

Abstract
Twenty eight new analogues were synthesized by optimizing the structure of MP-10 and their in vitro binding affinities towards PDE10A, PDE3A/B, and PDE4A/B were determined. Among these new analogues, 10a, 10b, 10d, 11a, 11b and 11d are very potent towards PDE10A and have IC50 values of 0.40 ± 0.02, 0.28 ± 0.06, 1.82 ± 0.25, 0.24 ± 0.05, 0.36 ± 0.03 and 1.78 ± 0.03 nM respectively; these six compounds displayed high selectivity for PDE10A versus PDE3A/3B/4A/4B. The promising compounds will be further validated in vivo to identify PDE10A imaging tracers.
Introduction
Phosphodiesterase 10A (PDE10A) is one of the 11 families of phosphodiesterase enzymes. PDE10A is a unique dual specificity enzyme; it is able to hydrolyze cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) to inactive adenosine monophosphate (AMP) and guanosine monophosphate GMP, respectively.1,2 PDE10A was identified independently by three groups in 1999 (ref. 2–4) and has the most unique expression with mRNA levels in the brain and testis.3 Within the brain, the highest expression of PDE10A protein is in the medium spiny neurons of the striatum (caudate and putamen), nucleus accumbens, and olfactory tubercle of mice, dogs, cynomolgus and humans.5 Due to the unique striatal localization of PDE10A and the function of PDE10A in the brain, inhibition of PDE10A represents a novel approach for treating neurological and psychiatric disorders such as schizophrenia, Parkinson’s disease, and Huntington’s disease (HD);6–8 this therapeutic approach may avoid unwanted side effects such as extrapyramidal symptoms of conventional therapeutics. For example, investigating the levels of PDE10A in the brain of patients with Huntington’s disease indicated that the level of PDE10A protein was decreased compared to that of age-matched healthy individuals;9 after treatingwithPDE10A inhibitor, 2-[4-[pyridin-4-yl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-3-yl]-phenoxymethyl]-quinoline (TP-10), an analogue of 2-((4-(1-methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl)phenoxy)-methyl)quinoline (MP-10) (Fig. 1), both neuropathology and symptoms in R6/2 mice were ameliorated; the behavioural symptoms of R6/2 mice, such as rotarod performance, were improved.10 In addition, chronically treating with TP-10 resulted in changes in gene expression11 in the striatum of engineered PDE10A knocked out mice; this provided more evidence that the PDE10A enzyme is a promising therapeutic target for psychotic and related neurodegenerative diseases.
Fig. 1.

Structures of [11C]papaverine, TP-10 and its C-11 or F-18 labeled analogue radiotracers.
Positron emission tomography (PET) provides a unique and novel non-invasive imaging tool to investigate the changes in levels of PDE10A in the brain of living subjects and to study the physiological function of PDE10A in the central nervous system (CNS). Investigators have made significant efforts to identify a clinically suitable PET probe for imaging the changes in levels of PDE10A in the human brain.8,12–14 In recent years, [11C]-papaverine and other radioligands were evaluated in vivo.15–20 However, due to the lack of suitable radiopharmaceutical properties of the reported radiotracers,15–20 the evaluation of PDE10A PET tracers is still in process. To date, no clinically suitable PET tracer was reported. Among the reported PET radiotracers, substituted pyrazole-containing ligands radio-labeled by introducing carbon-11, or fluorine-18 via N-alkylation of the pyrazole ring had attracted more attention16,18,19 because of the use of MP-10 as a PDE10A inhibitor in clinical trials for treatment of schizophrenia;21 PET radiotracers, [11C]MP-10, 18F-JNJ41510417, [18F]1, [18F]2 shown in Fig. 1, were made by introducing the [11C]methyl, [18F]fluoroethyl or [18F]fluoropropyl group on the N-atom of the pyrazole scaffold and further evaluated in vivo in rodents and monkeys.16,18,19,22 Our group had reported that the in vivo microPET studies of [11C] MP-10 in monkeys showed a very clear anatomic structure of monkey brain and the highest accumulation of radioactivity in the striatal area of the brain, higher target-to-non-target ratios, but the kinetics displayed an increasing trend post-injection of [11C]MP-10. Further metabolite analysis of monkey plasma, rodent plasma and brain tissues revealed that the radioactive metabolite formed post-injection of radiotracer [11C]MP-10 by O-dealkylation of [11C]MP-10 in vivo; this radioactive metabolite was able to cross the blood–brain-barrier and accumulate in the brain; radioactivity of the radioactive metabolite in the brain may lead to misinterpretation of the measurement of PDE10A levels in the brain. Thus, further optimization of the pyrazole-containing analogues to overcome the radioactive metabolite concern is imperative.
In our current studies, we had taken the following strategies to design the new analogues to overcome the radioactive metabolite concern: (1) introducing a methoxy into a quinonline fragment of the MP-10 structure, which creates a new position to label with carbon-11; we expect that the new radioactive metabolite obtained from the substituted 2-methylquinoline fragment via O-dealkylation of phenyl ether does not have the capability to cross the blood–brain-barrier (BBB) and enter into the brain; (2) replacing the oxygen (O) atom that links the substituted methyl-quinoline fragment and the substituted pyrazole fragment of MP-10 with a nitrogen (N) or sulfur (S) atom; we expect that the new derivatives in which the N or S serves as a linkage atom will be more stable. This may increase the metabolic stability of radiotracers in vivo or form new radioactive metabolites that do not enter into the brain and accumulate in the striatal area and other regions. In the current manuscript, we reported our efforts on synthesizing the new analogues based on the above design strategies and their in vitro binding affinity evaluation.
Results and discussion
Chemistry
The syntheses of compounds 10a–f and 11a–f were accomplished according to Scheme 1.
Scheme 1.

Synthesis of compounds 10a–f and 11a–f. Reagents and conditions: (a) CDI, N,O-dimethylhydroxylamine hydrochloride, NEt3, CH2Cl2, rt; (b) 4-picoline, LDA, −78 C to rt; (c) N,N-dimethylformamide-dimethyl acetal, reflux, then methylhydrazine; (d) H2, Pd/C, ethanol/EtOAc; (e) NBS, AIBN, CCl4, reflux; (f) NaH, DMF, rt.
The syntheses of several MP-10 analogues were achieved by a Mitsunobu reaction of heteroaromatic benzyl alcohols with 6 as an alkylating agent.21 Attempts to synthesize compounds 10a–f and 11a–f were very challenging due to similar retention factor values (Rf) of intermediates quinolin-2-yl-methanol and the final compounds 10a–f and 11a–f. To overcome this challenge, an alternative approach was taken; substituted phenols 6 or 7 were coupled with substituted 2-bromomethylquinolines 9a–f to afford target compounds for which the purification was easily achieved by column chromatography. Synthesis of target compounds began with commercially available 4-(benzyloxy) benzoic acid (3). Following the literature procedure,21 compounds 4 and 5 were afforded in three steps. Debenzylation of compounds 4 and 5 afforded substituted phenols 6 and 7. Commercially unavailable 4-methoxy-2-methylquinoline (8b) was synthesized by treating 4-chlorine-2-methylquinoline with sodium methoxide (NaOCH3);23,24 8-methoxy-2-methylquinoline (8f) was synthesized via O-alkylation of 2-methylquinolin-8-ol with iodomethane in dimethylformamide (DMF). Methoxy-2-bromomethylquinolines 9a–f were synthesized by bromination of methoxy substituted 2-methylquinolines 8a–f using N-bromosuccinimide (NBS). O-Alkylation of phenols 6 or 7 with substituted 2-bromomethylquinolines 9a–f afforded compounds 10a–f or 11a–f respectively, following the literature procedure with minor modification.21,25
The syntheses of the S-atom bridged analogues 10g–j and 11g–j were achieved according to Scheme 2. Synthesis began by reacting 4-bromobenzoic acid 12 with N,O-dimethylhydroxylamine hydrochloride and 1,1-carbonyldiimidazole (CDI) to afford Weinreb amide 13 in nearly quantitative yield. Treatment of 4-picoline with lithium diisopropyl amide (LDA) at −78 °C followed by adding this solution to the solution of 13 in tetrahydrofuran (THF) afforded compound 14. Treatment of compound 14 with dimethoxymethyl-dimethyl amine afforded the key intermediate enaminones 15 and 16. Compounds 15 or 16 upon treatment with sodium thiomethoxide in dimethylacetamide (DMA) at high temperature afforded intermediates 17 or 18 respectively. Coupling of thiophenols 17 or 18 with substituted 2-bromomethylquinolines 9b, 9d–f afforded thioether analogues 10g–j or 11g–j.
Scheme 2.

Synthesis of compounds 10g–j and 11g–j. Reagents and conditions: (a) CDI, N,O-dimethylhydroxylamine hydrochloride, NEt3, CH2Cl2, rt; (b) 4-picoline, LDA, −78 °C to rt; (c) N,N-dimethylformamide-dimethyl acetal, reflux, then NH2NHCH3; (d) NaSMe, DMA, 150 °C; (e) substituted 2-bromomethylquinoline, NaH, DMF, 0 °C to rt, overnight.
The syntheses of the N-atom bridged analogues 10k–n and 11k–n were achieved according to Scheme 3. The Gabriel synthesis procedure26–28 was followed by treating the aromatic bromides 15 or 16 with potassium phthalimide to afford the corresponding N-alkylphthalimide 19 or 21. Treating 19 or 21 with ethanolic hydrazine following the Ing–Manske procedure29 afforded aromatic amines 20 or 22. N-Alkylation of the aromatic amines 20 or 22 with substituted 2-bromomethylquinolines 9b, 9d–f afforded the desired N-atom bridged analogues 10k–n or 11k–n.
Scheme 3.
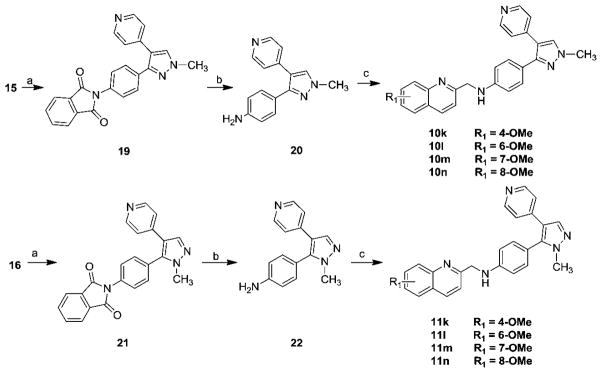
Synthesis of compounds 10k–n and 11k–n. Reagents and conditions: (a) potassium phthalimide, Cul, DMA, 150 °C; (b) hydrazine hydrate, EtOH, reflux; (c) substituted 2-bromomethylquinoline, Na2CO3, acetonitrile, 80 °C.
Biological binding studies
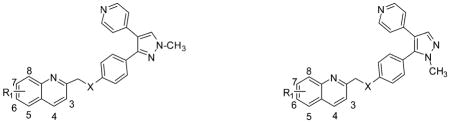
We explored the structure of MP-10 by introducing the methoxy group into the quinoline fragment of the MP-10. The strategy of introducing the methoxy group is to provide a new position for introducing the [11C]methoxy group if the new analogues have high binding affinity and selectivity for PDE10A. To test this strategy, we first synthesized compounds 10b, 10d–f and their regioisomers 11b, 11d–f by introducing the methoxy group at the 4, 6, 7 and 8 positions of the quinoline fragment. The in vitro PDE10A binding affinity data of compounds 10b, 10d–f displayed the decreasing order as: 4-methoxy > 6-methoxy > 7-methoxy ≈ 8-methoxy; the IC50 values (nM) of 10b and 10d–f were 0.28 ± 0.06, 1.82 ± 0.25, 46.0 ± 6, and 41.5 ± 6.5 nM respectively. Among compounds 10b, 10d–f, compounds 10b and 10d are very potent for PDE10A, particularly, for 10b, the in vitro IC50 value reached 0.28 nM.
To extend our structural modification studies, the regioisomeric counterparts 11b, 11d–f were also synthesized and their in vitro binding affinities were determined as shown in Table 1. The in vitro data suggested that compounds 11b, 11d–f and compounds 10b, 10d–f have similar decreasing binding affinity order; for compounds 11b, 11d, 11e, and 11f, the IC50 values (nM) were 0.36 ± 0.03, 1.78 ± 0.03, 95 ± 15, and 199 ± 21 nM; 4-methoxy substituted compound 11b, similar to its counterpart 10b, has the highest binding affinity for PDE10A. The next most potent compound is the 6-methoxy substituted 11d. Compounds 10b and 11b had similar subnanomolar affinities (0.1 nM < IC50 values < 0.5 nM). However, for the less potent compounds 11e and 11f, the IC50 values revealed that the 8-methoxy substituted 11f (199 nM) has a 2-fold higher IC50 value than that the 7-methoxy substituted compound 11e (95 nM).
Table 1.
PDE10A affinities of new analoguesa
| ||||
|---|---|---|---|---|
| IC50 value (nM)
|
||||
| Compd | X | R1 | PDE10A | Log Db |
| 10a | O | 3-OMe | 0.40 ± 0.02 | 3.87 |
| 10b | O | 4-OMe | 0.28 ± 0.06 | 3.79 |
| 10c | O | 5-OMe | 60 ± 26 | 3.92 |
| 10d | O | 6-OMe | 1.82 ± 0.25 | 3.61 |
| 10e | O | 7-OMe | 46 ± 6 | 3.77 |
| 10f | O | 8-OMe | 41.5 ± 6.5 | 3.28 |
| 10g | S | 4-OMe | 168 ± 52 | 3.89 |
| 10h | S | 6-OMe | 415 ± 65 | 3.72 |
| 10i | S | 7-OMe | 400 ± 60 | 3.88 |
| 10j | S | 8-OMe | 635 ± 15 | 3.39 |
| 10k | N | 4-OMe | 13 900 ± 1100 | 3.52 |
| 10l | N | 6-OMe | 7500 ± 700 | 3.36 |
| 10m | N | 7-OMe | 7400 ± 600 | 3.52 |
| 10n | N | 8-OMe | 10 900 ± 700 | 3.02 |
| 11a | O | 3-OMe | 0.24 ± 0.05 | 3.87 |
| 11b | O | 4-OMe | 0.36 ± 0.03 | 3.79 |
| 11c | O | 5-OMe | 81.4 ± 9.2 | 3.92 |
| 11d | O | 6-OMe | 1.78 ± 0.03 | 3.61 |
| 11e | O | 7-OMe | 95 ± 15 | 3.77 |
| 11f | O | 8-OMe | 199 ± 21 | 3.28 |
| 11g | S | 4-OMe | 1850 ± 450 | 3.89 |
| 11h | S | 6-OMe | 610 ± 80 | 3.72 |
| 11i | S | 7-OMe | 635 ± 55 | 3.88 |
| 11j | S | 8-OMe | 3050 ± 450 | 3.39 |
| 11k | N | 4-OMe | 645 ± 255 | 3.52 |
| 11l | N | 6-OMe | 4250 ± 650 | 3.36 |
| 11m | N | 7-OMe | 5150 ± 250 | 3.52 |
| 11n | N | 8-OMe | 4600 ± 400 | 3.02 |
IC50 is defined as the concentration of the inhibitor required to reduce the [3H]cAMP hydrolysis activity of recombinant human PED10A by 50% with scintillation proximity assay.
Calculated value at pH = 7.4 by ACD/I-Lab ver. 7.0 (Advanced Chemistry Development, Inc., Canada).
From the above structure–activity relationship analysis, we concluded that introduction of a methoxy group at an appropriate position of the quinoline fragment in the structure of MP-10 was able to retain the high PDE10A binding affinity. Thus, we further explored the structure of MP-10 by replacement of the O-atom link bridge with a N-atom or S-atom. For the S-atom bridged analogues, compounds 10g–j and 11g–j were synthesized; for the N-atom bridged analogues, 10k–n and 11k–n were synthesized. Our in vitro data revealed: (1) compounds 10g–j, containing a 4-(1-methyl-4-(pyridin-4-yl)-1H-pyrazol-5-yl)phenyl fragment, had higher affinity than their corresponding isomers 11g–j, containing a 4-(1-methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl)phenyl fragment; this observation was similar to the results of the O-atom bridged analogues 10b, 10d–f versus 11b, 11d–f; (2) even the S-atom bridged analogues 10g–j displayed higher binding affinity than that N-atom bridged analogues 10k–n, and the binding affinity of methoxy substituted analogues displayed the order as: 4-methoxy > 6-methoxy ≈ 7-methoxy > 8-methoxy, for which the IC50 values are 168 ± 52, 415 ± 65, 400 ± 60, 635 ± 15 nM for compounds 10g–j respectively. The PDE10A binding affinities of S-atom bridged analogues 10g–j were much lower than those of their corresponding O-atom bridged analogues 10b, 10d–f. Among 16 compounds 10g–n and 11g–n containing either S-atom or N-atom bridge, the most potent compound 10g had the IC50 value of 168 ± 52 nM, which was 600-fold less potent than that of 10b (0.28 ± 0.06 nM), the corresponding O-atom bridged analogue. The binding affinities of N-atom link bridge analogues 10k–n and 11k–n decreased dramatically (IC50 value > 645 nM). Thus, replacing the O-atom bridge linkage with a S-atom or N-atom did not generate PDE10A potent compounds.
Based on above in vitro data, only the O-atom bridge linkage was able to retain the high binding potency for the PDE10A enzyme. We further synthesized 10a, 10c, 11a, and 11c by introducing the methoxy group at 3 and 5 positions of the quinoline fragment; the in vitro data suggested that 3-methoxy substituted compounds 10a and 11a were very potent for PDE10A and the IC50 values are 0.40 ± 0.02 and 0.24 ± 0.05 nM, which had similar PDE10A binding potency to that of the 4-methoxy substituted compounds, 10b and 11b. However, 5-methoxy substituted compounds 10c and 11c displayed lower PDE10A binding affinities; the IC50 values were 60 ± 26 nM for 10c and 81.4 ± 9.2 nM for 11c.
Inhibition of PDE3A/B could lead to arrhythmia and increased mortality,30,31 and the role of phosphodiesterase 4 could be a regulator of central nervous system function and inhibition of PDE4A could increase heart and respiratory rates.32 The binding affinities of compounds 10a, 10b, 10d, 11a, 11b, and 11d, which have IC50 values less than 5 nM for PDE10A, were further evaluated in vitro for PDE3A/B and PDE4A/B; the results are shown in Table 2. The in vitro data suggested that these six compounds had very weak binding affinity for PDE3A/3B and PDE4A/4B with the IC50 value > 1500 nM. This suggested that six compounds 10a, 10b, 10d, 11a, 11b and 11d not only had high potency for PDE10A but also had high selectivity for PDE10A versus PDE3A/3B and 4A/4B.
Table 2.
PDE3A/B and PDE4A/B affinities of lead analogues
| IC50 value (103 × nM)
|
||||
|---|---|---|---|---|
| Compd | PDE3A | PDE3B | PDE4A | PDE4B |
| 10a | 123 ± 21 | 82.7 ± 10 | 3.85 ± 0.23 | 3.43 ± 0.21 |
| 10b | 27.5 ± 2.5 | 3.85 ± 0.95 | 2.56 ± 0.11 | 1.79 ± 0.10 |
| 10d | 78.0 ± 4.0 | 9.75 ± 1.25 | 3.37 ± 0.18 | 2.56 ± 0.09 |
| 11a | 18.7 ± 3.1 | 16.9 ± 2.1 | 199 ± 16.0 | 31.5 ± 3.60 |
| 11b | 29.0 ± 4.0 | 4.80 ± 1.20 | 4.18 ± 0.33 | 5.06 ± 0.40 |
| 11d | 1.50 ± 0.50 | 3.00 ± 0.14 | 5.60 ± 0.52 | 7.10 ± 0.58 |
Conclusion
In the present study, we optimized the structure of MP-10 to identify new compounds by introducing a methoxy group in the quinoline fragment. The in vitro data suggested that six compounds 10a, 10b, 10d, 11a, 11b, and 11d had high PDE10 binding potency with IC50 values of 0.40 ± 0.02, 0.28 ± 0.06, 1.82 ± 0.25, 0.24 ± 0.05, 0.36 ± 0.03, and 1.78 ± 0.03 nM respectively. These six compounds also displayed high selectivity for PDE10A against PDE3A/3B and PDE4A/4B with IC50 > 1500 nM. After further evaluating their PDE10A binding selectivity against another PDE enzyme family, it is worthwhile to label them with carbon-11 to conduct further in vivo validation. However, optimizing the structure of MP-10 by replacing the O-atom bridge with a S-atom or N-atom and introducing the methoxy group into the quinoline fragment led to a dramatic loss of the PDE10A binding affinities for the newly synthesized analogues. These new analogues and their in vitro binding affinities could provide helpful information for further structure– activity relationship studies.
Supplementary Material
Abbreviations
- Anal
Analysis
- BBB
Blood–brain-barrier
- CDI
1,1-Carbonyldiimidazole
- CNS
Central nervous system
- cAMP
cyclic Adenosine monophosphate
- cGMP
cyclic Guanosine monophosphate
- DCM
Dichloromethane
- DMF
N,N-Dimethylformamide
- EtOH
Ethanol
- EtOAc
Ethyl acetate
- HD
Huntington’s disease
- LDA
Lithium diisopropyl amide
- MeOH
Methanol
- MP-10
2-((4-(1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl)-phenoxy)-methyl)quinolone
- NBS
N-Bromosuccinimide
- PDE3A
Phosphodiesterase 3A
- PDE3B
Phosphodiesterase 3B
- PDE4A
Phosphodiesterase 4A
- PDE4B
Phosphodiesterase 4B
- PDE10A
Phosphodiesterase 10A
- PET
Positron emission tomography
- NaOCH3
Sodium methoxide
- THF
Tetrahydrofuran
- TEA
Triethylamine
- TP-10
2-[4-[Pyridin-4-yl-1-(2,2,2-trifluoro-ethyl)-1H-pyrazol-3-yl]-phenoxymethyl]-quinoline
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20239e
References
- 1.Bora RS, Gupta D, Malik R, Chachra S, Sharma P, Saini KS. Biotechnol Appl Biochem. 2008;49:129–134. doi: 10.1042/BA20070090. [DOI] [PubMed] [Google Scholar]
- 2.Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA. Gene. 1999;234:109–117. doi: 10.1016/s0378-1119(99)00171-7. [DOI] [PubMed] [Google Scholar]
- 3.Soderling SH, Bayuga SJ, Beavo JA. Proc Natl Acad Sci U S A. 1999;96:7071–7076. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K, Omori K. J Biol Chem. 1999;274:18438–18445. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- 5.Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, Strick CA, Schmidt CJ, Stephenson DT. J Histochem Cytochem. 2006;54:1205–1213. doi: 10.1369/jhc.6A6930.2006. [DOI] [PubMed] [Google Scholar]
- 6.Menniti FS, Faraci WS, Schmidt CJ. Nat Rev Drug Discovery. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 7.Menniti FS, Chappie TA, Humphrey JM, Schmidt CJ. Curr Opin Invest Drugs. 2007;8:54–59. [PubMed] [Google Scholar]
- 8.Siuciak JA. CNS Drugs. 2008;22:983–993. doi: 10.2165/0023210-200822120-00002. [DOI] [PubMed] [Google Scholar]
- 9.Hebb AL, Robertson HA, Denovan-Wright EM. Neuroscience. 2004;123:967–981. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleiman RJ, Kimmel LH, Bove SE, Lanz TA, Harms JF, Romegialli A, Miller KS, Willis A, des Etages S, Kuhn M, Schmidt CJ. J Pharmacol Exp Ther. 2011;336:64–76. doi: 10.1124/jpet.110.173294. [DOI] [PubMed] [Google Scholar]
- 12.Siuciak JA, Strick CA. Expert Opin Drug Discovery. 2007;2:1001–1009. doi: 10.1517/17460441.2.7.1001. [DOI] [PubMed] [Google Scholar]
- 13.Kehler J, Ritzen A, Greve DR. Expert Opin Ther Pat. 2007;17:147–158. [Google Scholar]
- 14.Kehler J, Kilburn JP. Expert Opin Ther Pat. 2009;19:1715–1725. doi: 10.1517/13543770903431050. [DOI] [PubMed] [Google Scholar]
- 15.Tu Z, Xu J, Jones LA, Li S, Mach RH. Nucl Med Biol. 2010;37:509–516. doi: 10.1016/j.nucmedbio.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu Z, Fan J, Li S, Jones LA, Cui J, Padakanti PK, Xu J, Zeng D, Shoghi KI, Perlmutter JS, Mach RH. Bioorg Med Chem. 2011;19:1666–1673. doi: 10.1016/j.bmc.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Lu X, Xu J, Rothfuss J, Mach RH, Tu Z. Eur J Med Chem. 2011;46:3986–3995. doi: 10.1016/j.ejmech.2011.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celen S, Koole M, De Angelis M, Sannen I, Chitneni SK, Alcazar J, Dedeurwaerdere S, Moechars D, Schmidt M, Verbruggen A, Langlois X, Van Laere K, Andres JI, Bormans G. J Nucl Med. 2010;51:1584–1591. doi: 10.2967/jnumed.110.077040. [DOI] [PubMed] [Google Scholar]
- 19.Plisson C, Salinas C, Weinzimmer D, Labaree D, Lin SF, Ding YS, Jakobsen S, Smith PW, Eiji K, Carson RE, Gunn RN, Rabiner EA. Nucl Med Biol. 2011;38:875–884. doi: 10.1016/j.nucmedbio.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Hu E, Ma J, Biorn C, Lester-Zeiner D, Cho R, Rumfelt S, Kunz RK, Nixey T, Michelsen K, Miller S, Shi J, Wong J, Hill Della Puppa G, Able J, Talreja S, Hwang DR, Hitchcock SA, Porter A, Immke D, Allen JR, Treanor J, Chen H. J Med Chem. 2012;55:4776–4787. doi: 10.1021/jm3002372. [DOI] [PubMed] [Google Scholar]
- 21.Verhoest PR, Chapin DS, Corman M, Fonseca K, Harms JF, Hou XJ, Marr ES, Menniti FS, Nelson F, O’Connor R, Pandit J, Proulx-LaFrance C, Schmidt AW, Schmidt CJ, Suiciak JA, Liras S. J Med Chem. 2009;52:5188–5196. doi: 10.1021/jm900521k. [DOI] [PubMed] [Google Scholar]
- 22.Andrés JI, de Angelis M, Alcázar J, Celen S, Bormans G. Curr Top Med Chem. 2012;12:1224–1236. doi: 10.2174/156802612800672853. [DOI] [PubMed] [Google Scholar]
- 23.Lister T, Prager RH, Tsaconas M, Wilkinson KL. Aust J Chem. 2003;56:913–916. [Google Scholar]
- 24.Andersen KE, Lundt BF, Jorgensen AS, Braestrup C. Eur J Med Chem. 1996;31:417–425. [Google Scholar]
- 25.Tu Z, Fan J, Li S, Jones LA, Cui J, Padakanti PK, Xu J, Zeng D, Shoghi KI, Perlmutter JS, Mach RH. Bioorg Med Chem. 2011;19:1666–1673. doi: 10.1016/j.bmc.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheehan JC, Bolhofer VA. J Am Chem Soc. 1950;72:2786. [Google Scholar]
- 27.Khan MN. J Org Chem. 1996;61:8063–8068. doi: 10.1021/jo961172a. [DOI] [PubMed] [Google Scholar]
- 28.Landini D, Rolla F. Synthesis. 1976:389–391. [Google Scholar]
- 29.Ing HR, Manske RHF. J Chem Soc. 1926:2348–2351. [Google Scholar]
- 30.Mager G, Klocke RK, Kux A, Hopp HW, Hilger HH. Am Heart J. 1991;121:1974–1983. doi: 10.1016/0002-8703(91)90834-5. [DOI] [PubMed] [Google Scholar]
- 31.Movsesian M, Stehlik J, Vandeput F, Bristow MR. Heart Fail Rev. 2009;14:255–263. doi: 10.1007/s10741-008-9130-x. [DOI] [PubMed] [Google Scholar]
- 32.Heaslip RJ, Evans DY. Eur J Pharmacol. 1995;286:281–290. doi: 10.1016/0014-2999(95)00457-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.