

Abstract
Catalytic enantioselective 1,2-diboration of 1,3-dienes followed by cascade allylborations with dicarbonyls provides rapid entry into carbocyclic reaction products. The stereochemical course of this reaction was studied along with its application in the synthesis of pumilaside aglycon.
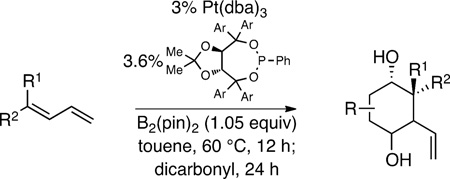
Catalytic enantioselective reactions that convert simple building blocks into complex structures are powerful tools for organic synthesis. Critical to the utility of such methods is that the product structures bear functionality and stereochemistry that is appropriate for important synthesis objectives. In this connection, we have been developing a series of catalytic enantioselective diboration reactions that provide reactive bis(boryl) intermediates that may engage in multiple bond-forming operations.1,2 One such diboration applies to 1,3-dienes: the 1,2-diboration of these substrates provides an α-chiral allylboron reagent (1, Scheme 1) that can engage in asymmetric carbonyl allylation (1→2→3).3 In considering how this transformation might apply to the construction of carbocyclic natural products, it was considered that if a second carbonyl was tethered to the first, the allylboron generated upon the first carbonyl allylation might participate in a subsequent intramolecular allylation (3→4).4 It was presumed that this second allylation would be subject to the conformational preferences of a chelated intermediate and might occur with useful levels of stereoinduction. The overall reaction cascade would therefore offer a single-pot reaction that converts a 1,3-diene and a dicarbonyl to a carbocyclic product bearing four contiguous stereocenters. Of critical consequence is the observation that the reaction products bear the structural hallmarks of terpene natural products. Accordingly, this tandem allylation strategy might offer an exceptionally rapid entry to these important compounds.
Scheme 1.

Study of the abovedescribed synthesis strategy commenced with the examination of the coupling between geranial-derived diene 5 and succinic dialdehyde.5 The general procedure is straightforward: Pt-catalyzed enantioselective diboration of the diene at 60 °C for 12 hours in toluene solvent was followed by addition of the dialdehyde. After stirring for 24 hours at 60 °C, the reaction was subject to alkaline work-up. Upon completion of the reaction sequence, cyclic diol 6 was isolated in 80% yield. Not only is this process efficient, but the stereoselection is remarkable: diol 6 was obtained in >15:1 diastereoselection and in 98:2 enantiomeric ratio. Single-crystal X-ray analysis of 6 showed it to be configured as depicted in Scheme 2.
Scheme 2.

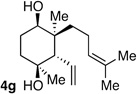
Following the conditions described above, a series of other dienes and dicarbonyls were examined in the tandem allylation reaction. As depicted in Table 1, reaction efficiency is generally good, and the derived cyclic diols can be isolated in good to excellent yields. Interestingly, and in contrast to the geranial-derived diene in Scheme 2, the isomeric neral-derived diene furnishes the trans 1,4-diol as the reaction product (entry 1, Table 1). As depicted in entries 2 and 3, the reaction extends to phthalic dialdehyde and provides enantiomerically enriched reduced benzoquinones as the reaction product. Because the 1,2-diboration also extends to simple cis dienes and exocyclic dienes, cyclic diols bearing tertiary centers and spirocycles can be accessed with excellent diastereo- and enantioselection (entry 5 and 6). Importantly, high levels of regioselectivity were observed with non-symmetric carbonyls. In these cases (entries 7–9), the first addition occurs to the less hindered aldehyde, with cyclization occurring on the remaining carbonyl. Importantly, when this reactivity extends to ketoaldehydes, tertiary alcohols are crafted in a highly selective fashion. Extension of this transformation to seven-membered ring synthesis has yet to be successful; use of valeric dialdehyde in place of succinic dialdehyde gave complex mixtures. Presumably other 1,5-dialdehydes with alternate conformational preferences may ameliorate this situation. As a last point of note, a general feature of the tandem allylation products is that the stereoisomers are often readily separable by chromatography such that isomerically pure compounds can be easily obtained even from moderately selective reactions; thus, given the level of functional group complexity that is quickly established in this reaction, even non-selective reactions may find use in complex molecule synthesis.
Table 1.
Tandem diene diboration/double allylation of dicarbonyls.a
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | dicarbonyl | product | d.r. e.r. yield |
| 1 | prenyl | Me | 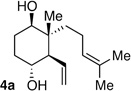 |
5:1 dr 6:4 er 76 % |
|
| 2 | Me | prenyl |  |
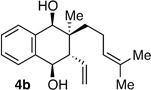 |
2.8:1 dr 96:4 er 83% |
| 3 | prenyl | Me | 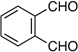 |
 |
1.2:1 dr 97:3 er 72% |
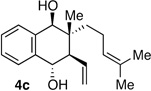
| 4b | Me | CH2OSiR3 | 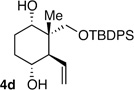 |
10:1 dr 97:3 er 71% |
|
| 5 | pentyl | H | 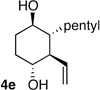 |
>20:1 dr 91:9 er 39% |
|
| 6 | -(CH2)5- | 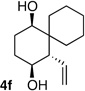 |
9:1 dr 88:12 er 72% |
||
| 7 | Me | prenyl | 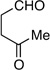 |
 |
11:1 dr 97:3 er 77% |
| 8 | Me | prenyl | 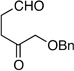 |
 |
>20:1 dr 97:3 er 60% |
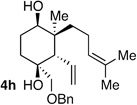
| 9 | Me | prenyl | 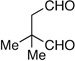 |
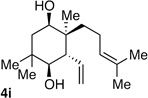 |
>20:1 dr 97:3 er 54% |
Entries 1, 4, and 6 employed 2 equivalents of dicarbonyl; entries 2, 3, 7, and 8 employed 1 equiv. dicarbonyl; entry 5 employed 3 equiv dicarbonyl; entry 9 employed 2 equivalents of diene/B2(pin)2. Diastereoselectivity determined by 1H NMR analysis; yield refers to isolated yield of the regioisomer mixture.
(S,S) ligand employed for this experiment.
The stereochemical outcome of the reactions in Table 1 can be readily understood on the basis of a trans-decalin-like transition state structure with intramolecular allylboration occurring through a chair-like six-membered array (Scheme 3).6 The outcome of entry 6 (Table 1), wherein the only stereogenic element in the intermediate resides at C-5, reveals an inherent preference for an axial orientation of the C5-OB(pin) group (A, Scheme 3). While this preference is indeed surprising, it is in line with observations about the axial preference of C4 alkoxy groups in cyclohexanone derivatives.7 This feature is thought to arise from either a) electrostatic attraction between the axial oxygen that bears a δ- and either the carbonyl carbon8 or axial hydrogens α to the carbonyl9, both of which bear δ+; b) hydrogen bonding between the axial C4 heteroatom and acidic axial hydrogens adjacent to the carbonyl10; or c) hyperconjugative interactions between the C-O σ* and an adjacent axial CH donor.11 In the context of this tandem reaction sequence, 1,4-syn selectivity is highest with geranial-derived substrates (i.e. Scheme 2 and entries 7–9 in Table 1) suggesting that the axial preference for an OB(pin), acting in concert with a preference for the larger R2 to reside in an equatorial site, enhances selectivity for reaction through structure A. In contrast, the influence of substituents at C4 in neralderived substrates and cis dienes (R1 larger than R2, entries 1 and 5, Table 1) counteracts the axial preference of the OB(pin) group sufficiently to turn over diastereoselection: reaction occurs through B and furnishes the trans 1,4-diol.
Scheme 3.

Model for stereoinduction in tandem allylations.
As suggested in the introduction, the rapid increase in molecular complexity that accompanies the tandem allylation reaction described herein should facilitate rapid construction of complex molecular structures. The validity of this assertion was probed by targeting the aglycon of the glycoterpene natural product pumilaside B.12 As shown in Scheme 4, a carbocyclic intermediate (8) bearing appropriate substitution and stereochemistry was readily constructed by diboration of 7 followed by addition to 4-oxovaleraldehyde. This delivered trans 1,5-diol 8 in good yield and excellent stereoselection. Ring-closing metathesis with the Hoveyda-Grubbs NHC catalyst13 followed by cyclopropanation furnished 10. Conversion of the dibromocyclopropane 10 to the dimethylcyclopropane target was easily accomplished by treatment with lithium dimethylcuprate followed by methyl iodide.14 The compound so obtained was spectroscopically identical with the compound reported by Kitajima and co-workers.15
Scheme 4.

Synthesis of pumilaside aglycon.
In conclusion, a strategy for constructing highly substituted and stereochemically-complex functionalized cyclohexanols from simple precursors has been established. These motifs should enable rapid construction of complex molecular targets and the utility of these methods will be further probed in due course.
Supplementary Material
ACKNOWLEDGMENT
The NIH (GM-59471) is acknowledged for financial support and AllyChem is acknowledged for a donation of B2(pin)2.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Enantioselective alkene diboration: Kliman LT, Mlynarski SN, Morken JP. J. Am. Chem. Soc. 2009;131:13210. doi: 10.1021/ja9047762. Trudeau S, Morgan JB, Shrestha M, Morken JP. J. Org. Chem. 2005;70:9538. doi: 10.1021/jo051651m. Miller SP, Morgan JB, Nepveuz FJV, Morken JP. Org. Lett. 2004;6:131. doi: 10.1021/ol036219a. Morgan JB, Miller SP, Morken JP. J. Am. Chem. Soc. 2003;125:8702. doi: 10.1021/ja035851w. Diene diboration: Hong K, Morken JP. J. Org. Chem. 2011;76:9102. doi: 10.1021/jo201321k. Schuster CH, Li B, Morken JP. Angew. Chem. Int. Ed. 2011;50:7906. doi: 10.1002/anie.201102404. Burks HE, Kliman LT, Morken JP. J. Am. Chem. Soc. 2009;131:9134. doi: 10.1021/ja809610h. Allene diboration: Burks HE, Liu S, Morken JP. J. Am. Chem. Soc. 2007;129:8766. doi: 10.1021/ja070572k. Woodward A, Burks HE, Chan LM, Morken JP. Org. Lett. 2005;7:5505. doi: 10.1021/ol052312i. Pelz N, Woodward AR, Burks HE, Sieber JD, Morken JP. J. Am. Chem. Soc. 2004;126:16328. doi: 10.1021/ja044167u. For related alkyne double hydroboration: Jung H-Y, Yun J. Org. Lett. 2012;14:2606. doi: 10.1021/ol300909k. Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928.
- 2.For reviews of catalytic diboration, see: Dang L, Lin Z, Marder TB. Chem. Commun. 2009:3987. doi: 10.1039/b903098k. Ramírez J, Lillo V, Segarra AM, Fernández E. C. R. Chemie. 2007;10:138. Burks HE, Morken JP. Chem. Commun. 2007:4717. doi: 10.1039/b707779c. Beletskaya IB, Moberg C. Chem. Rev. 2006;106:2320. doi: 10.1021/cr050530j. Ishiyama T, Miyaura N. Chem. Rec. 2004;3:271. doi: 10.1002/tcr.10068. Marder TB, Norman NC. Topics in Catalysis. 1998;5:63.
- 3.Kliman LT, Mlynarski SN, Ferris GE, Morken JP. Angew. Chem. Int. Ed. 2012;51:521. doi: 10.1002/anie.201105716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For intramolecular carbonyl allylboration, see: Koert U. Org. Lett. 2006;8:3829. doi: 10.1021/ol0614471. Ballard EC, Morken JP. Synthesis. 2004;9:1321. Kurihara K, Yamada A, Takahashi M, Takahashi Y, Miyaura N. Tetrahedron. 2003;59:537. Krüger J, Hoffman RW. J. Am. Chem. Soc. 1997;119:7499. Yamomoto Y, Ahiko T, Ishiyama T, Miyaura N. Chem. Lett. 1997:811. Hoffmann RW, Hense A. Liebigs Ann. 1996:1283. Hoffman RW, Münster I. Tet. Lett. 1995;36:1431. Suzuki A. J. Chem. Soc. Chem. Commun. 1994;4:467. Watanabe T, Miyaura N, Suzuki A. J. Organomet. Chem. 1993:4444. Bandur NG, Brückner D, Hoffmann RW, Watanabe T, Sakai M, Miyaura N, Hoffmann RW, Sander T, Hense A. Liebigs Ann. Chem. 1993:771. Hoffmann RW, Sander T, Hoffmann RW. Liebigs Ann. Chem. 1993:1193. Niel G. Liebigs Ann. Chem. 1991:1195.
- 5.(a) dos Santos C, Bahlaouan Z, Kassimi KE, Troufflard C, Hendra F, Delarue-Cochin S, Zahouily M, Cavé C, Joseph D. Heterocycles. 2007;73:751. [Google Scholar]; (b) Enkisch C, Schneider C. Eur. J. Org. Chem. 2009;32:5549. [Google Scholar]
- 6.(a) Brichford NL, Henderson JL. J. Org. Chem. 2002;67:4236. doi: 10.1021/jo0164002. [DOI] [PubMed] [Google Scholar]; (b) Omoto K, Fujimoto H. J. Org. Chem. 1998;63:8331. [Google Scholar]; (c) Gennari C, Fioavanzo E, Bernardi A, Vulpetti A. Tetrahedron. 1994;50:8815. [Google Scholar]; (d) Vulpetti A, Gardner M, Gennari C, Bernardi A, Goodman JM, Paterson I. J. Org. Chem. 1993;58:1711. [Google Scholar]; (e) Gajewski JJ, Bician W, Brown HC, Racherla US, Pellechia PJ. J. Org. Chem. 1990;55:1868. [Google Scholar]; (f) Li Y, Houk KN. J. Am. Chem. Soc. 1989;111:1236. [Google Scholar]
- 7.(a) Baldry KW, Gordon MH, Hafter R, Robinson MJT. Tetrahedron. 1976;32:2589. [Google Scholar]; (b) Stowlow RD, Giants TW. Chem. Comm. 1971;11:528. [Google Scholar]
- 8.(a) Kleinpeter E, Heydenreich M, Koch A, Linker T. Tetrahedron. 2012;68:2363. [Google Scholar]; (b) Woerpel KA. J. Org. Chem. 2011;76:7706. doi: 10.1021/jo200950s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Baghdasarian G, Woerpel KA. J. Org. Chem. 2006;71:6851. doi: 10.1021/jo060968z. [DOI] [PubMed] [Google Scholar]; (d) Dibble DJ, Ziller JW, Woerpel KA. J. Org. Chem. 2011;76:7706. doi: 10.1021/jo200950s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freitas MP, Tormena CF, Olivira PR, Rittner R. J. Mol. Struc. (Theochem) 2002;589–590:147. [Google Scholar]
- 10.Takahashi O, Yamasaki K, Kohno Y, Ueda K, Suezawa H, Nishio M. Bull. Chem. Soc. Jpn. 2009;82:272. [Google Scholar]
- 11.(a) Kihara M, Iwai Y, Nagao Y. Hetereocycles. 1995;41:2279. [Google Scholar]; (b) Nagao Y, Goto M. Hetereocycles. 1995;41:883. [Google Scholar]
- 12.Kitajima J, Kimizuka K, Tanaka Y. Chem. Pharm. Bull. 2000;48:77. doi: 10.1248/cpb.48.77. [DOI] [PubMed] [Google Scholar]
- 13.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168. [Google Scholar]
- 14.(a) Simmons B, Walji AM, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:4349. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rigby JH, Bellemin A. Synthesis. 1988:188. [Google Scholar]
- 15.While the 13C NMR and 1H NMR spectra of the enantiomerically-enriched material prepared by us are identical to that reported by Kitajima, the rotation is increased (+49.95° vs. +10°) and the melting point of the synthetic material is lower (108–110 °C vs 172–175 °C). We are currently working to understand this discrepancy.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.