

Abstract
Bacterial genomes and large-scale computer software projects both consist of a large number of components (genes or software packages) connected via a network of mutual dependencies. Components can be easily added or removed from individual systems, and their use frequencies vary over many orders of magnitude. We study this frequency distribution in genomes of ∼500 bacterial species and in over 2 million Linux computers and find that in both cases it is described by the same scale-free power-law distribution with an additional peak near the tail of the distribution corresponding to nearly universal components. We argue that the existence of a power law distribution of frequencies of components is a general property of any modular system with a multilayered dependency network. We demonstrate that the frequency of a component is positively correlated with its dependency degree given by the total number of upstream components whose operation directly or indirectly depends on the selected component. The observed frequency/dependency degree distributions are reproduced in a simple mathematically tractable model introduced and analyzed in this study.
Keywords: gene frequency, metabolic network, software dependency
Individual components of complex interconnected systems are used with vastly different frequencies. Examples include the frequency with which individual genes and their orthologs are encoded in genomes of different species (1); the frequency of local installations of individual software packages in multicomponent software projects (2); broad power-law distributions of the frequency of citations, visitations, or other measures of popularity of individual publications, Web pages, YouTube videos, Facebook, and Twitter pages, etc. (3–5); and power-law distribution of word use frequencies in text (6).
The explanations of the observed broad distribution of use frequency (or popularity) of individual components generally fall into two broad categories. The first category invokes random multiplicative processes (7, 8) recently exemplified by the preferential attachment model of growing networks (9, 10). These models, recently invoked to explain frequency distribution of genes in pan-genomes of bacterial species (11), largely ignore functional differences between components so that the ultimate popularity of a component is determined mostly by its age as well as random events in early phases of growth of the system. The second category of models invokes heterogeneity of functional roles of individual components (12, 13). It is reasonable to assume that the frequency of a component is mainly determined by the breadth of its functional role in the system. This explanation is especially applicable to biological and technological systems subject to natural and artificial selection, respectively. Indeed, the frequency of genes whose “popularity” is not matched by their functional importance will be quickly corrected by the evolution. In agreement with this explanation, genes encoding certain core enzymes of central metabolism or ribosomal components are present in genomes of virtually all species (figure 4 in ref. 14). However, genes encoding peripheral enzymes tend to have much lower frequency of appearance in genomes (14). The same rule applies to multicomponent software projects such as Linux, where the most frequently installed components (e.g., Python and gzip) are also among the most functionally important and reusable software libraries. Most other packages either directly or indirectly depend on these low-level components for their operation. As a result, these packages end up being installed on the vast majority (if not all) of individual Linux computers. In what follows, we present empirical results supporting this second, functional explanation of the power-law distribution of frequency of components in complex biological and technological systems.
Fig. 4.

Zipf’s plots of total dependency degree in real metabolic (blue symbols in A) and Linux (red symbols in B) systems fitted with a random dependency model with D = 2 and n = 1,500 (green symbols in A) or n = 10,000 (green symbols in B), respectively. C and E show Kdep vs. the layer number in the metabolic network and the best-fitting random model, respectively. D and F do the same for Linux dependency network and its best approximation with random model. Black dots show scatter plots of individual nodes, and color lines are binned averages.
Results
Empirical Distribution of Component Frequencies.
The eggNOG database (15) provided us with information about the presence or absence of genes from 45,000 orthologous gene families in genomes of more than 500 bacterial species. The Ubuntu popularity contest project quantified the frequencies of installation of about 200,000 Linux packages on more than 2 million individual computers (2) (see Materials and Methods for details). We found the distributions of components’ frequencies fi in both biological and technological systems to share multiple common features, including a power-law scaling regime P(f) ∼ f−γ with γ ∼ 1.5 (see Fig. 1B for genomes and Fig. 1E for Linux) terminating with a peak at the maximal frequency f ∼ 1 (Fig. 1 A and D). This peak, formed by components present in the vast majority of systems, also manifests itself as a broad plateau at f ∼ 1 in Zipf’s rank-frequency plots (Fig. 1 C and F). A broad distribution of gene frequencies has been previously reported in biological literature (1, 16–19); however, this study reports and explains its scaling exponent.
Fig. 1.

The histogram P(f) of the frequency f of bacterial genes present in genomes (A) or Linux software packages installed on computers (D) in semilogarithmic coordinates. Dashed lines show a piecewise linear fit used to define core (f > 0.95), character (0.95 ≥ f > 0.1), and accessory (f ≤ 0.1) components (1, 16). When plotted in log-log coordinates (B for genes and E for Linux), the histogram is consistent with the power law P(f) ∼ f−γ with the exponents γGenomes = 1.62, and γLinux = 1.42 (solid lines in B and E). In rank-frequency Zipf’s plots (C for genes and F for Linux), core components manifest themselves as plateaus at f ∼ 1. Straight lines in C and F are the best power-law fits used to determine γGenomes,γLinux, and the arrows point to 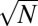 —the mathematically predicted number of core components.
—the mathematically predicted number of core components.
U-shaped P(f) distributions are sometimes plotted on semilogarithmic scale (1) with piecewise linear fit used to define three types of components dubbed “core” (f > 0.95), “character” (0.95 ≥ f > 0.1), and “accessory” (f ≤ 0.1) genes (16). In Fig. 1A we validate these previous observations and demonstrate them for Linux systems (Fig. 1D). We also confirm the existence and explain the origins of a sharp cross-over separating the core components with f ∼ 1 from the rest of the distribution. We mathematically predict the number of core components to be ∼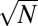 , where N is the total number of components with nonzero frequencies that are functionally connected to the core. The empirical data are in approximate agreement with this prediction. The separation between character and accessory genes is less well defined. Indeed, when plotted in log-log coordinates, the power-law scaling observed for f ≪ 1 directly crosses over into the core region at f ∼ 1 without an obvious intermediate region corresponding to character genes. In what follows, we argue that the power law is expected on purely theoretical grounds. Thus, “fractal organization of the gene Universe” (20, pp. 71–75) manifests itself both in the scale-free distribution of component frequencies (as reported in this study) as well as in qualitatively similar shapes of P(f) at different evolutionary timescales (as demonstrated in ref. 1).
, where N is the total number of components with nonzero frequencies that are functionally connected to the core. The empirical data are in approximate agreement with this prediction. The separation between character and accessory genes is less well defined. Indeed, when plotted in log-log coordinates, the power-law scaling observed for f ≪ 1 directly crosses over into the core region at f ∼ 1 without an obvious intermediate region corresponding to character genes. In what follows, we argue that the power law is expected on purely theoretical grounds. Thus, “fractal organization of the gene Universe” (20, pp. 71–75) manifests itself both in the scale-free distribution of component frequencies (as reported in this study) as well as in qualitatively similar shapes of P(f) at different evolutionary timescales (as demonstrated in ref. 1).
Component’s Frequency Is Positively Correlated with Its Dependency Degree.
It is reasonable to expect the frequency of a component (a gene or a software package) to be influenced by its importance or the breadth of its functional role in the system. For a given component, we quantify the latter by the number of other components whose operation critically depends on it either directly (referred to as the direct dependency degree kdep) or directly + indirectly (referred to as the total dependency degree Kdep). The difference between kdep and Kdep can be easily understood in the dependency network of Linux packages (21). Edges of this directed network connect a given package to packages it requests to install during its own installation process. Some of these packages have direct dependencies of their own. For example, Fig. S1 visualizes direct and indirect dependencies of the Firefox browser. This cascade of sequential installations continues until all downstream packages required for the operation of the chosen package are installed. So, though kdep(i) counts the packages that require installation of the package i at the first step of this multistep process, Kdep(i) counts the packages that do so at any step.
Though a similar interdependence of individual genes on each other certainly exists in biological systems, it is more difficult to quantify. Using the algorithm described in ref. 13, we calculated the dependency network for a subset of all gene families corresponding to metabolic enzymes (see Materials and Methods for details). Briefly, our algorithm derives upstream/downstream relations of enzymes reflecting their relative positions in metabolic pathways. The functioning of an anabolic enzyme requires the presence of enzymes in the smallest pathway necessary to synthesize all of its substrates from the minimal set of core metabolites (see Materials and Methods for our algorithm searching for such minimal pathway). The total dependency degree Kdep(i) of the enzyme i is given by the total number of enzymes in this minimal pathway located downstream from it for anabolic enzymes (or upstream from it for catabolic enzymes). However, the direct dependency degree, kdep(i), counts enzymes located one step below (or above) it in this hierarchy. The direct dependency degree of an enzyme is closely related to its degree in the adjacency matrix of the metabolic network previously studied in refs. 10 and 22. Fig. S2 visualizes dependencies among enzymes in a particular metabolic pathway.
The scatter plot of the frequency of a component vs. its total (direct + indirect) dependency degree Kdep clearly shows positive correlation between the two variables. The Spearman rank correlation 0.3 (metabolic enzymes) and 0.47 (Linux packages) is highly statistically significant (P < 10−16). A somewhat weaker correlation for metabolic enzymes can be attributed to an important difference between dependency networks in biological and computer systems. The dependencies of software packages in Linux are explicitly specified by their designers and thus totally unambiguous. The biological systems are designed in a more robust fashion and allow some flexibility in dependencies among their components. For example, in metabolic networks there is often more than one enzyme synthesizing a metabolite used by another enzyme, which makes the definition of dependency degree of an enzyme more ambiguous and weakens its correlation with its frequency. To verify this hypothesis, we constructed a dependency network of metabolites instead of metabolic enzymes and recomputed their use frequencies in metabolic networks of different organisms (Fig. S3). The correlation coefficient (0.45) was considerably better than for metabolic enzymes (0.3) and just slightly lower than that observed for Linux packages (0.47) (Fig. 2).
Fig. 2.

Components’ frequencies f (y axis) are positively correlated with their total (direct + indirect) dependency degrees Kdep (x axis) for both metabolic enzymes (A) (Spearman’s rs = 0.30) and Linux packages (B) (Spearman’s rs = 0.47). The black lines and symbols show the geometric averages of f in each logarithmic bin of Kdep.
Dependency Degrees Follow Power-Law Distributions.
The distributions of direct (kdep) and total (Kdep) dependency degrees for the metabolic enzymes as well as Linux packages are shown in Fig. 3; both have a power-law scaling region with exponents around −2 (kdep shown in Fig. 3A) and −1.5 (Kdep shown in Fig. 3B), correspondingly. In addition to the power-law region, Zipf’s rank degree plots of Kdep (Fig. 4 A and B), but not of kdep, have plateaus formed by the core components with the largest Kdep (compare with frequency plateaus Fig. 1 C and E). Direct dependency degrees in a variety of large software projects have been previously reported to have scale-free distribution with exponents around −2 (see ref. 23 for Linux as well as in-degree exponents in table 1 of ref. 24).
Fig. 3.

Probability distributions of direct (kdep; A) and total (Kdep; B) dependency degrees for metabolic enzymes (blue diamonds) and Linux packages (red circles). Power-law fits to direct degree cumulative distribution give −2.08 for metabolic enzymes and −1.91 for Linux packages, and are both consistent with the −2.0 scaling law (solid line in A). Power-law fits to direct degree cumulative distribution give −1.5 for metabolic enzymes and −1.56 for Linux packages, consistent with the mathematically derived −1.5 scaling (solid line in B).
Discussion
One of the intriguing results presented above is a remarkable similarity of distributions of frequencies (Fig. 1) as well as topological properties of dependency networks in biological (Fig. 3, red circles) and technological (Fig. 3, blue diamonds) systems. It is rather surprising to see near-perfect overlap of distributions in these two systems of very different origins: one is optimized by nature over billions of years of evolution, whereas the other is designed by a distributed population of human software engineers over the past several decades. In fact, we argue below that the functional form of P(Kdep) and P(f) observed in this study is a universal property of any multicomponent and multilayered complex system. Such systems grow by gradually acquiring new components whose operation extends the functions performed by previously acquired components. Dependency networks connecting components to each other in such systems tend to be multilayered as a direct consequence of the long history of growth and evolution (25). Metabolic and software dependency networks used in this study with 34 and >40 layers, respectively, are indeed multilayered. A slightly different version of the universal metabolic network has been estimated (25) to have up to 60 layers of enzymes gradually acquired over billions of years of biological evolution (see figures 6 and 7 in ref. 25).
One mathematically tractable example of a multilayered dependency network is provided by a critical random branching tree (26)—namely, a tree with the branching ratio b close to 1. Here the branching ratio b ≤ 〈kdep〉 counts nodes that directly depend on a given node and are located one layer above it. Indeed, in a branching tree with b significantly larger or smaller than 1, either the number of layers is logarithmically small (b ≫ 1) or the branches terminate prematurely (b ≪ 1), rendering a multilayered network impossible.
For a critical branching tree, one can show that 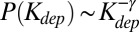 with γ = 1.5. Indeed, the part of the tree located upstream of a given node itself constitutes an instance of a critical branching process that is independent from the rest of the tree; therefore, its size is distributed with the Galton–Watson exponent γ = 1.5 (see ref. 26 for the mathematical derivation). Because no subtree can be larger than the parent tree, in a tree of size N, one expects to find
with γ = 1.5. Indeed, the part of the tree located upstream of a given node itself constitutes an instance of a critical branching process that is independent from the rest of the tree; therefore, its size is distributed with the Galton–Watson exponent γ = 1.5 (see ref. 26 for the mathematical derivation). Because no subtree can be larger than the parent tree, in a tree of size N, one expects to find 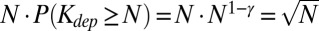 subtrees with sizes about N. Therefore, about
subtrees with sizes about N. Therefore, about 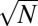 nodes located at the lowest layers of the dependency network will have the largest possible dependency degree Kdep ∼ N (see Materials and Methods for more details). These nodes constitute the plateau in Zipf’s plots (Figs. 1 C and E and 4 A and B) and the large-X peak in the U-shaped distribution of dependency degrees or frequencies of system’s components (Fig. 1 A and D).
nodes located at the lowest layers of the dependency network will have the largest possible dependency degree Kdep ∼ N (see Materials and Methods for more details). These nodes constitute the plateau in Zipf’s plots (Figs. 1 C and E and 4 A and B) and the large-X peak in the U-shaped distribution of dependency degrees or frequencies of system’s components (Fig. 1 A and D).
Though the distribution of dependency degrees in a critical branching tree is in excellent agreement with the empirically observed data, there is a conceptual difference between real-life dependency networks and trees. In a tree, each component directly depends on one, and only one, downstream component. However, in real-life networks this number, D, is certainly larger than one; it varies from component to component, but averages ∼2 for both metabolic networks and Linux packages. To describe real-life dependency networks with D > 1, we introduced and studied the following simple model. In our model, dependency networks start to grow from a few seed components. At each evolutionary time-step, one adds a new component depending on Di randomly selected existing components. For simplicity we assume Di to have a Poisson distribution with average 〈Di〉 = D. However, as shown in SI Materials and Methods, our results depend only on the average value of Di. We mathematically derive (see SI Materials and Methods for step-by-step calculations) that the total dependency degree Kdep in a dependency network of size N generated by this model has a power-law tail 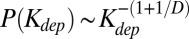 as well as a plateau in the Zipf’s plot composed of N(D−1)/D nearly universal components. Simulations of the model with D = 2 and n = 1,500 (green line in Fig. 4A) provides a reasonable fit to the metabolic dependency degree distribution (blue diamonds in Fig. 4A), whereas D = 2 and n = 10,000 (green line in Fig. 4B) is an excellent fit to the Linux dependency degree distribution (red circles in Fig. 4B).
as well as a plateau in the Zipf’s plot composed of N(D−1)/D nearly universal components. Simulations of the model with D = 2 and n = 1,500 (green line in Fig. 4A) provides a reasonable fit to the metabolic dependency degree distribution (blue diamonds in Fig. 4A), whereas D = 2 and n = 10,000 (green line in Fig. 4B) is an excellent fit to the Linux dependency degree distribution (red circles in Fig. 4B).
We see that the model with D ∼ 2 provides a rather good fit to dependency networks in both biological and technological systems. Metabolic enzymes usually have two substrates, and rarely one or three and more substrates. Hence in this case there is a good biophysical explanation for the observed value of Dmet = 1.7 ∼ 2. The situation is more complicated for the Linux dependency network where there are no geometrical limitations on the number of direct dependencies of a software package. This network is characterized by a large number of direct links between packages already indirectly connected on each other. Such shortcuts (known as feed-forward loops in the network jargon) do not change the overall (direct + indirect) network of package dependencies. Because our model does not contain feed-forward loops beyond those created by pure chance, we pruned them from the Linux direct dependency network as well. After removing all direct links short-circuiting any chain of direct links in the Linux dependency network, we were left with a direct dependency network with DLinux = 2.4 ∼ 2 and the same set of direct + indirect package dependency links as the original network. Admittedly in the case of Linux packages we have no ready explanation for this particular value of D beyond a vague notion that the easiest way to make a new package is to combine the outputs of two already existing ones.
The similarity between real-life dependency networks and those generated by our model with D = 2 extends beyond the shape of the total dependency degree distribution with the exponent γ = 1 + 1/D = 1.5 and 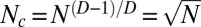 of the best-connected components forming the plateau in Zipf’s plots. Our model makes very specific predictions about how the dependency degree of a component depends on the time when it was first added to the dependency network. Unfortunately, obtaining system-wide information about these “creation” times is not easy for Linux, and downright impossible for metabolic enzymes. As advocated in ref. 25, the time of appearance of a metabolic enzyme in the metabolic pan-network can be estimated from its layer number obtained by the scope expansion algorithm. Using the layer number of a node in a real-life dependency network as a proxy of its acquisition/creation time, we investigated its correlations with its total dependency degree. It stands to reason that older nodes located at bottom layers will tend to have a systematically larger dependency degree both in model and real networks; this is indeed what was observed and shown Fig. 4 C–E.
of the best-connected components forming the plateau in Zipf’s plots. Our model makes very specific predictions about how the dependency degree of a component depends on the time when it was first added to the dependency network. Unfortunately, obtaining system-wide information about these “creation” times is not easy for Linux, and downright impossible for metabolic enzymes. As advocated in ref. 25, the time of appearance of a metabolic enzyme in the metabolic pan-network can be estimated from its layer number obtained by the scope expansion algorithm. Using the layer number of a node in a real-life dependency network as a proxy of its acquisition/creation time, we investigated its correlations with its total dependency degree. It stands to reason that older nodes located at bottom layers will tend to have a systematically larger dependency degree both in model and real networks; this is indeed what was observed and shown Fig. 4 C–E.
An important caveat in applying the 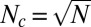 relationship is that N counts only those components that are directly or indirectly connected to the core by the functional dependency network; for biological systems, this allows us to reconcile the apparent paradox. Indeed, the pan-genome of all bacterial species is believed to be open (16). That is to say, N continues to increase without any hint at saturation as we sequence genomes of new bacterial species or even new strains of the same species (figure 1 in ref. 27). At the same time, the core bacterial genome remains relatively stable. Different methods result in somewhat different estimates of Nc, ranging from 250 in ref. 16 to 400 in refs. 17–19. To reconcile the apparent stability of Nc with unlimited growth of N, one recalls that continuing expansion of N is caused by either nonfunctional (prophages or transposable elements) or extremely niche-specific gene families—both are likely to be disconnected from the core and hence will not contribute to growth of Nc. Assuming Nc ≤ 500, one gets the upper bound on the number of gene families connected to the core at ∼250,000.
relationship is that N counts only those components that are directly or indirectly connected to the core by the functional dependency network; for biological systems, this allows us to reconcile the apparent paradox. Indeed, the pan-genome of all bacterial species is believed to be open (16). That is to say, N continues to increase without any hint at saturation as we sequence genomes of new bacterial species or even new strains of the same species (figure 1 in ref. 27). At the same time, the core bacterial genome remains relatively stable. Different methods result in somewhat different estimates of Nc, ranging from 250 in ref. 16 to 400 in refs. 17–19. To reconcile the apparent stability of Nc with unlimited growth of N, one recalls that continuing expansion of N is caused by either nonfunctional (prophages or transposable elements) or extremely niche-specific gene families—both are likely to be disconnected from the core and hence will not contribute to growth of Nc. Assuming Nc ≤ 500, one gets the upper bound on the number of gene families connected to the core at ∼250,000.
The frequency of a given component is expected to be strongly correlated with its total dependency degree. Indeed, the system using any of Kdep components located upstream of a given component is guaranteed to include this component itself. Hence, if every software package (metabolic enzyme) was equally likely to be initially selected (with probability 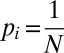 ) for local installation on a computer (incorporation into a bacterial genome), one would have
) for local installation on a computer (incorporation into a bacterial genome), one would have 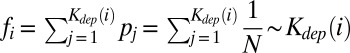 . Deviations from this idealized linear relationship between fi and kdep(i) in real data reflect among other things a nonuniform frequency of initial selection or installation of upstream components. Indeed, idiosyncratic differences in popularity pi of higher-level components will be translated into differences in installation frequencies of lower-level components required for their operation. By adjusting the values of pi—the initial popularity of components—we were able to increase the correlation coefficient between fi and
. Deviations from this idealized linear relationship between fi and kdep(i) in real data reflect among other things a nonuniform frequency of initial selection or installation of upstream components. Indeed, idiosyncratic differences in popularity pi of higher-level components will be translated into differences in installation frequencies of lower-level components required for their operation. By adjusting the values of pi—the initial popularity of components—we were able to increase the correlation coefficient between fi and 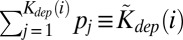 to 0.8 up from ∼0.5.
to 0.8 up from ∼0.5.
Comparison between biological and technological networks has been previously performed by Yan et al. (28), and a number of similarities as well as significant differences were reported. However, the biological and technological systems studied were rather different from the ones we used in this study. The focus of the analysis performed in ref. 28 was on regulation and control represented by transcriptional regulatory network in Escherichia coli and the call graph between subroutines within the Linux kernel. However, in this article we compare biological and technological systems with independently installable components represented by metabolic enzymes encoded in bacterial genomes and software packages installed on top of the Linux kernel. A more systematic analysis of similarities and differences between different versions of biological and technological complex systems will have to await future studies.
Materials and Methods
Our methods are briefly summarized here, and a more detailed description is provided in SI Materials and Methods.
Empirical Data for Frequencies of Use of Bacterial Genes.
The eggNOG database v3.0 (15) contains the mapping of orthologous gene families to 630 species with fully sequenced genomes. We included in our analysis 529 bacterial genomes and their gene families assigned based on the clusters of orthologous genes and universal nonsupervised orthologous groups, which together cover 44,283 prokaryotic orthologous gene families. The resulting table showing the presence or absence of individual gene families in genomes was then processed to obtain the gene frequency f, defined as the fraction of 529 genomes the family is represented by at least one gene.
Empirical Data for Frequencies and Mutual Dependencies of Linux Packages.
The package dependency network of Linux distribution Ubuntu 11.04 Natty was obtained by first getting a complete list of packages from http://packages.ubuntu.com/, and then running the command apt-rdepends to find all of the direct and indirect requirements for each package. The resulting network contains 33,473 packages, 157,667 direct, and 2,439,011 total (direct + indirect) dependency relations. The installation frequency data for 192,392 packages on 2,047,796 computers were downloaded from the package popularity contest project (http://popcon.ubuntu.com). In our analysis we used statistics for the whole archive sorted by the field institution.
Construction of the Dependency Matrices for the Metabolic Network.
We used the union of all reactions in the Kyoto Encyclopedia of Genes and Genomes database (29) to construct upstream/downstream relations between enzymes using the following algorithm related to the scope expansion algorithm of ref. 25. For every enzyme, the minimal metabolic pathway connecting the product(s) of this enzyme to the set of five core metabolites was constructed as described in ref. 13. The direct dependency links were then drawn between the selected enzyme and enzymes in the top layer of this pathway, and direct + indirect links connect it to all enzymes in the minimal pathway. The resulting dependency network contains 1,832 reactions/enzymes connected to each other by 3,118 direct and 49,168 direct + indirect dependencies.
Power-Law Fits to the Data.
Power-law fits to distributions were performed using MatLab package plfit.m developed by Aaron Clauset and collaborators, and downloaded from http://tuvalu.santafe.edu/~aaronc/powerlaws.
Supplementary Material
Acknowledgments
We thank P. Dixit for useful discussions, critical reading, and editing of the manuscript; and K. Dill and J. Peterson for useful comments and suggestions. This work was supported by US Department of Energy Office of Biological Research Grant PM-031.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217795110/-/DCSupplemental.
References
- 1.Koonin EV, Wolf YI. Genomics of bacteria and archaea: The emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36(21):6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pennarun A, Allombert B, Reinholdtsen P. Ubuntu Popularity Contest. Available at http://popcon.ubuntu.com/. Accessed September 5, 2011.
- 3.Sala A, Zheng H, Zhao BY, Gaito S, Rossi GP. Proceedings of the 29th ACM SIGACT-SIGOPS Symposium on Principles of Distributed Computing. New York: Assoc for Computing Machinery; 2010. pp. 400–401. [Google Scholar]
- 4.Price DJ. Networks of scientific papers. Science. 1965;149(3683):510–515. doi: 10.1126/science.149.3683.510. [DOI] [PubMed] [Google Scholar]
- 5.Java A, Song X, Finin T, Tseng B. Proceedings of the Ninth WebKDD and First SNA-KDD 2007 Workshop on Web Mining and Social Network Analysis. New York: Assoc for Computing Machinery; 2007. pp. 56–65. [Google Scholar]
- 6.Zipf GK. Selected Studies of the Principle of Relative Frequency in Language. 1st Ed. Cambridge, MA: Harvard Univ Press; 1932. [Google Scholar]
- 7.Yule U. A mathematical theory of evolution, based on the conclusions of Dr. J. C. Willis, F.R.S. Phil Trans R Soc B. 1925;213:21–87. [Google Scholar]
- 8.Simon HA. On a class of skew distribution functions. Biometrika. 1955;42(3-4):425–440. [Google Scholar]
- 9.Albert R, Barabási A-L. Statistical mechanics of complex networks. Rev Mod Phys. 2002;74(1):47–97. [Google Scholar]
- 10.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási A-L. The large-scale organization of metabolic networks. Nature. 2000;407(6804):651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 11.Haegeman B, Weitz JS. A neutral theory of genome evolution and the frequency distribution of genes. BMC Genomics. 2012;13:196. doi: 10.1186/1471-2164-13-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maslov S, Krishna S, Pang TY, Sneppen K. Toolbox model of evolution of prokaryotic metabolic networks and their regulation. Proc Natl Acad Sci USA. 2009;106(24):9743–9748. doi: 10.1073/pnas.0903206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pang TY, Maslov S. A toolbox model of evolution of metabolic pathways on networks of arbitrary topology. PLOS Comput Biol. 2011;7(5):e1001137. doi: 10.1371/journal.pcbi.1001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada T, Kanehisa M, Goto S. Extraction of phylogenetic network modules from the metabolic network. BMC Bioinformatics. 2006;7:130. doi: 10.1186/1471-2105-7-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell S, et al. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40(Database issue):D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lapierre P, Gogarten JP. Estimating the size of the bacterial pan-genome. Trends Genet. 2009;25(3):107–110. doi: 10.1016/j.tig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Danchin A, Fang G, Noria S. The extant core bacterial proteome is an archive of the origin of life. Proteomics. 2007;7(6):875–889. doi: 10.1002/pmic.200600442. [DOI] [PubMed] [Google Scholar]
- 18.Fang G, Rocha EP, Danchin A. Persistence drives gene clustering in bacterial genomes. BMC Genomics. 2008;9:4. doi: 10.1186/1471-2164-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danchin A. Bacteria as computers making computers. FEMS Microbiol Rev. 2009;33(1):3–26. doi: 10.1111/j.1574-6976.2008.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koonin EV. The Logic of Chance: The Nature and Origin of Biological Evolution. Upper Saddle River, NJ: FT Press; 2011. [Google Scholar]
- 21.LaBelle N, Wallingford E. 2004. Inter-package dependency networks in open-source software. arXiv:cs/0411096.
- 22.Barabási A-L, Oltvai ZN. Network biology: Understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 23.Maillart T, Sornette D, Spaeth S, von Krogh G. Empirical tests of Zipf’s law mechanism in open source Linux distribution. Phys Rev Lett. 2008;101(21):218701. doi: 10.1103/PhysRevLett.101.218701. [DOI] [PubMed] [Google Scholar]
- 24.Louridas P, Spinellis D, Vlachos V. 2008. Power laws in software. ACM Trans Softw Eng Methodol 18:2:1–2:26.
- 25.Handorf T, Ebenhöh O, Heinrich R. Expanding metabolic networks: Scopes of compounds, robustness, and evolution. J Mol Evol. 2005;61(4):498–512. doi: 10.1007/s00239-005-0027-1. [DOI] [PubMed] [Google Scholar]
- 26.Athreya KB, Ney PE. Branching Processes. New York: Dover; 2004. [Google Scholar]
- 27.Touchon M, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5(1):e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan K-K, Fang G, Bhardwaj N, Alexander RP, Gerstein M. Comparing genomes to computer operating systems in terms of the topology and evolution of their regulatory control networks. Proc Natl Acad Sci USA. 2010;107(20):9186–9191. doi: 10.1073/pnas.0914771107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.