

Abstract
Conventional synthesis of polymers by ATRP is relatively low throughput, involving iterative optimization of conditions in an inert atmosphere. Automated, high-throughput controlled radical polymerization was developed to accelerate catalyst optimization and production of disulfide-functionalized polymers without the need of an inert gas. Using ARGET ATRP, polymerization conditions were rapidly identified for eight different monomers, including the first ARGET ATRP of 2-(diethylamino)ethyl methacrylate and di(ethylene glycol) methyl ether methacrylate. In addition, butyl acrylate, oligo(ethylene glycol) methacrylate 300 and 475, 2-(dimethylamino)ethyl methacrylate, styrene, and methyl methacrylate were polymerized using bis(2-hydroxyethyl) disulfide bis(2-bromo-2-methylpropionate) as the initiator, tris(2-pyridylmethyl)amine as the ligand, and tin(II) 2-ethylhexanoate as the reducing agent. The catalyst and reducing agent concentration was optimized specifically for each monomer, and then a library of polymers was synthesized systematically using the optimized conditions. The disulfide-functionalized chains could be cleaved to two thiol-terminated chains upon exposure to dithiothreitol, which may have utility for the synthesis of polymer bioconjugates. Finally, we demonstrated that these new conditions translated perfectly to conventional batch polymerization. We believe the methods developed here may prove generally useful to accelerate the systematic optimization of a variety of chemical reactions and polymerizations.
INTRODUCTION
The optimization of reaction conditions is a routine and time-consuming challenge for most synthetic chemists and can often be rate-limiting in scientific progress. We developed a systematic approach to accelerate this process in the context of polymer science. The discovery of activator generated by electron transfer (AGET)1 atom transfer radical polymerization (ATRP),2–6 which enables polymerizations to be conducted without freeze–pump–thaw cycles,7 has greatly simplified the setup of ATRP and poised it to reach new areas of research.6,7 In addition to elimination of the inert gas requirement, activators regenerated by electron transfer (ARGET) ATRP8–10 has reduced the amount of required copper catalyst to ppm levels.11 However, the discovery of AGET and ARGET ATRP requires new polymerization conditions to optimize polymerization kinetics and control functionality. This process is typically iterative and relatively low throughput, thereby limiting the diversity and number of conditions that can be explored. Here we develop methods that can facilitate the rapid development of polymerization conditions for new monomers and accelerate the production of functional polymers of tunable length without the need of an inert gas.
Automated, high-throughput (HT) methods are important research tools for the synthesis and screening of small molecules12–17 and polymers.18–26 Such methods have resulted in the discovery of drugs such as sorafenib27 and polymerization catalysts, including new polyolefin catalysts.28 Efficient reaction optimization is a key component of chemical research, which requires significant time input. One important challenge is to adapt new chemistries such as ARGET ATRP to robotic platforms, thereby accelerating optimization. Combinatorial strategies are useful in a number of areas, including in polymer science.18–25,28–38 Anionic35 and cationic39 polymerization, reversible addition–fragmentation chain transfer (RAFT) polymerization,34 and ATRP40–43 methods have been developed for automated platforms. However, the limiting factor is that the synthesis of narrowly distributed macromolecules by living polymerization usually requires very clean reaction conditions and an inert atmosphere. Progress in this area so far has required customized reaction setups with a continuous stream of argon or physical placement of the robot inside of a glovebox.36,41,43
To address these challenges, we sought to develop automated ARGET ATRP with the following aims: (1) to rapidly optimize polymerization conditions without deoxygenation, including conditions for the polymerization of new monomers, (2) to synthesize polymers of increasing lengths with disulfide functionality, and (3) validate the optimized conditions by translating automated synthesis to conventional batch polymerization. The development of ARGET ATRP in the presence of limited amounts of air (in which the oxygen is eventually consumed) allowed for automation of ATRP in sealed vials without deoxygenation. Since all of the polymers contain disulfide bridges in the center of the chains, and can be cleaved to two thiol-terminated chains, they can potentially be conjugated to other molecules such as drugs, siRNA, and proteins. The conditions developed on the robot translated to conventional batch polymerization in a chemical fume hood, suggesting that this method of reaction optimization has potential to provide valuable conditions for general purpose ARGET ATRP of various monomers.
EXPERIMENTAL SECTION
Materials
Bis(2-hydroxyethyl) disulfide, bromo-2-methylpropionic acid, N,N′-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino)-pyridine (DMAP), DL-dithiothreitol (DTT), tin(II) 2-ethylhexanoate (Sn(Oct)2), copper(I) bromide (99.999% trace metals basis) (CuBr), copper(II) bromide (99.999% trace metals basis) (CuBr2), tetrahydrofuran (anhydrous) (THF), anisole (anhydrous), and ethanol (200 proof) were purchased from Aldrich and used as received. Tris(2-pyridylmethyl)amine (TPMA) was purchased from ATRP Solutions and used as received. Bis[2-(2-bromoisobutyryloxy)ethyl] disulfide ((BiBOE)2S2) was synthesized following a literature procedure.44 Butyl acrylate (BA), di(ethylene glycol) methyl ether methacrylate (DEGMEMA), oligo(ethylene glycol) methacrylate 300 (OEOMA300, poly(ethylene glycol) methyl ether methacrylate, Mn = 300 g/mol), oligo(ethylene glycol) methacrylate 475 (OEOMA475, poly(ethylene glycol) methyl ether methacrylate, Mn = 475 g/mol), 2-(dimethylamino)ethyl methacrylate (DMAEMA), 2-(diethylamino)-ethyl methacrylate (DEAEMA), styrene (Sty), and methyl methacrylate (MMA) were purchased from Aldrich and passed through a short column containing basic aluminum oxide to remove the inhibitor before use. Unless otherwise noted, all other chemicals were obtained from commercial sources and used without further purification.
HTP Synthesis
Robotic polymerizations were performed on a customized Symyx Core Module equipped with four different liquid dispensing elements (a single tip, a motor driven gripper with a heated piercing tip, a positive displacement tip, and a heated parallel 4-tip). Library Studio (Symyx Discovery Tools) was used to design the polymer libraries. Automation of this process accelerated not only material handling but also data manipulations and reaction calculations. The MW, density, drawn chemical structure, and dissolved concentration were entered into Library Studio to enable rapid calculation of reaction stoichiometry, taking into account the MW, density, structure, and concentration. Polymerizations were conducted in 4 mL vials equipped with a stir bar and sealed with a rubber septum. The reactants were transferred from stock solutions in 20 mL vials into 4 mL reaction vessels using the piercing tip. The total volume of all components per vessel was fixed to 3.7 mL (0.3 mL of air). All experiments followed a set algorithm: (1) Set stirring to 300 rpm, (2) transfer monomer, (3) transfer initiator, (4) transfer catalyst (inactive, oxidized form), (5) transfer reducing agent (starts polymerization by reducing the catalyst after consumption of air), (6) set temperature to 45 °C, (7) wait 16 h, and (8) cool down to room temperature and stop stirring.
A typical polymerization was as follows: For the synthesis of PMMA with a targeted DP of 135, 1.042 mL of MMA (9.82 mmol) was added to a 4 mL vial equipped with a stir bar and rubber septum. 319.0 μL of a 100 mg/mL solution of (BiBOE)2S2 in anisole (31.9 mg, 70.57 μmol of (BiBOE)2S2) was added upon stirring. 2.014 mL of a catalyst solution of CuBr2 (2.7 mmol/L, 1.19 mg, 5.32 μmol) and TPMA (6.5 mmol/L, 3.80 mg, 13.09 μmol) in anisole was transferred to the vial. 324.1 μL of a 136.4 mg/mL solution of Sn(Oct)2 (44.20 mg, 109.1 μmol) was added, and the stirred mixture was heated to 45 °C for 16 h.
Optimization of the Reducing Agent Concentration
For each of the eight monomers, a polymerization was carried out at 12 different concentrations of reducing agent. The catalyst concentration was held constant in all polymerizations, which was 0.037:1 (catalyst:initiator, mol:mol). The targeted degree of polymerization (DP) was 75 for OEOMA475, 110 for OEOMA300, and 150 all other monomers. The molar ratios of Sn(Oct)2 to the initiator for the OEOMA polymers were 0.30, 0.45, 0.60, 0.75, 0.90, 1.05, 1.20, 1.35, 1.50, 1.65, 1.80, and 1.95 to 1.00; for all other polymerizations 0.30, 0.60, 0.90, 1.20, 1.50, 1.80, 2,25, 3.15, 4.05, 4.95, 5.85, and 6.75 to 1.00. In total, 96 separate polymerizations were carried out to optimize the reducing agent concentration.
Optimization of the Catalyst Concentration
For each monomer, the polymerization was carried out at 12 different concentrations of catalyst. The reducing agent concentration was held constant in all reactions. The targeted DP was 75 for OEOMA475, 110 for OEOMA300, and 150 all other monomers. The molar ratios of copper(II) bromide to the initiator were 0.031, 0.037, 0.042, 0.050, 0.056, 0.062, 0.075, 0.087, 0.1, 0.112, 0.125, and 0.137 to 1.00. In total, 96 separate polymerizations were carried out to optimize the catalyst concentration.
Synthesis of Polymers with Different Chain Length
Using the results from the optimization for the concentration of catalyst and reducing agent, polymers were synthesized with different chain lengths. For POEOMA the targeted DP was 15, 30, 45, 60, 67.5, 75, 82.5, 90, 97.5, 105, 120, and 150; for all other polymers, it was 22.5, 45, 67.5, 90, 112.5, 135, 157.5, 180, 202.5, 225, 300, and 375. The concentrations of catalyst and reducing agent used are listed in Table 1. In total, 96 polymers were synthesized with various lengths.
Table 1.
Optimized Molar Ratios of Catalyst and Reducing Agent Used for the Synthesis of Polymers with Different Targeted Degree of Polymerization
| monomer | [CuBr2]/[I] | [Red]/[I] |
|---|---|---|
| MMA | 0.075 | 1.50 |
| DEGMEMA | 0.100 | 0.90 |
| OEOMA300 | 0.062 | 0.75 |
| OEOMA475 | 0.062 | 1.50 |
| DMAEMA | 0.050 | 1.20 |
| DEAEMA | 0.056 | 1.80 |
| BA | 0.062 | 1.50 |
| Sty | 0.050 | 2.25 |
Batch Polymerization of MMA in a Round-Bottom Flask
We equipped a standard 5 mL round-bottom flask with a standard 14/20 rubber septum and stir bar (VWR 58948-116 polygon spinbar, 1/2 in. × 5/16 in.) and measured that this flask can hold 6.6 mL of solution. We based the calculation such that the vial would contain 92.5% solution and 7.5% air (analogous to the automated polymerization condition), setting the total volume to be 6.105 mL. For the synthesis of PMMA with a targeted DP of 135, 1.695 mL of MMA (1.598 g, 15.97 mmol) was added via pipet to a 5 mL vial equipped with a stir bar. 534.6 μL of a 100 mg/mL solution of (BiBOE)2S2 in anisole (53.46 mg, 118.3 μmol of (BiBOE)2S2) was added via pipet. 3.347 mL of a catalyst solution of CuBr2 (2.7 mmol/L, 1.98 mg, 8.87 μmol) and TPMA (6.5 mmol/L, 6.31 mg, 21.76 μmol) in anisole was transferred to the flask via pipet. The flask was sealed with a rubber septum and secured tightly using electrical tape. 0.527 mL of a 136.4 mg/mL solution of Sn(Oct)2 (71.88 mg, 177.4 μmol) was injected via syringe to start the polymerization. The reaction was placed into a preheated oil bath set to 45 °C and stirred at 300 rpm for 16 h.
Batch Polymerization of DMAEMA in a Round-Bottom Flask
Similar to the polymerization of MMA, we set the total volume to be 6.105 mL for a standard 5 mL round-bottom flask. For the synthesis of PDMAEMA with a targeted DP of 135, 2.794 mL of DMAEMA (2.607 g, 16.58 mmol) was added via pipet to a 5 mL vial equipped with a stir bar. 555.2 μL of a 100 mg/mL solution of (BiBOE)2S2 in anisole (55.52 mg, 122.8 μmol of (BiBOE)2S2) was added via pipet. 2.317 mL of a catalyst solution of CuBr2 (2.7 mmol/L, 1.37 mg, 6.14 μmol) and TPMA (6.5 mmol/L, 4.36 mg, 15.06 μmol) in anisole was transferred to the flask via pipet. The flask was sealed with a rubber septum and secured tightly using electrical tape. 0.438 mL of a 136.4 mg/mL solution of Sn(Oct)2 (59.71 mg, 146.6 μmol) was injected via syringe to start the polymerization. The reaction was placed into a preheated oil bath set to 45 °C and stirred at 300 rpm for 16 h.
Reduction of the Disulfide Bond Using DTT
The cleavage of the disulfide bonds was performed in THF using DTT. 1 mL of the polymer solution (3 mg/mL in THF) was transferred into a 4 mL vial. 2 mL of a DTT solution (0.1 g/mL in THF) was added. The mixture was stirred in a sealed vial for 4 days at 50 °C, and samples for GPC measurements were taken every 24 h. In a parallel control without DTT, solutions of the same polymers were stirred, heated to 50 °C, and analyzed every 24 h.
Purification
The copper catalyst and the reducing agent were removed by passing the polymer solution through a neutral alumina oxide matrix. The simultaneous purification of 24 samples was performed using a Visiprep Vacuum Manifold (Supelco). After the polymerization was finished, 0.2 mL of polymer solution was transferred into an 8 mL glass vial and diluted with 3 mL of THF. Twenty-four neutral aluminum oxide solid-phase extraction (SPE) cartridges (Waters) were placed on the manifold and filled with the diluted polymer solution. The pressure in the chamber was reduced, and the purified polymer solution was collected in 8 mL glass vials. After filtration through a 0.2 μm PTFE syringe filter, the solution was used for GPC analysis without further dilution.
GPC Analysis
All polymers were analyzed using a Waters GPC system equipped with a 2400 differential refractometer, 515 pump, and 717-plus autosampler. The flow rate was 1 mL/min, and the mobile phase was THF. The Styragel columns (Waters) and detector were thermostated at 35 °C. Linear polystyrene standards were used for calibration.
RESULTS AND DISCUSSION
Monomer and Catalyst Selection
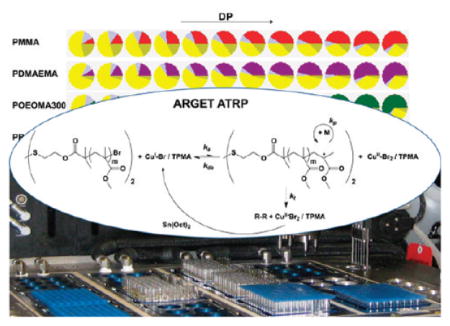
Catalyst optimization is an important element of ATRP. In order to conduct controlled polymerizations, the molar ratios and concentrations must be systematically varied in order to determine ideal polymerization conditions. Automated ARGET ATRP aided in this process, in terms of both the calculations in Library Studio and the physical dispensing of reactants. ARGET ATRP can control the polymerization of many different kinds of monomers, and we sought to investigate polymerization conditions for a range of monomers using automated methods (Figure 1). We selected eight monomers, which were representative of acrylate, methacrylate, styrenic, PEG mimic, and cationic classes (Figure S1). For development, we used copper(II) bromide (CuBr2), tris(2-pyridylmethyl)amine (TPMA) as the ligand, tin(II) 2-ethylhexanoate (Sn(Oct)2) as the reducing agent, and anisole as the solvent (except for the PEG-based monomers, where ethanol was used). First, the required concentrations of reducing agent and catalyst were systematically optimized. In the second phase, various narrowly distributed polymers were synthesized using the optimized conditions. Bis[2-(2-bromoisobutyryloxy)ethyl] disulfide ((Bi-BOE)2S2)44 was used as the initiator because it allows for the synthesis of disulfide-functionalized polymers that can be subsequently cleaved into two thiol-terminated chains. The choice of this initiator limited the selection of the reducing agent for ARGET ATRP. Sn(Oct)2 has an appropriate redox potential, where copper(II) is reduced to copper(I), but the disulfide bridge is not cleaved during the polymerization. Preliminary experiments verified that the disulfide bond is not reduced by Sn(Oct)2.
Figure 1.

Automated ARGET ATRP enabled rapid catalyst optimization and efficient synthesis of a polymer library that can be tailored for specific applications.
The process of optimization yielded information for appropriate concentrations of catalyst and reducing agent for various monomers. To the best of our knowledge, the polymerization of 2-(diethylamino)ethyl methacrylate (DEAE-MA) and diethylene glycol methyl ether methacrylate (DEGMEMA) by ARGET ATRP has not yet been reported in the literature. Oligo(ethylene glycol) methyl ether methacrylate (OEOMA) has been polymerized by ATRP, RAFT, and other methods and found applications in biomedical research as free-radically polymerizable PEG mimics.45–50 For all 288 polymerizations, the total volume was held constant at 3.7 mL (0.3 mL of air), and a single algorithm was used to standardize the results. All components were added to vials sealed with a rubber septum. After addition of the reducing agent via a piercing robotic tip, the polymerization could begin upon consumption of the air and reduction of CuII to CuI. The available amount of oxygen in the reaction vessel directly affects the required amount of reducing agent. At the beginning of the process, CuII is reduced by the reducing agent to CuI, which is immediately oxidized by oxygen in the air. However, additional reducing agent can resume the process. This cycle is repeated until all of the oxygen in the reaction vessel is completely consumed. After this induction period, the polymerization proceeds.
Optimization of the Reducing Agent Concentration
Twelve different concentrations of reducing agent were tested at constant catalyst concentration and fixed degree of polymerization (DP). All polymers were analyzed by gel permeation chromatography (GPC), and the distribution curves were plotted in the same diagram (Figure 2) (for clarity, the figure does not show all curves). The curve for the ideal concentration of reducing agent is red, where (1) the polydispersity index (PDI) was as low as possible, (2) the deviation between theoretical and experimental MW was as low as possible, and (3) the shape of the curve was most ideal (one maximum, no shoulders). (4) For similar results, the lower concentration of reducing agent was favored. In a typical ATRP process, PDI decreases with conversion and with degree of polymerization. This can be attributed to relatively slow exchange between active and dormant species (especially important in low Cu systems) and to slow initiation. However, PDI increases with conversion in the presence of chain breaking reactions (transfer and termination). Although conversion would be a good way to account for these differences, we focused on measurement of MW by GPC because it was more amenable to this high-throughput study. Overall, the results for these experiments were within the expected range of required reducing agent to consume all air in the vial.7,11 In all experiments, the air volume in each reaction vessel was 300 μL, which corresponds to 0.0028 mmol of oxygen. In addition to reducing CuII to CuI, 300 μL of air requires an additional 0.0056 mmol of Sn(Oct)2 because for each equivalent of oxygen, 4 equiv of CuI can be oxidized to CuII, but only 2 CuII equiv are reduced back by 1 equiv of Sn(Oct)2.
Figure 2.

Optimization of the reducing agent concentration. Selected traces for the polymerization of MMA, DEGMEMA, OEOMA300, OEOMA475, DMAEMA, DEAEMA, BA, and Sty show the effect of changing the reducing agent concentration. Optimal results are indicated in red.
The optimal ratio of reducing agent to initiator for methyl methacrylate (MMA) was determined to be 1.5:1 (mol:mol), where the resulting polymer Mn was close to theoretical and the PDI was 1.16. Increasing the reducing agent for MMA did not negatively affect the molecular weight distribution. In contrast, 1.5:1 for DEGMEMA resulted in a broad PDI and a shoulder in the GPC trace, indicating loss of control. The optimum value for DEGMEMA was 0.9:1. Polymerization of OEOMA300 showed a similar behavior, where 0.3:1 and below resulted in low Mn, and increasing the ratio to above 1.1:1 showed peak broadening. The optimum was found to be 0.8:1. When increasing the PEG side chain length of the PEG-based monomer OEOMA475, a 1.5:1 ratio was found to be optimum. Controlled polymerization of 2-(dimethylamino)ethyl methacrylate (DMAEMA) was observed over a wide range of reducing agent from 0.6 to 2.3. This may be due to the monomer’s ability to serve as an intrinsic reducing agent, thereby limiting the effect of Sn(Oct)2 addition.51 Less control was observed for polymerization of DEAEMA over this range, with the lowest PDI (1.46) achieved at a 1.8:1 ratio. For butyl acrylate (BA), a higher ratio of reducing agent to initiator (3.2:1) was required to obtain a DP close to the targeted DP. However, the doubling of MW suggests the possibility of polymer chain coupling at high reducing agent concentrations. Better control was observed at 1.5:1; however, the DP reached was lower than theoretical. Low degrees of polymerization were also observed for polymerization of styrene (Sty).
Optimization of the Catalyst Concentration
We next optimized the catalyst concentration. Stock solutions of preformed catalyst7 were used in all polymerizations with the molar ratio TPMA:CuBr2 = 2.4:1. Twelve different concentrations of catalyst at constant concentration of reducing agent and constant DP were tested (0.031–0.137 times the initiator mole concentration) for all eight monomers. All samples were analyzed by GPC, and the distribution curves for each polymer type are shown in Figure 3. The selection of the optimal concentration was done in accordance with those criteria listed above and is shown as the red trace in all plots. Ideal catalyst concentrations for seven of the eight monomers were between 0.05 and 0.075 times the initiator concentration (mol). Although the PDI remained low for PMMA, a slightly increasing PDI was observed at higher CuBr2 concentrations, with a broadening GPC trace. For DEGMEMA, the optimal value was 0.1. Nonideal curves containing shoulders were observed for values above and below 0.1. OEOMA300 and OEOMA475 polymerized similarly and required a lower amount of catalyst (0.062 or lower). Higher catalyst levels resulted in lower obtained MW. For DMAEMA and BA, the polymerizations were successful over a wide range of catalyst concentrations, with an optimal range of 0.05–0.062. DEAEMA was also polymerizable using a range of concentrations; however, the low and high concentrations of catalyst led to longer chains and higher PDI (>1.5). In theory, a catalytic amount of up to 1 mol % relative to monomer is required for ATRP.11 In these experiments, six of the eight monomers required 5–6 mol % of catalyst relative to the initiator. In comparison, a previous study in the presence of air without robotic automation used 2–3 times less catalyst.7 Our studies here have shown that slightly higher catalyst concentrations can lead to better controlled polymerization, at least in automated systems, where vented robotic needles may allow for more air transfer upon addition of the reducing agent.
Figure 3.

Optimization of the catalyst concentration for polymerization. Selected traces for the polymerization of MMA, DEGMEMA, OEOMA300, OEOMA475, DMAEMA, DEAEMA, BA, and Sty show the effect of changing catalyst concentration. Optimal results are indicated in red.
Synthesis of Disulfide-Bridged Polymers with Different Lengths
Using optimized concentrations of catalyst and reducing agent for each monomer (Table 1), we targeted 12 different DPs for each polymer type (from 15 to 375). Optimization of the polymerization conditions yielded conditions that allowed for the controlled synthesis of various polymers with increasing lengths. In the case of MMA, the experimental DP obtained was close to the theoretical DP, with a narrow MW distribution (PDI < 1.2). Polymerization of DEGMEMA resulted in the broadest distribution for polymers (PDI = 1.37–1.82). Decent control and increasing MW were observed for OEOMA300, OEOMA475, DEGMEMA, DMAEMA, and DEAEMA, which may have biomedical application. P(OEOMA475) had a PDI of 1.18–1.26 with close agreement between theoretical and experimental DP. P(OEOMA300), however, had higher PDI values and a shoulder in the curves above a length of 73 units, but all measured molecular weights were close to the theoretical. The shoulder might be due to chain transfer reactions, since we did not observe evidence for dimethacrylate impurities in LC-MS measurements. Polymerization of DMAEMA resulted in DPs from 16 to 213 units, where the PDI was 1.26–1.37. Similar results were obtained for PDEAEMA, but with slightly broader PDI. The highest experimental DPs obtained were DP 271 for PDEAEMA (Mn 51 400 g/mol; PDI 1.54) and DP 251 for PMMA (Mn 25 600 g/mol; PDI 1.16) (Figure 4). In all cases, the MW increased with higher targeted DP. A comparison between theoretical and measured Mn (calculated by GPC) verified this trend and showed that all monomers (except for Sty) could be polymerized to high DP due to successful optimization of the catalyst and reducing agent concentration. Overall, the optimization of catalyst and reducing agent concentrations allowed for the production of various polymers with increasing length and control over MW and MWD.
Figure 4.

Using optimized concentrations of catalyst and reducing agent, a series of polymers were synthesized by automated ARGET ATRP.
Polymer Cleavage to Thiol
In order to demonstrate the ability to cleave these disulfide-containing polymers into two thiol-terminated chains of half the MW, dithiothreitol (DTT) was used to cleave the disulfide bond. The resulting thiol-terminated chains have the potential for conjugation to (bio)molecules. Figure S2 shows GPC traces for selected reactions with DTT in THF at 50 °C. Cleavage of PMMA was complete within 24 h, while PBA was partially reduced after 96 h. The rate of cleavage for PSty was slower, but reduction was nevertheless observed. The MW of P(OEOMA475) also decreased, but the broader PDI and steric hindrance of this polymer resulted in slower reduction.
Translation of Automated Synthesis to Conventional Batch Synthesis
We have shown that automated methods can facilitate the rapid development of polymerization conditions for various monomers. However, most chemists synthesize polymers using traditional glassware in chemical fume hoods. To validate that these new conditions have practical utility, we investigated if these robotic conditions in glass vials would translate to conventional batch synthesis in a round-bottom flask. In our automated synthesizer, the 4 mL vials contained 92.5% solution and 7.5% air. We used that ratio of total solution volume to air when calculating the set volume for a standard 5 mL round-bottom flask. Optimized conditions for polymerization of MMA and DMAEMA (Table 1) were used in the calculation. As shown in Figure 5, the results from the automated and batch syntheses matched, proving that the automated conditions directly translated to conventional batch polymerization in standard glassware. Nearly identical experimental DP and MW were obtained.
Figure 5.

Using the optimized conditions determined by automated polymerization, conventional batch polymerization in round-bottom flasks closely matched robotic polymerization.
CONCLUSION
Efficient reaction optimization is a key component of chemical research. We developed a systematic approach to accelerate reaction optimization. ARGET ATRP conditions were rapidly identified for representative monomers of acrylate, methacrylate, styrenic, PEG mimic, and cationic classes, including the first ARGET ATRP of DEAEMA and DEGMEMA. Because a functional initiator was used, all polymers were end functionalized with thiol groups after cleavage of the disulfide bond. This thiol group can be utilized for attachment to other molecules and may be useful for the synthesis of bioconjugates to drugs, proteins, and siRNA. These conditions translated to conventional batch polymerization, suggesting that these new polymerization conditions will be useful for other scientists to follow when carrying out ARGET ATRP. We therefore believe that the methods developed here may prove generally useful to accelerate the systematic optimization of a variety of reactions across the chemical disciplines.
Supplementary Material
Acknowledgments
D.J.S. was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) for Individual Postdoctoral Fellows, Award F32EB011867 from the National Institutes of Health (NIH). M.L. was supported by the Studienstiftung des deutschen Volkes and the Graduate School of Excellence MAINZ. The research was partially funded by a grant from Alnylam Pharmaceuticals. We thank Arturo Vegas, Patrick Fenton, and Sean P. Collins for assistance with robotic automation and polymer purification.
Footnotes
Figures S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jakubowski W, Matyjaszewski K. Macromolecules. 2005;38:4139–4146. [Google Scholar]
- 2.Wang J-S, Matyjaszewski K. J Am Chem Soc. 1995;117:5614–5615. [Google Scholar]
- 3.Kato M, Kamigaito M, Sawamoto M, Higashimura T. Macromolecules. 1995;28:1721–1723. [Google Scholar]
- 4.Matyjaszewski K, Xia JH. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 5.Braunecker WA, Matyjaszewski K. Prog Polym Sci. 2007;32:93–146. [Google Scholar]
- 6.Matyjaszewski K, Tsarevsky N. Nature Chem. 2009;1:276–288. doi: 10.1038/nchem.257. [DOI] [PubMed] [Google Scholar]
- 7.Matyjaszewski K, Dong H, Jakubowski W, Pietrasik J, Kusumo A. Langmuir. 2007;23:4528–4531. doi: 10.1021/la063402e. [DOI] [PubMed] [Google Scholar]
- 8.Matyjaszewski K, Jakubowski W, Min K, Tang W, Huang J, Braunecker W, Tsarevsky N. Proc Natl Acad Sci U S A. 2006;103:15309–15314. doi: 10.1073/pnas.0602675103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakubowski W, Min K, Matyjaszewski K. Macromolecules. 2006;39:39–45. [Google Scholar]
- 10.Jakubowski W, Matyjaszewski K. Angew Chem, Int Ed. 2006;45:4482–4486. doi: 10.1002/anie.200600272. [DOI] [PubMed] [Google Scholar]
- 11.Pintauer T, Matyjaszewski K. Chem Soc Rev. 2008;37:1087–1097. doi: 10.1039/b714578k. [DOI] [PubMed] [Google Scholar]
- 12.Geysen H, Schoenen F, Wagner D, Wagner R. Nat Rev Drug Discovery. 2003;2:222–230. doi: 10.1038/nrd1035. [DOI] [PubMed] [Google Scholar]
- 13.Lowe G. Chem Soc Rev. 1995;24:309–317. [Google Scholar]
- 14.Thompson LA, Ellman JA. Chem Rev. 1996;96:555–600. doi: 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- 15.Rohrer SP, Birzin ET, Mosley RT, Berk SC, Hutchins SM, Shen DM, Xiong YS, Hayes EC, Parmar RM, Foor F, Mitra SW, Degrado SJ, Shu M, Klopp JM, Cai SJ, Blake A, Chan WWS, Pasternak A, Yang LH, Patchett AA, Smith RG, Chapman KT, Schaeffer JM. Science. 1998;282:737–740. doi: 10.1126/science.282.5389.737. [DOI] [PubMed] [Google Scholar]
- 16.Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 17.Ramstrom O, Lehn JM. Nat Rev Drug Discovery. 2002;1:26–36. doi: 10.1038/nrd704. [DOI] [PubMed] [Google Scholar]
- 18.Xiang XD, Sun XD, Briceno G, Lou YL, Wang KA, Chang HY, Wallacefreedman WG, Chen SW, Schultz PG. Science. 1995;268:1738–1740. doi: 10.1126/science.268.5218.1738. [DOI] [PubMed] [Google Scholar]
- 19.Brocchini S, James K, Tangpasuthadol V, Kohn J. J Am Chem Soc. 1997;119:4553–4554. [Google Scholar]
- 20.Tian J, Coates GW. Angew Chem, Int Ed. 2000;39:3626–3629. [PubMed] [Google Scholar]
- 21.Anderson DG, Lynn DM, Langer R. Angew Chem, Int Ed. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 22.Anderson DG, Levenberg S, Langer R. Nature Biotechnol. 2004;22:863–866. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 23.Hoogenboom R, Meier MAR, Schubert US. Macromol Rapid Commun. 2003;24:16–32. [Google Scholar]
- 24.Meier MAR, Schubert US. J Mater Chem. 2004;14:3289–3299. [Google Scholar]
- 25.Hook A, Anderson D, Langer R, Williams P, Davies M, Alexander M. Biomaterials. 2010;31:187–198. doi: 10.1016/j.biomaterials.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 26.Siegwart DJ, Whitehead KA, Nuhn L, Sahay G, Cheng H, Jiang S, Ma ML, Lytton-Jean A, Vegas A, Fenton P, Levins CG, Love KT, Lee H, Cortez C, Collins SP, Li YF, Jang J, Querbes W, Zurenko C, Novobrantseva T, Langer R, Anderson DG. Proc Natl Acad Sci U S A. 2011;108:12996–13001. doi: 10.1073/pnas.1106379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Nat Rev Drug Discovery. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 28.Boussie T, Diamond G, Goh C, Hall K, LaPointe A, Leclerc M, Lund C, Murphy V, Shoemaker J, Tracht U, Turner H, Zhang J, Uno T, Rosen R, Stevens J. J Am Chem Soc. 2003;125:4306–4317. doi: 10.1021/ja020868k. [DOI] [PubMed] [Google Scholar]
- 29.Bosman AW, Heumann A, Klaerner G, Benoit D, Frechet JMJ, Hawker CJ. J Am Chem Soc. 2001;123:6461–6462. doi: 10.1021/ja010405z. [DOI] [PubMed] [Google Scholar]
- 30.Hoogenboom R, Schubert US. J Polym Sci, Part A: Polym Chem. 2003;41:2425–2434. [Google Scholar]
- 31.Hoogenboom R, Fijten MWM, Abeln CH, Schubert US. Macromol Rapid Commun. 2004;25:237–242. [Google Scholar]
- 32.Fijten MWM, Meier MAR, Hoogenboom R, Schubert US. J Polym Sci, Part A: Polym Chem. 2004;42:5775–5783. [Google Scholar]
- 33.Tweedie CA, Anderson DG, Langer R, Van Vliet KJ. Adv Mater. 2005;17:2599–2604. [Google Scholar]
- 34.Fijten MWM, Paulus RM, Schubert US. J Polym Sci, Part A: Polym Chem. 2005;43:3831–3839. [Google Scholar]
- 35.Guerrero-Sanchez C, Abeln C, Schubert US. J Polym Sci, Part A: Polym Chem. 2005;43:4151–4160. [Google Scholar]
- 36.Webster D. Macromol Chem Phys. 2008;209:237–246. [Google Scholar]
- 37.Webster DC, Meier MAR. Adv Polym Sci. 2010;225:1–15. [Google Scholar]
- 38.Becer CR, Schubert US. Adv Polym Sci. 2010;225:17–62. [Google Scholar]
- 39.Hoogenboom R, Fijten MWM, Meier MAR, Schubert US. Macromol Rapid Commun. 2003;24:92–97. [Google Scholar]
- 40.Zhang HQ, Fijten MWM, Hoogenboom R, Reinierkens R, Schubert US. Macromol Rapid Commun. 2003;24:81–86. [Google Scholar]
- 41.Zhang HQ, Marin V, Fijten MWM, Schubert US. J Polym Sci, Part A: Polym Chem. 2004;42:1876–1885. [Google Scholar]
- 42.Zhang HQ, Abeln CH, Fijten MWM, Schubert US. e–Polym. 2006;8:1–9. [Google Scholar]
- 43.Nasrullah M, Webster D. Macromol Chem Phys. 2009;210:640–650. [Google Scholar]
- 44.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2005;38:3087–3092. [Google Scholar]
- 45.Oh JK, Min K, Matyjaszewski K. Macromolecules. 2006;39:3161–3167. [Google Scholar]
- 46.Oh JK, Tang CB, Gao HF, Tsarevsky NV, Matyjaszewski K. J Am Chem Soc. 2006;128:5578–5584. doi: 10.1021/ja060586a. [DOI] [PubMed] [Google Scholar]
- 47.Oh JK, Siegwart DJ, Matyjaszewski K. Biomacromolecules. 2007;8:3326–3331. doi: 10.1021/bm070381+. [DOI] [PubMed] [Google Scholar]
- 48.Oh JK, Siegwart DJ, Lee HI, Sherwood G, Peteanu L, Hollinger JO, Kataoka K, Matyjaszewski K. J Am Chem Soc. 2007;129:5939–5945. doi: 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- 49.Siegwart DJ, Oh JK, Gao HF, Bencherif SA, Perineau F, Bohaty AK, Hollinger JO, Matyjaszewski K. Macromol Chem Phys. 2008;209:2180–2193. [Google Scholar]
- 50.Siegwart DJ, Srinivasan A, Bencherif SA, Karunanidhi A, Oh JK, Vaidya S, Jin R, Hollinger JO, Matyjaszewski K. Biomacromolecules. 2009;10:2300–2309. doi: 10.1021/bm9004904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong H, Matyjaszewski K. Macromolecules. 2008;41:6868–6870. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.