

Abstract
Higher order folding of chromatin fibre is mediated by interactions of the histone H4 N-terminal tail domains with neighbouring nucleosomes. Mechanistically, the H4 tails of one nucleosome bind to the acidic patch region on the surface of adjacent nucleosomes, causing fibre compaction. The functionality of the chromatin fibre can be modified by proteins that interact with the nucleosome. The co-structures of five different proteins with the nucleosome (LANA, IL-33, RCC1, Sir3 and HMGN2) recently have been examined by experimental and computational studies. Interestingly, each of these proteins displays steric, ionic and hydrogen bond complementarity with the acidic patch, and therefore will compete with each other for binding to the nucleosome. We first review the molecular details of each interface, focusing on the key non-covalent interactions that stabilize the protein–acidic patch interactions. We then propose a model in which binding of proteins to the nucleosome disrupts interaction of the H4 tail domains with the acidic patch, preventing the intrinsic chromatin folding pathway and leading to assembly of alternative higher order chromatin structures with unique biological functions.
Keywords: chromatin structure, nucleosome-binding proteins, higher order folding
1. Introduction
The DNA of eukaryotic genomes is organized as a nucleoprotein complex termed chromatin. A long-standing challenge for both biological and physical scientists has been to understand how the protein components of chromatin regulate genome structure and the accessibility of the underlying DNA. The subunit of chromatin is the nucleosome, which consists of approximately 146 bp of DNA wrapped around an octamer of core histones [1]. Each core histone has a short N-terminal ‘tail’ domain that is unstructured and extends into solution. The remaining residues of each core histone are largely α-helical and collectively form the body of the nucleosome. Nucleosomes spaced repetitively along the DNA are referred to as nucleosomal arrays. The DNA that connects adjacent nucleosomes in an array is called linker DNA. Nucleosomal arrays are intrinsically dynamic. In low salt conditions, nucleosomal arrays adopt an extended beads-on-a-string conformation [2]. Under physiological ionic conditions, nucleosome–nucleosome interactions cause nucleosomal arrays to condense into folded and oligomeric structures. The core histone tail domains mediate array condensation, with the H4 tail domain contributing a particularly vital role [3–6]. If the H4 tail is removed by proteolytic or recombinant means [4,7–9], or modified by acetylation [6,10,11], nucleosomal arrays no longer condense normally. Central to the function of the H4 tail domain during chromatin condensation is its interaction with a specific region on the surface of neighbouring nucleosomes termed the ‘acidic patch’ [1,12]. The acidic patch is formed from six H2A and two H2B residues, which together create a highly contoured and negatively charged binding interface on the nucleosome surface. This review focuses on the role of the acidic patch in mediating the higher order structure of the chromatin fibre. This subject should interest biological scientists because control of chromatin fibre structure is linked to regulation of fibre function. It should appeal to the physical scientist because of its emphasis on molecular mechanism. Key questions that we address are what is the physico-chemical basis for how the acidic patch mediates nucleosome–nucleosome interactions and the higher order structure of the chromatin fibre? At the atomic level, why is one relatively small region of the nucleosome surface so central to mediating genomic structure and function?
Genomic chromatin in vivo is composed of nucleosomal arrays and many different bound proteins, e.g. architectural proteins, transcription factors and remodelling enzymes. Many chromatin-associated proteins interact directly with the nucleosome. These nucleosome-binding proteins provide a means to manipulate the intrinsic structure and function of the underlying chromatin fibre. In other words, the higher order architecture in any given region of the genome will be dictated by the specific properties of the nucleosome-binding protein(s) in question. For example, yeast Sir3 interacts with nucleosomes and organizes the chromatin at telomeres and the silent mating loci into a specialized architecture that is repressive to transcription [13]. When Sir3p binds to nucleosomal arrays in vitro, it assembles a hypercondensed chromatin structure that is fundamentally different than that formed by the parent arrays [14,15]. The fact that there are so many different nucleosome-binding proteins presumably reflects the existence of an equally large number of specific chromatin architectures, each of which is tailored to a specific functional state of the chromatin fibre.
Much recent effort has gone towards obtaining atomic-level information about protein–nucleosome complexes. Results to date have revealed that all five of the nucleosome-binding proteins for which there are experimental or modelled co-structures interact with the acidic patch on the nucleosome surface. This surprising result raises a key mechanistic question: why is the same region on the nucleosome surface a common binding site for both the H4 tail domain and many nucleosome-binding proteins? The answer to this question must, in some way, be related to the biological functions of the different nucleosome-binding proteins. Here, we review the molecular details of the various protein–acidic patch interactions. We then propose a model in which proteins that bind to the nucleosome compete with the H4 tail domain for the acidic patch, preventing the nucleosome–nucleosome interactions that govern intrinsic chromatin condensation and leading to alternate chromatin architectures dictated by the specialized properties of the bound proteins. In this fashion, we propose that nucleosome-binding proteins lead to structural and functional remodelling of the chromatin fibre.
2. The nucleosome acidic patch and higher order chromatin structure
2.1. The nucleosome acidic patch
The arrangement of the core histones within the histone octamer produces a nucleosome surface that is highly contoured and asymmetrically charged (figure 1). A particularly striking feature of the nucleosome surface is the cluster of eight acidic residues (E56, E61, E64, D90, E91, E92 of H2A and E102, E110 of H2B) that forms a negatively charged ‘acidic patch’ (figure 1). The acidic patch has the shape of a narrow groove. The edges of the groove are formed by the α1-helix and the C-terminal αC-helix extension of H2B. The bottom of the groove is formed from the α2-helix and C-terminal extension of H2A (figure 1). The bottom of the groove also is contoured, with the H2A α2-helix forming a shallow ridge. Residues E102 and E110 project into the groove from the H2B αC extension. H2A residues E56, E61 and E64 come from the α2-helix, while residues D90, E91 and E92 are part of the H2A C-terminal extension. The acidic patch also has non-polar character, with H2A residues Y50, V54 and Y57 forming a hydrophobic pocket along the bottom of the groove. Altogether the acidic patch is a complex interface, with distinct shape and non-polar features together with pronounced negative surface charge density. In terms of size, the acidic patch is sufficiently large to be able to accommodate a range of binding motifs (loops, extended strands, hairpin, α-helix), as will be discussed in §§2.2 and 2.3.
Figure 1.

The nucleosome and its acidic patch. (a) Electrostatic potential view of the nucleosome surface. The electrostatic rendering was performed using APBS tools in PyMOL (http://www.poissonboltzmann.org/apbs/examples/visualization/apbs-electrostatics-in-pymol#TOC-Generating-the-PQR). A pqr file was generated from the 1KX5 histone octamer (no DNA), using pdb2pqr software. APBS was then run through PyMOL. The electrostatic potential displayed is between −25 (red) and +25 (blue) kT e–1. (b) Close-up view of the acidic patch. Histone H2A is shown in yellow and H2B in red. The α1- and αC-helices of H2B, α2- and αC-helices of H2A and the eight residues that make up the acidic patch are labelled. PDB 1KX5.
2.2. H4 tail interaction with the acidic patch during higher order chromatin folding
Under physiological ionic conditions in vitro, short arrays of nucleosomes are in equilibrium between unfolded, folded and oligomeric conformational states [2]. The unfolded conformation is characterized by maximum extension between nucleosomes, leading to a beads-on-a-string structure. Cations facilitate the short- and long-range nucleosome–nucleosome interactions that cause array condensation, acting through a complex mechanism [16]. Short-range nucleosome–nucleosome interactions induce local array compaction [2,17], often referred to as ‘higher order’ chromatin folding. Early studies that used trypsin to remove all of the core histone tail domains showed that one or more of the tails was essential for folding [7,9,18]. Studies of recombinant nucleosomal arrays lacking or containing single tail domains identified the H4 tail as a primary mediator of the folding transition [3]. The H4 tail domain therefore is presumed to be an important determinant of chromatin fibre architecture in vivo.
Similar to all core histone NTDs, the H4 tail domain is highly basic, and for many years was thought to bind to DNA [19,20]. However, a mechanism involving interaction with the nucleosome acidic patch surfaced from the initial X-ray crystallography studies of the nucleosome. An interaction between nucleosomes in the crystal lattice was observed, in which residues 16–24 of the H4 tail domain were bound to the acidic patch of a neighbouring nucleosome [1]. As indicated above, the central role of the H4 tail domain in mediating higher order chromatin folding was beginning to emerge at this time. It was therefore hypothesized that an interaction between the H4 tail domain and the acidic patch of neighbouring nucleosomes might mediate folding into 30 nm fibres [1,3,21]. Strong evidence for the involvement of the acidic patch in higher order chromatin folding came from mutational studies of the acidic patch residues in H2A.Z nucleosomes. Tremethick and co-workers [12] noted that the acidic patch region of nucleosomes containing the H2A.Z variant was expanded by several residues, and speculated that if the acidic patch was involved in higher order chromatin folding, H2A.Z-containing chromatin fibres would fold more readily than regular H2A fibres. They observed just that. If the additional acidic patch residues in H2A.Z were mutated back to the residues in H2A, the mutated chromatin fibres folded identically as regular H2A fibres [12]. Together with identification of the H4 tail domain as a key mediator of local nucleosome–nucleosome interactions [3], these studies established that H4 tail domain interactions with the acidic patch are required for higher order chromatin folding.
The details of the H4 tail–acidic patch interaction remain an open question. The H4 NTD adopts an extended structure when interacting with the acidic patch in the crystal lattice (figure 2a). Consistent with this result, V21 of the H4 tail is in proximity to H2A E64 in folded nucleosomal arrays [21]. Yang & Arya [22] recently performed molecular dynamic simulations of the H4 tail domain docked to the nucleosome. Their computations suggested that amino acids 1–13 of the H4 tail domain are unfolded, but that residues 16–22 have a propensity for α-helix formation. The modelled α-helix fits snugly in the acidic patch groove (figure 2b). H4 K16, R19, K20 and R23 are on the same face of the helix, and form extensive non-covalent interactions with acidic patch residues. H4 K16 is a particularly key residue, forming a strong salt bridge with H2A E61, and other salt bridges with H2A E64, D90, E92 and H2B E102. The modelling studies found that acetylation of H4 K16 disrupts all of these salt bridges, while H4 K16 acetylation has been shown experimentally to disrupt higher order chromatin folding [6,10,11]. In the modelled structure, H4 R19 interacts with H2A E56, H4 K20 interacts with H2B E110 and H4 R23 with H2A E56 and H2B E110. Finally, both H4 R17 and H18 interact with H2A E64. However, we note that other modelling studies suggest that free H4 NTD is a hairpin [23], and that acetylation promotes α-helix formation [24]. Moreover, the modelling results of Allahverdi et al. [6] suggest that H4 K16 may interact with H2B residues R96-L99. The structure of the H4 tail in a chromatin context is an area that will benefit from experimental studies in the future.
Figure 2.
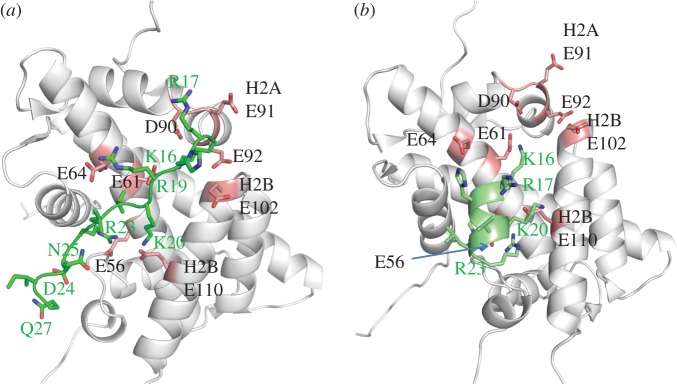
H4 tail domain–acidic patch interactions. (a) Close-up view of the H4 tail–acidic patch interaction observed in the crystal structure [1]. The H4 tail is shown in lime green. Histones are in grey, with acidic patch residues shaded pink. PDB 1A01. (b) Close-up view of the modelled H4 peptide–acidic patch interaction taken from [22]. The H4 peptide is shown in green. Histones are in grey, with acidic patch residues shaded pink. PDB provided courtesy of Dr G. Arya.
3. The acidic patch and nucleosome-binding proteins
This section discusses the five proteins for which there are co-structures or models with the nucleosome. These proteins have a variety of unrelated cellular functions, yet they all bind to the same acidic patch region of the nucleosome as the H4 tail domain.
3.1. Latency-associated nuclear antigen
In 2006, Barbera et al. [25] characterized the binding of a short fragment of the Kaposi's sarcoma herpes virus (KSHV) latency-associated nuclear antigen (LANA) to the nucleosome using biochemical and X-ray crystallography approaches. KSHV preserves its genetic material in episomes, which become associated with the host chromosomes and are replicated by the host cell machinery. LANA is the protein that tethers the KSHV episomes to mitotic chromosomes, and the LANA–chromosome interaction is essential for virus survival [26]. The first 22 N-terminal amino acids of LANA comprise the chromosome association domain [27]. Scanning alanine mutagenesis studies [28] determined that residues 5–16 of the chromosome association domain are required for LANA binding to chromosomes and for episome persistence. Electrophoretic mobility shift assay experiments showed that a peptide comprising amino acids 1–23 of LANA interacts with isolated nucleosomes [25]. Pull-down experiments determined that the LANA 1–23 peptide binds to the structured domains of isolated H2A–H2B dimers [25]. The biochemical studies indicated that the nucleosome is the chromosomal target for LANA. The crystal structure of the nucleosome complexed to the 1–23 peptide (figure 3a) revealed the molecular basis for LANA binding to nucleosome [25].
Figure 3.

Nucleosome-binding protein co-structures. (a) Close-up view of the LANA peptide–acidic patch interaction. The LANA peptide is shown in dark green. Histones are in grey, with acidic patch residues shaded pink. PDB 1ZLA. (b) RCC1–acidic patch interaction. RCC1 is shaded in cyan. Histones are in grey, with acidic patch residues shaded pink. PDB 3MVD. (c) Sir3–acidic patch interaction. Sir3 BAH domain is shaded in magenta. Histones are in grey, with acidic patch residues shaded pink. PDB 3TU4. (d) Modelled HMGN2–acidic patch interaction. HMGN2 is shown in black, with key residues labelled in red. Histones are in grey, with acidic patch residues shaded pink. PDB provided courtesy of Dr Y. Bai.
The LANA 1–23 peptide forms a hairpin structure and shows a great deal of charge and shape complementarity with the acidic patch of the nucleosome. The LANA 1–23 hairpin fits between the C-terminal αC-helix and the α1-helix of H2B, and extensively interacts with H2A along its α2-helix. The LANA peptide fits tightly in the acidic pocket, with the non-polar M6 and L8 residues inserted into a hydrophobic region formed by residues V54, Y50 and Y57 on the H2A α2-helix. There are many non-covalent interactions between the LANA hairpin and the acidic patch residues. In particular, R9 of LANA forms salt bridges with E61, D90 and E92 of H2A, similar to modelled interaction of K16 with the H4 tail [22]. Notably, R9 of LANA is essential for chromatin association [25,28], just as K16 of the H4 tail is essential for chromatin compaction [6,10,11]. R9 of LANA also forms hydrogen bonds with H2A E61, D90 and E92. S10 of LANA forms a hydrogen bond with H2A E64, and R7 of LANA forms a salt bridge with H2B E110. Altogether, the crystal structure showed that residues 5–16 of the LANA peptide are the only amino acids that are involved in interactions with the nucleosome acidic patch. These are the same residues that are required for tethering episomes to the chromosomes [28], indicating that the LANA chromosome associate domain evolved to use the nucleosome acidic patch for episome attachment.
3.2. Interleukin-33
Interleukin-33 (IL-33) is a member of the IL-1 family. It functions both as a cytokine and as an intracellular nuclear factor [29]. In 2007, Carriere et al. [30] showed that IL-33 is an abundant chromatin-associated factor in endothelial cells in vivo and it has roles in transcription regulation as well. Roussel et al. [29] demonstrated that residues 40–58 of IL-33 are sufficient for chromatin binding and called this region the chromatin binding motif (CBM). Roussel et al. [29] performed in vivo fluorescence experiments followed up by in vitro glutathione S-transferase pull-down experiments to demonstrate association with chromatin and isolated histones H2A–H2B. Through alanine scanning mutations they showed that there are six residues essential for chromatin and histone interactions: M45, L47, R48, S49, G50 and I53. Residues 44–52 of IL-33 have homology to residues 5–14 of LANA (both contain a MXLRSG sequence). Based on this conserved motif, Roussel et al. [29] suggested that the IL-33 CBM binds to the same location on the nucleosome as the LANA chromosome association domain.
Roussel et al. [29] modelled the CBM–nucleosome complex using the crystal structure of LANA 1–23 peptide bound to the nucleosome as a guide. Modelling suggested that IL-33 CBM forms a tight hairpin similar to the LANA peptide. Not surprisingly, the shape and charges of the IL-33 CBM were complementary with the acidic patch on the nucleosome surface. R48 of IL-33 formed salt bridges with H2A E61, D90 and D92, replacing R9 of LANA. S49 forms hydrogen bonds with E61 and E64, in place of S10 of LANA. A hydrophobic pocket formed by H2A Y50, V54 and Y57 allows the insertion of IL-33 M45, replacing LANA M6. Residues L51 and I53 in IL-33 replace R12 and T14 from LANA, hence the IL-33 residues create a denser overall network of hydrophobic contacts. Although the IL-33 CBM has bulkier N- and C-termini compared with LANA, modelling revealed that these bulky ends could be accommodated on the nucleosome surface above the acidic patch without steric constraints. The modelling results were supported by experiments showing that the IL-33 CBM failed to bind to H2A/H2B with a mutated acidic patch. Altogether, the modelling results suggested that the IL-33 CBM may bind to the acidic patch region of the nucleosome.
3.3. Regulator of chromosome condensation 1
Regulator of chromosome condensation 1 (RCC1) is a guanine-exchange factor also known as RanGEF. RCC1 is a nucleosome-binding protein that recruits and activates Ran GTPase, creating a Ran concentration gradient in the vicinity of chromosomes [31–33]. The Ran concentration gradient is central for nuclear function, regulating nucleo-cytoplasmic transport [34], assembly of mitotic spindles [31,33] and nuclear envelope formation after mitosis [32]. RCC1 consists of a seven-bladed β-propeller domain and a disordered N-terminal tail [35]. The crystal structure of the RCC1–nucleosome complex resolved at 2.9 Å resolution (figure 3b) revealed that about three quarters of the contacts between the RCC1 β-propeller domain and the nucleosome are with histones H2A and H2B, with the remaining involving nucleosomal DNA interactions [36]. Several unique loops of the β-propeller mediate RCC1 binding to the nucleosome. Of note, the ‘switchback’ loop binds to the acidic patch on the nucleosome surface, consistent with biochemical experiments showing that RCC1 binds to isolated H2A–H2B dimers, and that LANA competes with RCC1 in binding assays [37].
As with LANA and IL-33, RCC1 interacts with key acidic patch residues via an extensive network of electrostatic interactions. R223 of RCC1 forms hydrogen bonds and salt bridges with H2A residues E92, D90 and E61, similar to R9 of LANA. R223 is critical for RCC1 nucleosome binding [37], exactly as R9 of LANA. H2A E61 also is hydrogen bonded to RCC1 R216, which forms hydrogen bonds with H2A E64. S214 and S217 of RCC1 also are hydrogen bonded to H2A E64, while S217 forms an additional hydrogen bond with H2B V45. The RCC1 acidic patch interaction has shape complementarity; there are extensive van der Waals contacts between the switchback loop residues and H2B E102, L103, H106 and V45, on the walls of the acidic patch groove.
3.4. SIR3
Saccharomyces cerevisiae does not have the typical heterochromatin structures found in higher eukaryotes. Rather, it assembles transcriptionally ‘silent’ chromatin at the telomeres and silent mating loci [13]. The silent information regulator (SIR) proteins, SIR1p, SIR2p, SIR3p and SIR4p, initiate and maintain the silenced chromatin state. Of the Sir proteins, Sir3 functions by modulating chromatin architecture. For example, when Sir3 is overexpressed in vivo, silent chromatin spreads outside its normal boundaries, even without the normal complement of the other Sirs [38–40]. In vitro, Sir3 is a nucleosome-binding protein [14] that assembles uniquely condensed higher order chromatin structures [14,15].
Many of the Sir3 mutations that affect silencing occur in approximately 200 residue bromo-associated homology (BAH) domain located at the Sir3 N-terminus. Armache et al. [41] recently obtained a crystal structure of the Sir3BAH domain complexed with the nucleosome at 3.0 Å resolution (figure 3c). The BAH–nucleosome interface is extensive. Twenty-eight Sir3 residues are involved in the interaction. The regions of contact between the BAH domain and the nucleosome observed in the crystal structure match very well with those identified in genetic screens [41,42]. Most of the interactions are with the core histones, although some are with nucleosomal DNA. The BAH domain interacts with the core histones in multiple locations; the BAH domain contacts the H4 tail domain, binds many H3 and H4 residues on the nucleosome surface, and also interacts with the acidic patch. BAH residues 17–37 are involved in the acidic patch interaction. These residues are disordered in the structure of BAH alone [43,44], but form loop 1 when BAH is bound to the nucleosome. As can be seen in figure 3c, BAH residues 28–34 fit snugly in the acidic patch groove. Although the electron density in this region is poor, R28, R29, R30, R32, K33 and R34 all appear to be able to make salt bridges and hydrogen bonds with acidic patch residues. In particular, BAH R32 is positioned to interact with H2A E61, D90 and E92, analogous to key arginine residues in each of the other nucleosome-binding proteins discussed above. Several of these basic residues affect silencing when mutated [45,46], suggesting that the BAH–acidic patch interaction is physiologically relevant.
3.5. HMGN2
The HMGN family of chromatin architectural proteins binds to nucleosomes, competes with linker histone H1 for nucleosomal sites, and causes chromatin decompaction in vitro [47]. HMGNs also cause decondensation of nucleosomal arrays lacking H1 [48,49]. HMGN proteins have diverse cellular effects. In particular, they act to modulate transcription [47,50,51] and DNA repair [52]. The N-terminal region of the HMGN proteins contains a 30 residue nucleosome-binding domain (NBD). Within the NBDs of all five HMGNs is a stringently conserved core sequence, RRSARLSA [47]. Mutational analyses indicate that the NBD core is solely responsible for mediating HMGN–nucleosome interactions [53]. Insight into the molecular basis of the NBD–nucleosome interaction has come from the studies of Kato et al. [54], who used methyl transverse relaxation optimized NMR (methyl-TROSY) to characterize the structure of HMGN2 bound to a 167 bp nucleosome. The methyl-TROSY experiments followed the methyl groups of Val, Leu and Ile, allowing characterization of the nucleosome-binding sites for HMGN2.
HMGN2 binding affects the methyl groups of H2A L65 and H2B V45 and L103, while the methyl groups on H3 and H4 remain unperturbed. The affected methyl groups are close to the H2A E61, E64, D90, E91 and E92 and H2B E102, suggesting that HMGN2 binds to the nucleosome near the acidic patch. The location of HMGN2 was verified using paramagnetic relaxation enhancement experiments. Several HMGN2 residues were mutated to cysteines, and cross-linked to paramagnetic compound to monitor the distance between residues on the nucleosomes and the HMGN2 paramagnetic centres. The reduction of peak intensities of methyl groups on the nucleosome in proximity to the acidic patch confirmed the HMGN2–acidic patch interaction.
The involvement of the acidic patch in HMGN2 binding was further investigated by mutational analysis and isothermal titration calorimetry [54]. The H2A E61K, E64K, D90S/E91T/E92T and H2B E102T mutations all disrupt HMGN2–nucleosome interactions, confirming that the NBD binds to acidic patch residues. NBD R22 and R26 were shown to be necessary for binding, highlighting the importance of these HMGN2 residues. Kato et al. [54] also performed computational modelling of the NBD complexed to the nucleosome (figure 3d). HMGN2 residues 22–29, comprising the conserved core sequence were modelled to form an extended strand that extensively interacts with the acidic patch along its entire length. NBD R26 is positioned to make salt bridges and hydrogen bonds with H2A E61, D90 and D92, while R23 and S24 are poised to interact electrostatically with H2A E61 and E64. Finally, it is interesting to note that phosphorylation of NBD S24 and S28 disrupt HMGN2–chromatin interactions in vitro, and S24,28E mutation disrupts HMGN2–chromatin interaction in vivo [55,56], presumably because of charge repulsion with the acidic patch residues [47,54].
3.6. Overlapping complementarity
LANA, IL-33, RCC1, Sir3 and HMGN2 each display the precise hydrogen bond complementarity, ionic complementarity and steric complementarity found at any protein–protein interface (figure 3). However, the interfaces physically overlap (figure 4). This phenomenon extends to the atomic level; LANA, IL-33, RCC1, Sir3 and HMNGN2 all have a strategically located arginine residue that forms charged hydrogen bonds with H2A E61, D90 and D92 of the acidic patch. An important ramification of these findings is that only a single nucleosome-binding protein will be able to interact with the surface of a particular nucleosome at any time. As will be described in §10, this brings into play competition and the law of mass action.
Figure 4.
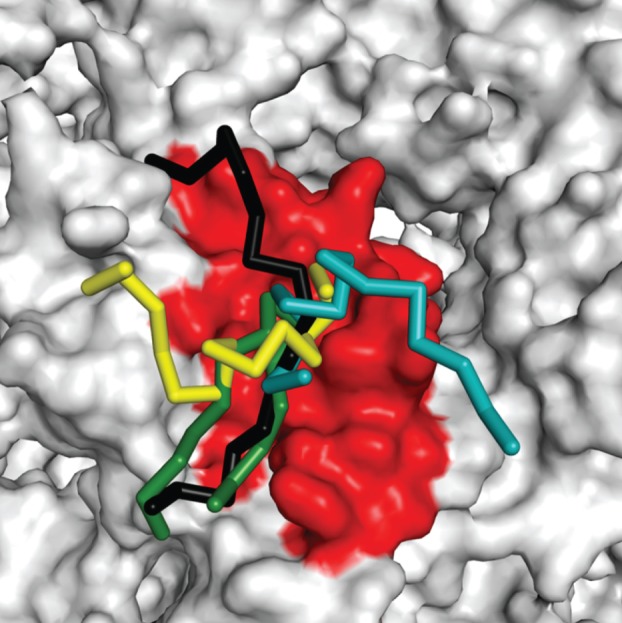
Nucleosome-binding proteins display overlapping complementarity with the acidic patch. The regions of LANA (green), RCC1 (yellow), Sir3 (cyan) and HMGN2 (black) that interact with the acidic patch are overlaid together. Peptides are shown in backbone view in which the α-carbons are connected by straight tubes. The nucleosome is shown in surface mode. The acidic patch region is shaded red.
4. Hypothesis: nucleosome-binding proteins interact with the acidic patch to trigger remodelling of higher order chromatin structure
In the absence of nucleosome-binding proteins, the higher order structure of the chromatin fibre in any given region of the genome will be dictated by intrinsic nucleosome–nucleosome interactions mediated by H4 tail–acidic patch contacts (§2). Does this ‘ground state’ structure become altered when nucleosome-binding proteins associate with the chromatin fibre? We believe the answer is yes. Now, there are five proteins for which co-structures with the nucleosome have been determined experimentally or obtained by computational methods. We find it striking that each of these proteins also binds to the nucleosome in part through interactions with the surface acidic patch (§3). One could argue that this simply reflects the importance of the acidic patch as a universal chromatin ‘tethering’ domain, if not for the fact that the H4 tail also interacts with the acidic patch during higher order chromatin folding. In this section, we present a speculative model in which nucleosome-binding proteins compete with the H4 tail domain for binding to the acidic patch, triggering remodelling of the higher order structure and functionality of the chromatin fibre.
The key premise of our model is that when a regulatory protein occupies the acidic patch of a given nucleosome, the H4 tail domains from neighbouring nucleosomes will be occluded. In this manner, binding of the regulatory protein will disrupt the ground state nucleosome–nucleosome interactions in the vicinity of the bound protein(s) (figure 5). Of the five nucleosome-binding proteins highlighted in §3, each uses only a small portion of its sequence (e.g. hairpin, loop, extended strand) to interact with the acidic patch. Thus, a direct prediction of our model is that peptides corresponding to the acidic patch binding regions of these proteins will inhibit the formation of higher order chromatin structure in chromatin folding experiments in vitro. Competition for the acidic patch is envisioned as a molecular trigger that initially leads to transient chromatin unfolding. In our hypothesis, once the ground state chromatin structure has been disrupted, the final fibre architecture will be specified by the unique properties of the full-length protein bound to the nucleosome (figure 5). The extremes of the chromatin structures that can be generated through this common mechanism are nicely illustrated by HMGN2 and Sir3. HMGN2 is a small disordered protein that unfolds the chromatin fibre [47–49]. Computational studies suggest that the full-length protein binds to the acidic patch via its nucleosome-binding domain (§3.5; figure 3d), leaving the C-terminal portion of the polypeptide chain in a position to disrupt H1 binding to the nucleosome [54]. The combination of an occupied acidic patch (thereby blocking H4 tail binding), together with the absence of H1, will ensure that the HMGN2-bound chromatin fibre will remain in a fully decondensed state, as has been observed experimentally. Sir3 is a large (approx. 110 kDa) multi-domain protein that helps establish and maintain a transcriptionally silent state at specific regions of the yeast genome [13]. Opposite of HMGN2, Sir3 induces a hypercondensed structure upon binding to chromatin in vitro [14,15]. Like many other nucleosome-binding proteins [57], Sir3 self-associates [58], leading to fibre–fibre cross-linking and extensive reconfiguration of higher order chromatin structure [57]. Based on the available data, we envision that each different nucleosome-binding protein will direct formation of a different specific chromatin architecture that supports its biological function (figure 5). This aspect of our model also is experimentally accessible. For example, we predict that the morphologies of chromatin fibres complexed with purified nucleosome-binding proteins in vitro will range from extended to highly decondensed and will differ for each specific protein studied.
Figure 5.

A model for the mechanism of remodelling of chromatin higher order structure by nucleosome-binding proteins. KGS is the equilibrium constant for the ground state H4 tail–acidic patch interaction. K1 is the equilibrium constant for the interaction of nucleosome-binding protein 1 with the acidic patch. K2 is the equilibrium constant for the interaction of nucleosome-binding protein 2 with the acidic patch. The specific chromatin structure induced by a particular nucleosome-binding protein can range from completely decondensed to hypercondensed. See text for details.
Another important implication of our model is that nucleosome-binding proteins will compete not only with the H4 tail, but also with themselves, for binding to the acidic patch. As shown in figure 4, the interfaces for LANA, IL-33, RCC1, Sir3 and HMGN2 physically overlap, and therefore only a single nucleosome-binding protein will be able to interact with the surface of a particular nucleosome at any time. What are the ramifications of ‘overlapping complementarity’ for the structure of genomic chromatin? Given that there are two acidic patches per nucleosome and many nucleosomes in any given region of a chromatin fibre, genomic chromatin fibre in effect is a complex array of binding sites for proteins that interact with the acidic patch. This brings mass action into play (figure 5). Each nucleosome-binding protein, as well as the H4 tail, will have an equilibrium constant that describes the strength of its interaction with the acidic patch. The local concentration of the H4 tails always will be very high, setting up the ground state chromatin structure. A nucleosome-binding protein will outcompete the H4 tail if it has a much higher equilibrium constant, or if its local concentration becomes very high, leading to structural and functional remodelling of the fibre. As such, the interplay between equilibrium constants and relative protein concentrations will determine which nucleosome-binding proteins are localized to any given region of genomic chromatin in vivo. Considering that each nucleosome-binding protein is predicted to assemble a different specific higher order chromatin structure, it seems likely that genomic chromatin is not structurally homogeneous, but rather is partitioned into many different micro-domains depending on the specific protein composition of the region in question.
Acknowledgements
This work was supported by NIH grant nos GM045916 and GM066834 to J.C.H., GM088409 to K.L. and an International Rett Syndrome post-doctoral fellowship to A.K. K.L. is supported by the Howard Hughes Medical Institute.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 7, 251–260 [DOI] [PubMed] [Google Scholar]
- 2.Hansen JC. 2002. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31, 361–392 10.1146/annurev.biophys.31.101101.140858 (doi:10.1146/annurev.biophys.31.101101.140858) [DOI] [PubMed] [Google Scholar]
- 3.Dorigo B, Schalch T, Bystricky K, Richmond TJ. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327, 85–96 10.1016/S0022-2836(03)00025-1 (doi:10.1016/S0022-2836(03)00025-1) [DOI] [PubMed] [Google Scholar]
- 4.Gordon F, Luger K, Hansen JC. 2005. The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nucleosomal arrays. J. Biol. Chem. 280, 33 701–33 706 10.1074/jbc.M507048200 (doi:10.1074/jbc.M507048200) [DOI] [PubMed] [Google Scholar]
- 5.McBryant S, Klonoski J, Sorensen TC, Norskog SS, Williams S, Resch MG, Toombs JA, Hobdey SE, Hansen JC. 2009. Determinants of histone H4 N-terminal domain function during nucleosomal array oligomerization: roles of amino acid sequence, domain length, and charge density. J. Biol. Chem. 25, 16 716–16 722 10.1074/jbc.M109.011288 (doi:10.1074/jbc.M109.011288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu C-F, Nordenskiold L. 2011. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 39, 1680–1691 10.1093/nar/gkq900 (doi:10.1093/nar/gkq900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fletcher T, Hansen J. 1995. Core histone tail domains mediate oligonucleosome folding and nucleosomal DNA organization through distinct molecular mechanisms. J. Biol. Chem. 270, 25 359–25 362 10.1074/jbc.270.43.25359 (doi:10.1074/jbc.270.43.25359) [DOI] [PubMed] [Google Scholar]
- 8.Schwarz P, Felthauser A, Fletcher T, Hansen J. 1996. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry 35, 4009–4015 10.1021/bi9525684 (doi:10.1021/bi9525684) [DOI] [PubMed] [Google Scholar]
- 9.Tse C, Hansen JC. 1997. Hybrid trypsinized nucleosomal arrays: identification of multiple functional roles of the H2A/H2B and H3/H4 N-termini in chromatin fiber compaction. Biochemistry 36, 11 381–11 388 10.1021/bi970801n (doi:10.1021/bi970801n) [DOI] [PubMed] [Google Scholar]
- 10.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 10.1126/science.1124000 (doi:10.1126/science.1124000) [DOI] [PubMed] [Google Scholar]
- 11.Robinson P, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 2008. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol. 381, 816–825 10.1016/j.jmb.2008.04.050 (doi:10.1016/j.jmb.2008.04.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan JY, Rangasamy D, Luger K, Tremethick DJ. 2004. H2A.Z alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol. cell 16, 655–661 10.1016/j.molcel.2004.10.023 (doi:10.1016/j.molcel.2004.10.023) [DOI] [PubMed] [Google Scholar]
- 13.Rusche LN, Kirchmaier AL, Rine J. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Ann. Rev. Biochem. 72, 481–516 10.1146/annurev.biochem.72.121801.161547 (doi:10.1146/annurev.biochem.72.121801.161547) [DOI] [PubMed] [Google Scholar]
- 14.Georgel PT, Palacios DeBeer MA, Pietz G, Fox CA, Hansen JC. 2001. Sir3-dependent assembly of supramolecular chromatin structures in vitro. Proc. Natl Acad. Sci. USA 98, 8584–8589 10.1073/pnas.151258798 (doi:10.1073/pnas.151258798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBryant SJ, Krause C, Woodcock CL, Hansen JC. 2008. The silent information regulator 3 protein, SIR3p, binds to chromatin fibers and assembles a hypercondensed chromatin architecture in the presence of salt. Mol. Cell. Biol. 28, 3563–3572 10.1128/MCB.01389-07 (doi:10.1128/MCB.01389-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korolev N, Allahverdi A, Lyubartsev A, Nordenskiöld L. 2012. The polyelectrolyte properties of chromatin. Soft Matter 8, 9322–9333 10.1039/c2sm25662b (doi:10.1039/c2sm25662b) [DOI] [Google Scholar]
- 17.Thoma F, Koller T, Klug A. 1979. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 83, 403–426 10.1083/jcb.83.2.403 (doi:10.1083/jcb.83.2.403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allan J, Harborne N, Rau D, Gould H. 1982. Participation of core histone‘ tails’ in the stabilization of the chromatin solenoid. J. Cell Biol. 93, 285–297 10.1083/jcb.93.2.285 (doi:10.1083/jcb.93.2.285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ausio J, Dong F, van Holde KE. 1989. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone ‘tails’ in the stabilization of the nucleosome. J. Mol. Biol. 206, 451–463 10.1016/0022-2836(89)90493-2 (doi:10.1016/0022-2836(89)90493-2) [DOI] [PubMed] [Google Scholar]
- 20.Hong L, Schroth G, Matthews H, Yau P, Bradbury E. 1993. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 ‘tail’ to DNA. J. Biol. Chem. 268, 305–314 [PubMed] [Google Scholar]
- 21.Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ. 2004. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306, 1571–1573 10.1126/science.1103124 (doi:10.1126/science.1103124) [DOI] [PubMed] [Google Scholar]
- 22.Yang D, Arya G. 2011. Structure and binding of the H4 histone tail and the effects of lysine 16 acetylation. Phys. Chem. Chem. Phys. 13, 2911–2921 10.1039/c0cp01487g (doi:10.1039/c0cp01487g) [DOI] [PubMed] [Google Scholar]
- 23.Potoyan D, Papoian G. 2011. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J. Am. Chem. Soc. 133, 7405–7415 10.1021/ja1111964 (doi:10.1021/ja1111964) [DOI] [PubMed] [Google Scholar]
- 24.Potoyan D, Papoian G. 2012. Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc. Natl Acad. Sci. USA 109, 17 857–17 862 10.1073/pnas.1201805109 (doi:10.1073/pnas.1201805109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311, 856–861 10.1126/science.1120541 (doi:10.1126/science.1120541) [DOI] [PubMed] [Google Scholar]
- 26.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644 10.1126/science.284.5414.641 (doi:10.1126/science.284.5414.641) [DOI] [PubMed] [Google Scholar]
- 27.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75, 3948–3959 (doi:10.1128/JVI.75.8.3948-3959.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbera A, Ballestas M, Kaye K. 2004. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J. Virol. 78, 294–301 10.1128/JVI.78.1.294-301.2004 (doi:10.1128/JVI.78.1.294-301.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roussel L, Erard M, Cayrol C, Girard J-P. 2008. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. 9, 1006–1012 10.1038/embor.2008.145 (doi:10.1038/embor.2008.145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. 2007. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl Acad. Sci. USA 104, 282–287 10.1073/pnas.0606854104 (doi:10.1073/pnas.0606854104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carazo-Salas RE, Guarguaglini G, Gruss OJ, Segref A, Karsenti E, Mattaj IW. 1999. Generation of GTP-bound Ran by RCC1 is required for chromatin-induced mitotic spindle formation. Nature 400, 178–181 10.1038/22133 (doi:10.1038/22133) [DOI] [PubMed] [Google Scholar]
- 32.Clarke P, Zhang C. 2008. Spatial and temporal coordination of mitosis by Ran GTPase. Nat. Rev. Mol. Cell Biol. 9, 464–477 10.1038/nrm2410 (doi:10.1038/nrm2410) [DOI] [PubMed] [Google Scholar]
- 33.Kalab P, Heald R. 2008. The RanGTP gradient—a GPS for the mitotic spindle. J. Cell Sci. 121, 1577–1586 10.1242/jcs.005959 (doi:10.1242/jcs.005959) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izaurralde E, Kutay U, Kobbe CV, Mattaj IW, Görlich D. 1997. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 16, 6535–6547 10.1093/emboj/16.21.6535 (doi:10.1093/emboj/16.21.6535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renault L, Nassar N, Vetter I, Becker J, Klebe C, Roth M, Wittinghofer A. 1998. The 1.7 Å crystal structure of the regulator of chromosome condensation (RCC1) reveals a seven-bladed propeller. Nature 392, 97–101 10.1038/32204 (doi:10.1038/32204) [DOI] [PubMed] [Google Scholar]
- 36.Makde RD, England JR, Yennawar HP, Tan S. 2010. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 10.1038/nature09321 (doi:10.1038/nature09321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.England JR, Huang J, Jennings MJ, Makde RD, Tan S. 2010. RCC1 uses a conformationally diverse loop region to interact with the nucleosome: a model for the RCC1–nucleosome complex. J. Mol. Biol. 398, 518–552 (doi:10.1016/j.jmb.2010.03.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rusché L, Kirchmaier A, Rine J. 2002. Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 2207–2222 10.1091/mbc.E02-03-0175 (doi:10.1091/mbc.E02-03-0175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hecht A, Strahl-Bolsinger S, Grunstein M. 1996. Spreading of transcriptional represser SIR3 from telomeric heterochromatin. Nature 383, 92–96 10.1038/383092a0 (doi:10.1038/383092a0) [DOI] [PubMed] [Google Scholar]
- 40.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 11, 83–93 10.1101/gad.11.1.83 (doi:10.1101/gad.11.1.83) [DOI] [PubMed] [Google Scholar]
- 41.Armache K-J, Garlick JD, Canzio D, Narlikar GJ, Kingston RE. 2011. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science 334, 977–982 10.1126/science.1210915 (doi:10.1126/science.1210915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchberger JR, Onishi M, Li G, Seebacher J, Rudner AD, Gygi SP, Moazed D. 2008. Sir3-nucleosome interactions in spreading of silent chromatin in Saccharomyces cerevisiae. Mol. Cell. Biol. 28, 6903–6918 10.1128/MCB.01210-08 (doi:10.1128/MCB.01210-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Connelly J, Yuan P, Hsu H, Li Z, Xu RM, Sternglanz R. 2006. Structure and function of the Saccharomyces cerevisiae Sir3 BAH domain. Mol. Cell. Biol. 26, 3256–3265 10.1128/MCB.26.8.3256-3265.2006 (doi:10.1128/MCB.26.8.3256-3265.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou Z, Danzer JR, Fox CA, Keck JL. 2006. Structure of the Sir3 protein bromo adjacent homology (BAH) domain from S. cerevisiae at 1.95 Å resolution. Protein Sci. 15, 1182–1186 10.1110/ps.052061006 (doi:10.1110/ps.052061006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norris A, Bianchet MA, Boeke JD. 2008. Compensatory interactions between Sir3p and the nucleosomal LRS surface imply their direct interaction. PLoS Genet. 4, e1000301. 10.1371/journal.pgen.1000301 (doi:10.1371/journal.pgen.1000301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone E, Reifsnyder C, McVey M, Gazo B, Pillus L. 2000. Two classes of sir3 mutants enhance the sir1 mutant mating defect and abolish telomeric silencing in Saccharomyces cerevisiae. Genetics 155, 509–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Postnikov Y, Bustin M. 2010. Regulation of chromatin structure and function by HMGN proteins. Biochim. Biophys. Acta 1799, 62–68 10.1016/j.bbagrm.2009.11.016 (doi:10.1016/j.bbagrm.2009.11.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vestner B, Bustin M, Gruss C. 1998. Stimulation of replication efficiency of a chromatin template by chromosomal protein HMG-17. J. Biol. Chem. 273, 9409–9414 10.1074/jbc.273.16.9409 (doi:10.1074/jbc.273.16.9409) [DOI] [PubMed] [Google Scholar]
- 49.Trieschmann L, Alfonso PJ, Crippa MP, Wolffe AP, Bustin M. 1995. Incorporation of chromosomal proteins HMG-14/HMG-17 into nascent nucleosomes induces an extended chromatin conformation and enhances the utilization of active transcription complexes. EMBO J. 14, 1478–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bustin M, Reeves R. 1996. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol. 54, 35–100 10.1016/S0079-6603(08)60360-8 (doi:10.1016/S0079-6603(08)60360-8) [DOI] [PubMed] [Google Scholar]
- 51.Bustin M. 1999. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19, 5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerlitz G. 2010. HMGNs, DNA repair and cancer. Biochim. Biophys. Acta 1799, 80–85 10.1016/j.bbagrm.2009.10.007 (doi:10.1016/j.bbagrm.2009.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ueda T, Catez F, Gerlitz G, Bustin M. 2008. Delineation of the protein module that anchors HMGN proteins to nucleosomes in the chromatin of living cells. Mol. Cell. Biol. 28, 2872–2883 10.1128/MCB.02181-07 (doi:10.1128/MCB.02181-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato H, van Ingen H, Zhou B-R, Feng H, Bustin M, Kay LE, Bai Y. 2011. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl Acad. Sci. USA 108, 12 283–12 288 10.1073/pnas.1105848108 (doi:10.1073/pnas.1105848108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prymakowska-Bosak M. 2001. Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol. Cell. Biol. 21, 5169–5178 10.1128/MCB.21.15.5169-5178.2001 (doi:10.1128/MCB.21.15.5169-5178.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spaulding S, Fucile N, Bofinger DP, Sheflin LG. 1991. Cyclic adenosine 3′, 5′-monophosphate-dependent phosphorylation of HMG 14 inhibits its interactions with nucleosomes. Mol. Endocrinol. 5, 45–50 10.1210/mend-5-1-42 (doi:10.1210/mend-5-1-42) [DOI] [PubMed] [Google Scholar]
- 57.McBryant SJ, Adams VH, Hansen JC. 2006. Chromatin architectural proteins. Chromosome Res. 14, 39–51 10.1007/s10577-006-1025-x (doi:10.1007/s10577-006-1025-x) [DOI] [PubMed] [Google Scholar]
- 58.McBryant SJ, Krause C, Hansen JC. 2006. Domain organization and quaternary structure of the Saccharomyces cerevisiae silent information regulator 3 protein, Sir3p. Biochemistry 45, 15 941–15 948 10.1021/bi061693k (doi:10.1021/bi061693k) [DOI] [PubMed] [Google Scholar]