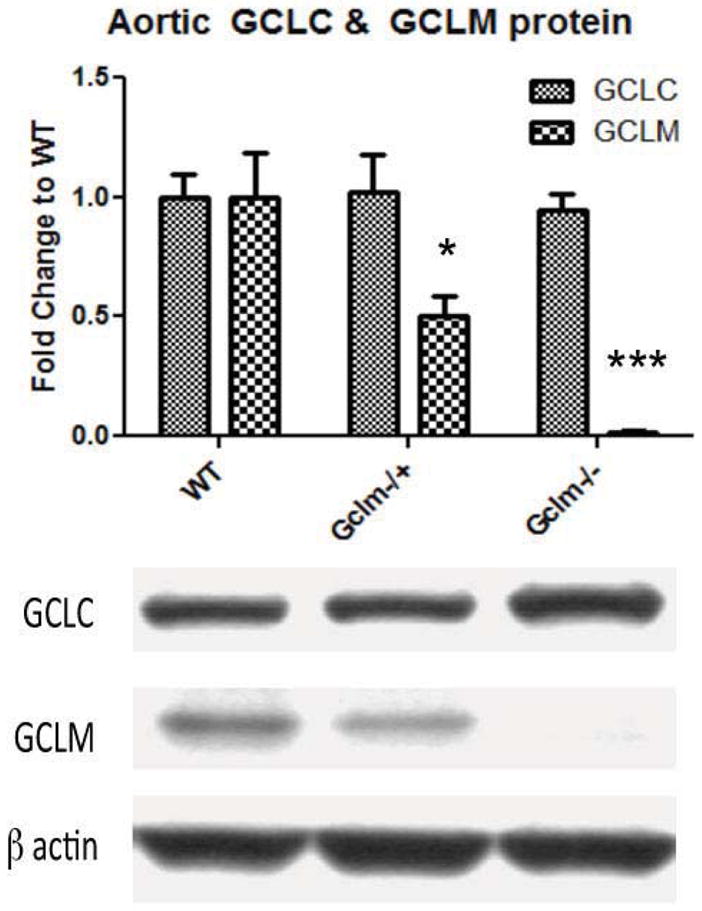
Protein level of aortic GCLC and GCLM normalized to β actin as measured by western blot within WT, Gclm−/+, and Gclm−/− mice. Nine aortas were collected from each genotype, 3 aortas from each genotype were combined and homogenized together. Bars represent means from an n of 3, each n representing 3 aortas. All error bars in figures represent standard error of the mean (SEM). * & *** = Significant difference from the matched control at p-values of < 0.05 and 0.001, respectively.
Aortic GSH and GSH:GSSG ratio in WT, Gclm−/+, and Gclm−/− mice
Because the total GSH content is a critical component to the maintenance of intracellular thiol redox status, we measured both reduced (GSH) and oxidized (GSSG) forms of GSH in aortas from WT, Gclm−/+, and Gclm−/− mice. GSH was 12.9 nmol/mg protein in the aortas of WT mice, and in Gclm−/+ and Gclm−/− mice, aortic GSH was 9.3 and 1.6 nmol/mg protein (72.4 % and 12.4% of WT, respectively; Figure 2a). Thus, as expected, Gclm−/− mice have an extremely low level of GSH within the aorta, and importantly Gclm−/+ mice were found to have a slight but significantly lower level of aortic GSH compared to WT.
FIGURE 2.

a) Total aortic GSH (GSH+GSSG) measured by HPLC and normalized to protein level within WT, Gclm−/+, and Gclm−/− mice. b) Total aortic glutathione disulfide (GSSG) measured by HPLC and normalized to protein level within WT, Gclm−/+, and Gclm−/− mice. c) %GSSG of total aortic GSH (GSH+GSSG) measured by HPLC and normalized to protein level within WT, Gclm−/+, and Gclm−/− mice. d) ΔEGSSG/2GSH calculated from aortic tissue, assuming pH of 7.4 and temperature of 37°C. Each GSH and GSSG measure was made in a homogenate of 3 combined aortas from the same genotype. An n of 3 WT, 3 Gclm−/+, and 5 Gclm−/− was reached. All error bars in figures represent standard error of the mean (SEM). * & *** = Significant difference from the matched control at p-values of < 0.05 and 0.001, respectively.
Aortic GSSG contents were 0.42, 0.38, and 0.14 nmol/mg protein in WT, Gclm−/+, and Gclm−/− mice, respectively (Figure 2b). When expressed as a percentage of total GSH, we observed that GSSG was low across all genotypes (3.2, 3.9, and 7.9% of total aortic GSH in WT, Gclm−/+, and Gclm−/− mice, respectively; Figure 2c). Although the %GSSG level in Gclm−/− aorta appeared to be nearly double that observed in WT and Gclm−/+ mice, this only approached statistical significance (p=0.09). Thus, even if %GSSG is possibly elevated in the aorta of Gclm−/− mice, the actual aortic GSSG content was found to be significantly less in Gclm−/− mice compared to that of WT mice.
Biological reductive potential is an important determinant of intracellular oxidative conditions. Although it would be difficult to measure the actual intracellular redox potential in these aortas, measuring the relative concentrations of the reduced and oxidized forms of this redox couple (i.e. GSSG/2GSH) can be used to provide an estimate. GSSG/2GSH is widely used to estimate redox potential and has certain advantages over NADP+/NADPH (Dalton et al., 2004). By using GSSG and GSH concentrations, we calculated the redox potential of GSSG/2GSH (ΔEGSSG/2GSH) in the aortas of WT, Gclm−/+, and Gclm−/− mice using the approach previously taken by Dalton and colleagues (Dalton et al., 2004). Assuming cytosolic conditions of pH 7.2 and 37°C, we calculated the aortic ΔEGSSG/2GSH to be -162mV (±10.8 SD) in WT mice (Figure 2d). This value increased to -148 (±3.6 SD) and -118mV (±15.3 SD) in Gclm−/+ and Gclm−/− mice respectively (Figure 2d). ΔEGSSG/2GSH is significantly greater in Gclm−/− mice compared to WT (p=0.008), and although it does not reach significance (p=0.20), there is a trend for increased ΔEGSSG/2GSH in the Gclm−/+ mice relative to that of WT mice.
ACh-stimulated relaxation of aortic rings from WT, Gclm−/+, and Gclm−/− mice
As expected, increasing ACh concentrations produced relaxation of aortic rings in all 3 genotypes (Figure 3a). Relaxation was assessed at ACh concentrations ranging from 1 nM to 10 μM, and maximum relaxation was reached at 1 μM ACh across all genotypes. Importantly, ACh-mediated vessel relaxation was significantly compromised in the aortic rings from Gclm−/+mice (Figure 3a). Interestingly, Gclm−/− mice had a unique relaxation response, being compromised at lower ACh concentrations (0.001-0.1 μM ACh), yet elevated at higher concentrations (1-10 μM ACh), reaching a statistically significant increase in maximal relaxation when compared to aortic rings from Gclm−/+ mice (Figure 3a). This is a peculiar observation, as aortic rings from Gclm−/−mice have a relaxation curve that ‘crosses over’ the relaxation curve from WT aortic rings. No differences among the genotypes were observed after treatment with sodium nitroprusside (Figure 3b), suggesting these Gclm genotype-dependent differences are endothelium dependent.
FIGURE 3.

Acetylcholine- (a) and sodium nitroprusside- (b) stimulated aortic ring relaxation in 10 WT, 12 Gclm−/+, and 9 Gclm−/− mice. Vascular reactivity was analyzed by repeated-measurement 2-way ANOVA. Concentration-response curves were fitted with a nonlinear regression program (GraphPad Prism) to obtain values of maximal effect, which were compared by 1-way ANOVA.
Detection of aortic NO• by Fe(DETC)2 spin trap and ESR spectroscopy
Although ACh is believed to mediate vessel relaxation primarily via the stimulation of NO• production, others have postulated that ACh can also mediate vessel relaxation by means other than NO• (Leo et al., 2008). In order to determine if these differences in ACh-mediated relaxation are NO• dependent, we measured the level of NO• produced by aortas of WT, Gclm−/+, and Gclm−/− mice using the Fe(DETC) spin trap and ESR spectroscopy following 90 minute stimulation with 5 μM ACh as previously described (Khoo et al., 2004). The levels of NO• production by the aortic rings followed trends that are consistent with ACh-mediated vessel relaxation across Gclm genotypes (Figure 4). As day-to-day variability in data collection masked some of the effect, after normalizing samples to the same day WT control, we observe a significant increase (p=0.04) in NO• production between Gclm−/− and Gclm−/+ (Figure 4b). We found that aortas from Gclm−/+ mice exhibited a 17% reduction in total NO• compared to WT aortas (Figure 4b) while the NO• level produced by Gclm−/− aortas was roughly 37% greater than aortas from WT mice.
FIGURE 4.

Production of nitric oxide (NO•) as measured by NO-Fe(DETC)2 spin trap and ESR spectroscopy and further normalized to protein following 5 μM ACh-stimulation for 90 min in aortas from WT, Gclm−/+, and Gclm−/− mice in both raw data (a) and as fold change to WT by assay (b). Three aortas were combined together for each n, and an n of 5 was obtained for each genotype. Statistical significance was determined by T-Test between fold change by day between Gclm−/+ and Gclm−/− aortas.
Measurement of Aortic cGMP
To determine if these changes in ACh-stimulated vessel relaxation are also accompanied by impairments in soluble guanylate cyclase (sGC) activity and cGMP production, we measured cGMP in aortas taken from WT, Gclm−/+, and Gclm−/− mice under basal conditions. As shown in Figure 5, we observed a trend for less cGMP content in the aortas of Gclm−/+ and Gclm−/− mice relative to that of WT mice, but this only approached statistical significance (WT vs. Gclm−/−, P=0.10).
FIGURE 5.
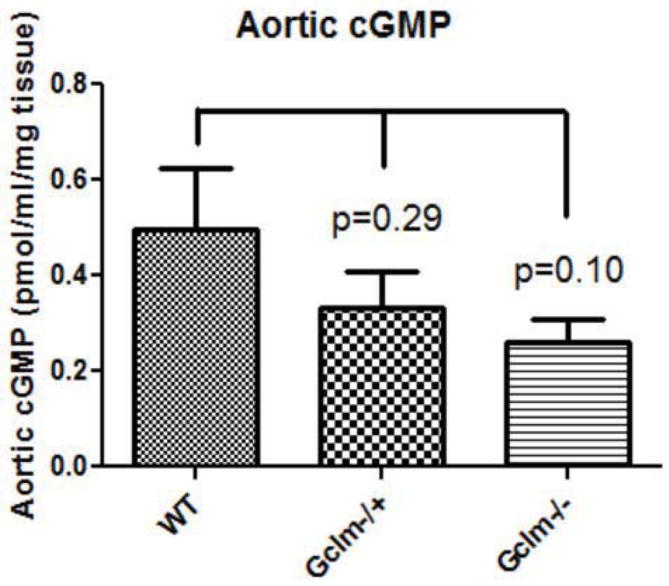
Production of aortic cGMP as measured by enzyme immunoassay (EIA) normalized to tissue weight. Whole aortas were collected from male WT, Gclm−/+, and Gclm−/− mice and one cGMP measure was made per aorta, and an n of 8 was obtained per genotype.
Assessment of aortic nitrotyrosine modification by immunofluorescence
As a means to investigate aortic oxidative and nitrosative stress, we measured aortic nitrotyrosine modification by immunofluorescence microscopy on frozen sections prepared from WT, Gclm−/+, and Gclm−/− mice. We observed a significant increase in mean fluorescence intensity representing nitrotyrosine staining in aortas from Gclm−/+ mice compared to aortas from WT mice (p=0.04) (Figure 6). Although there was a trend for aortas from Gclm−/− mice to have an intermediate phenotype, there was no statistical difference when compared to either Gclm−/+ or WT mice.
FIGURE 6.

a) Brightfield and fluorescent images of representative cross sections taken from the aortas of WT, Gclm−/+, and Gclm−/− mice. Red fluorescence, detected by immunofluorescence in the emission range of 600-700nm, represents positive nitrotyrosine staining. a) Quantification of mean fluorescence intensity, averaging 4 sections per aorta, in 3 aortas per genotype. Statistical significance was observed by T-Test comparing WT to Gclm−/+ aortas.
Determination of aortic ROS production using L-012 chemiluminescence
To increase our understanding of the production of ROS within the aorta of WT, Gclm−/+, and Gclm−/− mice, we measured ROS using the luminol-derived ROS reporter L-012. After incubating cleaned aortic rings in L-012, or L-012 plus the NOS inhibitor L-NAME, chemiluminescence was measured. We did not observe any difference in L-012 chemiluminescence among the 3 genotypes (Supplemental Figure 1a). L-NAME treatment appeared to increase L-012 chemiluminescence by roughly 10-40% (Supplemental Figure 1b), but this effect did not reach statistical significance nor did it appear to be greatly influenced by genotype.
PE-stimulated constriction of aortic rings from WT, Gclm−/+, and Gclm−/− mice
PE increased the contractile force of aortic rings in all 3 genotypes (Figure 7). Aortic rings from WT and Gclm−/+ mice were not significantly different in their response to PE at any concentration tested. This was true when analyzed as total contractile response (as measured in mN force) (Figure 7a), or when analyzed as a percentage of maximum contraction induced by potassium-physiological saline solution (K-PSS) (Figure 7b). Interestingly, aortic rings from the Gclm−/− mice had a dysregulated response to PE. When analyzed as total contractile force, there was a significantly greater response in aortic rings from Gclm−/− mice when compared to Gclm−/+ mice at 10 nM PE (Figure 7a). When analyzed as a percentage of total K-PSS-induced contraction, we observe that aortic rings from Gclm−/− mice have an increased contractile response at the lower concentrations of PE (1-100 nM), but interestingly a compromised contractile response at higher concentrations of PE (10-100 μM) (Figure 7b).
FIGURE 7.

Phenylephrine (PE)-stimulated contraction of aortic rings from 10 WT, 12 Gclm−/+, and 9 Gclm−/− mice as both total force (a) and as % of total K-PSS contraction (b). Vascular reactivity was analyzed by repeated-measurement 2-way ANOVA. Concentration-response curves were fitted with a nonlinear regression program (GraphPad Prism) to obtain values of maximal effect, which were compared by 1-way ANOVA.
PE-stimulated constriction following NOS inhibition and endothelium removal
Because the contraction to PE is balanced by the continual production on EDRFs, when EDRF synthesis is abrogated, enhanced contraction can occur. As such, it is important to examine the role of NO• production as well as any potential factor released from the endothelium to moderate PE-induced contraction. In order to examine these factors, we either pre-incubated aortic rings with L-NAME, or we removed the endothelium and tested PE-stimulated contraction in WT, Gclm−/+, and Gclm−/− mice. When comparing the effects of L-NAME treatment or endothelium removal (No-Endo) within a genotype, we do not observe any differences between treatments in either aortic rings from WT (Figure 8a) or Gclm−/− (Figure 8c) mice. This evidence supports the supposition that NO• is the principle EDRF balancing PE-contraction in these genotypes. However, we did observe a significant increase in the effect of L-NAME treatment over endothelium removal in the aortas from Gclm−/+ mice (Figure 8b), suggesting a role for alternative EDCFs in mediating PE-contraction. When comparing among genotypes, we observed that with prior L-NAME treatment, contraction increased to a maximum of 32.6% greater than K-PSS-induced contraction in WT aortic rings (Figure 9a). This increased to an even greater level in Gclm−/+ mice (p=0.039 Two-way ANOVA) with the maximum contraction reaching 48.4% of K-PSS-induced contraction (Figure 9a). Alternatively, L-NAME treatment in Gclm−/− mice produced an increase of only 15.9% of the K-PSS-induced contraction (Figure 9b), a significantly reduced effect compared to WT (p=0.0004). After endothelium removal, a similar trend is observed (Figure 9c and 9d). Endothelium removal produced a maximal increase in contraction of 39.5 and 51.4% of K-PSS-induced contraction in the aortic rings from WT and Gclm−/+ mice, respectively (not statistically different). But, similar to the trends observed following L-NAME treatment, the effect of endothelium removal was severely impaired in the aortic rings from Gclm−/− mice (p<0.0001, Gclm−/− vs WT)(Figure 9d).
FIGURE 8.
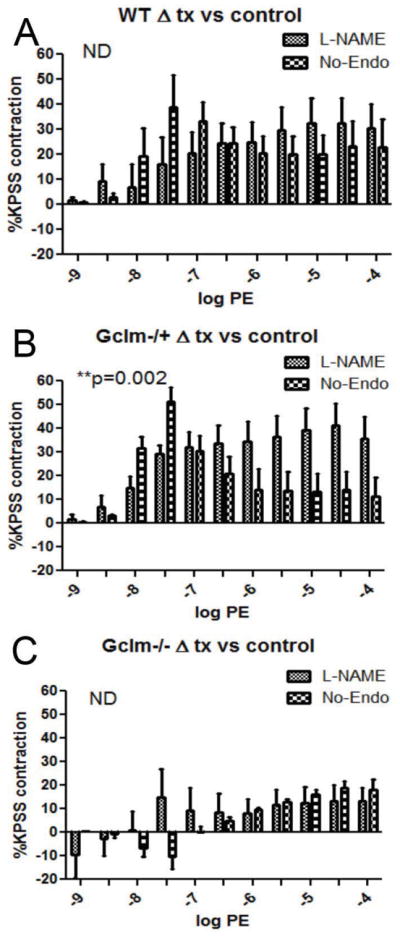
Differences between untreated aortic rings and either L-Name treated or endothelium removed aortic rings in phenylephrine (PE)-stimulated contraction of aortas from 5 WT (a), 7 Gclm−/+ (b), and 5 Gclm−/− (c) mice, as measured by %KPSS total contraction. Statistical analysis between L-NAME and endothelium removed effects were determined by Two-way ANOVA (GraphPad Prism).
FIGURE 9.

Differences between untreated aortic rings and either L-Name treated (panels a and b) or endothelium removed aortic rings (panels c and d) following phenylephrine (PE)-stimulated contraction. a) comparing aortas from 5 WT and 7 Gclm−/+ following L-NAME treatment, b) 5 WT and 5 Gclm−/− following L-NAME treatment, c) 5 WT and 7 Gclm−/+ following endothelium removal, and d) 5 WT and 5 Gclm−/− following endothelial removal as measured by %KPSS total contraction. Statistical analysis between L-NAME and endothelium removed effects were determined by Two-way ANOVA (GraphPad Prism).
Whole genome microarray of aortic mRNA transcripts from WT, Gclm−/+, and Gclm−/− mice
To better understand possible gene regulation that may be taking place to compensate for low GSH content within Gclm−/− mice, we performed a whole genome microarray comparing mRNA transcript levels in the aortas of WT, Gclm−/+, and Gclm−/− mice. Using selection criteria of an unadjusted p-value < 0.05 and a |fold| difference > 1.5, the comparison between aortic mRNA isolated from WT and Gclm−/− mice reveals 789 genes that were significantly altered using these criteria. Using similar criteria for comparison between WT and Gclm−/+ mice, 588 genes were selected. Figure 10 is a Venn diagram showing that in these comparisons, 129 genes were dysregulated in common in WT vs Gclm−/− and WT vs Gclm−/+aortas. Tables 1 and 2 show the top 25 genes upregulated in WT vs Gclm−/− and WT vs Gclm−/+ comparisons. Interestingly, the most upregulated gene in the WT vs Gclm−/− comparison was beta-defensin 4 (44 fold upregulated). Importantly, many of the genes most upregulated in the aorta of Gclm−/− mice are also upregulated in the aortas of Gclm−/+ mice, but not to the same magnitude (Table 1). Moreover, it is clear that transcriptional changes present in Gclm−/+ mice are not nearly as dramatic as those observed in Gclm−/− mice. The gene upregulated to the greatest magnitude in the WT vs Gclm−/+ comparison is recombination activating gene 1 (4.5 fold upregulated) (Table 2). Interestingly, when comparing the magnitude of upregulation in the top 25 genes in Gclm−/+ mice to the magnitude of change observed for these same genes in Gclm−/− mice, there is little concordance, suggesting that the modest loss of GSH in Gclm−/+ mice is associated with a unique compensatory response relative to that observed in Gclm−/− mice.
FIGURE 10.

Venn Diagram indicating the number of gene selected for by microarray using criteria of an unadjusted p-value of <0.05 and a |fold| difference >1.5. The intersection of these circles represents the number of genes that have both been selected for in Gclm−/− vs WT comparison and Gclm−/+ vs WT comparison.
TABLE 1.
List of the top 25 genes observed to be upregulated in the Gclm−/− vs WT comparison. Fold change values are listed for both the Gclm−/− vs WT and Gclm−/+ vs WT comparison.
| Gene Code | Full name of gene | Fold Change | |
|---|---|---|---|
| Gclm−/− to WT | Gclm−/+ to WT | ||
| Defb4 | defensin beta 4 | 44.2 | 3.3 |
|
| |||
| Serpinb3a | serine (or cysteine) peptidase inhibitor, clade B (ovalbumin), member 3A | 26.0 | 3.0 |
|
| |||
| Krt4 | keratin 4 | 20.8 | 2.6 |
|
| |||
| Serpinb12 | serine (or cysteine) peptidase inhibitor, clade B (ovalbumin), member 12 | 20.6 | 2.8 |
|
| |||
| Krtdap | keratinocyte differentiation associated protein | 16.3 | 2.7 |
|
| |||
| Crct1 | cysteine-rich C-terminal 1 | 16.1 | 2.6 |
|
| |||
| NA | NA | 15.5 | 2.3 |
|
| |||
| Sprr3 | small proline-rich protein 3 | 15.3 | 2.4 |
|
| |||
| Lor | loricrin | 13.7 | 2.3 |
|
| |||
| Rptn | repetin | 12.9 | 2.4 |
|
| |||
| Gm94 | predicted gene 94 | 12.5 | 2.5 |
|
| |||
| Krt13 | keratin 13 | 11.5 | 2.2 |
|
| |||
| Dsg1a | desmoglein 1 alpha | 10.3 | 2.6 |
|
| |||
| Asprv1 | aspartic peptidase, retroviral-like 1 | 10.0 | 2.1 |
|
| |||
| Lce3a | late cornified envelope 3A | 9.8 | 2.4 |
|
| |||
| 4833423E24Rik | RIKEN cDNA 4833423E24 gene | 9.0 | 2.4 |
|
| |||
| Serpinb5 | serine (or cysteine) peptidase inhibitor, clade B, member 5 | 8.8 | 2.6 |
|
| |||
| Dsc1 | desmocollin 1 | 8.4 | 2.4 |
|
| |||
| Lce3c | late cornified envelope 3C | 8.1 | 2.2 |
|
| |||
| I1I0032A04Rik | RIKEN cDNA 1110032A04 gene | 8.1 | 2.2 |
|
| |||
| Tgm3 | transglutaminase 3, E polypeptide | 8.0 | 2.0 |
|
| |||
| Mt4 | metallothionein 4 | 7.8 | 1.9 |
|
| |||
| Lce1a1 | late cornified envelope 1A1 | 7.7 | 2.1 |
|
| |||
| Ada | adenosine deaminase | 7.6 | 2.8 |
|
| |||
| Them5 | thioesterase superfamily member 5 | 7.6 | 2.3 |
TABLE 2.
List of the top 25 genes observed to be upregulated in the Gclm−/+ vs WT comparison. Fold change values are listed for both the Gclm−/− vs WT and Gclm−/+ vs WT comparison.
| Gene Code | Full name of gene | Fold Change | |
|---|---|---|---|
| Gclm−/− to WT | Gcim−/+ to WT | ||
| Rag1 | recombination activating gene 1 | 1.4 | 4.5 |
|
| |||
| Themis | thymocyte selection associated | 1.4 | 4.5 |
|
| |||
| Tnnc2 | troponin C2, fast | 5.6 | 4.0 |
|
| |||
| Myh1 | myosin, heavy polypeptide 1, skeletal muscle, adult | 6.3 | 4.0 |
|
| |||
| Tnnt3 | troponin T3, skeletal, fast | 5.7 | 3.9 |
|
| |||
| Myot | myotilin | 6.0 | 3.9 |
|
| |||
| Ly6d | lymphocyte antigen 6 complex, locus D | 5.1 | 3.8 |
|
| |||
| Neb | nebulin | 5.9 | 3.8 |
|
| |||
| Myh8 | myosin, heavy polypeptide 8, skeletal muscle, perinatal | 5.9 | 3.7 |
|
| |||
| Ccr9 | chemokine (C-C motif) receptor 9 | 1.2 | 3.6 |
|
| |||
| Dntt | deoxynucleotidyltransferase, terminal | 1.3 | 3.5 |
|
| |||
| Myoz1 | myozenin 1 | 4.4 | 3.5 |
|
| |||
| Satbl | special AT-rich sequence binding protein 1 | 1.3 | 3.5 |
|
| |||
| Atp2a1 | ATPase, Ca++ transporting, cardiac muscle, fast twitch 1 | 5.2 | 3.4 |
|
| |||
| H19 | HI9 fetal liver mRNA | 5.3 | 3.4 |
|
| |||
| Deft4 | defensin beta 4 | 44.2 | 3.3 |
|
| |||
| Dsc3 | desmocollin 3 | 5.3 | 3.3 |
|
| |||
| Ighm | immunoglobulin heavy constant mu | 1.1 | 3.2 |
|
| |||
| Acta1 | actin, alpha 1, skeletal muscle | 4.9 | 3.2 |
|
| |||
| Actn3 | actinin alpha 3 | 3.9 | 3.2 |
|
| |||
| My13 | myosin, light polypeptide 3 | 3.7 | 3.2 |
|
| |||
| Cd3g | CD3 antigen, gamma polypeptide | 1.0 | 3.1 |
|
| |||
| Serpinb3a | serine (or cysteine) peptidase inhibitor, clade B (ovalbumin), member 3A | 26.0 | 3.1 |
|
| |||
| Cd8b1 | CD8 antigen, beta chain 1 | 1.1 | 3.1 |
|
| |||
| Mir181b-1 | microRNA 181 b-1 | 1.4 | 3.0 |
To better understand how these transcriptional regulatory changes may influence certain canonical biological pathways, we analyzed these microarray data using Ingenuity Pathway Analysis. Figure 11 shows the top 10 canonical pathways modified in WT vs Gclm−/− (Figure 11a) and WT vs Gclm−/+ comparisons (Figure 11b) ranked on p-value significance. Strikingly, we observed that the most significantly altered pathway in the WT vs Gclm−/− comparison is Calcium Signaling. Alternatively, the most significantly altered pathway in the WT vs Gclm−/+ comparison is T Cell Receptor Signaling. Calcium signaling is a fundamental biological pathway that regulates both vascular constriction as well as vascular dilation. Supplemental Figure 2 presents the Calcium Signaling pathway and highlights those genes within this pathway that are modified in the WT vs Gclm−/− comparison. In viewing this pathway, it is apparent that upregulation of genes such as calmodulin, ryanodine receptor, troponins, and SERCA are driving this highly significant finding.
FIGURE 11.

Ingenuity Pathway Analysis results. The top ten canonical pathways dysregulated for the (a) Gclm−/− vs WT and (b) Gclm−/+ vs WT comparisons are shown. Pathways were ranked by p-value significance using genes selected by pre-established selection criteria.
We had previously hypothesized that the Nrf2-mediated Oxidative Stress Response canonical pathway would be most upregulated in the aorta of Gclm−/− mice to compensate for the dramatic loss of GSH content. We observed that although the Nrf2-mediated Oxidative Stress Response pathway is significantly altered in the aorta of Gclm−/− mice, significance is just barely past our threshold of –log(p-value) of 1.3 (Supplemental Figure 3). Fitting with this low level of significance, we observe this trend to be driven by only modest modification of eight genes (Supplemental Figure 3b), one of which is Gclm. In this same assessment in the aorta of Gclm−/+ mice, we observe a slightly greater significance value (Supplemental Figure 4), but again this is a weak association driven by only four genes other than Gclm (Supplemental Figure 4b).
Discussion
In this report, we investigated the influence of genetic disruption of the GSH synthesis gene Gclm on aortic GSH and GSSG content, NO• synthesis, nitrotyrosine protein modification, vascular relaxation of aortic rings following ACh treatment, aortic cGMP levels, vascular contraction following PE treatment of untreated, L-NAME treated, and endothelium denuded aortic rings, and whole genome gene expression changes as measured by microarray. The major findings of this study are: 1) Disruption of Gclm results in an aortic GSH content of 72% and 12% of WT in Gclm−/+ and Gclm−/− mice, respectively; 2) aortic rings from Gclm−/+ mice have an impaired ACh-mediated vessel relaxation and increased aortic nitrotyrosine modification in comparison to WT; 3) aortic rings from Gclm−/− mice do not have an impaired ACh-mediated relaxation but instead have an enhanced maximum relaxation and increased NO• production in comparison to Gclm−/+ mice; 4) aortic rings from Gclm−/− mice have an enhanced PE-induced contraction; 5) L-NAME treatment and endothelium removal enhances PE-induced contraction in aortic rings from WT and Gclm−/+ mice, but this response is severely compromised in aortic rings from Gclm−/− mice; and 6) DNA microarray analysis showed that Calcium Signaling is the most significantly altered canonical pathway in the aorta between Gclm−/− and WT mice. Together these data suggest that a slight reduction in GSH content (as occurs in Gclm−/+ mice) impairs ACh-mediated vessel function via the loss of bioavailable NO• and enhanced oxidative stress. However, severely depleted GSH (as occurs in Gclm−/− mice) results in some compensatory responses that may ameliorate oxidative stress and prevent the loss of bioavailable NO•. However, loss of GSH in Gclm−/− mice nonetheless compromises vascular function in response to PE, which may be more highly dependent on GSH and as well as alterations in calcium signaling due to compensatory responses.
GSH depletion can impair vascular thiol redox status and this may have important implications for the development of vascular disease and the onset of acute cardiovascular events. ROS are capable of inactivating NO• as well as compromising the function of critical enzymes that mediate vascular tone (i.e. eNOS, SERCA, RhoA/ROCK), and the dynamic maintenance of GSH levels and the GSH:GSSG ratio have been hypothesized to be important factors in preventing these adverse clinical outcomes. Several reports have investigated the role of GPx’s (Espinola-Klein et al., 2007, Jin et al., 2011, Weiss et al., 2001) and glucose-6-phosphate dehydrogenase (G6PD) the primary source to cellular NADPH (Leopold et al., 2007), in vascular dysfunction. Although these studies have placed an emphasis on GSH redox cycling, no reports to date have investigated the influence of genetic manipulation of de novo GSH synthesis genes on vascular reactivity.
We found that aortic rings from Gclm−/+ mice have an impaired ACh-mediated relaxation (Figure 3a). Alternatively, aortic rings from Gclm−/− mice do not, and surprisingly we observed aortic rings from Gclm−/− mice to have an enhanced relaxation at higher ACh concentrations when compared to Gclm−/+. These effects seem to be due to alterations in bioavailable NO• as we demonstrated a significant increase in NO• synthesis following ACh stimulation in aortas from Gclm−/− mice compared to Gclm−/+ mice (Figure 4b). Our findings that the basal levels of aortic cGMP are perhaps decreased in Gclm−/+ and Gclm−/− mice, would suggest that soluble guanylate cyclase (sGC) activity is impaired with decreased GSH levels (Figure 5). Nitrosothiols and GSH have been shown to influence sGC posttranslational modifications and activity (Oppermann, 2011). Although this trend did not reach significance, these trends may explain observations of impaired ACh-stimulated vessel relaxation in aortic rings from both Gclm−/+ and Gclm−/− mice at the lowest ACh concentrations (Figure 3). Of course this impaired response at lower ACh concentrations is lost in aortic rings from Gclm−/− mice as concentrations of ACh increase, potentially suggesting a compensatory mechanism whereby full sGC activity is restored when it is fully activated by NO•.
In addition, we observed that aortas from Gclm−/+ mice have enhanced nitrotyrosine protein modification as measured by immunohistochemistry of aortic cross-sections (Figure 6). This is an important observation as it indicates that lacking one copy of GCLM leads to enhanced oxidative and nitrosative conditions, even though it only results in modest reductions in aortic GSH and does not influence %GSSG or the GSH reductive potential (Figure 2). Although our aortic ROS measurements by L-012 did not demonstrate any increased ROS within the aortas of Gclm−/+ mice (Supplemental Figure 1), L-012 may not be specific or sensitive enough to detect subtle changes in ROS generation across genotypes. Nonetheless, our observation of enhanced aortic nitrotyrosine protein modification (Figure 6), compromised ACh-stimulated vessel relaxation (Figure 3), and reduced ACh-stimulated NO• production (Figure 4) in aortas from Gclm−/+ mice strongly suggest enhanced oxidative stress and compromised eNOS function within the aortas of these mice.
These findings are in agreement with observations by Nakamura and colleagues regarding an impairment in coronary blood flow after ACh infusion in patients with the GCLM -588C/T SNP (Nakamura et al., 2003). At higher ACh concentrations, we did not find impairment in ACh-mediated relaxation in aortic rings from Gclm−/− mice, suggesting that an adaptation has taken place in these animals that prevents overt oxidative stress and loss of eNOS function. Works by Rush and colleagues (Denniss et al., 2011, Ford et al., 2006) have demonstrated that a 10-day treatment with the GCL inhibitor BSO caused depletion of GSH, which led to impairment in ACh-mediated relaxation of aortic rings but not the common carotid artery in rats. This 10-day treatment with BSO resulted in a 50–60% reduction in GSH within the liver, a level that does not reach the extent of GSH depletion found in Gclm−/− mice, but far exceeds the GSH depletion found in Gclm−/+ mice. These results indeed suggest that the rapid loss of GSH by pharmacologic inhibition of its synthesis can lead to enhanced vascular oxidative stress and impaired NO• synthesis, but these data provide little insight into the human condition whereby 5’ promoter SNPs in GCL genes have a lifelong influence on vascular health. The advantages of the Gclm−/+ mouse model are that it targets a gene that has high frequency SNP in humans and, with the remaining Gclm copy providing sufficient GCLM protein for nearly normal GCL function, we suggest that it is roughly comparable to that of humans with the 5’ promoter SNP in GCLM.
In addition to the clinical implications of impaired vessel relaxation, there are significant concerns regarding increased sensitivity to vessel constriction following stimulation of α-adrenergic receptors. GSH, oxidative stress, and ROS have been shown to influence vessel constriction (Adachi et al., 2004, Tong et al., 2010, Kajimoto et al., 2007, Nunes et al., 2010, Oka et al., 2008). Due to the potential influence of ROS and GSH levels on vessel constriction, we measured contractile response to PE in aortic rings from WT, Gclm−/+, and Gclm−/− mice by wire myography. We observed that rings from Gclm−/+ mice did not have any altered response to PE-induced contraction, however, rings from Gclm−/− mice exhibited enhanced contraction to PE (Figure 7). Ford and coworkers (2006) demonstrated that BSO treatment produced an enhanced sensitivity to PE-induced contraction in the aortic rings of rats, which were attributed to an increase in oxidative stress in the vascular wall. Although there is significant evidence indicating that oxidative stress can enhance vessel constriction, our results did not support this contention. In the aortas of Gclm−/+ mice, we demonstrate reduced ACh-relaxation, trends for reduced bioavailable NO•, and enhanced aortic oxidative stress as measured by nitrotyrosine protein modification, but we did not observe an enhanced PE-induced contraction. Dramatic loss of GSH in Gclm−/− mice may not result in overt oxidative stress or loss of bioavailable NO•, as compensatory mechanisms have taken place, but our observation of enhanced PE-stimulated contraction may point to the unique role of GSH in vascular function. This may support the notion that oxidative stress within the vessel can very well impair NO• production and vessel relaxation, but this enhanced contraction may have more to do with alterations in GSH content, possibly supporting an important role for GSNO or protein S-glutathiolation in vasomotor activity.
To understand the role of NO• and endothelium derived relaxing and constricting factors (EDRF/EDCF) in mediated PE-contraction, we treated aortic rings with the NOS inhibitor L-NAME or mechanically removed the endothelium prior to PE treatment. In WT mice, removal of the endothelium produces an increase in contraction that exceeds NOS inhibition at lower PE concentrations. However, at higher PE concentrations, NOS inhibition produces an enhanced contraction that seems to match endothelium removal (Figure 8a). This suggests that EDRFs other than NO• may be playing a role in moderating PE-induced contraction, at least at lower PE concentrations. However, at higher PE-concentrations, this is apparently entirely due to NOS activity. In Gclm−/+ mice, endothelium removal produced a much more significant increase in PE-induced contraction relative to that of NOS inhibition (Figure 8b) at the lower concentrations of PE. This result suggests that in Gclm−/+ mice, NOS inhibition at lower PE concentrations does little to balance the PE-induced contraction, thus fitting with our prior data indicating a slight impairment in NOS function in these mice, and potential reduction in sGC activity. Interestingly, in aortic rings from Gclm−/+ mice, as PE concentrations increase, inhibition of NOS increases contraction to a level that well exceeds endothelium removal (Figure 8b). This may indicate that Gclm−/+ mice do have altered endothelial function in response to PE. Nonetheless, the fact that Gclm−/+ mice do not have altered PE-contraction under basal conditions suggests that these alterations confer protection against the slight loss of GSH.
The enhanced contraction seen at higher PE concentrations in Gclm−/+ aortas after NOS inhibition (as compared to endothelium removal) suggests that endothelial factors are contributing to vessel constriction by synthesizing EDCFs. Thus when the endothelium is removed, the increase in contraction previously conferred by endothelium dependent processes is lost, resulting in only modest increases in contraction. Although the role of EDCFs in mediating vessel constriction in response to PE has not been well established, Denniss et al. (2011) demonstrated that GSH depletion by BSO treatment did not influence an ACh-stimulated contractile response under conditions of NOS inhibition. The ACh-mediated contractile response, found to be COX-1 mediated, was no different in the common carotid artery of control or BSO treated rats and was not modified by the superoxide quenching agent Tempol, suggesting that the contractile response is not mediated by ROS or GSH. The lack of an association observed by Denniss et al. (2011) may be due to compensatory upregulation of alternative antioxidant response genes following a 10-day treatment with BSO, whereas the Gclm−/+ mouse has only slight reduction in GSH which is not expected to cause a similar compensatory response. Nonetheless, it is possible that Gclm−/+ mice are unable to maintain sufficient levels of GSH in certain tissues that are subject to elevated levels of oxidation under normal physiological conditions.
The fact that NOS inhibition and endothelium removal in Gclm−/− mice produced an enhancement in PE-induced vessel constriction at levels dramatically lower than that observed in WT or Gclm−/+ mice is striking (Figure 9b and 9d). It may be that the endothelium in these mice does not have a significant moderating influence on vessel constriction following PE treatment. This suggests that NO• and NOS activity play significantly smaller roles in moderating PE-contraction in these mice. However, this is in direct opposition to our results, where we demonstrate that aortas from Gclm−/− mice produce more NO• and are more responsive to ACh-stimulated relaxation than aortas from Gclm−/+ mice. After NOS inhibition by L-NAME, the enhanced contraction would be expected to exceed that observed in WT or Gclm−/+ mice. Catecholamine-stimulated vessel contraction caused by α-adrenoceptor stimulation is counteracted by the simultaneous activation of β-adrenoceptors, activation of eNOS, and subsequent NO• release from the endothelium (Figueroa et al., 2009). Our data suggest that significant GSH depletion seen in Gclm−/− mice and the compensatory action that takes place protects or even enhances ACh-mediated eNOS activation (mediated by Ca2+ influx and calmodulin dependent eNOS activation), but it may result in compromised PE or β-adrenergic receptor-mediated eNOS activation (dependent on eNOS phosphorylation). The activation of eNOS and NO•-mediated compensatory relaxation following catecholamine-stimulated vessel constriction is critical in preventing overt vessel constriction and increases in blood pressure. Our findings suggest that GSH specifically has an important role in providing this protection. The relationship between GSH synthesis and β-adrenergic receptor-dependent eNOS activation may have important implications and is the subject of future research.
We demonstrate here that although there is nearly a 90% loss of GSH within the aorta of Gclm−/− mice, there is no loss of bioavailable NO• or increased vascular oxidative stress as measured by aortic protein nitrotyrosine modification. This is in stark contrast to our observation that a modest 27% loss of aortic GSH in Gclm−/+ mice, results in compromised NO•, vessel relaxation, and enhanced vascular oxidative stress. These observations point to the conclusion that there are substantial compensatory responses that take place in Gclm−/− mice that provide adequate protection against the loss of GSH. In an assessment of whole genome transcriptional changes within the aorta, we observe that 789 genes are significantly changed in the comparison between Gclm−/− and WT mice using a selection criteria of an unadjusted p-value of <0.05 and an |fold| >1.5 (Figure 10). Interestingly, although we hypothesized that the Nrf2-mediated Oxidative Stress Response canonical pathway would be principally upregulated and responsible for the conferred protection, we do not observe this. Although not selected as a gene regulated by Nrf2, the most notable ‘antioxidant response’ gene that is within the top 25 genes upregulated is metallothionein 4, upregulated 7.8 fold in the aortas of Gclm−/− mice compared to WT. Interestingly, the most upregulated gene in the aortas of Gclm−/− mice was defensin beta 4. Upregulated 44 fold above aortas from WT mice, defensin beta 4 is a member of the small host-defense peptide family of defensins. This was an unexpected observation. However, as copy number and genetic polymorphisms of β-defensins are common in human populations, there have been previous reports of associations of β-defensin copy number in humans with vasculitis associated with SLE (Zhou et al., 2012) and ischemic stroke (Tiszlavicz et al., 2011). There has been one report that β-defensin can inhibit PE-stimulated vessel contraction in aortic rings of rats (Nassar et al., 2002), but there are no published works to date showing a direct role for β-defensins in mediating vascular reactivity. It is noteworthy that these defensins have high cysteine residue content. Although it is classically understood that these cysteine residues are cross-linked to provide structure, as defensin beta 4 is the highest upregulated gene in the aorta of Gclm−/− mice, it suggests that in addition to its antimicrobial role, it may have antioxidant and/or vascular protective properties.
In terms of classical antioxidant responses, although there are significant alterations in the Nrf2-mediated Oxidative Stress Response canonical pathway in both the Gclm−/− vs WT and Gclm−/+ vs WT comparisons, these associations are barely significant and are driven by only a few genes, modestly dysregulated. Although this significance is slightly greater in that of the Gclm−/+ vs WT comparison, any protective response is likely not sufficient considering our observations of enhanced nitrotyrosine protein modification and loss of bioavailable NO• following ACh stimulation. That we observed only minimally modified Nrf2 signaling in the aorta of Gclm−/− compared to WT mice suggests that they are capable of adapting to the low GSH level by alternative pathways without ‘turning on’ the classical Nrf2 antioxidant pathway. Understanding how these Gclm−/− mice are capable of adapting to the dramatic loss of GSH is a valuable area of study and is subject to future research.
Although we do not see any dramatic modification of the canonical Nrf2 antioxidant pathway in the aortas of Gclm−/− mice, in a fascinating observation, we do see that Ca2+ signaling in the most significantly modified canonical pathway in the Gclm−/− vs WT comparison (Figure 11 and Supplemental Figure 2). It is well known that oxidative stress and protein S-glutathiolation can modulate Ca2+ signaling in a dynamic fashion. Thus it is reasonable to believe that if GSH is depleted, and typical dynamic regulation of the Ca2+ is lost, compensatory transcriptional regulatory responses may occur to attempt to maintain normal vascular tone. As shown in Supplemental Figure 2, we observed many genes within certain families that participate in Ca2+ signaling and vascular reactivity were upregulated, these include but are not limited to, myosin, heavy polypeptide 1, skeletal muscle, adult (6.3 fold), myotilin (6.0 fold), myosin, heavy polypeptide 8, skeletal muscle, perinatal (6.0 fold), troponin T3, skeletal, fast (5.7 fold), troponin C2, fast (5.6 fold), ATPase, Ca++ transporting, cardiac muscle, fast twitch 1 (5.2 fold), and calmodulin 4 (4.0 fold). Although this is not causative data, this is strong suggestive evidence that these compensatory actions contribute to the enhanced PE-stimulated contraction we observed in our aortic ring studies. In addition, increases in genes such as calmodulin 4 may also contribute to our observations of enhanced ACh-stimulated relaxation and NO• production.
Conclusions
In the studies reported here, we found that lowering GSH content by targeted disruption of the GSH synthesis gene Gclm influences vascular reactivity in mouse aortic rings. Gclm−/+ aortas have an impaired response to ACh-stimulated relaxation and enhanced nitrotyrosine protein modification. Gclm−/− aortas have an enhanced ACh-relaxation in aortic rings, increased NO• production compared to Gclm−/+ aortas, increased sensitivity to PE-stimulated contraction, and altered expression of genes within the canonical Ca2+ signaling pathway. In addition, by testing PE-contraction following NOS inhibition and endothelium removal, we found that Gclm−/+ aortas seem to balance PE-contraction with NOS activity to a greater extent than WT mice. Also Gclm−/+ aortas have a decreased contractile response after endothelium removal when compared to L-NAME treatment, possibly suggesting a role for EDCFs. Interestingly, we observed that PE-contraction in Gclm−/− mice is not strongly influenced by NOS activity or endothelium derived factors, suggesting that GSH depletion and the compensatory reactions that take place in these mice, may influence PE-stimulated eNOS activity but not ACh-stimulated eNOS activity. This may highlight the unique role GSH has in balancing vessel contraction by EDRFs. By testing not only Gclm−/− mice, but also Gclm−/+ mice, we were able to investigate the effects of only slightly compromised GSH synthesis, and compare this to effects seen in Gclm−/−mice that have dramatically decreased GSH levels. Overall, we conclude that the relative expression of the GSH synthesis gene Gclm is an important determinant of vascular reactivity in mice. Because the Gclm−/+ mouse likely mimics the vascular effects previously observed in humans with 5’ promoter SNPs in GCLM, we believe they represent a suitable model for further investigation of the mechanisms underlying this compromised vascular reactivity.
Supplementary Material
Supplemental Figure 1. Measurement of aortic ROS production using L-012 chemiluminescence. Aortic rings (4mm in length) were incubated in L-012 (100 μM) (a) or with L-012 in the presence of L-NAME (b) for twenty minutes. N equals 3 for each genotype, with each n representing two rings from the same mouse. Error bars in figures represent standard error of the mean (SEM).
Supplemental Figure 2. Schematic diagram of the Ca2+ Signaling canonical pathway with the Gclm−/− vs WT comparison in the aorta, as generated from Ingenuity Pathway Analysis. Intensity of red color indicates an increase in gene expression observed in this microarray analysis.
Supplemental Figure 3. Analysis of the Nrf2-mediated Oxidative Stress canonical pathway in the Gclm−/− vs WT comparison of the aorta by microarray. a) Bar represents –log(p-value) whereas yellow box represents ratio of positively vs. negatively regulated genes. b) Table of the genes responsible for the observed changes in Nrf2 canonical pathway.
Supplemental Figure 4. Analysis of the Nrf2-mediated Oxidative Stress canonical pathway in the Gclm−/+ vs WT comparison of the aorta by microarray. a) Bar represents –log(p-value) whereas yellow box represents ratio of positively vs. negatively regulated genes. b) Table of the genes responsible for the observed changes in Nrf2 canonical pathway.
Highlights.
Mice lacking the GSH synthesis gene Gclm have compromised vascular reactivity
Gclm−/+ mice have impaired ACh-stimulated vessel relaxation but Gclm−/− mice do not
Gclm−/− mice have enhanced PE-stimulated vessel contraction but Gclm−/+ do not
NOS inhibition/endothelium removal revealed differences in PE-contraction
These data provide insight regarding the role of GSH synthesis in vascular function
Acknowledgments
This study was supported by NIH grants 5P50ES015915, 5P30ES007033, 5T32ES007032, 5R01DK073878 and 5U24DK076126.
Abbreviations
- GSH
glutathione
- GPx
glutathione peroxidase
- GRx
glutathione disulfide reducatase
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphism
- GCLC
glutamate cysteine ligase catalytic subunit
- GCLM
glutamate cysteine ligase modifier subunit
- WT
wild type
- NO•
nitric oxide
- ACh
acetylcholine
- PE
phenylephrine
- L-NAME
N3-nitro-L-arginine methyl ester
- EDRF
endothelium derived relaxing factor
- eNOS
endothelial nitric oxide synthase
- BH4
tetrahydrobiopterin
- O2
oxygen
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced
- O2•−
superoxide radical
- G6PD
glucose-6-phosphate dehydrogenase
- GSNO
S-nitrosoglutathione
- H2O2
hydrogen peroxide
- GSSG
glutathione disulfide
- γ-GC
γ-glutamylcysteine
- GCL
glutamate cysteine ligase
- GS
glutathione synthase
- MI
myocardial infarction
- BSO
L-buthionine-[S,R]-sulfoximine
- cGMP
cyclic guanosine monophosphate
- SPF
specific pathogen free
- HPLC
high pressure liquid chromatography
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- EDCF
endothelium derived constricting factor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- K-PSS
potassium-physiological salt solution
- ESR
electron spin resonance
- Fe(DETC)2
non-colloidal iron diethyldithiocarbamate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Chad S. Weldy, Email: weldyc@u.washington.edu.
Ian P. Luttrell, Email: luttri@u.washington.edu.
Collin C. White, Email: ccwhite@u.washington.edu.
Vicki Morgan-Stevenson, Email: vmorgan@u.washington.edu.
Theo K. Bammler, Email: tbammler@u.washington.edu.
Richard P. Beyer, Email: dbeyer@u.washington.edu.
Zahra Afsharinejad, Email: zafshari@u.washington.edu.
Francis Kim, Email: fkim@u.washington.edu.
Kanchan Chitaley, Email: kanchanc@u.washington.edu.
Terrance J. Kavanagh, Email: tjkav@u.washington.edu.
References
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–7. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- Botta D, White CC, Vliet-Gregg P, Mohar I, Shi S, Mcgrath MB, Mcconnachie LA, Kavanagh TJ. Modulating GSH synthesis using glutamate cysteine ligase transgenic and gene-targeted mice. Drug Metab Rev. 2008;40:465–77. doi: 10.1080/03602530802186587. [DOI] [PubMed] [Google Scholar]
- Brocq ML, Leslie SJ, Miliken P, Megson IL. Endothelial Dysfunction: From Molecular Mechanisms to Measurement, Clinical Implications, and Therapeutic Opportunities. Antioxidants and Redox Signaling. 2008;10:1631–1673. doi: 10.1089/ars.2007.2013. [DOI] [PubMed] [Google Scholar]
- Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2005;102:9056–61. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-A, Wang T-Y, Varadharaj S, Reyes LA, Hemann C, Talukder MaH, Chen Y-R, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–8. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney A, Storey J. Bioconductor’s qvalue package 2006 [Google Scholar]
- Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–26. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Denniss SG, Levy AS, Rush JW. Effects of Glutathione-Depleting Drug Buthionine Sulfoximine and Aging on Activity of Endothelium-Derived Relaxing and Contracting Factors in Carotid Artery of Sprague Dawley Rats. Journal of cardiovascular pharmacology. 2011 doi: 10.1097/FJC.0b013e3182239f36. [DOI] [PubMed] [Google Scholar]
- Eaton DL, Hamel DM. Increase in gamma-glutamylcysteine synthetase activity as a mechanism for butylated hydroxyanisole-mediated elevation of hepatic glutathione. Toxicol Appl Pharmacol. 1994;126:145–9. doi: 10.1006/taap.1994.1100. [DOI] [PubMed] [Google Scholar]
- Espinola-Klein C, Rupprecht HJ, Bickel C, Schnabel R, Genth-Zotz S, Torzewski M, Lackner K, Munzel T, Blankenberg S, Investigators A. Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am J Cardiol. 2007;99:808–12. doi: 10.1016/j.amjcard.2006.10.041. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Poblete I, Fernández R, Pedemonte C, Cortés V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–43. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- Ford RJ, Graham DA, Denniss SG, Quadrilatero J, Rush JWE. Glutathione depletion in vivo enhances contraction and attenuates endothelium-dependent relaxation of isolated rat aorta. Free Radic Biol Med. 2006;40:670–8. doi: 10.1016/j.freeradbiomed.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–35. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- Haque JA, Mcmahan RS, Campbell JS, Shimizu-Albergine M, Wilson AM, Botta D, Bammler TK, Beyer RP, Montine TJ, Yeh MM, Kavanagh TJ, Fausto N. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab Invest. 2010;90:1704–17. doi: 10.1038/labinvest.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Zeng H, Li F, Feng J, Liu S, Liu J, Yu J, Mao J, Hong T, Chen AF, Wang X, Wang G. Homocysteine impairs coronary artery endothelial function by inhibiting tetrahydrobiopterin in patients with hyperhomocysteinemia. AJP: Endocrinology and Metabolism. 2010;299:E1061–E1065. doi: 10.1152/ajpendo.00367.2010. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jin RC, Mahoney CE, Coleman Anderson L, Ottaviano F, Croce K, Leopold JA, Zhang Y-Y, Tang S-S, Handy DE, Loscalzo J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 2011;123:1963–73. doi: 10.1161/CIRCULATIONAHA.110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E, Wesselkamper SC, Shertzer HG, Leikauf GD, Dalton TP, Chen Y. Glutathione deficient C57BL/6J mice are not sensitized to ozone-induced lung injury. Biochem Biophys Res Commun. 2010;396:407–12. doi: 10.1016/j.bbrc.2010.04.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AW, Durante W, Korthuis RJ. Heme Oxygenase-1 Deficiency Leads to Alteration of Soluble Guanylate Cyclase Redox Regulation. Journal of Pharmacology and Experimental Therapeutics. 2010;335:85–91. doi: 10.1124/jpet.110.169755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto H, Hashimoto K, Bonnet SN, Haromy A, Harry G, Moudgil R, Nakanishi T, Rebeyka I, Thébaud B, Michelakis ED, Archer SL. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: a newly recognized mechanism for sustaining ductal constriction. Circulation. 2007;115:1777–88. doi: 10.1161/CIRCULATIONAHA.106.649566. [DOI] [PubMed] [Google Scholar]
- Khoo JP, Alp NJ, Bendall JK, Kawashima S, Yokoyama M, Zhang Y-H, Casadei B, Channon KM. EPR quantification of vascular nitric oxide production in genetically modified mouse models. Nitric Oxide. 2004;10:156–61. doi: 10.1016/j.niox.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Koide S-I, Kugiyama K, Sugiyama S, Nakamura S-I, Fukushima H, Honda O, Yoshimura M, Ogawa H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J Am Coll Cardiol. 2003;41:539–45. doi: 10.1016/s0735-1097(02)02866-8. [DOI] [PubMed] [Google Scholar]
- Krejsa CM, Franklin CC, White CC, Ledbetter JA, Schieven GL, Kavanagh TJ. Rapid activation of glutamate cysteine ligase following oxidative stress. J Biol Chem. 2010;285:16116–24. doi: 10.1074/jbc.M110.116210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo MDM, Siddegowda YKB, Kumar D, Tandan SK, Sastry KVH, Prakash VR, Mishra SK. Role of nitric oxide and carbon monoxide in N(omega)-Nitro-L-arginine methyl ester-resistant acetylcholine-induced relaxation in chicken carotid artery. Eur J Pharmacol. 2008;596:111–7. doi: 10.1016/j.ejphar.2008.07.058. [DOI] [PubMed] [Google Scholar]
- Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–46. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res. 2001;88:756–62. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- Maron BA, Zhang Y-Y, Handy DE, Beuve A, Tang S-S, Loscalzo J, Leopold JA. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284:7665–72. doi: 10.1074/jbc.M809460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcconnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, Chatterton-Kirchmeier S, White CC, Pierce RH, Kavanagh TJ. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99:628–36. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- Nakamura S-I, Kugiyama K, Sugiyama S, Miyamoto S, Koide S-I, Fukushima H, Honda O, Yoshimura M, Ogawa H. Polymorphism in the 5'-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation. 2002;105:2968–73. doi: 10.1161/01.cir.0000019739.66514.1e. [DOI] [PubMed] [Google Scholar]
- Nakamura S-I, Sugiyama S, Fujioka D, Kawabata K-I, Ogawa H, Kugiyama K. Polymorphism in glutamate-cysteine ligase modifier subunit gene is associated with impairment of nitric oxide-mediated coronary vasomotor function. Circulation. 2003;108:1425–7. doi: 10.1161/01.CIR.0000091255.63645.98. [DOI] [PubMed] [Google Scholar]
- Nassar T, Akkawi SE, Bar-Shavit R, Haj-Yehia A, Bdeir K, Al-Mehdi A-B, Tarshis M, Higazi Aa-R. Human alpha-defensin regulates smooth muscle cell contraction: a role for low-density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. Blood. 2002;100:4026–32. doi: 10.1182/blood-2002-04-1080. [DOI] [PubMed] [Google Scholar]
- Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67:3823–36. doi: 10.1007/s00018-010-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Fagan KA, Jones PL, Mcmurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. British Journal of Pharmacology. 2008;155:444–454. doi: 10.1038/bjp.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, et al. Regulation of vascular guanylyl cyclase by endothelial nitric oxide-dependent posttranslational modification. Basic Res Cardiol. 2011;106:539–549. doi: 10.1007/s00395-011-0160-5. [DOI] [PubMed] [Google Scholar]
- Shi ZZ, Osei-Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM, Lukin DJ, Danney CM, Matzuk MM, Lieberman MW. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc Natl Acad Sci USA. 2000;97:5101–6. doi: 10.1073/pnas.97.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Stamler J, Cunningham M, Loscalzo J. Reduced thiols and the effect of intravenous nitroglycerin on platelet aggregation. Am J Cardiol. 1988;62:377–80. doi: 10.1016/0002-9149(88)90962-9. [DOI] [PubMed] [Google Scholar]
- Thompson SA, White CC, Krejsa CM, Diaz D, Woods JS, Eaton DL, Kavanagh TJ. Induction of glutamate-cysteine ligase (gamma-glutamylcysteine synthetase) in the brains of adult female mice subchronically exposed to methylmercury. Toxicology Letters. 1999;110:1–9. doi: 10.1016/s0378-4274(99)00133-2. [DOI] [PubMed] [Google Scholar]
- Tiszlavicz Z, Somogyvári F, Szolnoki Z, Sztriha LK, Németh B, Vécsei L, Mándi Y. Genetic polymorphisms of human 3-defensins in patients with ischemic stroke. Acta Neurol Scand. 2011 doi: 10.1111/j.1600-0404.2011.01613.x. [DOI] [PubMed] [Google Scholar]
- Tong X, Evangelista A, Cohen RA. Targeting the redox regulation of SERCA in vascular physiology and disease. Curr Opin Pharmacol. 2010;10:133–138. doi: 10.1016/j.coph.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N, Zhang YY, Heydrick S, Bierl C, Loscalzo J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc Natl Acad Sci USA. 2001;98:12503–8. doi: 10.1073/pnas.231428998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldy CS, White CC, Wilkerson H-W, Larson TV, Stewart JA, Gill SE, Parks WC, Kavanagh TJ. Heterozygosity in the glutathione synthesis gene Gclm increases sensitivity to diesel exhaust particulate induced lung inflammation in mice. Inhal Toxicol. 2011;23:724–35. doi: 10.3109/08958378.2011.608095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–8. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- Xie H-H, Zhou S, Chen D-D, Channon KM, Su D-F, Chen AF. GTP Cyclohydrolase I/BH4 Pathway Protects EPCs via Suppressing Oxidative Stress and Thrombospondin-1 in Salt-Sensitive Hypertension. Hypertension. 2010;56:1137–1144. doi: 10.1161/HYPERTENSIONAHA.110.160622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X-J, Cheng F-J, Lv J-C, Luo H, Yu F, Chen M, Zhao M-H, Zhang H. Higher DEFB4 genomic copy number in SLE and ANCA-associated small vasculitis. Rheumatology (Oxford, England) 2012 doi: 10.1093/rheumatology/ker419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Measurement of aortic ROS production using L-012 chemiluminescence. Aortic rings (4mm in length) were incubated in L-012 (100 μM) (a) or with L-012 in the presence of L-NAME (b) for twenty minutes. N equals 3 for each genotype, with each n representing two rings from the same mouse. Error bars in figures represent standard error of the mean (SEM).
Supplemental Figure 2. Schematic diagram of the Ca2+ Signaling canonical pathway with the Gclm−/− vs WT comparison in the aorta, as generated from Ingenuity Pathway Analysis. Intensity of red color indicates an increase in gene expression observed in this microarray analysis.
Supplemental Figure 3. Analysis of the Nrf2-mediated Oxidative Stress canonical pathway in the Gclm−/− vs WT comparison of the aorta by microarray. a) Bar represents –log(p-value) whereas yellow box represents ratio of positively vs. negatively regulated genes. b) Table of the genes responsible for the observed changes in Nrf2 canonical pathway.
Supplemental Figure 4. Analysis of the Nrf2-mediated Oxidative Stress canonical pathway in the Gclm−/+ vs WT comparison of the aorta by microarray. a) Bar represents –log(p-value) whereas yellow box represents ratio of positively vs. negatively regulated genes. b) Table of the genes responsible for the observed changes in Nrf2 canonical pathway.