

Abstract
Diffuse large B-cell lymphoma (DLBCL) with an activated B-cell (ABC) gene-expression profile has been shown to have a poorer prognosis compared with tumors with a germinal center B-cell type. ABC cell lines have constitutive activation of STAT3; however, the mechanisms regulating STAT3 signaling in lymphoma are unknown. In studies of class-I histone deacetylase (HDAC) expression, we found overexpression of HDAC3 in phospho STAT3-positive DLBCL and the HDAC3 was found to be complexed with STAT3. Inhibition of HDAC activity by panobinostat (LBH589) increased p300-mediated STAT3Lys685 acetylation with increased nuclear export of STAT3 to the cytoplasm. HDAC inhibition abolished STAT3Tyr705 phosphorylation with minimal effect on STAT3Ser727 and JAK2 tyrosine activity. pSTAT3Tyr705-positive DLBCLs were more sensitive to HDAC inhibition with LBH589 compared with pSTAT3Tyr705-negative DLBCLs. This cytotoxicity was associated with downregulation of the direct STAT3 target Mcl-1. HDAC3 knockdown upregulated STAT3Lys685 acetylation but prevented STAT3Tyr705 phosphorylation and inhibited survival of pSTAT3-positive DLBCL cells. These studies provide the rationale for targeting STAT3-positive DLBCL tumors with HDAC inhibitors.
Keywords: lymphoma, HDAC3, LBH589, STAT3, Mcl-1
INTRODUCTION
Gene expression profiling studies have distinguished three molecular subtypes of diffuse large B-cell lymphoma (DLBCL) known as ‘germinal center B-cell-like’ (GCB), ‘activated B-cell-like’ (ABC)1 and primary mediastinal B-cell type.2,3 GCB DLBCLs arise from normal germinal center B cells, whereas ABC DLBCLs originate from post-germinal center B cells that are arrested during plasmacytic differentiation.2 In general, the GCB group responds better to conventional chemotherapy, whereas patients with ABC type tend to be more refractory.4 Compared with the current understanding of GCB-type DLBCL, the biology and pathogenic mechanisms of ABC-type DLBCL are less understood. The signal transducers and activators of transcription (STATs) belong to a family of cytoplasmic transcription factors. Phosphorylated STATs form homo- or heterodimers that then translocate to the nucleus, where they bind DNA and regulate gene transcription.5,6 Phosphorylation and acetylation are crucial posttranslational modifications that regulate the activities of various proteins.7 With respect to lymphoma, NF-kB along with STAT3 is activated and highly expressed in ABC DLBCL cell lines.8,9 The mechanism of NF-kB regulation has been extensively studied in DLBCL, whereas the mechanisms that regulate STAT3 in these tumors are unknown. Site-specific acetylation is an important regulatory modification that influences protein–protein interaction. In solid tumor cancer cells, Yuan et al.,10 demonstrated that STAT3 dimerization is regulated by reversible acetylation of lysine at 685 in the SH2 domain of STAT3. Two classes of enzymes, histone acetyltransferases and histone deacetylases (HDACs), reversibly regulate the extent of protein acetylation. In humans, HDACs are divided into three classes: class-I (HDAC1, HDAC2, HDAC3 and HDAC8); the class-II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10); and the class-III SIR2-like proteins.11 Class-I HDACs are widely expressed, whereas the expression of many class-II HDACs are tissue-specific. HDAC1, 2 and 8 are predominantly nuclear proteins, whereas HDACs 3, 4, 5, 7 and 9 shuttle between the nucleus and cytoplasm.12 Different studies have recently demonstrated that histones are not the only proteins under the control of HDACs.13 For example, HDACs can act on non-histone substrates, such as tubulin and the transcription factors p53 and NF-kB.14,15 Increasing evidence suggests that the regulation of gene expression by STATs requires HDAC activity. The HDAC inhibitor tricostatin A inhibits STAT3-dependent transcriptional activity induced by platelet-derived growth factor in NIH 3T3 cells and blocks STAT1 activation in interferon gamma-stimulated carcinoma cells.16 In this present study, we investigated the molecular mechanism of STAT3 regulation in DLBCL and demonstrate that targeting pSTAT3-positive DLBCL with HDAC inhibitors is a potential therapeutic option for these patients.
MATERIALS AND METHODS
Reagents
LBH589 (Panobinostat) was a gift from Novartis Pharmaceuticals (Basel, Switzerland). Annexin V fluorescein isothiocyanate was obtained from BD Biosciences (San Diego, CA, USA). Phospho specific antibodies for STAT3 (Tyrosine 705), STAT3 (Serine 727), STAT1 (Tyrosine 701), STAT5 (Tyrosine 694) and JAK2 (Tyrosine 1007/1008) were obtained from Cell Signaling Technology (Beverly, MA, USA). Phospho-tyrosine antibody was from Millipore (Billerica, MA, USA). Antibodies for acetylated STAT3 (Lysine 685), STAT3, STAT1, STAT5, JAK2, JAK1, HDAC1, HDAC3, HDAC2, acetylated H3, acetylated H4, Bcl-2, Bcl-XL, Mcl-1, Bim, Bak, Bax, cMyc, cyclin D1 and p21 were all purchased from Cell Signaling Technology. Normal rabbit IgG control antibody was also from Cell Signaling Technology. Normal mouse IgG control antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). SHP1 and SHP2 antibodies were purchased from Cell Signaling Technology. Recombinant human IL-10 was purchased from R&D Systems (Minneapolis, MN, USA).
Lymphoma samples
Tumor samples of DLBCL were obtained through the University of Iowa/Mayo Lymphoma SPORE Biospecimens Core and the Predolin Foundation Biobank. Patients signed informed consent to provide excess tissue for research on a protocol approved by the Mayo Clinic Institutional Review Board. A total of 12 primary patient samples of DLBCL were used for this study; in 6 of these patients cryopreserved tumor cell suspensions were available. All 12 DLBCL cases were sub-grouped into GCB or ABC subtype based on the Hans immunohistochemistry algorithm applied to paraffin-embedded tumor samples.17 CD19 + B-cells from the peripheral blood from normal healthy blood donors were used as normal B-cell controls. The SUDHL-6 (DHL6), OCI-Ly7 (Ly7), OCI-Ly19 (Ly19), OCI-Ly1 (Ly1), OCI-Ly3 (Ly3), SUDHL-2 (DHL2), U2932 and HBL1 DLBCL cell lines were a kind gift from Dr Louis Staudt. All cell lines were cultured in Iscove’s modified Dulbecco’s medium supplemented with 20% human serum. DLBCL lines were serum starved with 0.5% bovine serum albumin overnight and then treated with rIL-10.
Immunohistochemistry and Immunoflorescence
Sections (4-μm thick) were cut from formalin-fixed paraffin-embedded tissue blocks and stained as previously described with antibodies to HDAC3, HDAC1 and pSTAT3.18 For immunoflorescence microscopy, cytospins were prepared from LBH589-treated and -untreated cells, dried and fixed with 10% neutral-buffered formalin. Cells were permeabilized with 0.25% Triton X and antibody to STAT3 was added to respective slides. Biotinylated anti-rabbit secondary antibody was added followed by streptavidin-conjugated Alexa fluorchrome (SA-AF594). Slides were mounted with mounting media containing DAPI.
Cell-survival and -proliferation assay
Cells (1 × 106/well) were cultured in the presence of various concentrations of LBH for 48 h or otherwise indicated and flow-cytometry assay was performed as previously described.18 Cell proliferation was determined by the tritiated-thymidine method.18
Western-blotting and co-immunoprecipitation assay
5 × 106 cells were incubated with various concentrations of LBH at 37 °C for various lengths of time, as indicated.18 To the lysates, 2–5 μg of specific antibodies were added and complexes were allowed to form by incubating with rotation at 4 °C. A 50% slurry (25 μl) of protein A-Sepharose or protein G-Sepharose was then added and the incubation continued for 2 h. Immunoprecipitates captured with Sepharose beads were washed four times with RIPA buffer and analyzed by immunoblotting.
Transfection
For small interfering RNA (siRNA) experiments, cells were transfected with human B cell Nucleofector kit (Amaxa Biosystems Lonza, Gaithersrburg, MD, USA). Briefly, 10 × 106 cells were transiently transfected with 250 or 500 nM of HDAC3 siRNA (Santa Cruz Biotechnology) using the U-15 program. For plasmid transfection, 10 × 106 cells were transfected with HDAC3 or HDAC1 plasmid by electroporation. HDAC3 and HDAC1 plasmids were provided by Dr Hungwen Chen (National Taiwan University).
Nuclear and cytoplasmic analysis
Nuclear and cytoplasmic extracts were separated with NE-PER nuclear and cytoplasmic extraction kit from Thermo Scientific (Kalamazoo, MI, USA) as instructed.
RESULTS
Co-expression of class-I HDAC and pSTAT3Tyr705 in subtypes of DLBCL tumors
Increasing evidence suggests that the regulation of gene expression by STATs requires class-I HDAC activity.19 DLBCL human samples typed as ABC and GCB by immunohistochemistry were stained for tyrosine pSTAT3 and HDAC class-I (HDAC1 and HDAC3) expression. Both ABC and GCB tumors were HDAC1 positive (Figure 1a). All five ABC tumors were HDAC3 positive compared with only one of five GCB tumors (Figure 1a). Furthermore, all ABC tumors showed very strong nuclear positivity for phosphorylated STAT3 at tyrosine 705 residue; GCB cells were negative or only weakly positive (Figure 1a). On immunoblotting, all three ABC DLBCLs tested expressed HDAC3 protein (Figure 1b), whereas only 1/3 GCB samples expressed HDAC3. HDAC1 expression was observed in four of six cases (2/3 ABC and 2/3 GCB). All cases expressed total STAT3, with higher amounts in ABC-type DLBCL. pSTAT3Tyr705 was only found in ABC-type DLBCL patient samples (3/3) (Figure 1b). The above studies were repeated in DLBCL cell lines and produced similar results. The ABC-type DLBCL cell lines Ly3 and Ly10 showed higher HDAC3 expression than the GCB lines Ly7 and DHL-6 (Figure 1c). Both types expressed HDAC1 with slightly higher levels in ABC; both ABC and GCB cells expressed HDAC2 to a comparable level (Figure 1c). The ABC lines showed positivity for pSTAT3Tyr705 and expressed higher total STAT3 compared with GCB cells. Normal peripheral blood CD19 + B cells expressed HDACs 1 and 2, but not HDAC3 (Figure 1d). These results suggest that HDAC3 is aberrantly co-expressed with pSTAT3Tyr705 in ABC DLBCL cells but not in normal B-cells or GCB DLBCL.
Figure 1.
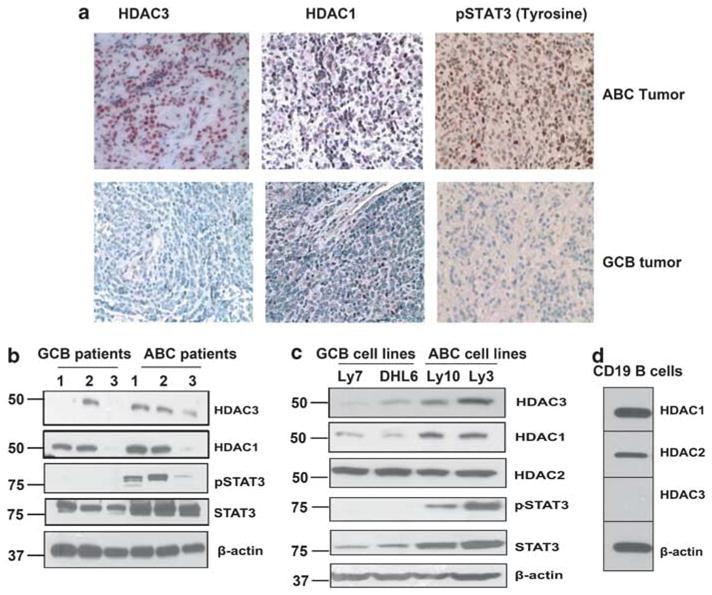
Expression of HDACs and pSTAT3 in ABC and GCB diffuse large B-cell lymphoma: (a) Expression of HDAC1, HDAC3 and pSTAT3Tyr705 were assessed in ABC and GCB patient samples by immunohistochemistry. A representative example (ABC = 7; GCB = 5) is shown (400 ×). (b, c) Expression of HDAC1, 2, 3, pSTAT3 Tyr705 and STAT3 were assessed at protein level by immunoblotting in ABC and GCB lymphoma cells from patients (b); in ABC and GCB human cell lines (c). (d) Expression of HDAC1, 2 and 3 in CD19 + B (n = 3) cells by immunoblotting.
HDAC3 accumulates in the nucleus and forms complex with STAT3 Nuclear and cytoplasmic analysis of pSTAT3Tyr705-positive Ly3 cell line showed that HDAC3 predominantly resides in the nucleus (Figure 2a). STAT3 was found in both cytoplasm and nucleus; whereas activated STAT3 Tyr705 was restricted to the nucleus.
Figure 2.
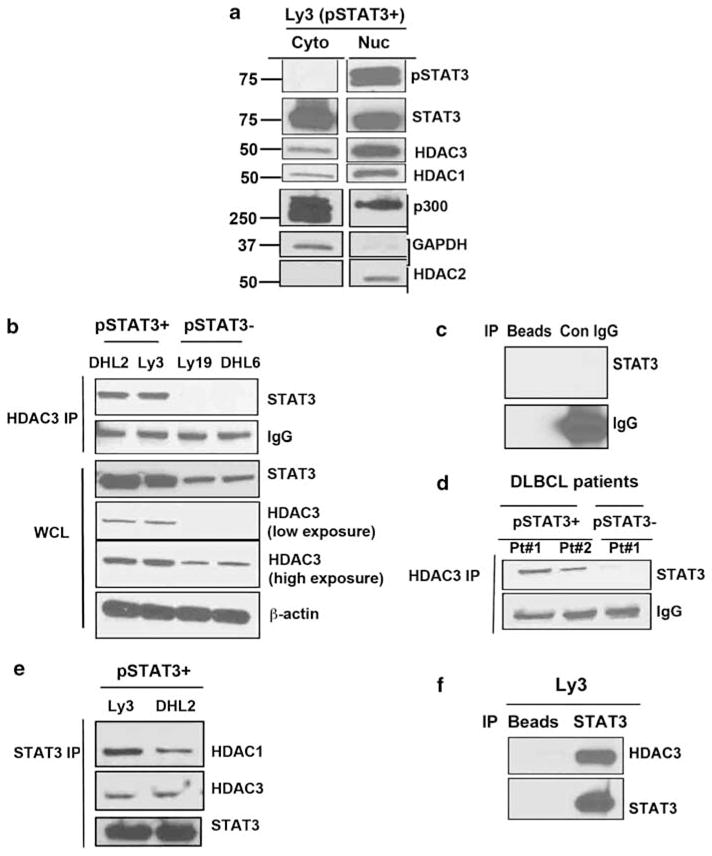
Interaction between STAT3 and HDAC3 in subtypes of DLBCLs: (a) Nuclear and cytoplasmic distribution of HDAC3/HDAC1 and STAT3 (both phosphorylated and unphosphorylated) in Ly3 cells. (b) HDAC3 was immunoprecipitated from lysates of two ABC and two GCB cell lines and the immuno-complex was examined for STAT3. (c) The immunoprecipitation control with normal rabbit IgG and beads. (d) HDAC3 was immunoprecipitated from two STAT3 + and one STAT3-DLBCL patient cells and immuno-complex were blotted for STAT3. (e) STAT3 was pulled down from two ABC cell lines (Ly3 and DHL2) and blotted with HDAC1, HDAC3 and STAT3 antibodies. (f) The immunoprecipitation control with beads alone.
We investigated whether STAT3 and HDAC3 could physically interact. First, we pulled down endogenous HDAC3 from whole-cell lysates from two pSTAT3Tyr705-positive (Ly3, DHL2) and two pSTAT3Tyr705-negative (Ly19, DHL6) DLBCL lines and blotted with a specific STAT3 antibody. Precipitation of HDAC3 revealed a strong association with STAT3 in both DHL2 and Ly3 cells (Figure 2b). We did not observe any association of HDAC3 with STAT3 in either Ly19 or DHL6 (pSTAT3-negative) cell lines (Figure 2b). Whole-cell lysates (bottom panel Figure 2b) showed that both Ly19 and DHL6 express low amounts of STAT3 and HDAC3. These complexes were not detectable in the immunocomplexes obtained using a control IgG antibody or beads alone (Figure 2c). We tested whether this association also is found in pSTAT3Tyr705-positive DLBCL patient samples. Indeed, in both pSTAT3-positive DLBCL tumors HDAC3 and STAT3 were complexed (Figure 2d).
Furthermore, STAT3 was found to be associated with HDAC3 suggesting a reciprocal relationship between these two proteins (Figure 2e). These complexes were not detected in precipitates using beads alone (Figure 2f).
HDAC inhibition with LBH acetylates STAT3 at Lys685
As acetylation of STAT3 alters the distribution rather than the functional status of STAT3,20 we next examined the effect of HDAC inhibition with LBH on endogenous STAT3 acetylation and localization of STAT3 in pSTAT3Tyr705-positive DLBCL cells. STAT3 was immunoprecipitated from Ly3 whole-cell extracts and blotted for acetylated STAT3 with acetyl lysine antibody. The basal acetylation level of endogenous STAT3 was low but increased after LBH treatment in a dose-dependent manner in early (Figure 3a) and late time points (Figure 3b). Immunoprecipitation with control IgG failed to demonstrate these complexes (Figure 3c). Even low doses of LBH (10 nM) were able to increase STAT3 acetylation. We have also seen some increase in STAT3 acetylation in response to LBH treatment in whole cell lysates (Figure 3a–lower panel). Similar results were obtained when total STAT3 was pulled down from Ly3 and DHL2 cells and probed with acetyl lysine antibody (Figure 3d). CREB-binding protein and its homologue p300 is a transcriptional co-activator that regulates STAT3 activity.21 Wang et al.22 have shown that STAT3 is acetylated by p300 both in vitro and in vivo. To demonstrate that p300 is complexed with STAT3 in lymphoma cells, we pulled down endogenous p300 with a p300-specific antibody and probed for STAT3. p300 and STAT3 were shown to physically interact and form complexes in both pSTAT3 + DLBCL cell lines (Figure 3e) compared with IgG control (Figure 3f). Further experiments were performed to learn if STAT3 in DLBCL cells could be acetylated by p300 and if the level of acetylation at lysine 685 could be increased with the HDAC inhibitor LBH. As indicated in Figure 3g, a low level of acetylated STAT3 was found in the p300 immunoprecipitate from untreated cells that increased with LBH treatment in both Ly3 and DHL2 cells.
Figure 3.
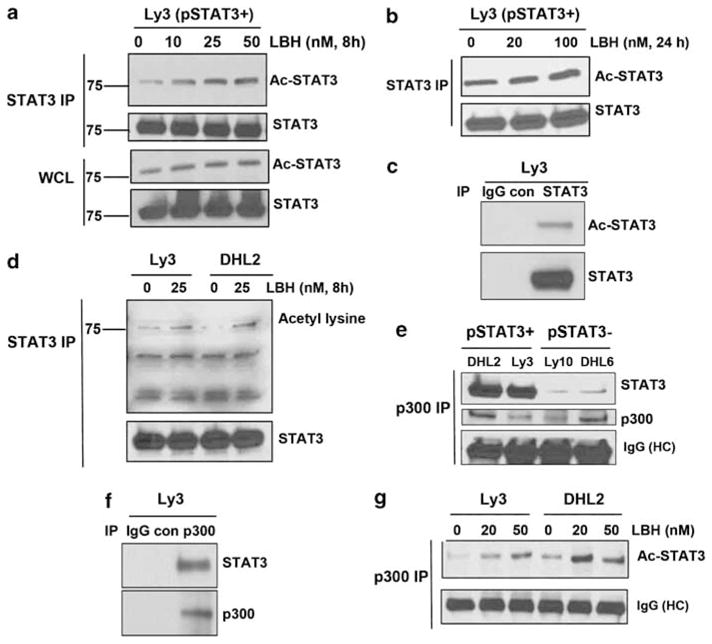
Effect of HDAC inhibition on STAT3 acetylation: (a, b) Ly3 cells were treated with various concentrations of LBH for 8 h (a) and 24 h (b) and then lysed. Lysates were immunoprecipitated with STAT3 antibody followed by western blot using STAT3Lys685 antibody. (c) Mouse IgG was used as control antibody for STAT3 immunoprecipitation. (d) Ly3 and DHL2 cells were treated with LBH for 8 h, lysed and immunoprecipitated with STAT3 antibody. Blot was then probed with acetyl lysine antibody. (e) Lysate from two ABC and two GCB lines were pulled down with p300 antibody and blotted with STAT3 and p300 antibodies. (f) Rabbit IgG was used as control for p300 immunoprecipitation. (g) Ly3 and DHL2 cells were treated with LBH lysed and then immunoprecipitated with p300 and examined for acetylated STAT3.
HDAC activity is required for cytokine-stimulated STAT3 nuclear translocation
We next examined by immunoblot and immunofluorescence microscopy whether HDAC inhibition altered the nuclear versus cytoplasmic localization of STAT3. Immunoblot analysis showed that in serum-starved cells STAT3 was distributed in both the cytosol and nucleus (Figure 4). A time-course analysis of IL-10 (a known inducer of the STAT3 pathway) in serum-starved Ly3 cells resulted in an increase in both the nuclear and cytoplasmic content of pSTAT3 (Supplementary Figure S1). However, IL-10 was able to increase Ac STAT3 predominantly in cytoplasm. Acetylated STAT3 was upregulated as early as 15 min with IL-10 treatment and the level was sustained up to 2 h (Supplementary Figure S1). However, phoshorylated-STAT3 levels were sustained up to 30 min and tended to decline over time (Supplementary Figure S1).
Figure 4.
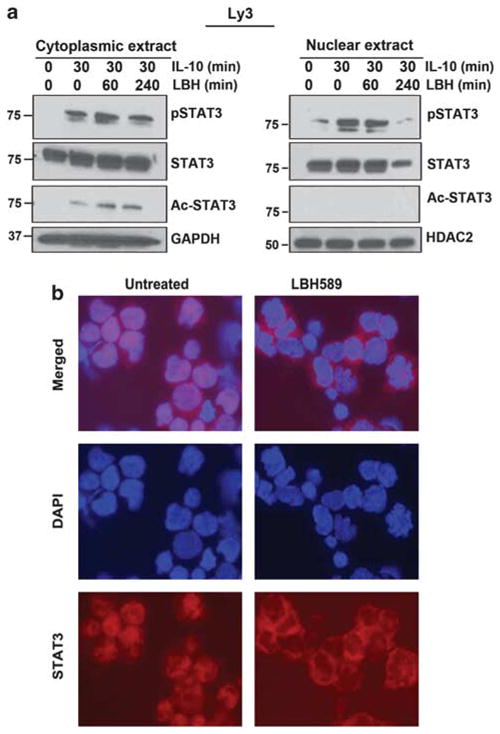
Effects of HDAC inhibition on STAT3 nuclear and cytoplasmic localization: (a) Serum-starved Ly3 cells were pretreated with LBH for 60 and 240 min and then treated with IL-10 (100 ng/ml) for 30 min. Nuclear and cytoplasmic extracts were separated and blotted for antibodies as indicated. (b) Localization of STAT3 was examined by immunoflorescence in LBH-treated Ly3 cells as described in materials and methods section.
Pretreatment with LBH for 240 min blocked IL-10-induced (30-min treatment) STAT3 nuclear translocation with a slight increase in cytosolic STAT3 (Figure 4). Although, there was no pSTAT3Tyr705 in either the cytosol or nuclear compartment at baseline, IL-10 induced pSTAT3Tyr705 within 30 min in the cytosol and nucleus. LBH was able to inhibit nuclear pSTAT3Tyr705 at 240 min, with little effect on cytosolic pSTAT3 (Figure 4). Interestingly, IL-10 was able to induce STAT3 acetylation at lysine 685 only in the cytoplasm, which was slightly increased by LBH addition.
Ly3 cells endogenously expressing STAT3 were treated with LBH and immuno-stained with an STAT3 antibody. At baseline, Ly3 cells showed STAT3 in both nuclear and cytoplasmic cell compartments (Figure 4b); with LBH treatment, nuclear STAT3 staining was abolished and STAT3 was found exclusively in the cytoplasm.
HDAC inhibition specifically abolishes tyrosine phosphorylation of STAT3
STAT3 is modified in vivo at amino- and carboxy-terminal sites by posttranslational modifications, including phosphorylation and acetylation.23 To determine whether HDAC inhibition leads to impaired activation of STAT3, we selected two pSTAT3Tyr705 DLBCL lines (DHL-2 and Ly3), along with pSTAT3-positive DLBCL patient samples. The samples were treated with LBH and the effect on activated and total STAT3 was examined. LBH was able to dephosphorylate STAT3 tyrosine phosphorylation in a dose-dependent manner in Ly3 cell within 24 h (Figure 5a). LBH had very little inhibitory effect on pSTAT3 serine and total STAT3 levels (Figure 5a). We have seen similar results when pSTAT3 DLBCL patient samples were treated with LBH (Figure 5b). We also investigated the effect of LBH using short-term (8 h) treatment in Ly3 and DHL2 cells. LBH caused a dose-dependent decrease in phosphorylation of STAT3 protein at tyrosine residue without much effect on pSTAT3 serine levels (Figure 5c). The effect of LBH on the phosphorylation of STAT1 and STAT5 was unable to be evaluated because they were not constitutively activated in Ly3 cells (Figure 5d). As JAK2 is a known upstream activator of STAT3 signaling, we investigated the effect of LBH on JAK2 phosphorylation by western blotting. LBH was not able to inhibit JAK2 phosphorylation in the Ly3 and DHL2 cell lines (data not shown). Collectively, these results demonstrate that LBH is specifically targeting STAT3 signaling in DLBCL tumors that express pSTAT3.
Figure 5.
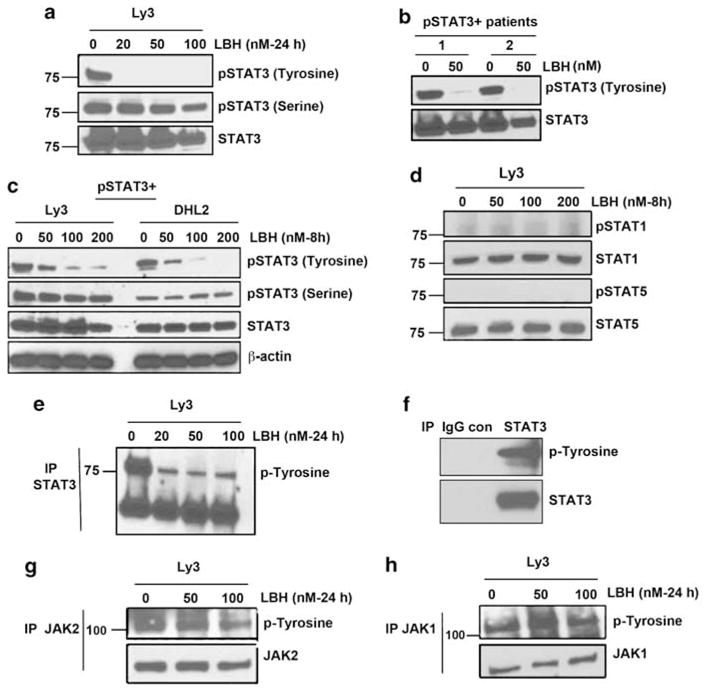
Effects of HDAC inhibition on STAT3 signaling: (a) Ly3 cells were treated for 24 h with various concentrations of LBH. Whole-cell lysates were then subjected to SDS--PAGE followed by immunoblotting using pSTAT3 Tyr705 and pSTAT3 Ser727 antibodies. (b) Cells from ABC and GCB patients were treated with LBH for 8 h and blotted for pSTAT3 Tyr705 antibody. (c) Ly3 and DHL2 cells were treated with various concentrations of LBH for 8 h and and then blotted with pSTAT3 Tyr705 and pSTAT3 Ser727 antibodies. (d) Ly3 cells were treated with various concentrations of LBH and examined for pSTAT1Tyr701 and pSTAT5Tyr694 levels. (e–h) After a 24-h treatment with LBH, STAT3 immunoprecipitates (e), JAK2 immunoprecipitates (g) and JAK1 immunoprecipitates (h) were probed with phospho-tyrosine antibody. (f) Immunoprecipitation control with mouse IgG antibody is shown.
To further confirm the effect of LBH on JAK/STAT signaling, we determined the effect of LBH on tyrosine activity of STAT3 and its upstream targets JAK2 and JAK1 (Figures 5e–h). We immunoprecipitated endogenous STAT3, JAK2 and JAK1 with antibodies to STAT3, JAK2, JAK1 and normal IgG control and blotted with phospho-tyrosine antibody. Figure 5e showed that LBH treatment results in significant reduction of the phosphotyrosine levels of STAT3. Immunoprecipitation with control IgG did not show baseline STAT3 tyrosine activity (Figure 5f). However, LBH treatment did not suppress phosphotyrosine levels of JAK2 and JAK1 (Figures 5g–h). These data suggest that STAT3-upstream pathway components are not targets of deacetylase activity. SHP1 and SHP2 are known to be involved in the dephosphorylation of STAT3 in keratinocytes.24 Next, we determined that whether HDAC inhibition through LBH dephosphorylate STAT3 by activating STAT3-specific phosphatases, such as SHP1 and SHP2. Our data in Ly3 cells demonstrate that LBH was not able to activate SHP1 and SHP2 protein expression (Supplementary Figure S2) indicating that these phosphatases are not involved in LBH-mediated STAT3 dephosphorylation.
pSTAT3-positive DLBCLs are more sensitive to HDAC inhibition than pSTAT3-negative DLBCLs
We investigated the effect of LBH on cell viability in pSTAT3-positive DLBCL lines- Ly3, HBL1 and U2932 and the pSTAT3-negative DLBCL lines- DHL-6, Ly1 and Ly19. As shown in Figures 6a–c, significant inhibition of survival was observed in all three pSTAT3 + DLBCL cell lines with LD90 values in the 25-nM range. In contrast, the pSTAT3− lines DHL6, Ly1 and Ly19 cells were less sensitive to LBH-induced inhibition of survival at the indicated doses under the same experimental conditions (Figures 6d–f). To further investigate the nature of pSTAT3-positive DLBCL cell death induced by LBH, we measured the levels of PARP cleavage in Ly3 cell lines, following LBH treatment. As shown in Figure 6g, LBH treatment induced PARP cleavage in Ly3 cells at low concentrations (10 nM). We have also seen that in Ly3 cells, the basal level of acetylated-histones H3 and H4 was low but increased significantly in a dose-dependent manner with LBH treatment (Figure 6h).
Figure 6.

Differential effects of LBH on pSTAT3-positive versus pSTAT3-negative DLBCL survival: (a–e) pSTAT3-positive DLBCL lines Ly3, HBL1 and U2932 (a–c) and pSTAT3-negative DLBCL lines DHL6, Ly1 and Ly19 (d–f) were treated with various concentrations of LBH for 48 h and survival was assessed by flow cytometry. Data represent the mean±s.d. of three independent experiments. (g, h) pSTAT3 + (Ly3) and pSTAT3− (DHL6) cells were treated with various concentrations of LBH and lysates were probed for PARP (g) and acetylated H3 and H4 (h) antibodies. (i, j) pSTAT3 + (Ly3) and pSTAT3− (DHL6) cells were treated with LBH for 24 h and lysates were probed with indicated antibodies.
We next evaluated the effect of LBH on survival proteins downstream of STAT3. STAT3 has been previously shown to regulate Bcl-2 family anti-apoptotic protein expression, including Bcl-xL, Mcl-1 and Bcl-2.25,26 DLBCL cells that are pSTAT3 positive expressed higher levels of the anti-apoptotic protein Mcl-1 but similar levels of Bcl-2 and Bcl-xL (Supplementary Figure S3). In pSTAT3 positive cells, treatment with LBH depleted the levels of the Mcl-1 compared with pSTAT3-negative cells. We observed no effect on Bcl-2 and Bcl-xL in either pSTAT3-positive or -negative cells (Figure 6i). Next, we evaluated the effect of LBH on pro-apoptotic proteins BAK, BAX and Bim. When compared with untreated controls, exposure to LBH resulted in slight increase in the expression of Bim, particularly BimEL in Ly3 cells, however, no changes were noted in the expression of BAX and BAK in both pSTAT3-positive versus pSTAT3-negative DLBCL cells (Supplementary Figure S3).
Both cyclin D1 and c-Myc are STAT3 transcriptional targets and promote cell-cycle progression. LBH inhibited c-Myc expression in both pSTAT3-positive and -negative DLBCL cells; however, no detectable effect was observed on cyclin D1 expression (Figure 6j). HDACi have been shown to induce p21 expression.27 LBH treatment was able to induce p21 in a dose-dependent manner in both pSTAT3-positive and -negative DLBCL cells (Figure 6j). LBH effects on the transcriptional regulation of Mcl-1 were then examined using reverse transcriptase-PCR analysis. LBH resulted in moderate transcriptional inhibition of Mcl-1 gene expression at 24 h (data not shown).
HDAC3 has a role in STAT3 acetylation/deacetylation
Type-I HDACs regulate transcription activation by STATs.28 To determine whether HDAC3 contributes to STAT3 deacetylation, we tested the effect of overexpression of HDAC3 on the acetylation state of endogenous STAT3 at Lys-685. Ly3 cells were transfected with or without an increasing amount of HDAC3. The transfected cells were lysed and immunoprecipitated with anti-STAT3 antibody, and immunoblotting was performed with anti-AcSTAT3 Lys685 or anti-STAT3 antibody. Overexpression of HDAC3 resulted in a decreased acetylation state of STAT3 (Figure 7a). In a similar experiment, HDAC1 over-expression did not have much effect on STAT3 acetylation (Figure 7b). Having established that HDAC3 physically associates with STAT3 and that LBH treatment increases STAT3 acetylation in ABC DLBCL cells, we then assessed the ability of HDAC3 to deacetylate STAT3. For this, we employed siRNA to deplete endogenous HDAC3 in Ly3 cells. As shown in Figure 7c HDAC3 knockdown upregulated STAT3 acetylation, indicating that HDAC3 negatively regulates the STAT3 acetylation in vivo. HDAC3 inhibition also inhibits constitutive STAT3 tyrosine phosphorylation without effect on total STAT3 (Figure 7c).
Figure 7.
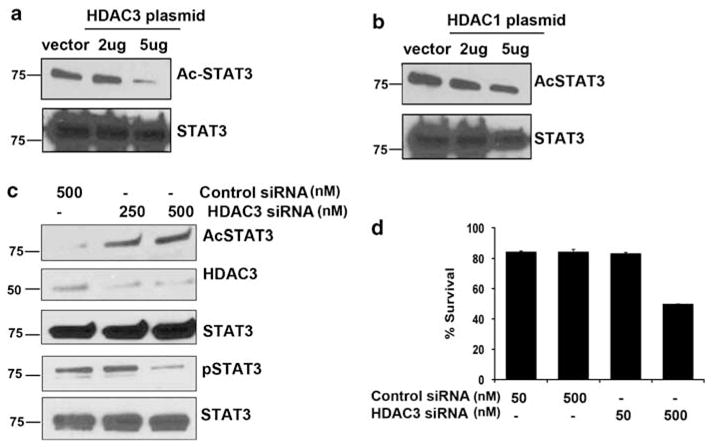
Effects of HDAC3 overexpression and inhibition on STAT3 acetylation, phosphorylation and survival: (a, b) Forty-eight hours after transfection with HDAC3 (a) and HDAC1 (b) plasmid, Ly3 cells were harvested and subjected to immunoprecipitation with STAT3 antibody. Blots were then examined for acetylated STAT3 and STAT3. (c) Forty-eight hours after siRNA transfection targeting HDAC3; Ly3 cell were harvested and lysed and immunoblotted for antibodies indicated. (d) Effect on HDAC3 inhibition through HDAC3 siRNA was assessed on Ly3 cell survival.
HDAC class-1 histone (specifically, HDAC3) is required for cell growth and is involved in the apoptotic process via the regulation of pro-apoptotic genes.29,30 To evaluate whether this was true for malignant lymphoma, we treated Ly3 cells with either HDAC3 or control siRNA for 96 h. HDAC3 transfected cells at 500 nM showed a 50% reduction in survival as detected by Annexin/PI staining compared with control (Figure 7d).
DISCUSSION
DLBCL tumors can have different molecular signatures that can predict survival. 1 Delineating which oncogenic signaling pathways are implicated in DLBCL sensitivity and resistance are important to the development of new therapeutic strategies. In order to improve outcome in patients with ABC DLBCL, we explored how these tumors are regulated from a signaling perspective. Recently, Ding et al.8 showed that STAT3 is constitutively activated in the ABC group of DLBCL. Constitutive STAT3 activation may result from the expression of upstream tyrosine kinases or from autocrine stimulation by cytokines or growth factors. These mechanisms may not be generally sufficient for lymphomagenesis but may be important for the growth and resistance of DLBCL. The present study uncovered a regulatory pathway of STAT3 in ABC DLBCL that has implications for therapy. In leukemia, regulation by HDACs can result in aberrant gene transcription.31 We have shown in this study that HDAC3 is not expressed in normal blood B --cells, but is highly expressed in DLBCL tumors that have high levels of STAT3 (total and phosphorylated). However, we did find one patient sample out of five studied that was GCB and HDAC3 + that did not express pSTAT3 indicating that other factors can interfere with the HDAC3/STAT3 interaction and hence block STAT3 signaling. Further studies in larger numbers of patient samples of DLBCL are required to understand the frequency of this abnormality and the clinical significance.
HDACs typically exist as components of large, multi-subunit complexes that interact with other cellular proteins.32 Indeed, we found that HDAC3 and pSTAT3 are primarily located in the nucleus; whereas unphosphorylated STAT3 was found in both compartments. HDACs can function as enzymes to deacetylate histones and regulate transcription. Although histones represent a primary target for the physiological function of HDACs, the antitumor effect of HDAC inhibitors might also work by modulating the acetylation status of a series of non-histone proteins.33,34 In our studies, treatment of pSTAT3 DLBCL cells with the HDAC inhibitor LBH increased the level of acetylated STAT3 at lysine 685. Histone acetyltransferase CBP/p300 is an important transcriptional coactivator capable of regulating acetylation of a wide range of transcription factors, including p53, E2F, AP-1 and NF-κB.35,36 Our data show that p300 and STAT3 physically associate with each other in ABC cells (and to a lesser extent in GCB cells). Our pull-down assay further demonstrated that LBH also increases the p300-mediated acetylation of STAT3. After ligand stimulation, latent STATs in the cytoplasm shuttle to and accumulate in the nucleus by crossing through nuclear pore complexes.37 STAT3 predominately resides in the cytoplasm of unstimulated cells; upon stimulation, it quickly translocates into the nucleus and binds to the DNA of target genes, inducing their expression.18 Treating cells with HDACi LBH increases STAT3 acetylation and results in decreased STAT3 protein in the nucleus along with a significant decrease in nuclear phosph-STAT3 at tyrosine. This suggests that HDAC inhibition may increase STAT3 export from the nucleus to the cytoplasm or block STAT3 nucleus entry. It may be possible that this nuclear export interferes with the STAT3 reactivation cycle and results in reduced STAT3 phosphorylation.
STAT3 is a novel non-histone substrate of HDAC3 in ABC-type DLBCL. Furthermore, we showed that HDAC3-mediated deacetylation functions as a molecular switch that may initiate a series of events in the activation of the STAT3 transcriptional response. Our data suggest that HDAC inhibition is able to inhibit constitutive STAT3 tyrosine phosphorylation in whole-cell lysates of ABC cells. This effect is mediated through STAT3 because pSTAT1 and pSTAT5 are not present in these cells and LBH did not inhibit pJAK1 and pJAK2 levels. The mechanism(s) underlying HDAC-mediated regulation of STAT3 tyrosine phosphorylation and acetylation remains elusive. It has been shown that acetylated lysine 685 is situated on the SH2 domain of STAT3 where tyrosine residue 705 is located.23 These two posttranslational modifications may work mutually. One possibility is that LBH-induced STAT3 lysine acetylation prevents its tyrosine phosphorylation, thereby shutting down STAT3 activation. A recent study showed that STAT1 acetylation counteracts IFN-induced STAT1 phosphorylation,38 supporting the hypothesis that STAT3 acetylation regulates its phosphorylation. Both phosphorylation and acetylation are modulators of protein functions and interactions and obviously are a large potential area of research. By using STAT3K685R mutants, Yuan et al.10 showed that STAT3 lys685 acetylation is critical for STAT3 dimers required for cytokine-stimulated transcriptional regulation. Additional studies are required to elucidate the mechanisms by which HDAC3 regulate the activation of STAT3 in ABC DLBCL. The effects of HDACi on gene expression are thought to be highly selective and lead to transcriptional activation of genes, including the cyclin-dependent kinase inhibitor p21. They also repress other genes important in control of cell growth. Our results clearly show that LBH can inhibit the STAT3-regulated anti-apoptotic protein Mcl-1 without much effect on Bcl-2 and Bcl-XL in ABC cells. However, the effect of LBH on cell-cycle proteins p21, cMyc and cyclin D1 was found to be very similar in pSTAT3-positive and -negative cells. Taken together, it appears that Mcl-1 overexpression through STAT3 is a mechanism of resistance in pSTAT3 DLBCL cells.
These results have therapeutic importance for DLBCL. Our data show that LBH has potent activity in DLBCL, especially when the DLBCL cells are dependent on STAT3 for survival. We demonstrate that HDACi anti-apoptotic activity is not dependent on primary histone targets but rather on targeting the non-histone protein STAT3 and ultimately constitutive STAT3 signaling. Our data also clearly shows that HDAC3 is the important HDAC to be targeted in DLBCL. This suggests the potential to select patients for LBH therapy that have tumors that overexpress pSTAT3. Currently, a number of HDAC inhibitors are in clinical trials for the treatment of various malignancies. Specifically, LBH589 is in clinical trials for the treatment of lymphoma and multiple myeloma.39,40
Supplementary Material
Acknowledgments
This work is supported by Lymphoma SPORE (P50 CA097274) Career Development Award to MG (from NCI/University of Iowa/Mayo Clinic); R01CA127433 to TEW and the Predolin Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
MG and TEW designed the research. MG interpreted and analyzed all the data, made the figures and wrote the manuscript. MG, MJS, JJH and LEW performed the experiments. TEW provided clinical samples and wrote the manuscript.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 2.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 4.Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol. 2011;29:200–207. doi: 10.1200/JCO.2010.30.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 6.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 7.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 8.Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111:1515–1523. doi: 10.1182/blood-2007-04-087734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 11.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370 (Part 3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 13.Bannister AJ, Miska EA, Gorlich D, Kouzarides T. Acetylation of importin-alpha nuclear import factors by CBP/p300. Curr Biol. 2000;10:467–470. doi: 10.1016/s0960-9822(00)00445-0. [DOI] [PubMed] [Google Scholar]
- 14.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1) Mol Cell Biol. 2006;26:2782–2790. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catania A, Iavarone C, Carlomagno SM, Chiariello M. Selective transcription and cellular proliferation induced by PDGF require histone deacetylase activity. Biochem Biophys Res Commun. 2006;343:544–554. doi: 10.1016/j.bbrc.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–282. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- 18.Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114:2926–2935. doi: 10.1182/blood-2009-05-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klampfer L, Huang J, Swaby LA, Augenlicht L. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem. 2004;279:30358–30368. doi: 10.1074/jbc.M401359200. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Holloway MP, Ma L, Cooper ZA, Riolo M, Samkari A, et al. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129–36137. doi: 10.1074/jbc.M110.152777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284:479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- 22.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–11534. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- 23.O’Shea JJ, Kanno Y, Chen X, Levy DE. Cell signaling. Stat acetylation-a key facet of cytokine signaling? Science. 2005;307:217–218. doi: 10.1126/science.1108164. [DOI] [PubMed] [Google Scholar]
- 24.Kim DJ, Tremblay ML, Digiovanni J. Protein tyrosine phosphatases, TC-PTP, SHP1, and SHP2, cooperate in rapid dephosphorylation of Stat3 in keratinocytes following UVB irradiation. PLoS One. 2010;5:e10290. doi: 10.1371/journal.pone.0010290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zushi S, Shinomura Y, Kiyohara T, Miyazaki Y, Kondo S, Sugimachi M, et al. STAT3 mediates the survival signal in oncogenic ras-transfected intestinal epithelial cells. Int J Cancer. 1998;78:326–330. doi: 10.1002/(SICI)1097-0215(19981029)78:3<326::AID-IJC12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 26.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 27.Lavelle D, Chen YH, Hankewych M, DeSimone J. Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am J Hematol. 2001;68:170–178. doi: 10.1002/ajh.1174. [DOI] [PubMed] [Google Scholar]
- 28.Ray S, Lee C, Hou T, Boldogh I, Brasier AR. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008;36:4510–4520. doi: 10.1093/nar/gkn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 30.Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redner RL, Wang J, Liu JM. Chromatin remodeling and leukemia: new therapeutic paradigms. Blood. 1999;94:417–428. [PubMed] [Google Scholar]
- 32.Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 34.McLaughlin F, La Thangue NB. Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 35.Schuringa JJ, Schepers H, Vellenga E, Kruijer W. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 2001;495:71–76. doi: 10.1016/s0014-5793(01)02354-7. [DOI] [PubMed] [Google Scholar]
- 36.Zhang JJ, Vinkemeier U, Gu W, Chakravarti D, Horvath CM, Darnell JE., Jr Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc Natl Acad Sci U S A. 1996;93:15092–15096. doi: 10.1073/pnas.93.26.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharya S, Schindler C. Regulation of Stat3 nuclear export. J Clin Invest. 2003;111:553–559. doi: 10.1172/JCI15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12:4628–4635. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 40.Prince HM, Bishton MJ, Johnstone RW. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009;5:601–612. doi: 10.2217/fon.09.36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.